
Abstract
Plantago ovata (Plantaginaceae) is an economically and medicinally important species, however, least is known about its genomics and evolution. Here, we report the first complete plastome genome of P. ovata and comparison with previously published genomes of related species from Plantaginaceae. The results revealed that P. ovata plastome size was 162,116 bp and that it had typical quadripartite structure containing a large single copy region of 82,084 bp and small single copy region of 5,272 bp. The genome has a markedly higher inverted repeat (IR) size of 37.4 kb, suggesting large-scale inversion of 13.8 kb within the expanded IR regions. In addition, the P. ovata plastome contains 149 different genes, including 43 tRNA, 8 rRNA, and 98 protein-coding genes. The analysis revealed 139 microsatellites, of which 71 were in the non-coding regions. Approximately 32 forward, 34 tandem, and 17 palindromic repeats were detected. The complete genome sequences, 72 shared genes, matK gene, and rbcL gene from related species generated the same phylogenetic signals, and phylogenetic analysis revealed that P. ovata formed a single clade with P. maritima and P. media. The divergence time estimation as employed in BEAST revealed that P. ovata diverged from P. maritima and P. media about 11.0 million years ago (Mya; 95% highest posterior density, 10.06–12.25 Mya). In conclusion, P. ovata had significant variation in the IR region, suggesting a more stable P. ovata plastome genome than that of other Plantaginaceae species.
Similar content being viewed by others


Introduction
Chloroplasts carry out photosynthesis along with other crucial functions including the biosynthesis of starch, fatty acids, amino acids, and pigments in plants and various other eukaryotic lineages1,2. Typically, maternally inherited angiosperm plastome genomes are extremely conserved and have a quadripartite structure ranging from 115 to 165 kb in length and comprising a small-single-copy region (SSC) and large-single-copy region (LSC) parted by an inverted repeat (IR)3,4. The gene order and content are mostly conserved; however, at genome and gene levels, a number of variations have been reported in various plastome genomes5. The plastome genome is a useful tool in evolutionary studies and genomics because of its non-recombinant nature6. Furthermore, the plastome genome is highly conserved mostly in angiosperms, evolutionary hotspots such as addition and deletion of genes7, contraction and expansion of IR region8, inversion of various genomic regions, copy number variations in tandem repeats9, and single nucleotide polymorphisms (SNPs)7. SNPs and InDels are valuable molecular markers in these conserved plastomes within the species level10. In brief, various characteristics including uniparental inheritance, absence of recombination, and a haploid state11,12 make plastome genome valuable for phylogeographic and phylogenetic studies to understand the history of most plant lineages13,14. At present, there are over 2700 plastid genome sequences submitted to the National Centre for Biotechnology Information (NCBI), including all of the major plant lineages. Altogether, plastome genome sequences possess sufficient information, and sequencing of these genomes play a vital role in diversification and to facilitate comparisons among various plant species15,16.
Plantago is an important genus of Plantaginaceae17, commonly known as Plantains and is usually a perennial or annual herb or sub-shrub. The genus has a worldwide distribution with ca. 200 species, among which only two species namely P. psyllium and P. ovata have been widely studied for seed husk production18. Morphologically, the seed husk of P. ovata is enclosed by a thin white membrane, usually known as Blonde Psyllium in English. The genetic diversity of Plantago species is important for breeding various cultivars, which are valuable additions that are resistant to different diseases19. The meiotic system and chromosomal structure of P. ovata have various features which account for its narrow genetic base such as a small 621-Mb genome size with 4 (2n = 2x = 8) chromosomes20.
In an comprehensive phylogenetic study of the family Plantaginaceae based on different chemical, embryological, and morphological data, researchers were unable to identify a sister-family for this monophyletic group17. However, Dhar et al.21 and Ronsted et al.22 have determined various molecular markers that are useful for inferring the phylogenetic relationships of the genus Plantago. The ITS and 5S rRNA regions depicted on the sequence data showed that P. ovata is closely related to P. arenaria23. In recent time with the advancement in molecular techniques, different molecular markers were used to access the intra and interspecific relationship among Plantago species19,24. However, such studies investigated smaller intraspecific diversity than the interspecific diversity.
Taking into account the taxonomic and phylogenetic complications for the genus, and lack of concentrated evidences, here in the present study, we sequenced and performed a comparative analysis of the complete plastome of P. ovata with the plastomes of six related species (P. media, P. maritima, Veronica nakaiana, V. presica, V. veronicstrum, and Digitalis lanata). We aimed to elucidate and compare the regions of high sequence divergence, IR expansion, intron contents, and phylogenomics of P. ovata with plastomes of related species. In addition, we employed divergence time estimation to reconstruct the phylogenetic relationship and divergence time of P. ovata, P. media, and P. maritima based on whole plastomes.
