
Abstract
Pea is one of the most important grain legume crops in temperate regions worldwide. Improving pea yield is a critical breeding target. Nine inter-connected pea recombinant inbred line populations were evaluated in nine environments at INRAE Dijon, France and genotyped using the GenoPea 13.2 K SNP array. Each population has been evaluated in two to four environments. A multi-population Quantitative Trait Loci (QTL) analysis for seed weight per plant (SW), seed number per plant (SN), thousand seed weight (TSW) and seed protein content (SPC) was done. QTL were then projected on the multi-population consensus map and a meta-analysis of QTL was performed. This analysis identified 17 QTL for SW, 16 QTL for SN, 35 QTL for TSW and 21 QTL for SPC, shedding light on trait relationships. These QTL were resolved into 27 metaQTL. Some of them showed small confidence intervals of less than 2 cM encompassing less than one hundred underlying candidate genes. The precision of metaQTL and the potential candidate genes reported in this study enable their use for marker-assisted selection and provide a foundation towards map-based identification of causal polymorphisms.
Similar content being viewed by others


Introduction
Grain legumes play a central role in sustainable agriculture and food security. They produce protein-rich seeds and thanks to their special nitrogen nutrition largely based on a symbiosis with nitrogen-fixing soil bacteria, they allow for the reduction of nitrogen fertilizer use in cropping systems thus lowering agriculture energy costs and greenhouse gas production1,2. Pea (Pisum sativum L.) is one of the most cultivated grain legumes in temperate areas3. It has a high nutritional value and is used for human food and animal feed. With a demand for plant proteins rising worldwide, increasing pea seed yield and protein content are important breeding targets.
Seed yield and seed protein content are complex traits and highly quantitative. Numerous loci controlling seed yield and seed quality have been identified in pea4,5,6,7,8,9,10,11,12,13,14. The r and rb loci encoding Starch-Branching Enzyme 115 and ADP glucose-pyrophosphorylase16, respectively, and which control the wrinkled seed phenotype in pea have long been known to impact seed development, yield and seed protein content17,18. Burstin et al.7 suggested that seed yield and protein content QTL corresponded either to (i) genes controlling the plant source capacity to produce and fill seeds, or (ii) genes controlling seed sink strength such as genes involved in seed formation and storage products’ synthesis. Genes controlling plant architecture such as Le which determines inter-node length or Afila (Af) which determines leaf type would more likely correspond to source capacity loci while rugosus genes or a gene encoding a subtilase shown to be associated with a seed size QTL19 would correspond to sink strength loci. Bourgeois et al.8 have further shown that most protein quality QTL were co-localized with genes encoding major storage proteins.
Identifying causal polymorphisms for these traits has been challenging in pea. Most QTL-based research studies have considered a few bi-parental populations. As a result, the QTL detection was limited to phenotypic variability of both parents and the QTL confidence intervals were generally large, probably due to the limited population size or low-density linkage maps. To narrow QTL confidence intervals and provide a foundation to marker-assisted breeding for yield components and seed protein content in pea, we investigated these traits in nine inter-connected recombinant inbred line populations (RIL) derived from parents showing contrasted phenotypes20 providing a wide phenotypic variability. We used a high-density single nucleotide polymorphism (SNP) based genotyping platform, namely GenoPea 13.2 K SNP array20, to ensure high-quality dense genetic maps. We performed QTL mapping taking advantage of the multi-cross design21 to define QTL locations and genotypic effects. We then integrated all QTL results through a meta-analysis approach22. The wide phenotypic variability and the high-density linkage map allowed the identification of metaQTL for seed weight per plant (SW), seed number per plant (SN), thousand seed weight (TSW) and seed protein content (SPC). This refined confidence intervals and pinpointed some candidate genes thanks to the recently published pea genome sequence23.
