
Abstract
Tuber starch content (TSC) is a very important trait in potato (Solanum tuberosum L.). This study is the first to use expression quantitative trait loci (eQTL) mapping of transcript-derived markers for TSC in potato. Thirty-four differentially expressed genes were selected by comparing the RNA-seq data of contrasting bulked segregants. For the 11 candidate genes, we determined their relative expression levels across the segregating diploid potato population using RT-qPCR. We detected 36 eQTL as candidate genes distributed on all twelve potato chromosomes, and nine of them overlapped with QTL for TSC. Peaks for two eQTL, eAGPaseS-a and ePGRCRURSE5, were close to the corresponding loci of the large subunit of ADP-glucose pyrophosphorylase (AGPaseS-a) and the 12S globulin cruciferin gene (PGCRURSE5), respectively. The eQTL peaks for AGPaseS-a and PGRCRURSE5 explained 41.0 and 28.3% of the phenotypic variation at the transcript level. We showed the association of the DNA markers for AGPaseS-a and PGRCRURSE5 with QTL for TSC, and significant correlation between the expression level of PGRCRURSE5 and TSC. We did not observe a significant correlation between the expression level of AGPaseS-a and TSC. We concluded that the cruciferin gene PGRCRURSE5 is a novel candidate involved in the regulation of starch content in potato tubers.
Similar content being viewed by others


Introduction
Most agronomic traits of crop plants are affected by multiple loci, the environment and their interactions. Quantitative trait loci (QTL) mapping is a routine approach for studying the genetic architecture of complex traits. It yields information and the approximate genomic positions of the factors controlling a quantitative trait, but it does not identify its molecular basis1. Combining QTL analysis with genome-wide expression profiling has been termed expression QTL (eQTL) mapping or genetic genomics and creates great opportunities for dissecting quantitative traits2. eQTL data can be used to examine genome-wide gene expression levels and find candidate genes for a trait of interest. eQTL are empirically divided into two classes: cis and trans. A cis-eQTL represent a polymorphism physically located near the gene itself, and trans-eQTL reside at locations distant from the genes, frequently on different chromosomes3. The identification of genes underlying a trait can be more effective when the bulked sample analysis (BSA) method is applied. In the BSA protocol, plants with contrasting phenotype from a segregating population are pooled and then commonly screened to identify specific markers4,5. In potato (Solanum tuberosum L.), RNA pools that consist of genotypes based on contrasting phenotypic or marker data were used to select candidate genes for tuber flesh colour and cooking type6.
Potato is one of the most important crop plants in the world. Among numerous characteristics that are subject to selection in potato breeding, tuber starch content (TSC) is one of the most agronomically important. The starch content of potato tubers ranges from 10 to 25% of the tuber fresh weight7, and starch biosynthesis and breakdown in potato tubers are fairly well characterized metabolic networks8,9. Linkage mapping of QTL for specific gravity or TSC has been performed in numerous experimental populations of diploid potatoes10,11,12 in addition to association studies8,13.
Several enzymes involved in starch metabolism have been identified and characterized at the biochemical and molecular levels14,15. ADP-glucose pyrophosphorylase (AGPase) is the key enzyme in the regulation of starch metabolism content and quality7,16. All higher plant AGPases, including potato, are heterotetramers composed of two large and two small subunits17. For the large subunit AGPaseS, three loci were identified on potato chromosomes I, IV and VIII and for the small subunit AGPaseB, two loci on chromosomes VII and XII1,8. The locus AGPaseS-a on chromosome I co-localizes with QTL for tuber starch and/or sugar content18,19. In our previous paper, we described 12 QTL for starch content located on seven potato chromosomes in the diploid potato population 12-3. The gene encoding AGPaseS-a was localized within the most important QTL on chromosome I that accounted for 15.2% of the variance in tuber starch content12.
Recent advances in ‘omics’ technologies have made great progress in phenotypic variations and genotypic diversity for complex traits in plant sciences. The application of RNA-sequencing (RNA-seq) technologies has changed transcriptome analyses and gene expression studies20. High-throughput RNA-seq technology was used to identify eQTL associated with diverse biological processes in tomato21 and eQTL related to quantitative trait variation in maize22 and to identify gene networks involved in Verticillium dahliae disease resistance in potato23.