Results
General features of P. ovata plastome sequence and its comparison with related species
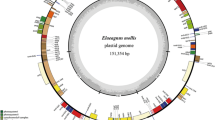
The genome size of P. ovata plastome is 162,116 bp and has a typical quadripartite structure containing an LSC (82,084 bp) and SSC (5,272 bp) and separated by a pair of identical IRs (37,380 bp) each. The GC content (38.1%) and LSC region of P. ovata is consistent with the previously reported plastomes belonging to the family Plantaginaceae (Table 1). Compared to the typical angiosperm genome structure, for example, that of P. media and P. maritima, P. ovata had considerably higher number of IRs measuring up to 37.4 kb in size (Table 1, Fig. 1) than other angiosperms. Additionally, in all the sequenced plastomes from Plantaginaceae, P. ovata has the largest plastome, except P. media (164,130 bp; Table 1). The plastome of P. ovata contains 147 different genes, including 43 tRNA, 8 rRNA, and 96 protein-coding genes (12 small and 9 large ribosomal subunits, 4 DNA-dependent RNA polymerases, 33 photosynthesis-related proteins, and 10 genes encoding other proteins) (Table 2, Fig. 1). About, 15.40% of the functional genes contains introns, including 7 tRNA and 16 protein-coding genes; the two genes clpP and ycf3 contain two introns (Table 3, Fig. 1). The lengths of these introns range from 483 bp (trnV-UAC) to 2,434 bp (trnK-UUU). The rps12 gene (small ribosomal protein 12) is trans-spliced and contains one intron; furthermore, its 5′ end exon is located in the LSC region, whereas the 3′ end exon is located in the IRb regions and also duplicated in the IRa region (Fig. 1).

Gene map of the P. ovata plastome genome. Genes drawn inside the circle are transcribed clockwise, and those outside the circle are transcribed counterclockwise. The red and green colour asterisks indicate intron-containing and trans-spliced genes respectively. Genes belonging to different functional groups are colour-coded. The darker grey in the inner circle corresponds to GC content, and the lighter grey corresponds to AT content.
Overall, the protein coding, rRNA and tRNA genes contain 47.96%, 5.57%, and 1.97%, respectively, of the P. ovata plastome sequence (Table S1). The GC content for tRNA (52.10%) and rRNA (55.20%) are the highest, followed by protein-coding genes (39%) in the coding regions. Similarly, GC content within the protein-coding genes at the first, second, and third positions of codon is 55.60%, 47.40%, and 36.70%, respectively (Table S1). Codon usage and codon–anticodon recognition pattern of P. ovata plastome are summarised in Table S2, in which a total of 24,322 codons from a genome size of 72,968 bp have been represented. Based on tRNA and protein-coding genes, RSCU frequency was calculated (Table S1). The most common amino acid was leucine (10.90%), whereas the least common one was cysteine (1.30%, Table S2). Codon usage is biased towards a high representation of A and T at the third position (82.60%), revealing a pattern similar to angiosperm plastid genomes (Table S1).
Comparative analysis of P. ovata plastome with the plastomes of related species
The synteny of P. ovata plastomes with seven other species from Plantaginaceae was analysed by mVISTA. The results showed high sequence similarities among the plastomes of several species, especially in protein-coding and IR regions (Fig. 2). The highest level of divergence was detected in intergenic regions, including atpH-atpI, rpoC1-rpoC2, ycf1-rps15, accD-psaI, psaA-ycf3, and trnL-rrn5. Besides these regions, some divergence was observed in protein-coding genes, viz. accD, clpP, ndhA, ndhF, rpl16, petD, matK, rpl16, ycf2, ycf1, and rpl2 (Fig. 2). In a pairwise sequence divergence analysis, P. ovata exhibited highest divergence (0.20) with V. persica and showed lowest divergence with P. media (0.048) (Table S3). The most divergent genes were clpP, accD, psaJ, rps3, ccsA, and matK. The highest pairwise divergence was detected in clpP gene (0.67) and accD gene (0.56) (Fig. 3). Furthermore, the synonymous (Ks) and non-synonymous (Ka) values of plastomes were calculated. The results revealed that P. ovata exhibited highest Ka/Ks value of plastome with V. nakiana (0.198/0.2506) and lowest with P. maritima (0.05/0.06), respectively. However, the most divergent genes, accD and clpP, showed variable results. The highest Ka/Ks value was exhibited by V. nakiana for accD gene, whereas the highest Ka/Ks value was exhibited by P. maritima for clpP gene (Figure S1). The length of the most divergent gene, accD, was 1,356 bp (452 aa), 1,347 bp (449 aa), and 1,257 bp (425 aa) in P. ovata, P. media, and P. maritima, respectively. In the other four species, the length of accD gene ranged from 1,470 to 1,497 bp (Fig. S2). Variation in the intron content of clpP gene was found in P. maritima, in which there was a complete loss of both introns (Fig. S3). This forms the basis for the highest divergence of P. maritima with P. ovata genome for the clpP gene. A comparative analysis of the P. ovata plastome revealed a varied number of SNPs and InDel substitution. The highest number of SNPs was detected in V. persica (53,660), whereas the lowest number of SNPs was observed in P. maritima (16,386). The highest number of InDel substitutions was detected in P. maritima (74,448) plastome (Table S4).

Alignment visualization of the P. ovata plastome genome sequences. VISTA-based identity plot showing sequence identity among the seven-species using P. ovata as a reference. The vertical scale indicates percent identity, ranging from 50% to 100%. The horizontal axis indicates the coordinates within the chloroplast genome. Arrows indicate the annotated genes and their transcription direction. The thick black lines show the inverted repeats (IRs).

Pairwise sequence distance of P. ovata, 72 genes with related species.