Results
Phenotypic variation for SW, SN, TSW and SPC in multiple populations
SW, SN, TSW and SPC measured in the field and the glasshouse for 1213 RIL from nine bi-parental populations revealed highly-significant line, year, and population effects (Supplementary Table S1). The traits exhibited continuous distributions in the nine populations indicating a polygenic inheritance (Supplementary Fig. S1). Transgressive lines were observed for all traits but TSW in Pop11, indicating that favourable alleles are brought by both parents in most cases. Highly-significant genotype effects (P < 0.0001) and high heritabilities were detected for the four traits in almost all populations and environments, except for Pop3, Pop4 and Pop5 in the field experiment in 2004 due to the impact of diseases on plants and Pop11 in the glasshouse in 2008 related to the F3 state of the population (Supplementary Table S1). The range of the data of the multi-population lines phenotyped in field environment was extensive: in spring sowing, SW varied from 1.7 to 47.9 g per plant; SN from 9.9 to 300.0, TSW from 78.6 to 317.3 g per plant, and seed protein content from 18.3 to 32.2% of seed dry weight. In winter sowing (i.e. Pop9 in 2008, 2009 and 2010), SW varied from 5.1 to 119.1 g per plant; SN from 23.8 to 586.4, TSW from 101.4 to 258.7 g per plant, and seed protein content from 19.3 to 30.3% of seed dry weight. The environment showed a highly significant effect on all traits in all populations, except SPC in Pop9. Genotype-by-environment (GxE) interactions were significant for Pop3, Pop5, and Pop9 (Supplementary Table S2) but not for Pop4. These effects were partly associated with different responses to environmental conditions such as diseases impacting plants in 2004 or the sowing period for Pop9 (Supplementary Table S2 and Table S1). However, a major determinant of these interactions was that the population composition was changed in Pop3, Pop5, and Pop9 in 2011 as compared to other years since tall plants carrying the Le allele were discarded for this trial (see “Material and methods”; Supplementary Fig. S3). In the glasshouse, SN and SW were significantly lower than in the field, especially for Pop10, unlike TSW and SPC that were stable across environments (Supplementary Table S1). Several parents of RIL populations were chosen for their high seed protein content i.e. Caméor, VavD265 and China. Progenies of the crosses involving two of these parents (i.e. Pop3, Pop9) showed high range of SPC up to > 30%. Transgressive segregants were notable for SPC, even in progenies where parents were not chosen for their high SPC such as in the Pop8 ‘Kazar’ x ‘Melrose’ where some of the RILs show SPC values up to 29% (Supplementary Table S1 and supplementary Fig. S1).
Correlations among yield components and seed protein content
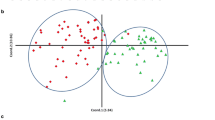
A principal component analysis of mean values for each line in each environment highlighted the wide phenotypic variability among the pea populations (Fig. 1). Axis 1 explained 48.3% of the phenotypic variance based mainly on SN and SW, and in a lesser extent, on TSW and SPC. Axis 2 explained 28% of the phenotypic variance and represented mainly TSW and SPC variability (Fig. 1a), and SW in a lesser extent. Axis 3 explained 21.7% of the phenotypic variance and opposed SPC and TSW (Fig. 1b). Accordingly, the overall correlation coefficients between SW, SN and TSW revealed that SW was highly correlated with SN (Pearson r = 0.89). Besides, TSW was significantly negatively correlated with SN (r = − 0.35) (Fig. 2). However, correlations between SW, SN and TSW slightly differed among populations and environments: SN was in all cases highly positively correlated with SW; TSW was positively correlated with SW in most cases except for Pop9 for which the correlation was negative in 2009 in autumn sowing, not significant in 2008 and 2010, and positive in 2011 in spring sowing, like the other populations. In Pop9, seed weight and seed number were lower in spring sowing than in autumn sowings (Supplementary Table S1). In most cases, SN and TSW were negatively correlated except in Pop8 in 2011 (Supplementary Fig. S2). The correlation between SPC and SW was slightly negative overall (Pearson r = − 0.11) (Fig. 2) but inconsistent across populations and environments. It ranged from significantly negative for Pop9 in 2008 (r = − 0.47) to significantly positive for Pop3 in 2011 and Pop10 in 2008 (r = 0.33). Likewise, SPC was significantly positively correlated with TSW (r = 0.16) except for Pop6, Pop8, and Pop9 in 2011 (Supplementary Fig. S2). TSW were lower in these populations (i.e. Pop6, Pop8 and Pop9) in 2011. In general, SPC was significantly negatively correlated with SN (r = − 0.16), except for Pop9 in 2011 and Pop10 in 2009.

(a, b) Principal component analysis of phenotypic traits of Pop3 to Pop10 observed on the field trials between 2004 and 2011 at INRAE Dijon (a axis 1&2, b axis 3&4). SN seed number per plant, SW seed weight per plant (g), TSW thousand seed weight (g) and SPC seed protein content (% of seed dry weight).

Pearson correlation coefficients between phenotypic traits recorded from 2004 to 2011 in Pop3 to Pop10 on the field trials at INRAE Dijon. SN seed number per plant, SW seed weight per plant (g), TSW thousand seed weight (g) and SPC seed protein content (% seed dry weight). *, ** and *** significant correlation at the P < 0.05, P < 0.01 and P < 0.001 probability level, respectively.