Here, we used a combination of BSA, comprehensive transcriptome analysis of differentially expressed genes (DEGs) and QTL/eQTL mapping to confirm possible candidates involved in the regulation of the starch content in potato tubers.
Materials and methods
Plant material
The plant material consisted of the potato diploid population 12-3 (F1 progeny, N = 175) from a cross of the seed parent DG 00-683 and the pollen parent DG 08-28/13. In the F1 progeny, the TSC segregated and was estimated from the ratio of tuber weight in air (g) to that in water (g) as described by Lunden24. The mean TSC values (percent fresh weight), evaluated in years 2012–2014, for DG 00-683 and DG 08-28/13 were 20.8% (± 3.5) and 11.8% (± 0.1), respectively12. In our previous study, we used population 12-3 for DArT map construction and QTL analysis of the TSC and sucrose content in potato leaves12. The F1 individuals were grown in three replications in a random pattern and scored for TSC directly after being harvested. Data set of TSC is presented in Supplementary Table S1. Samples (5 g) of tubers harvested in 2014 were collected in three replications per genotype, immediately frozen in liquid nitrogen and stored at − 80 °C.
Isolation of total RNA
Total RNA was isolated according to the protocol of Chomczyński and Sacchi25 using TRIZOL reagent. Briefly, frozen tubers were ground in liquid nitrogen, and 1 g of ground tissues was taken prior to the addition of 4 ml of TRIZOL reagent. After incubation at room temperature and centrifugation, the supernatants were transferred to fresh tubes. The extraction was performed twice in 3 ml of chloroform. The RNA was precipitated 15 min after the addition of 0.6 ml of salt solution (0.8 M sodium citrate and 1.2 M sodium chloride) and 0.6 ml of isopropanol. The RNA concentration and quality were determined using a biophotometer (Eppendorf) at 260 nm, 280 nm and 230 nm. The RNA was used for reverse transcription polymerase chain reaction (RT-PCR) and quantitative real-time PCR (RT-qPCR) experiments.
Construction of bulk RNA and Illumina sequencing
For the RNA-seq study, the quality and quantity of the total RNA were established using Bioanalyzer 2100 (Agilent). Four bulk RNA samples were constructed, each with two biological replicates. For each RNA bulk sample, equal amounts of total RNA (1 µg) from the tubers of six plants were pooled together. Bulks H1 and H2 consisted of high TSC genotypes, ranging from 19.0 to 23.4%; bulks L1 and L2 were made of low TSC genotypes, ranging from 12.5 to 15.0%. Plants in bulks H1 and L1 strongly expressed AGPaseS-a, whereas those in bulks H2 and L2 exhibited low levels; AGPaseS-a expression was determined as described in12. The mRNA was isolated using the NEBNext Poly(A) mRNA magnetic Isolation Module (New England Biolabs, E7490), and cDNA libraries were prepared using the NEBNext Ultra Direction RNA Library Kit for Illumina (New England Biolabs, E7420S). The established cDNA libraries were sequenced on the Illumina HiSeq 4000 sequencing platform (Illumina Inc., San Diego, CA, USA) to generate 100-bp paired-end reads (PE100). RNA-seq reads were conducted by Genomed S.A. (Warsaw, Poland). Quality control (QC) was done using Trimmotatic software (FASTQ-Illumina Phred + 33) for the raw data, which were trimmed by removing all empty and low-quality reads (Q < 30 and length < 50 bp), as well as all adaptor sequences, in order to obtain clean reads. The QC data are shown in Supplementary Table S2. Then, the index of the reference genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000226075.1) was built using Bowtie v2.1.0, and paired-end clean reads obtained for bulks H and L were aligned to the reference genome using TopHat v2.0.9 (Broad Institute, Boston, MA). Next, HTSeq v0.5.3 was used to count the number of reads mapped to each gene. The DEGs were identified by DESseq package. A comparison of DEGs from bulks H and L is shown in Supplementary Table S3. A scheme of the methodology used in this study is shown in Supplementary Fig. S1.