Simple sequence repeat (SSR) analysis
In the present study, we determined SSRs in the plastome of P. ovata as well as in the plastomes of other seven other related species (Fig. 4). We examined the type, distribution, and occurrence of SSRs in plastomes. Based on SSR analysis, a total of 139 microsatellites were detected in P. ovata plastome. Among these, 71 SSRs were identified in non-coding regions, 63 in coding regions, four in rRNA-coding region, and 1 in tRNA-coding region. Similarly, in LSC, IRs, and SSC, 76, 58 and 5 SSRs were detected, respectively (Fig. 4). In P. ovata plastome, a majority of SSRs consisted of tri- (69, 49%) and di-nucleotide (36, 25.80%) repeats. This pattern is similar to that in the related genomes analysed in this study. We did not detect penta-, hexa-, and heptanucleotides in P. ovata plastome. However, in P. maritima and P. media plastomes, two penta-nucleotides were observed, whereas one hexa- and heptanucleotide were detected in P. maritima plastomes (Fig. 4; Table S5). In P. ovata, almost 100% of the mononucleotides contain an A motif, whereas a majority of di-nucleotide SSRs were A/G (21, 58.30%) and A/T (11, 30.50%), respectively. A similar pattern of SSR motif was observed in related plastomes (Fig. 4).

Analysis of simple sequence repeats (SSR) in the seven Plantaginaceae plastomes; (A) SSR numbers detected in the seven species; (B) Frequency of identified SSR motifs in different repeat class types; (C) Frequency of identified SSRs in coding, Non-coding, rRNA and tRNA regions; (D) Frequency of identified SSRs in LSC, SSC and IR regions.
Repeat sequence analysis
Repeat sequence analysis of P. ovata plastome with related species revealed the presence of 32 forward repeats, 34 tandem repeats, and 17 palindromic repeats (Fig. 5). Among these repeats, 7 of the palindromic repeats were 30–44 bp in length, while 6 repeats were >90 bp in length. Similarly, 12 and 14 forward repeats were 30–44 bp and >90 bp in length, respectively, whereas approximately 21 tandem repeats were identified to have a length of 15–29 bp (Fig. 5). Overall, 83 repeats were detected in P. ovata plastome, which is lower than those in P. maritima plastome (89) and higher than those in V. persica (63) and D. lanata (68) plastomes. Approximately 25% palindromic repeats, 17.60% forward repeats, and 26.60% tandem repeats were distributed in the protein-coding regions of P. ovata plastome (Tables S6, S7). Moreover, a higher number of palindromic repeats (29), forward repeats (32), and tandem repeats (39) were detected in V. nakaiana, P. ovata and P. maritima plastomes (Fig. 5).

Analysis of repeated sequences in the seven Plantaginaceae plastomes. (A) Totals numbers of three repeat types; (B) Number of palindromic repeats by length; (C) Number of tandem repeats by length; (D) Number of forward repeats by length.
Contraction and expansion of IRs
IR regions are considered to be the most conserved regions in a chloroplast genome. The larger plastome sizes correlate with larger IR length; similar to the plastomes of two previously described Plantago species (P. maritima and P. media), P. ovata also contained IRs that significantly increased in size up to 37.40 kb as compared with IRs in the other related species (Table 1). Repeat expansion occurred in the SSC, resulting in the transfer of 9 former SSC genes (rps15, ndhI, ndhD, psaC ndhA, ndhH, ndhG, ndhE, and ycf1) into the IR regions (Fig. 1). Consistent with the observation in P. media and P. maritima plastomes, P. ovata also showed large-scale inversion of 13.80 kb within the expanded IR regions. For example, in P. media and P. ovata the breakpoint is inferred to be at trnL-ndhB and trnN-trnR, which are ycf1-rps15 and trnL-ndhB in P. maritima (Fig. 1). The gene arrangement in the IR region of P. ovata is more similar to that of P. media than that of P. maritima, where sixteen protein-coding genes (rpl2, rpl23, ycf15, rps12, rps7, ndhB, ycf1, rps15, ndhH, ndhA, ndhI, ndhG, ndhE, psaC and ndhD) are duplicated. However, in P. maritima the duplicated protein coding genes are eleven and five former SSC genes transferred into IR region. Another noteworthy variation among Plantago plastomes IR regions was an additional small-scale inversion related with ycf1 gene detected only in P. maritima plastome (Fig. S4).
The IR/LSC and IR/SSC borders of P. ovata were compared with related plastomes (Fig. 6). The rps19 gene was separated from the LSC/IRb region by 108 bp, and the rpl2 gene extended to the LSC region and was duplicated in IR regions. Contrastingly, in other plastomes, rpl2 did not extend up to the LSC region and was completely duplicated in IRs. Similarly, in P. ovata, owing to an extension of rpl2 gene at LSC/IRb borders, IRa ended up with the truncated copy of rpl2 gene. The position of ndhF gene in the SSC varied in these plastomes. In P. ovata, P. media, and P. maritima, ndhF gene was located 350, 14, and 59 bp away from IRb/SSC in the SSC, whereas in other related members of Plantaginaceae family, it extended up to the IRb region (Fig. 6). Furthermore, in P. ovata, the ccsA gene was 69 bp away from SSC/IRa border in the SSC, whereas ndhD gene was 194 bp away from this region and located in the IRa region. In case of P. media, ccsA extended to the IR region. Because the IR length in P. maritima is smaller than P. ovata and P. media, IR junction varied. Here, the ndhI was located 292 bp away from the IRb/SSC in the IRb region, whereas ndhG was 59 bp away from the SSC/IRa border in the SSC. In other Plantaginaceae members (D. lanata, V. persica, V. nakaiana, and V. veronicstrum), ndhH gene extended up to 24 bp, 51 bp, 57 bp, and 41 bp, respectively, into the IRb region at the IRb/SSC border. The psbA gene in all species was located in the LSC region and separated from the IRa/LSC border by 232–371 bp.