Multi-population QTL and metaQTL identification
A total of eighty-nine multi-population QTL were identified: 17 QTL for SW explaining from 4 to 30% of SW variance, 16 QTL for SN explaining from 4 to 39% of SN variance, 35 QTL for TSW explaining from 5 to 32% of TSW variance and 21 QTL of SPC explaining from 4 to 22% of SPC variance (Supplementary Table S3). For all traits, alleles having positive effects had different parent origin, no parent bringing all positive effect for a given trait. The meta-analysis of these QTL defined a number of metaQTL regions encompassing from one to fifteen multi-population QTL and reducing their confidence intervals: 27 metaQTL were identified for yield-related traits and seed protein content (Table 1, Fig. 3 and supplementary Table S4). 4 out of 7 metaQTL detected for SW were consistently detected in two to five environments (mQTL1.5, mQTL2.1, mQTL3.1, mQTL3.4), 10 out of 16 for TSW in two to five environments (mQTL1.1, mQTL1.3, mQTL1.4, mQTL2.2, mQTL3.1, mQTL3.4, mQTL4.4, mQTL5.1, mQTL5.3, mQTL6.3, mQTL7.2), 6 out of 12 for SPC in two to four environments (mQTL1.1, mQTL3.2, mQTL3.4, mQTL4.2, mQTL4.3, mQTL7.1) and only 2 out of 10 for SN in three to five environments (mQTL3.1, mQTL3.4). Two metaQTL (mQTL3.1 and mQTL3.4) controlling all the traits were consistently detected across environments. mQTL3.1 mapped at 24.73 cM on LG3-Chr5 with a confidence interval (CI) of 1.69 cM controlled the phenotypic variation of SN, SW, TSW and SPC in five environments. On the same chromosome, mQTL3.4 at 131.8 cM with a CI of 0.03 cM controlled SN, SW, TSW and SPC in five environments. MetaQTL mQTL1.4, mQTL1.5, mQTL2.1, mQTL2.3 and mQTL7.2 controlled yield and its components. Six metaQTL controlled a specific yield trait and the seed protein content: mQTL4.2, mQTL4.4, mQTL5.3, mQTL6.2 controlled TSW and SPC, mQTL3.2 controlled SW and SPC and mQTL7.1 controlled SN and SPC. Four metaQTL specific of TSW were detected in two to three environments: mQTL2.2 on the LG2-Chr6 at 52.70 cM (CI = 9.45 cM) in three environments, mQTL1.3 (LG1-Chr2 at 25.54 cM, CI = 5.66 cM), mQTL5.1 (LG5-Chr3 at 18.84 cM, CI = 10.29 cM) and mQTL6.3 (LG6-Chr1 at 86.17 cM, CI = 12.84 cM) in two environments. Conversely, mQTL4.3 (LG4-Chr4 at 50.63 cM, CI = 7.64 cM) and mQTL4.5 (LG4-Chr4 at 105.40 cM, CI = 10.50 cM) controlled exclusively SPC in two and one environments respectively.

Mapping of 27 metaQTL detected for seed weight (SW), seed number (SN), thousand seed weight (TSW) and seed protein content (SPC). Position (cM) and the maximum of phenotypic variance explained for each metaQTL (mQTL) are indicated to the right of the linkage groupe (LG). Confidances intervals are represented by the color on the linkage group. The lines on the left on the linkage group indicate the each QTL position on the consensus map. Color of the line indicates membership to the metaQTL.
MetaQTL were located on all linkage groups (Fig. 3). Taking advantage of the newly published genome sequence of pea23 we could identify the position and the genes underlying metaQTL and highlight potential candidate genes. For example, 17 genes were identified in the confidence interval of mQTL3.4 (CI = 0.03 cM), 51 genes in the confidence interval of mQTL5.3 (CI = 0.37 cM) and 56 genes in the confidence interval of mQTL1.5 (CI = 1.47 cM) (Table 1). However, even for the shortest confidence intervals, the number of underlying genes is still high: 126 genes were identified in the confidence interval of mQTL7.2 (CI = 0.96 cM), 135 genes for mQTL3.1 (CI = 1.69 cM), 141 genes are underlying for mQTL1.1 (CI = 2.19 cM) and 199 genes for mQTL7.1 (CI = 2.87 cM).
Discussion
Diversity and correlation of seed traits in pea
Our study shows a large range of genetic variation for seed productivity and quality traits carried by cultivated pea varieties and their progenies, suggesting avenues for breeding (Supplementary Table S1). Our results confirm that the seed weight per plant is highly positively correlated with the seed number per plant, and generally positively correlated with the thousand seed weight (Fig. 2, Supplementary Fig. S2). The relationship between seed weight per plant and seed protein content is varied: for example, the correlation was slightly positive within Pop4 and was negative within Pop9. Our study reveals that in pea, by contrast to cereals24,25, the relationship between seed yield per plant and seed protein content is not necessarily negative but depends on the environment and the genetic background.
Genetic control of traits
QTL detection is impacted by many factors, including the genetic background, the population size, the quality and density of the genetic map and the statistical method used for detection. Meta-analysis of QTL initially developed by Goffinet and Gerber26 and implemented by Veyrieras et al.22 and Sosnowski et al.27 is an efficient approach to identify consensus QTL positions among experiments and reduce their confidence intervals. The method has been used in a wide range of species28,29,30,31,32,33,34,35 and was applied in the present study for a large set of phenotypic data obtained from nine RIL populations and seven environments (Table 2). A total of 89 QTL explaining a part of phenotypic variation were detected across the seven pea chromosomes (Table 1).