Selection of differentially expressed genes
DEGs were identified based on the RNA-seq data by comparing H1 vs. L1 and H2 vs. L2, H1 and H2 were control samples. Data from these comparison represents up-regulated and down-regulated genes in bulks L1 and L2. A false discovery rate (FDR) of 0.05 and absolute values of log2 ratios ≥ 1.5 and ≤ − 1.5 for up- and downregulated genes were used as the threshold for determining the significance of gene expression differences. To confirm RNA-seq data and develop the transcript-derived markers, semi-quantitative RT-PCR assays were performed on bulked samples. Reverse transcription was performed using the PrimeScript Master Mix (TaKaRa, no. #RR036A) cDNA synthesis kit, and 2 µg of total RNA was used for each reaction. Semi-quantitative RT-PCR was performed using DreamTaq DNA polymerase (Thermo Fisher Scientific, No. EP0703) and products were visualized in 2% agarose gel with ethidinum bromide. The primer sequences and PCR condition applied in semi-quantitative RT-PCR are presented in Supplementary Table S4.
Construction of the genetic map with transcript-derived markers
Standard PCR markers were developed using the selected candidate gene sequences. The primer sequences and PCR parameters for amplification of cleaved amplified polymorphic sequence (CAPS) and one sequence characterized amplified region (SCAR) markers are described in Supplementary Table S5. JoinMap 4 software26 was used for mapping on the previously constructed DArT map, as described in12.
eQTL mapping
The expression of the ten selected candidate genes and AGPaseS-a was examined in the F1 progeny of the 12-3 population by RT-qPCR. SYBR Green PCR Master Mix (Roche, Switzerland) and 96-well plates with a LightCycler 480 II system (Roche, Switzerland) were used. The RT-qPCR was performed as previously described in Śliwka et al.12. The 1 μl of cDNA corresponding to 50 ng of total RNA was taken for analysis of each sample. Potato α-tubulin was used as the reference gene. Thermal cycling conditions were: 4 min denaturation at 95 °C followed by 55 cycles of 10 s at 90 °C, 20 s at temperature for primer annealing, and 30 s at 72 °C. To confirm amplification of gene-specific products, PCR product melting point was determined in the range of 65–97 °C. The primer sequences and RT-qPCR parameters are shown in Supplementary Table S6. Four technical replicates of the parents and F1 progeny of population 12-3 were performed. Relative expression levels were calculated in Microsoft Excel 2010. T tests for ΔΔCt cycle threshold values27 and calculation of standard errors of the mean (SE) were performed with Statistica software (Stat Soft Inc.). For eQTL mapping, MapQTL 6 software was utilized28, with internal mapping and a logarithm of odds (LOD) ≥ 3.0 as the threshold of significance. The Pearson correlation coefficient (r) and the probability value (p) were used to display correlations and the significance of differences in expression levels between the candidate genes and TSC. A probability value of p < 0.05 was considered to indicate statistical significance.
Additionally, the expression profiles of PGRCRURSE5 and AGPaseS-a were examined in tubers of the parental clones DG 00-683 and DG 08-28/13 at three developmental stages: stage I, the beginning of tuber formation (1 cm diameter tubers); stage II, tuber building, tuber approximately 2 cm diameter; and stage III- tuber maturity. The experiment was carried out in the same way as that for candidate gene expression in the F1 progeny of the 12-3 population.
Cloning and sequencing of PGCRURSE5 amplicons
PCR-based amplicons 1587 bp in size for the marker PGCRURSE5 were obtained from both parents and purified using a Clean-Up Kit (A&A Biotechnology, Gdynia, Poland) according to the manufacturer’s protocol. The amplicons were blunted using a Fast DNA End Repair Kit (Thermo Fisher Scientific) and cloned into a blunt pCRScript Amp SK cloning vector (Promega, Madison, Wisconsin, USA). E. coli Top10 chemocompetent cells were used for transformation, and colonies with inserts of interest were picked and sequenced bidirectionally. Sequencing reactions were performed using the BigDye Terminator v3.1 kit (Life Technologies Polska Ltd., Warsaw, Poland), and products were resolved on an ABI3730XL genetic analyser at the Laboratory of DNA Sequencing and Oligonucleotide Synthesis (Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw, Poland).
Results
Sequencing data, differential expression gene analysis and genetic mapping
A total of eight bulk samples of tuber RNA were analysed by Illumina sequencing. Altogether, over 497 million reads were generated, with the number of RNA-seq reads per library ranging from 29 to 41 million after filtering impurities (Supplementary Table S2). All raw and processed data have been deposited in the GEO database (GSE153031) under the link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE153031.