Comparison of border distance between adjacent genes and junctions of the LSC, SSC, and two IR regions among the plastome genomes of P. ovata and its relatives. Boxes above or below the main line indicate the adjacent border genes. The figure is not to scale with respect to sequence length and only shows relative changes at or near the IR/LSC or IR/SSC borders.
Phylogenetic analysis and Divergence time of P. ovata with related species
Here, the phylogenetic position of P. ovata within the order Lamiales was established by multiple alignment analysis of the complete plastome, 72 shared genes, matK gene, and rbcL gene of Lamiales members representing 8 families and 22 genera (Figs. 7; S5). Phylogenetic analyses were performed using ML, NJ, MP, and BI methods. ML analysis revealed that 28 out of 30 nodes had a bootstrap value of ≥99%, while the remaining had a value of 100% (Figs. 7; S5). The phylogenetic trees constructed based on the sequences of the complete genome, 72 shared genes, matK gene, and rbcL gene of P. ovata formed a clade with P. maritima and P. media via bootstrap and BI support. In the analysed data sets, D. lanata was inferred to be closest to the Plantago than to Veronica species. Furthermore, Gesneriaceae and Phrymaceae were found to be closely related families on the basis of the sequences of their plastomes, 72 shared genes, matK gene, and rbcL gene (Figs. 7; S5). The divergence time was estimated with Baysean approach as implemented in BEAST showed that P. ovata has diverged from the common ancestor of P. media and P. maritima at 11.0 million years ago (Mya; 95% HPD, 10.06–12.25 Mya) (Fig. S6). Additionally, the tree implemented in BEAST resulted a congruent topology with those generated by ML, NJ, and MP.

Phylogenetic trees were constructed for thirty-five species from eight families representing 22 genera using different methods, and tree is shown for the whole genome sequence data sets constructed by ML method. The whole genome sequence data set was used with four different methods, Bayesian inference (BI), maximum likelihood (ML), maximum persimony (MP) and neighbour-jouining (NJ). Numbers above the branches are the posterior probabilities of BI and bootstrap values of ML, MP and NJ respectively. Black dots represent the position for P. ovata.
Discussion
In this study, we sequenced and annotated the complete plastome of P. ovata and compared it with the plastomes of related Plantaginaceae species. Plantago ovata plastome exhibited a typical quadripartite structure of 162,116 bp, correlated with other Plantago species (Table 1)25. Though gene content and organisation in P. ovata plastome found to be similar with other species in Plantaginaceae, however, the genome size and IRs of P. ovata was markedly different from P. media and P. maritima25. Similar to P. media and P. maritima, P. ovata plastome exhibited a higher level of sequence and structural divergence as well as showed some rearrangements in its plastome in comparison with those for typical angiosperms plastomes25,26,27. We observed a similar number of intron-containing protein-coding genes with 7 tRNA genes (with an exception of ycf3 and clpP which contained two introns) in P. media plastome25. Conversely, in P. maritima, V. persica, V. sibiricum, D. lanata, and V. nakaiana, the number of intron-containing genes were 13, 14, 15, 15, and 15, respectively (Table 1)25,28. In synergy with other angiosperms, rps12 gene was unevenly divided with its 5′ terminal exon located in the LSC region, whereas the 3′ terminal exon and intron were located in the IRb region and also duplicated in the IRa region29,30. The trnK-UUU had the longest intron (2,434 bp)26,31. According to a previous study, introns play a vital role in gene expression regulation; improve exogenous gene expression and transformational efficiency32. This intronic effect has been widely described in various plants33,34,35 and termed as intron-mediated enhancement34,35,36. In fact, because some introns are markedly efficient in triggering gene expression, they are regularly included to constructs for maintaining a high expression level37. Cysteine and leucine were the least and the most common amino acids in P. ovata plastome, respectively (Table S2). These findings were also supported by the analysis of previous plastomes of other related species belonging to Plantaginaceae family25,28. The presence of high AT content at the third codon position is consistent with that in the plastomes of various terrestrial plants31,38,39,40.