The meta-analysis of these QTL revealed 27 consensus QTL, or metaQTL; each metaQTL corresponding to one to 15 initial QTL (Table 1). Most metaQTL were consistently detected in different environments, in spite of significant environmental and GxE effects. Fifteen metaQTL controlled more than one trait. When different traits were controlled by the same metaQTL, allelic effects were of the same sign for SN and SW QTL (5 metaQTL out of 5) and of opposite sign for SN or SW and TSW (5 metaQTL out of 6), similarly to the correlation study of these traits. Allelic effects of SPC QTL were of the same sign with SN and SW QTL in one case out of 5.
A survey of the literature allowed, when common markers where used or when markers’ genomic positions were known, to identify likely correspondence between the metaQTL from this study and QTL previously detected for seed yield, seed yield components and seed protein content in different pea populations and in different environments (supplementary Table S5). For example, mQTL1.1 was shown to control SPC and TSW in several environments in the present study, and was also detected by Moreau et al.12 as a locus controlling SW and by Gali et al.13 as a region controlling SPC; mQTL2.1 controlled SW in 3 environments in the present study as well as in Tar’an et al.5; mQTL3.1 controlled TSW in 5 environments in the present study as well as in Klein et al.8, Burstin et al.7 and Gali et al.13; mQTL3.2. controlled SPC in three environments in the present study as well as in Gali et al.13; mQTL3.4 controlled SW in 5 environments in the present study and also in Klein et al.8, Burstin et al.7 and Gali et al.13; Some metaQTL only controlled one trait in one environment in the present study but were also detected in other studies, such as mQTL5.2, a QTL of SPC also in Burstin et al.7 and Gali et al.13 (Table S4). Because the genetics of seed yield and seed protein content have been widely studied in soybean, we also searched for any related QTL to the pea metaQTL. Orthologous genes for the ones harbouring the peak markers of the metaQTL were identified and QTL within 50 kb around these genes were searched. This further reinforced the interest of the SW QTL mQTL2.1 and mQTL4.1, of the TSW QTL mQTL6.2 and mQTL2.2 and the SPC QTL mQTL4.3 and mQTL4.5 which corresponded to QTL of the same traits in soybean34,36,37,38,39,40 (Supplementary Table S5).
Candidate genes
The meta-analysis method allowed to locate with more confidence QTL regions associated with seed productivity and quality traits. In some cases, the number of the genes underlying the metaQTL confidence intervals was narrow. The confidence intervals of metaQTL mQTL3.4, mQTL5.3, mQTL1.5, mQTL7.2, mQTL3.1 and mQTL1.1 include 17, 51, 56, 126, 135, 141 genes, respectively. mQTL3.4 and mQTL1.5 encompass, respectively, the Le and Afila regions previously described as controlling a number of traits in pea7. Le is Psat5g299720 and encodes a 2OG-Fe(II) oxygenase involved in Gibberrelin biosynthesis. Selecting short plants for Pop3, 5, and 9 in 2011 did not prevent to detect this QTL because this gene also segregates in Pop6 and Pop8. The region also includes Psat5g299400, a gene belonging to the AUX/IAA family putatively involved in early response to auxin. The mQTL1.5 region also contains several genes encoding transcription factors expressed in flowers (Psat2g173120, Psat2g173160) or seeds (Psat2g173360, Psat2g173520, Psat2g173880) and genes encoding an aminotransferase (Psat2g172800) and a malate transporter (Psat2g173480) that could be relevant candidates for this metaQTL. The mQTL5.3 region includes two genes encoding phosphatidylethanolamine-binding proteins which expression peaks in the upper leaves as revealed by the Pea Gene Atlas41. The phosphatidylethanolamine-binding proteins in soybean and Arabidopsis thaliana are involved in flowering time, plant architecture and seed germination42. The mQTL1.1 region includes the locus AGPS2, a gene encoding ADP-glucose pyrophosphorylase (Psat2g005160) previously reported to be associated with seed size QTL in pea16, as well as a gene encoding a subtilase (Psat2g005680) expressed in flowers and pods41 different to subtilase gene (Psat0s1712g0120) in D’Erfurth et al.19. The mQTL3.1 region contains a gene encoding a Phosphoenolpyruvate carboxylase (Psat5g031640) expressed in above ground vegetative and reproductive tissues41 that could impact C assimilation and partitioning in the plant43,44. The mQTL3.1 region encompass two tandem genes (Psat7g127600, Psat7g127680) putatively encoding Kelch motif proteins. Interestingly, Kelch Motif-containing serine/threonine protein phosphatase was associated with a seed size QTL in rice45.