Two pairwise comparisons were performed. When H1 was compared to L1, ten genes were upregulated and 13 genes were downregulated significantly (P value < 0.05 and fold change ≥ 1.5 or ≤ − 1.5). The corresponding values for H2 vs. L2 data were 4 and 7. For only 15 candidate genes, differences in transcript-intensities between the bulks were observed using semi-quantitative RT-PCR (data not shown). The selected genes are listed in Table 1. DNA markers were developed for eight gene sequences from the comparison of H1 vs. L1 and for the SWEET12-like gene from the comparison of H2 vs. L2. The CAPS and SCAR markers were scored in population 12–3 and incorporated into the existing genetic map (Table 2, Supplementary Table S7). The DNA marker AGPaseS-a was mapped in our previous study12. Markers for UnCh865 and WAT1 were not mapped in population 12-3. The positions of the gene WAT1 and the uncharacterized gene UnCh865 were deduced from their positions on the physical map of the reference genome DM1-3 v4.03 and the positions of the closest DArT markers that were common to genetic map 12-3 and the physical reference map (Table 2).
Genetic positions of the candidate gene markers relative to their eQTL and QTL for TSC
For 11 candidate genes, we determined the relative expression levels in the tubers of all F1 individuals of population 12-3 by RT-qPCR (Supplementary Table S8) and used the results for eQTL analysis. The expression of the SWEET12-like gene was not measured in the F1 individuals (Table 2). Four candidate gene markers, PGCRURSE5, AGPaseS-a, R1B-23 and ANR, were mapped within the regions corresponding to the eQTL controlling their expression (cis-eQTL). However, only the markers PGCRURSE5 and AGPaseS-a were significantly associated with QTL for TSC and explained 18.8 and 18.5% of the variation in TSC, respectively. The PGCRURSE5 marker, 1587 bp in size, was cloned, sequenced and compared to the database with the BLASTN programme (NCBI database). We found two sequences in the DG 00-683 parent (GenBank accessions MT274591 and MT274592) and one sequence for this marker in the parent DG 08-28/13 (MT274590). This marker from the DG 00-683 parent shared 95% sequence identity with the Solanum tuberosum 12S seed storage protein CRD-like (alternative name: Cruciferin D; UniProt accession Q9ZWA9-1, GenBank accession XM_006349369.2). The HpaII recognition site was diagnostic of the PGCRURSE5 marker allele in parent DG 00-683. In contrast, none of the DG 08-28/13-derived PGCRURSE5 sequences contained the HpaII site. The marker 9-DES, although also mapped on chromosome I, was located outside the eQTL for the 9-DES region but within the QTL for TSC, and accounted for 18.4% of the variance in this trait. The locus of the gene UnCh865 was on chromosome VIII within the QTL with a moderate effect on TSC (LOD = 3.34; R2 = 8.1%) but not within the eQTL for UnCh865. The markers Pat3-k1, MLP34 and WAT1 were located in regions not affecting the expression of their genes or TSC (Table 2).
eQTL analyses
In our previous study, we performed QTL analysis of TSC using a phenotypic mean dataset (2012–2014) in population 12-312. In the current research, the TSC linkage map was enriched by a set of 9 DNA markers, for which we found polymorphism in parents and F1 individuals (Table 2, Supplementary Table S7). For 11 genes, expression products were obtained in RT-qPCR (Table 3 and Supplementary Fig. S2). The number of eQTL detected for particular candidate genes ranged from one (AGPaseS) to seven (ANR). We found eQTL located both close to the loci encoding the genes (cis-eQTL) and at independent locations (trans-eQTL). In total, 36 eQTL were mapped in population 12-3 (Table 3).
Colocalization of eQTL with QTL for TSC
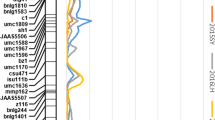
Nine of the 36 eQTL identified for candidate genes overlapped with QTL for TSC (Table 3). On chromosome I, two eQTL, for AGPaseS-a and PGRCRURSE5, overlapped with the strongest QTL for TSC (Fig. 1). The eQTL peaks for AGPaseS and PGRCRURSE5 were found at 99.3 and 84.3 cM, explaining 41.0 and 28.3% of the variance in the expression of these genes, respectively (Table 3). Both peaks were located near the loci encoding those genes (99.6 and 80.4 cM, Table 2).