The present results revealed that P. ovata shared a high sequence similarity with all the analysed species. However, relatively lower identity was detected for these plastomes in various comparable genomic regions. Previously in angiosperm genomes, the SSC and LSC regions showed more divergence than the IR regions25,27,29,41. These results are in agreement with previous reports which showed a higher sequence divergence because of copy correction for gene conservation in IR regions42. The plastome of P. ovata showed a high divergence level in non-coding and intergenic spacer regions (atpH-atpI, rpoC1-rpoC2, ycf1-rps15, accD-psaI, psaA-ycf3, and trnL-rrn5) as opposed to the coding regions (Fig. 2)26,39,43. However, some divergent protein-coding genes (accD, clpP, ndhA, ndhF, rpl16, petD, matK, rpl16, ycf2, ycf1, and rpl2) were also detected in P. ovata plastome. This is in correlation with previously reported findings for the plastomes of angiosperms31,43. Manezes et al.42 concluded that divergent plastome genes are predominantly detected in the LSC regions, suggesting a more rapid evolution trend44. Our results are based on divergence time estimation that showed a rapid divergence trend in genus Plantago and revealed that P. ovata has diverged from P. media and P. maritima at 11.0 million years (Mya; 95% HPD, 10.06–12.25 Mya; Fig. S6). Similar results were reported by Iwanycki Ahlstrand et al.45 on the basis of various genes that subgenus Plantago diverged about 8.8 Mya (95% HPD, 17.4–2.5 Mya). These results are also consistent with the divergence time estimation of family Plantaginaceae where its crown age was estimated 31.42 Mya46. The highest average pairwise distances were detected in clpP and accD genes of the P. ovata plastome (Fig. 3). This divergence was observed in accD gene because the plastomes of three Plantago species had different gene lengths. Previous studies have also observed varied lengths of accD gene within an angiosperm plastome26,47. In legume, this variation in length was partially explained by the presence of various tandemly repeated sequences47. Knockout experiments showed important functions of accD gene (encodes acetyl-CoA)48. However, in various relatively distant lineages (for example, Fabaceae, Poaceae, Lamiaceae, Geraniaceae, Campanulaceae, and Ericaceae), the accD gene was identified as pseudogene and completely absent which shows that the deletion or pseudogenisation even of accD occur independently26,49,50,51. The second most divergent gene, clpP, showed highest divergence with P. maritima, which can be a result of introns loss in P. maritima genome as reported previously by Zhu et al.25.
SSRs, on the other hand, are valuable markers to detect a high-degree variation with the same species and have been used to investigate polymorphisms and population genetics52. We have detected 139 microsatellites in P. ovata plastome and about 71 were found in non-coding regions. This is consistent with angiosperm plastomes in which the highest number of SSRs are detected mostly in non-coding regions29,39,53. Approximately, 147, 177, 146, 155, 157, and 152 SSRs were detected in P. maritima, P. media, D. lanata, V. persica, V. nakaiana, and V. sibiricum plastomes, respectively (Fig. 4). Tri- and dinucleotide repeats were predominantly present in P. ovata. This pattern was also observed in a related genome described previously28. An angiosperm plastome mostly contains mononucleated A motif, whereas most di-nucleotides contain A/G motif40,53,54. Mononucleotide, pentanucleotide, and hexanucleotide repeats contain “A” or “T” bases at higher frequencies, suggesting a biased base composition with an overall A-T richness in the plastomes55,56. Similar previous reports have revealed an uneven distribution of SSRs in Plantago species, owing to which it may be possible to procure more valuable information regarding effective molecular markers for intraspecific polymorphism57,58. We determined 32 forward, 34 tandem, and 17 palindromic repeats in P. ovata plastome. Repeat sequences have been reported in the plastomes of various plant lineages28,44,59. These types of sequences are very important and used extensively for a range of evolutionary and population genetic studies60,61. Additionally, repeat sequences play a central role in plastome rearrangement62,63. The length of palindromic and direct repeats in P. ovata were considerably long, ranging from 340 bp to 532 bp (Table S7); similar results have been previously reported for the plastomes of Poaceae, Fabaceae, Clematis, and Ranunculus species39,44,64. As reported previously, genome rearrangement and sequence diversity occur owing to an incorrect recombination of these repeat sequences and slipped strand mispairing65,66. Furthermore, the occurrence of these repeats indicate that the region is a critical hotspot for the reconfiguration of plastome genome66. Additionally, these repeats are an informative source for developing genetic markers for Plantago species, which can be further applied in phylogenetic and population studies62.