The present study pinpointed several robust metaQTL of seed yield and seed protein content in pea and proposed some candidate genes. This useful knowledge for marker assisted breeding, highlights the position and function of underlying genes to discover the causal polymorphisms of QTL in pea.
Materials and methods
Plant material and field experiments
A total of 1213 recombinant inbred lines (RIL) derived from nine mapping populations (Pop3 to Pop11) were developed by single seed descent (SSD). Pop3 to Pop10 are eight advanced inter-connected biparental RIL populations with six of them having Cameor as a common parent. Pop11 is an F3 population obtained from a cross between Cameor and Cerise. Passport data and phenotypic information relative to the parental lines of mapping population are described in Table S6 and in Tayeh et al.20. The population size, the generation, the number of lines genotyped and phenotyped used for QTL meta-analysis are indicated in Table 2. Phenotypic variability of morphological traits of RIL populations are given in supplementary Fig. S3. Pop3 to Pop10 and parental lines were evaluated in six field environments at INRAE Dijon, Domaine d’Epoisses, Bretenière, France (47°14′N, 5°05′E, altitude 210 m) between 2004 and 2011 in spring sowing, except for Pop9 in 2008, 2009, 2010 evaluated in winter sowing (Table 2). In 2011, a subset of the populations was sown. In Pop3, Pop5, and Pop9 where the Le gene controlling internode length segregates, only short lines were sampled for the trial in order to limit the shading of tall plant plots on their neighbours (supplementary Fig. S3). In Pop6 and Pop8, the Le gene also segregates but the lines Le type information was lacking before sowing. Furthermore, Burstin et al.7 have shown the major effect of this gene in the variation of seed traits and sampling only short plants intended to improve the detection of other QTL regions. Field experiments were carried out using a randomized complete block design. Each plot consisted of twenty-five seeds sown in a row of two meters long, with one meter spacing between two adjacent rows. Plants were grown against trellises. Weeds, insects and diseases were controlled chemically. At maturity, a sample of ten plants per line was harvested and Seed Number per plant (SN), Seed Weight per plant (SW, gram), Thousand Seed Weight (TSW, gram) were measured and Seed Protein Content (SPC, percentage of seed dry weight) was analysed by near-infrared spectrometry as described in Burstin et al.7.
Pop10 in 2008–2009 and Pop11 in 2008, 2009 and 2011 were phenotyped in glasshouses at INRAE Dijon, France (47° 32′ N, 5°07′ E, altitude 245 m) (Table 2). Two replicates of two plants per RIL were grown in 4-L pots filled with a 1:1 (v/v) mixture of sterilized atapulgite and clay balls (2–6 mm diameter) with a nitrate content (10 mM). The temperature and minimal day length were controlled (22° C/16 °C, 16-h photoperiod). SN, SW and TSW were measured at maturity per plant.
Statistical analysis
Statistical analysis of each environment dataset was carried out using R software v3.3 and v3.646. ANOVA were performed using the “aov” function in R, to determine the significance levels of the genotype and replication effects. The statistical model was: Yijk = µ + gi + rj + bk/j + eijk where Yijk is the value of the trait for genotype i in block k of the replicate j, µ the general mean, gi the genotypic effect, rj the replicate effect, bk/j the block k effect in the replicate j and eijk the residual. In the case of the populations phenotyped in several environments (Table 2), environment and genotype-by-environment interaction effects were added to the linear model of ANOVA. Broad sense heritability (h2) was estimated from ANOVA by h2 = σ2g / [σ2g + (σ2e/n)] with σ2g the genetic variance, σ2e the residual variance and n the number of replicates. Normality of residuals and homogeneity of variances were checked using Shapiro–Wilk and Bartlett’s test (P ≥ 0.05). RILs’ adjusted means calculated using the “lsmeans” library were used for QTL analysis. Pearson correlation coefficients between the traits for all environments were calculated from RILs adjusted means using “chart.correlation” function and “PerformanceAnalytics” library. Frequency distributions from RILs adjusted means using “car” library and the “hist” function. Principal component analysis from RILs adjusted means was performed using “fviz_pca” function and “factoextra” library.
Genotyping and QTL analyses
The 1213 RIL derived from nine mapping populations (Pop3 to Pop11) were previously genotyped using the GenoPea 13.2 K SNP Array and used for the construction of individual genetic maps and a consensus map as described in Tayeh et al.20. The framework consensus map included 1869 markers and had a total length of 794.9 cM Haldane. The references of these population8,10,20,47,48,49,50 are listed in Table 2.