LOD charts of QTL detected by interval mapping for the 3-year mean (2012–2014) tuber starch content and eQTL for AGPaseS and PGCRURSE5 candidate genes in the diploid potato mapping population 12-3. Threshold LOD = 3, marked by a line parallel to the x-axis.
On chromosome II, two eQTL were detected within the QTL for TSC 35.0–68.0 cM: eQTL located at 50.0–52.7 cM and explaining 17.2% of the variance in eWAT1 and located at 62.5–62.6 cM and explaining 8.4% of the variance in eIRL. The second eQTL for PGRCRURSE5, found on chromosome III, partially overlapped with the QTL for TSC mapped at 65.1–71.1 cM and explained 10.9% of the variance in ePGRCRURSE5. On chromosome III, we also detected the eQTL for MLP34 located at 124.4–130.5 cM and explained 9.4% of the variance in the expression of MLP34 that partially overlapped with the QTL for TSC at 129.4–130.5 cM. Chromosome IV possessed eQTL for R1B-23 that covered 44.3–95.1 cM (R2 = 25.1%) and overlapped with the QTL for TSC at 49.5–50.2 cM. On chromosome V, we detected a QTL for TSC at 46.0–50.7 cM that explained 8.6% of the variance in this trait. It overlapped with an eQTL for UnCh865 located at 40.5–55.6 cM that explained 14.8% of the variance observed in the expression of this gene. An eQTL for UnCh835 at 40.3–41.6 cM on chromosome VIII overlapped slightly with the QTL for TSC at 26.0–40.6 and explained up to 23.6% of the variance in eUnCh835 (Table 3). In addition, we detected 20 eQTL on chromosomes I, III, IV, V, VIII, IX and XII that were outside the QTL for TSC (Table 3). Seven eQTL were identified on chromosomes VI, VII and IX, whereas no QTL for TSC were found on these chromosomes (Table 3). Significant positive correlations were found between the expression levels of a few pairwise combinations of the candidate genes. The eQTL for PGRCRURSE5, MLP34, IRL and Pat3-k1 exhibited significant positive correlations with TSC (Supplementary Table S9).
AGPaseS-a and PGRCRURSE5 expression assay during major stages of tuber development
The markers PGCRURSE5 and AGPaseS-a were significantly associated with QTL for TSC (Table 2). In addition, eQTL for AGPaseS-a and PGRCRURSE5 overlapped with the strongest QTL for TSC on chromosome I (Fig. 1). Therefore, we evaluated the expression of AGPaseS-a and PGRCRURSE5 during major stages of tuber development. The expression level of AGPaseS-a was similar (not significantly different) in both parents at stage I of tuber development and significantly higher in the high-starch parent DG 00-683 than in the other parent at stages II and III. In the case of PGRCRURSE5, differential expression was observed between DG 00-683 and DG 08-28/13 at all three stages. PGRCRURSE5 expression in DG 00-683 was highest at stage II, while in the low-starch parent DG 08-28/13, its transcript level increased during tuber development and was highest at stage III in DG 08-28/13 (Fig. 2).

Relative expression levels of the AGPaseS-a and PGRCURSE5 genes in the high-starch parent DG 00-683 and the low-starch parent DG 08-28/13 at three tuber developmental stages: stage I—tuber formation; stage II—tuber building, tubers approximately 2 cm diameter; and stage III—tuber maturity. The levels of relative transcript accumulation are shown on the y-axis (logarithmic scale); values are presented as the means ± SD of three biological replicates. Asterisks indicate significant differences between high- and low-starch parents (Student’s t-test).
Discussion
Starch is the most abundant storage compound in plants. As in other higher plants, starch synthesis in potato is under transcriptional control, circadian and redox control, and phosphorylation regulation29,30. The AGPaseS loci, in particular, the locus AGPaseS-a on chromosome I, colocalized with QTL for TSC, and the data indicated a small effect on this trait in the mapping populations18,31. In association studies, the amplicons AGPsS-9a and AGPsS-10a, both derived from the AGPaseS-a locus, were correlated either positively or negatively with TSC32. Previously, we showed the large QTL region for TSC on potato chromosome I that overlapped with the AGPaseS-a locus. This result potentially means that the chromosomal segment also includes other genes that either directly or indirectly affect the starch content. The expression of AGPaseS-a was significantly higher in the high-TSC parent DG 00-683 than in the low-TSC parent DG 08-28/13 in the potato population 12-312. The AGPaseS-a allele contributed significantly to but was not necessary for a high TSC12.