Regarding IR regions, contraction and expansion at borders are the key motives underlying size variations in plastome genomes, and thereby play an important role in evolution48,67,68. Compared with a typical angiosperm genome, in P. ovata plastome, the IR region significantly increased in size up to 37.4 kb, which indicates the transfer of nine SSC genes to the IR regions. Similar results were observed in the previously reported plastomes of P. media and P. maritima, where 9 and 5 gene transfers occurred into the IR region, respectively25. An inversion of 13.8 kb was detected in P. ovata plastome, which was 21 kb and 14 kb in P. maritima and P. media plastomes, respectively (Fig. S4). The increase in total genome size can be explained by an increase in the length of IRs, as reported previously in various plastomes25,41,69. Previously, larger repeats ranging from >25 to 30 kb in size were also reported in angiosperms, and small size-related changes of a few hundred base pairs commonly occur in IR27,70. However, some larger and rare changes in the size of IR regions, ranging from 43,864 bp in Buchnera to 63,240 bp in S. forbessii have been reported71. The largest known repeat of 75,741 bp was reported in Geranium hortorum by Chumley, et al.72. During land plant evolution, there have been multiple instances of IR expansion and contraction which caused the displacement of an entire gene from the SC regions into the IR regions or from the IR to SSC regions25,71. In P. ovata plastome, the terminal IR gene adjacent to the SSC is highly conserve as reported previously25,71. In most species, trnN-GUU is a full-length IR gene present at the IR/SSC boundary, which provides a strong evidence for ancestral IR/SSC endpoint that has been conserved in most lineages25. However, similar to P. media and P. maritima, some degree of extension into the SSC was observed in P. ovata plastome which to some extent occurred in other angiosperms25,73. Similar to the other two Plantago species, in P. ovata plastome, IR expansion has distinct features as opposed to an enlarged IR lineage such as Berberis, Trochodenfron, and N. acuminate25. These features include an extensive genomic rearrangement, which suggest that a different mechanism of IR expansion may be involved. A similar mechanism was observed in Pelargonium IR expansion, which suggested a model involving multiple inversion promoted by these enlarged repeats72. In Plantago plastome, these repeats indicate that they may be involved in inversion events as reported previously25. Moreover, a detailed comparison between IR/LSC and IR/SSC border of P. ovata, which was subsequently compared with the borders in other related plastomes (Fig. 6). We carefully analysed and compared the exact IR border position and its adjacent genes among the plastomes of other Plantaginaceae species. The study revealed that the LSC/IRb was located between the rps19 and rpl2 genes in all Plantaginaceae plastomes. However, the rpl2 gene extended into the IRb regions ranging from 22 bp (V. persica) to 91 bp (V. veronicastrum) in all the analysed plastomes, except P. ovata, which had a 38-bp extension into the LSC regions. This distance was 57 bp and 73 bp away from the LSC/IRb border in P. media and P. maritima, respectively. Due to the smaller IR length of P. maritima among Plantago species, IR junctions vary. Previous studies have revealed that there is an expansion of the IR and LSC regions in angiosperm plastomes during evolution25,70,72.
The Plantago genus comprises approximately of 200 species17, most of which are mainly cross-pollinated in nature74. Some of these species are medicinally important and various wild species reportedly possess important genes that play a vital role in isabgol production when introgressed to cultivated species18. Continued studies have extended our knowledge to distinguish and understand the genomic structure of and phylogenetic relationships among Plantago species23,74,75. Taxonomy and phylogeny of the Plantago genus within Lamiales have been widely examined at the genus level20,22. Previous evolutionary relationships among different Plantago species were estimated by analysing nuclear polymorphisms, random amplified polymorphic DNA (RAPD) profiles, chloroplast DNA restriction fragment-length polymorphisms (RFLP), SSR region, and ITS region as well as the sequences of 5 S rRNA genes, rps14 gene, and plastid trnL-F20,22,75,76. However, complete genome sequencing provides more detailed insight31,77,78. In this regard, the complete genome sequence of P. ovata has been overlooked; therefore, the current dataset will provide more detailed insights into the role of various genes for understanding the plant’s life in a better manner. The plastome genomes have shown considerable applicability in phylogenetic studies and molecular and evolution systematics. During the recent years, various analyses have been conducted at deep nodes to answer phylogenetic questions based on the entire plastome genome and compared with multiple protein-coding genes79,80; this facilitates better understanding of complex evolutionary relationships among angiosperms81. Therefore, in this study, the phylogenetic position of P. ovata within Plantago and Lamiales was established by utilizing the complete plastome, 72 shared genes, matK gene, and rbcL gene among the members of 8 families representing 22 genera (Figs. 7 and S4). Phylogenetic analysis using BI and ML methods were performed. The results revealed that complete genome sequences (Fig. 7), 72 shared genes, matK gene, and rbcL gene (Fig. 4) from all the analysed species generated a phylogenetic tree with the same topology. In these phylogenetic trees (Figs. 7 and S4) constructed by employing ML, MP, NJ, and BI methods, P. ovata formed a single clade with P. maritima and P. media with high bootstrap (100%) and BI support. Moreover, the tree topology enabled inference of the relationship based on the phylogenetic studies conducted by Nina (2002)22 and Zhu (2015)25. The position of P. ovata within Lamiales confirms the previously published phylogeny described by Schäferhoff, et al.82. It was stated that Plantaginaceae is more closer to Gesneriaceae and Scrophulariaceae than Lamiaceae82. However, the chloroplast (cp) genome from Scrophulariaceae was not included owing to missing data in the NCBI database. Therefore, the present phylogeny revealed that Plantaginaceae is closer to Gesneriaceae and Phrymaceae on the basis of a complete plastome data set (Fig. 7). Similar results were observed on the basis of rbcL and matK genes (Figure S4), and were also consistent with the findings reported by Bastian et al.82. The results obtained here will help to recognise the evolutionary history of the Plantaginaceae. Furthermore, these results suggest that Plantaginaceae germplasm-related genetic resources are valuable and informative material for species identification, taxonomy elucidation, and phylogenetic inference of Plantaginaceae species. Moreover, phylogenetic inferences within Lamiales and Plantaginaceae could be improved if plastid genomes are made available, potentially providing dozens of valuable molecular markers for further research.