QTL composite interval mapping was carried out using the iterative QTL mapping method (iQTLm) of the MCQTL software v5.2.421. Cofactor selection and QTL detection Pvalue thresholds were determined after 1000 permutation tests on all traits, for a global genome-wide type I risk of 10% for cofactor selection, and 5% for QTL detection. Cofactors were searched by forward regression, using a threshold of P value = 3.50. QTL were searched by iQTLm, using a threshold of P value = 3.80. For each environment, MultiPop detection was performed using the genotyping from GenoPea 13.2 K SNP Array and the consensus genetic map (1869 markers—794.9 cM) developed by Tayeh et al.20. Model additive and interpop connected was used in Multipop function for the populations in the same environment. The Pvalue, global R2, individual R2, confidence interval and allelic effect at each QTL were estimated for each trait and used for metaQTL analyses.
MetaQTL analyses
Meta-analyses were performed using BioMercator version 4.2 software27. Meta-analysis was implemented on each chromosome to estimate the number, the position, the probability of individual QTL belonging to the metaQTL and 95% confidence interval (CI) of the each metaQTL. QTLProj command enabled the homothetic projection of the positions and the confidence intervals of the individual QTL onto the consensus map. QTLClust command performed the clustering of the projected QTL referring to the same trait on a given chromosome into all possible numbers of hypothetic clusters. This command determined the best clustering model based on the following criteria22: AIC (Akaike Information Criterion), AICc, AIC3, BIC (Bayesian information criterion) and AWE (Average Weight of Evidence). The best QTL model was selected when values of the model selection criteria were the lowest in at least three of the five models. It corresponds to the optimal number of clusters that best explain the observed QTL distribution along the consensus chromosome map. Finally, the QTLClustInfo command provided the number of metaQTL for each chromosome, the better position, the confidence interval and the contribution of each individual QTL20. The metaQTL map was drawn using BioMercator software27.
Candidate genes, functional annotation and expression
QTL flanking markers were positioned on ‘Cameor’ genome sequence23 through BLAST search and annotated genes in the QTL interval were retrieved and listed. QTL and orthologous genes in soybean were identified using USDA-ARS Soybean Genetics Database, SoyBase36 (https://www.soybase.org). Pea gene expression was obtained from Pea RNA-seq Gene Atlas41 (https://bios.dijon.inra.fr/FATAL/cgi/pscam.cgi).
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
References
Nemecek, T. et al. Environmental impacts of introducing grain legumes into European crop rotations. Eur. J. Agron. 28, 380–393 (2008).
Poore, J. & Nemecek, T. Reducing food’s environmental impacts through producers and consumers. Science 360, 987–992. https://doi.org/10.1126/science.aaq0216 (2018).
Rawal, V. & Navarro, D. K. The Global Economy of Pulses (FAO, Rome, 2019).
Page, D., Aubert, G., Duc, G., Welham, T. & Domoney, C. Combinatorial variation in coding and promoter sequences of genes at the Tri locus in Pisum sativum accounts for variation in trypsin inhibitor activity in seeds. Mol. Genet. Genomics 267, 359–369. https://doi.org/10.1007/s00438-002-0667-4 (2002).
Tar’an, B. et al. Identification of quantitative trait loci for grain yield, seed protein concentration and maturity in field pea (Pisum sativum L.). Euphytica 136, 297–306 (2004).
Timmerman-Vaughan, G. M. et al. Linkage mapping of QTL for seed yield, yield components, and developmental traits in pea The research was funded by the New Zealand Foundation for Research Science and Technology. Crop Sci. 45, 1336–1344. https://doi.org/10.2135/cropsci2004.0436 (2005).
Burstin, J. et al. Developmental genes have pleiotropic effects on plant morphology and source capacity, eventually impacting on seed protein content and productivity in pea. Plant Physiol. 144, 768–781. https://doi.org/10.1104/pp.107.096966 (2007).
Bourgeois, M. et al. A PQL (protein quantity loci) analysis of mature pea seed proteins identifies loci determining seed protein composition. Proteomics 11, 1581–1594. https://doi.org/10.1002/pmic.201000687 (2011).
Krajewski, P. et al. QTL for yield components and protein content: a multienvironment study of two pea (Pisum sativum L.) populations. Euphytica 183, 323–336 (2011).
Klein, A. et al. QTL analysis of frost damage in pea suggests different mechanisms involved in frost tolerance. Theor. Appl. Genet. 127, 1319–1330. https://doi.org/10.1007/s00122-014-2299-6 (2014).
Ferrari, B. et al. Association of SNP markers with agronomic and quality traits of field pea in Italy. Czech J. Genet. Plant Breed. 52, 83–93. https://doi.org/10.17221/22/2016-cjgpb (2016).
Moreau, C. et al. Recombinant inbred lines derived from cultivars of pea for understanding the genetic basis of variation in breeders’ traits. Plant Genet. Resour. 16(5), 424–436. https://doi.org/10.1017/s1479262118000345 (2018).
Gali, K. K. et al. Construction of high-density linkage maps for mapping quantitative trait loci for multiple traits in field pea (Pisum sativum L.). BMC Plant Biol. 18, 172. https://doi.org/10.1186/s12870-018-1368-4 (2018).