Here, we detected the differences between the parental clones in the expression levels of AGPaseS-a in tubers at different growth stages. The highest expression was detected in the tuber building stage, when high AGPase activity is required, as the tuber is a sink organ accumulating large amounts of starch33. Our results confirmed that AGPase activity remained high even when starch synthesis was inhibited in potato tubers detached from the mother plant34. In the current sequencing experiments, AGPaseS-a was difficult to analyse by RNA-seq, which was potentially due to the small transcript size restricted by the constructed RNA libraries and/or sequence overlap with other transcripts35,36.
We mapped the cruciferin (12S globulin) gene PGCRURSE5 to chromosome I and demonstrated that it also had a significant effect on TSC. In Arabidopsis thaliana and other crucifers, cruciferin is a main seed storage protein. Seed storage proteins serve as a source of nitrogen and amino acids that are necessary for germination and plant growth37,38. In potato tubers, the inhibition of starch synthesis was accompanied by a massive reduction in the expression of storage proteins, suggesting that the expression of storage protein genes is involved in starch metabolism in potato tubers39. Our study revealed higher expression of PGCRURSE5 in the high-starch parent DG 00-683 than in the low-starch parent DG 08-28/13 during the tuber formation and building stage, and we therefore concluded that the cruciferin protein can affect starch metabolism. Among the 9 eQTL identified for the selected candidate genes that overlapped with QTL for TSC, the peaks for two eQTL, AGPaseS-a and PGRCRURSE5, were close to the loci encoding those genes. In the case of ePGRCRURSE5, the presence of a trans-eQTL on chromosomes III and XII showed that PGCRURSE5 expression is influenced by trans-acting factors.
The cis-eQTL are likely mediated by polymorphisms within the corresponding genes, including the promoter regions, or by mRNA stability40. The overlap of QTL and eQTL may indicate a strong association between the genetic variation in the phenotypic trait and the gene transcript level41. The genes for AGPaseS-a and PGRCRURSE5 accounted for 18.5 and 18.8% of the variance in TSC, respectively. The eQTL peaks for AGPaseS-a and PGRCRURSE5 explained 41.0 and 28.3% of the phenotypic variance at the transcript level, respectively. The high association values between eQTL peaks and the explained expression variations could account for genetic sources of variation associated with dominance and epistasis as well as for non-genetic influences, such as developmental and environmental factors42. Colocalization of cis-eQTL and QTL seems to be more informative than that of trans-eQTL. Trans-eQTL are interpreted as evidence for trans-acting regulatory proteins such as transcription factors and other signalling proteins or small RNAs that may control the expression of a number of genes elsewhere in the genome43.
The role of AGPase as the first rate-limiting enzyme in starch biosynthetic pathways is well known8,44,45. We showed the association of the DNA marker for AGPaseS-a with QTL for TSC as well as the relationship between this marker and gene transcription. However, we did not observe a significant correlation between the expression level of AGPaseS-a and TSC. The abundance of mRNA transcripts only partially correlates with protein abundances, and these relationships are complex46,47. Therefore, in the case of the enzyme AGPase, their subunit structure and transcriptional regulation can affect the net activity of this enzyme complex.
Our study demonstrates the association between the marker PGCRURSE5 and QTL for total starch content, the relationship between this marker and the eQTL for PGRCRURSE5, and significant correlation between PGCRURSE5 expression and starch content in potato tubers. Recently, Sueng et al.48 has shown that non-enzymatic protein, termed Protein Targeting to Starch (PTST), is involved in starch synthesis in Arabidopsis. Our results identified the gene cruciferin as a novel candidate involved in the regulation of starch metabolism in potato tubers. It suggests that cruciferin may be a novel PTST protein in potato tubers.
References
Chen, X., Salamini, F. & Gebhardt, C. A potato molecular-function map for carbohydrate metabolism and transport. Theor. Appl. Genet. 102, 284–295 (2001).
Jansen, R. C. & Nap, J. P. Genetical genomics: The added value from segregation. Trends Genet. 17, 388–391 (2001).
Druka, A. et al. Expression quantitative trait loci analysis in plants. Plant Biotechnol. J. 8, 10–27 (2010).