Conclusion
The current findings reveal detailed insights of complete plastome genome of P. ovata for the first time through sequencing on Illumina HiSeq-2000 platform. The structure and gene contents of P. ovata plastome was found in synergy with related species in Plantaginaceae. Contrarily, the genome size and IRs of P. ovata were different from P. media and P. maritima. Through detailed bioinformatic analysis and comparative assessments, we retrieved essential genetic features such as repetitive sequences, SSRs, codon usage, IR contraction and expansion, Ka/Ks ratio, sequence divergence, divergence time and phylogenomic placement. More interestingly, we noticed the IR regions were found significantly increased in size (up to 37.4 kb), indicating transfer of nine SSC genes to the IR regions. Whilst, an inversion was comparatively similar between P. ovata and P. media but higher in P. maritima. The present phylogeny revealed that Plantaginaceae is closer to Gesneriaceae and Phrymaceae based on complete plastome datasets. The divergence time estimates showed that P. ovata diverge from common ancestor P. media and P. maritima around 11.18 million years ago (Mya). Current plastome genomic dataset and the detailed analysis of P. ovata and related species and their comparative analysis provides a powerful genetic resource for the future molecular phylogeny, evolution, population genetics and biological functions of genus Plantago.
Materials and Methods
Plastome sequencing and assembly
The fresh leaves of P. ovata were collected from District Dir Pakistan, and the collected sampled were immediately placed in liquid nitrogen and subsequently stored at −80 °C until DNA extraction. Young leaves were used to extract plastome DNA by following the protocol described by Shi et al.83 with numerous modifications, as described by Al-Dous et al.84. The Illumina HiSeq-2000 platform (San Diego, CA, USA) at Macrogen (Seoul, Korea) was used to sequence the resultant DNA. A total of 24,100,324 raw reads were generated for P. ovata, and CLC Genomics Workbench v7.0 (CLC Bio, Aarhus, Denmark) was used to trim and filter reads for the de novo genome assembly. Trimmomatic 0.36 was used for filtering the reads and trailing and leading nucleotide with a Phred score of <20 or when the Phred score dropped below 20 on implementing a 4-bp sliding-window approach. Similarly, reads of <50 bp were discarded after quality filtering and adaptor trimming. The first assembly was formed using SPADES v3.9.0, with an additional switchover to SOAPdenovo v2.04.
The resulting contigs were compared against the chloroplast genomes of P. maritima and P. media using BLASTN with an E-value cut-off of 1e-5. The regions which were uncertain in these genomes, such as IR junction’s region, were selected from the already published genome mentioned above to adjust the sequence length using the iteration method and by employing the Geneious v11.1.2 software85. Primers were procured from Macrogen Inc., South Korea to execute PCR amplification and Sanger sequencing to fill the gaps in previously reported data29. After incorporating the results of Sanger sequencing, the entire plastome was used as reference, and the initial short reads were remapped to refine the assembly and to get maximum coverage. From all the available data, only high quality reads were mapped back by using Bowtie2 in Geneious 11.1.285.
Genome annotation
The software, Dual organellar Genome Annotator (DOGMA)86, was used for annotating the P. ovata plastome genome. BLASTX and BLASTN were utilised to determine ribosomal RNAs, transfer RNAs, and the positions of coding genes. The tRNA genes were annotated by employing tRNAscan-SE, version 1.21, software under default settings87. Boundaries of genes, coding regions, exons, and introns were confirmed by using BLAST versus reference sequences. Furthermore, for manual adjustment, the start and stop codons and intron boundaries were manually adjusted with the help of the reference genome using Geneious (v. 11.1.2)85. Furthermore, for structural description, OGDRAW was used88. MEGA 6 software89 was used to determine the relative synonymous codon usage by avoiding the effect of amino acid composition. Finally, the divergence of the new P. ovata plastome from related species of family Plantaginaceae was assessed using mVISTA90 in Shuffle-LAGAN mode and by employing the new P. ovata genome as reference.
Characterisation of repetitive sequences and SSRs
REPuter was used to determine the repetitive sequences (direct, reverse, and palindromic repeats within these plastomes91. For repeat identification via REPuter, the following settings were used: (1) a minimum repeat size of 30 bp, (2) ≥90% sequence identity, and (3) Hamming distance of 1. Tandem Repeats Finder version 4.07 b was used to find tandem repeats by using default settings92. Similarly, for finding SSRs, the search parameters were set to ≥3 repeat units for pentanucleotide and hexanucleotide repeats, ≥4 repeat units for trinucleotide and tetranucleotide repeats, ≥8 repeat units for dinucleotide repeats, and ≥10 repeat units for mononucleotide repeats by employing Phobos version 3.3.1293.