Gali, K. K. et al. Genome-wide association mapping for agronomic and seed quality traits of field pea (Pisum sativum L.). Front. Plant Sci. 10, 20. https://doi.org/10.3389/fpls.2019.01538 (2019).
Bhattacharyya, M. K., Smith, A. M., Ellis, T. H. N., Hedley, C. & Martin, C. The wrinkled seed character of pea described by Mendel is caused by a transposon-like insertion in a gene encoding starch branching enzyme. Cell 60, 115–121 (1990).
Smith, A. M., Bettey, M. & Bedford, I. D. Evidence that the <em>rb</em> locus alters the starch content of developing pea embryos through an effect on ADP glucose pyrophosphorylase. Plant Physiol. 89, 1279–1284. https://doi.org/10.1104/pp.89.4.1279 (1989).
Wang, T. & Hedley, C. Seed development in peas: knowing your three ‘r’s’ (or four, or five). Seed Sci. Res. 1(1), 3–14. https://doi.org/10.1017/S096025850000057X (1991).
Perez, M. et al. Seed protein content and composition of near-isogenic and induced mutant pea lines. Seed Sci. Res. 3(3), 187–194. https://doi.org/10.1017/S096025850000177X (1993).
D’Erfurth, I. et al. A role for an endosperm-localized subtilase in the control of seed size in legumes. New Phytol. 196, 738–751 (2012).
Tayeh, N. et al. Development of two major resources for pea genomics: the GenoPea 13.2K SNP Array and a high-density, high-resolution consensus genetic map. Plant J. 84, 1257–1273. https://doi.org/10.1111/tpj.13070 (2015).
Jourjon, M. F., Jasson, S., Marcel, J., Ngom, B. & Mangin, B. MCQTL: multi-allelic QTL mapping in multi-cross design. Bioinformatics 21, 128–130 (2005).
Veyrieras, J.-B., Goffinet, B. & Charcosset, A. MetaQTL: a package of new computational methods for the meta-analysis of QTL mapping experiments. BMC Bioinform. 8, 49 (2007).
Kreplak, J. et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 51, 1411–1422. https://doi.org/10.1038/s41588-019-0480-1 (2019).
Simmonds, N. W. The relation between yield and protein in cereal grain. J. Sci. Food Agric. 67, 309–315. https://doi.org/10.1002/jsfa.2740670306 (1995).
Oury, F.-X. et al. Yield and grain protein concentration in bread wheat: a review and a study of multi-annual data from a French breeding program. J. Genet. Breed. 57, 59–68 (2003).
Goffinet, B. & Gerber, S. Quantitative trait loci: a meta-analysis. Genetics 155, 463–473 (2000).
Sosnowski, O., Charcosset, A. & Joets, J. BioMercator V3: an upgrade of genetic map compilation and quantitative trait loci meta-analysis algorithms. Bioinformatics 28, 2082–2083. https://doi.org/10.1093/bioinformatics/bts313 (2012).
Guo, B. et al. QTLs associated with resistance to soybean cyst nematode in soybean: meta-analysis of QTL locations. Crop Sci. 46, 595–602. https://doi.org/10.2135/cropsci2005.04-0036-2 (2006).
Hanocq, E., Laperche, A., Jaminon, O., Lainé, A. L. & Le Gouis, J. Most significant genome regions involved in the control of earliness traits in bread wheat, as revealed by QTL meta-analysis. Theor. Appl. Genet. 114, 569–584. https://doi.org/10.1007/s00122-006-0459-z (2007).
Rong, J. et al. Meta-analysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 176, 2577–2588. https://doi.org/10.1534/genetics.107.074518 (2007).
Lanaud, C. et al. A meta–QTL analysis of disease resistance traits of Theobroma cacao L. Mol. Breed. 24, 361–374. https://doi.org/10.1007/s11032-009-9297-4 (2009).
Löffler, M., Schön, C.-C. & Miedaner, T. Revealing the genetic architecture of FHB resistance in hexaploid wheat (Triticum aestivum L.) by QTL meta-analysis. Mol Breed. 23, 473–488. https://doi.org/10.1007/s11032-008-9250-y (2009).
Truntzler, M. et al. Meta-analysis of QTL involved in silage quality of maize and comparison with the position of candidate genes. Theor. Appl. Genet. 121, 1465–1482. https://doi.org/10.1007/s00122-010-1402-x (2010).
Qi, Z.-M. et al. A meta-analysis of seed protein concentration QTL in soybean. Can. J. Plant Sci. 91, 221–230. https://doi.org/10.4141/cjps09193 (2011).
Hwang, S. et al. Meta-analysis to refine map position and reduce confidence intervals for delayed-canopy-wilting QTLs in soybean. Mol Breed. 36, 91. https://doi.org/10.1007/s11032-016-0516-5 (2016).