Michelmore, R. W., Paran, I. & Kesseli, R. V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 88, 9828–9832 (1991).
Zou, C., Wang, P. & Xu, Y. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotech. J. 14, 1941–1955 (2016).
Kloosterman, B. et al. From QTL to candidate gene: Genetical genomics of simple and complex traits in potato using a pooling strategy. BMC Genomics 11, 158 (2010).
Ahmed, S., Zhou, X., Pang, Y., Jin, L. & Bao, J. Improving starch-related traits in potato crops: Achievements and future challenges. Starch 70, e1700113 (2018).
Schreiber, L. et al. SNPs in genes functional in starch-sugar interconversion associate with natural variation of tuber starch and sugar content of potato (Solanum tuberosum L.). G3 Genes Genomes Genet. 4, 1797–1811 (2014).
Van Harsselaar, J. K. et al. Genome-wide analysis of starch metabolism genes in potato (Solanum tuberosum L.). BMC Genomics 18, 37 (2017).
Schäfer-Pregl, R. et al. Analysis of quantitative trait loci (QTLs) and quantitative trait alleles (QTAs) for potato tuber yield and starch content. Theor. Appl. Genet. 97, 834–846 (1998).
Werij, J. S. et al. A limited set of starch related genes explain several interrelated traits in potato. Euphytica 186, 501–516 (2012).
Śliwka, J. et al. Mapping of quantitative trait loci for tuber starch and leaf sucrose contents in diploid potato. Theor. Appl. Genet. 129, 131–140 (2016).
Li, L. et al. Natural DNA variation at candidate loci is associated with potato chip color, tuber starch content, yield and starch yield. Theor. Appl. Genet. 116, 1167–1181 (2008).
Sowokinos, J. R. Biochemical and molecular control of cold-induced sweetening in potatoes. Am. J. Potato Res. 78, 221–236 (2001).
Solomos, T. & Mattoo, A. K. Starch-sugar metabolism in potato (Solanum tuberosum L.) tubers in response to temperature variations. In Genetic Improvement of Solanaceous Crops. Potato Vol. 1 (eds Razdan, M. K. & Mattoo, A. K.) 209–234 (Science Publishers Inc., New York, 2005).
Bahaji, A. et al. Starch biosynthesis, its regulation and biotechnological approaches to improve crop yields. Biotechnol. Adv. 32, 87–106 (2014).
Okita, T. W. et al. The subunit structure of potato tuber ADP-glucose pyrophosphorylase. Plant Physiol. 93, 785–790 (1990).
Gebhardt, C. et al. Genomic approaches for the improvement of tuber quality traits in potato. Acta Hort. 684, 85–91 (2005).
Sołtys-Kalina, D. et al. Quantitative trait loci for starch-corrected chip color after harvest, cold storage and after reconditioning mapped in diploid potato. Mol. Gen. Genom. 295, 209–219 (2020).
Klepikova, V. A. & Penin, A. A. Gene expression maps in plants: Current state and prospects. Plants 8, 309 (2019).
Ranjan, A. et al. eQTL regulating transcript levels associated with diverse biological processes in tomato. Plant Physiol. 172, 328–340 (2016).
Liu, H. et al. Distant eQTLs and non-coding sequences play critical roles in regulating gene expression and quantitative trait variation in maize. Mol. Plant 10, 414–426 (2017).
Tai, H. H. et al. Verticillium dahliae disease resistance and the regulatory pathway for maturity and tuberization in potato. Plant Genome 11, e170040 (2018).
Lunden, P. A. Underldokerd over forholder mellom popetens spesifikka vekt og deres torvstoff og Stivelsesinhold Forhl. Forsok Landbruket. 7, 81–107 (1956).
Chomczyński, P. & Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159 (1987).
Van Ooijen, J. W. JoinMap 4, Software for the Calculation of Genetic Linkage Maps in Experimental Populations (Kyazma B.V., Wageningen, 2006).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 (2008).
Van Ooijen, J. W. MapQTL 6, Software for Mapping of Quantitative Trait Loci in Experimental Populations of Diploid Species (Kyazma BV, Wageningen, 2009).
Nazarian-Firouzabadi, F. & Visser, R. G. F. Potato starch synthases: Functions and relationships. Biochem. Biophys. Rep. 10, 7–16 (2017).
Thalmann, M. & Santelia, D. Starch as a determinant of plant fitness under abiotic stress. New Phytol. 214, 943–951 (2017).
Schönhals, M. E. et al. Identification and reproducibility of diagnostic DNA markers for tuber starch and yield optimization in a novel association mapping population of potato (Solanum tuberosum L.). Theor. Appl. Genet. 129, 767–785 (2016).
Li, L. et al. Validation of candidate gene markers for marker-assisted selection of potato cultivars with improved tuber quality. Theor. Appl. Genet. 126, 1039–1052 (2013).
Aksenova, N. P. et al. Regulation of potato tuber dormancy and sprouting. Russ. J. Plant Physiol. 60, 301–312 (2013).
Tiessen, A. et al. Starch synthesis in potato tubers is regulated by post-translational redox modification of ADP-glucose pyrophosphorylase: A novel regulatory mechanism linking starch synthesis to the sucrose supply. Plant Cell 14, 2191–2213 (2002).
Gálvez, J. H. et al. Understanding potato with the help of genomics. AIMS Agric. Food. 2, 16–39 (2017).
Zhu, F. et al. Comparative performance of the BGISEQ-500 and Illumina HiSeq4000 sequencing platforms for transcriptome analysis in plants. Plant Methods 14, 69 (2018).
Wan, L. et al. Phosphorylation of the 12 S globulin cruciferin in wild-type and abi1-1 mutant Arabidopsis thaliana (thale cress) seeds. Biochem. J. 404, 247–256 (2007).
Gacek, K., Bartkowiak-Broda, I. & Batley, J. Genetic and molecular regulation of seed storage proteins (SSPs) to improve protein nutritional value of oilseed rape (Brassica napus L.) Seeds. Front. Plant Sci. 9, 890 (2018).
Müller-Röber, B., Sonnewald, U. & Willmitzer, L. Inhibition of the ADP-glucose pyrophosphorylase in transgenic potatoes leads to sugar-storing tubers and influences tuber formation and expression of tuber storage protein genes. EMBO J. 11, 1229–1238 (1992).
Wang, L. et al. Identification of QTLs with additive, epistatic and QTL x development interaction effects for seed dormancy in rice. Planta 239, 411–420 (2014).
Lima, R. P. M. et al. QTLs and eQTLs mapping related to citrandarins’ resistance to citrus gummosis disease. BMC Genomics 19, 516 (2018).
Kirst, M. et al. Genetic architecture of transcript-level variation in differentiating xylem of eucalyptus hybrid. Genetics 169, 2295–2303 (2005).
Baker, R. L. et al. Integrating transcriptomic network reconstruction and eQTL analyses reveals mechanistic connections between genomic architecture and Brassica rapa development. PLoS Genet. 15(9), e1008367 (2019).
Sonnewald, U. & Kossmann, J. Starches-from current models to genetic engineering. Plant Biotechnol. J. 11, 223–232 (2013).
Miao, H. et al. The AGPase family proteins in banana: genome-wide identification, phylogeny, and expression analyses reveal their involvement in the development, ripening, and abiotic/biotic stress responses. Int. J. Mol. Sci. 18, 1581 (2017).
Vogel, C. & Marcotte, E. M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232 (2012).
Williams, R. B. et al. The influence of genetic variation on gene expression. Genome Res. 17, 1707–1716 (2007).
Seung, D. et al. Protein targeting to starch is required for localising granule-bound starch synthase to starch granules and for normal amylose synthesis in Arabidopsis. PLoS Biol. 13, e1002080 (2015).
Acknowledgements
The research was supported by The National Science Centre in Poland, Grant UMO-2015/19/B/NZ9/00776.
Author information
Authors and Affiliations
Contributions
D.S.-K. performed most of the experiments and cowrote the manuscript; K.S., E.S., P.S.-D. performed the RT-qPCR experiments; J.S. performed the genetic mapping, QTL analyses and writing of the manuscript; and W.M. conceived and coordinated the project and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sołtys-Kalina, D., Szajko, K., Stefańczyk, E. et al. eQTL mapping of the 12S globulin cruciferin gene PGCRURSE5 as a novel candidate associated with starch content in potato tubers. Sci Rep 10, 17168 (2020). https://doi.org/10.1038/s41598-020-74285-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-74285-5
- Springer Nature Limited