Grant, D., Nelson, R. T., Cannon, S. B. & Shoemaker, R. C. SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res. 38, D843–D846. https://doi.org/10.1093/nar/gkp798 (2009).
Karikari, B. et al. Genome-wide detection of major and epistatic effect QTLs for seed protein and oil content in soybean under multiple environments using high-density bin map. Int. J. Mol. Sci. 20, 979 (2019).
Li, S., Xu, H., Yang, J. & Zhao, T. Dissecting the genetic architecture of seed protein and oil content in soybean from the Yangtze and Huaihe River valleys using multi-locus genome-wide association studies. Int. J. Mol. Sci. 20, 3041 (2019).
Zhang, T. et al. A combined linkage and GWAS analysis identifies QTLs linked to soybean seed protein and oil content. Int. J. Mol. Sci. 20, 5915 (2019).
Zhang, D. et al. The genetic architecture of water-soluble protein content and its genetic relationship to total protein content in soybean. Sci. Rep. 7, 5053. https://doi.org/10.1038/s41598-017-04685-7 (2017).
Alves-Carvalho, S. et al. Full-length de novo assembly of RNA-seq data in pea (Pisum sativum L.) provides a gene expression atlas and gives insights into root nodulation in this species. Plant J. 84, 1–19. https://doi.org/10.1111/tpj.12967 (2015).
Wang, Z. et al. Functional evolution of phosphatidylethanolamine binding proteins in soybean and arabidopsis. Plant Cell 27, 323–336. https://doi.org/10.1105/tpc.114.135103 (2015).
O’Leary, B., Park, J. & Plaxton, W. C. The remarkable diversity of plant PEPC (phosphoenolpyruvate carboxylase) recent insights into the physiological functions and post-translational controls of non-photosynthetic PEPCs. Biochem. J. 436, 15–34. https://doi.org/10.1042/bj20110078 (2011).
Radchuk, R. et al. Ectopic expression of phosphoenolpyruvate carboxylase in Vicia narbonensis seeds: effects of improved nutrient status on seed maturation and transcriptional regulatory networks. Plant J. 51, 819–839. https://doi.org/10.1111/j.1365-313X.2007.03196.x (2007).
Hu, Z. et al. A Kelch Motif-containing serine/threonine protein phosphatase determines the large grain QTL trait in rice. J. Integr. Plant Biol. 54, 979–990. https://doi.org/10.1111/jipb.12008 (2012).
Team, R. C. R: A language and environment for statistical computing (R Foundation for Statistical Computing, Vienna, Austria, 2019).
Bordat, A. et al. Translational genomics in legumes allowed placing in silico 5460 unigenes on the pea functional map and identified candidate genes in Pisum sativum L.. G3-Genes Genomes Genet. 1, 93–103 (2011).
Deulvot, C. et al. Highly-multiplexed SNP genotyping for genetic mapping and germplasm diversity studies in pea. BMC Genom. 11, 468 (2010).
Duarte, J. et al. Transcriptome sequencing for high throughput SNP development and genetic mapping in Pea. BMC Genom. 15, 126 (2014).
Bourion, V. et al. Genetic dissection of nitrogen nutrition in pea through a QTL approach of root, nodule, and shoot variability. Theor. Appl. Genet. 121, 71–86 (2010).
Acknowledgements
This study was supported by the ANR Genopea (project ANR-09-GENM-026) of the French National Research Agency. The authors thank the staff of the experimental unit of INRAE Dijon Domaine d’Epoisses, Bretenière and INRAE Dijon serres4PMI for the technical assistance in the phenotyping. We thank Emilie Vieille, Karen Boucherot and Chantal Martin for the DNA material production and Jonathan Kreplak for genomic data. We thank Gregoire Aubert and Nadim Tayeh for reviewing the manuscript.
Author information
Authors and Affiliations
Contributions
J.B. conceived the study. A.K. performed the statistical analysis, the QTL and metaQTL analyses. H.H., C.R.C., M.N.H., M.T., P.M. and A.K. produce the phenotypic data. All authors reviewed the manuscript and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Klein, A., Houtin, H., Rond-Coissieux, C. et al. Meta-analysis of QTL reveals the genetic control of yield-related traits and seed protein content in pea. Sci Rep 10, 15925 (2020). https://doi.org/10.1038/s41598-020-72548-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72548-9
- Springer Nature Limited
This article is cited by
-
Meta QTL analysis for dissecting abiotic stress tolerance in chickpea
BMC Genomics (2024)
-
Meta-QTL analysis reveals the important genomics regions for biotic stresses, nutritional quality and yield related traits in pearl millet
CABI Agriculture and Bioscience (2024)
-
Innovations in functional genomics and molecular breeding of pea: exploring advances and opportunities
aBIOTECH (2024)
-
Meta-QTL analysis enabled identification of candidate genes and haplotypes for enhancing biotic stress resistance in chickpea
Journal of Plant Biochemistry and Biotechnology (2024)