Sequence divergence, phylogenetic analyses and divergence time
Entire plastomes genomes and an isolated partition comprising 72 shared genes were used to examine the average pairwise sequence divergence for six species (P. media, P. maritima, V. nakaiana, V. presica, V. veronicastrum, and D. lanata) from the Plantaginaceae family. Ambiguous and missing gene annotations were checked by conducting comparative sequence analysis after assembling a multiple sequence alignment and comparing the gene order. These datasets were aligned using MAFFT v7.22294 under default parameters, and Kimura’s two-parameter (K2P) model was used to calculate pairwise sequence divergence95. Similarly, DnaSP v5.10.0196 was used to indemnify InDel polymorphisms among these plastomes, and for identifying single-nucleotide polymorphisms, a custom Python script (https://www.biostars.org/p/119214/) was employed. To resolve the phylogenetic position of P. ovata within the family Plantaginaceae and order Lamiales, 34 published plastome sequences of Lamiales species were downloaded from the NCBI database for phylogenetic analysis. First, on the basis of a conserved structure and gene order, a multiple alignment of the complete plastomes was created97, and the following four methods were applied to construct phylogenetic trees by employing the settings described previously: Bayesian inference (BI) was employed in MrBayes 3.1.298; maximum likelihood (ML) and neighbour-joining (NJ) were as implemented in MEGA 689; and maximum parsimony (MP) by using PAUP41,99. The best substitution model GTR + G was tested by jModelTest version v2.1.02100 according to the Akaike information criterion (AIC) for Bayesian posterior probabilities (PP) in BI analyses. The Markov Chain Monte Carlo (MCMC) method was run using four incrementally heated chains across 1,000,000 generations, starting from random trees and sampling 1 out of every 100 generations. To estimate the posterior probabilities, the values of first 30% of trees were discarded as burn-in. Maximum parsimony run was based on a heuristic search with 1000 random addition of sequence replicates with the tree-bisection-reconnection (TBR) branch-swapping tree search criterion. Similarly, the parameters for ML analysis were optimised using a BIONJ tree101 as the starting tree with 1000 bootstrap replicates by employing the Kimura 2-parameter model with invariant sites and gamma-distributed rate heterogeneity.
In the second, third, and fourth tiers of phylogenies, a set of 72 shared genes, matK gene, and rbcL gene from the plastome genomes of 34 Lamiales species were aligned using MAFFT version 7.22294 under default parameters and by making various manual adjustments to preserve and improve reading frames. The above two aforementioned phylogenetic inference models (ML and BI) were employed to construct trees using 72 concatenated genes, matK gene, and rbcL gene as mentioned above and suggested by Asaf et al.29.
To determine the divergence time of Plantago with those of other 34 species, we used the concatenated data matrix. Briefly general time reversible (GTR + G) substitution model was used with four rate categories, and a Yule tree speciation model with lognormal relaxed clock model in BEAST102 with a substitution rate prior. We used an average substitution rate of 3.0 × 10−9 substitutions per site per year (s/s/y) and a fossil-based method to calibrate the molecular divergence. To root the calibration time, we included five outgroups species as Dolichandra cyanachoides, Tanaecium tetragonolobum, Adenocalymma acutissimum, Adenocalymma biternatum and Adenocalymma marginatum from family Bignoniaceae. We selected these outgroups as all these species are closely related to our study model species and have fossil records older than genus Plantago103. The fossil records were employed through Log-normal distributed priors root set to the node ages of family Bignoniaceae 49.5 Mya (Offset = 45.0, Mean = 1.5 and SD = 0.5)104,105,106. The dating analyses involved 3 independent MCMC runs of 15 million generations. LOGCOMBINER was used to combine the tree files from all the three runs. Convergence and effective sample sizes were assessed in TRACER 1.5107. From each analysis we removed 25% of trees as burn-in. Lastly, the tree was calculated using TREEANNOTATOR and tree with 95% highest posterior density (HPD) was visualized in FIGTREE1.4.
References
Gray, M. W. The evolutionary origins of organelles. Trends in Genetics 5, 294–299 (1989).
Poczai, P. & Hyvönen, J. The complete chloroplast genome sequence of the CAM epiphyte Spanish moss (Tillandsia usneoides, Bromeliaceae) and its comparative analysis. PloS one 12, e0187199 (2017).
Palmer, J. D. Plastid chromosomes: structure and evolution. The molecular biology of plastids 7, 5–53 (1991).
Henry, R. J. Plant diversity and evolution: genotypic and phenotypic variation in higher plants. (Cabi Publishing, 2005).
Lee, H.-L., Jansen, R. K., Chumley, T. W. & Kim, K.-J. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Molecular Biology and Evolution 24, 1161–1180 (2007).
Graham, S. W. & Olmstead, R. G. Utility of 17 chloroplast genes for inferring the phylogeny of the basal angiosperms. American Journal of Botany 87, 1712–1730 (2000).
McNeal, J. R., Kuehl, J. V., Boore, J. L. & de Pamphilis, C. W. Complete plastid genome sequences suggest strong selection for retention of photosynthetic genes in the parasitic plant genus Cuscuta. BMC Plant Biology 7, 57 (2007).
Saski, C. et al. Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant molecular biology 59, 309–322 (2005).
Jheng, C.-F. et al. The comparative chloroplast genomic analysis of photosynthetic orchids and developing DNA markers to distinguish Phalaenopsis orchids. Plant science 190, 62–73 (2012).
Shaw, J. et al. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. American Journal of Botany 101, 1987–2004 (2014).
Jansen, R. K. et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proceedings of the National Academy of Sciences 104, 19369–19374 (2007).
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G. & Soltis, D. E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proceedings of the National Academy of Sciences 107, 4623–4628 (2010).
Fawcett, J. A. & Van de Peer, Y. Angiosperm polyploids and their road to evolutionary success. Trends in Evolutionary Biology 2, 3 (2010).
McKinnon, G. & Henry, R. Reticulate evolution in higher plants. Plant diversity and evolution: Genotypic and phenotypic variation in higher plants, 81–96 (2005).
Nock, C. J. et al. Chloroplast genome sequences from total DNA for plant identification. Plant biotechnology journal 9, 328–333 (2011).
Straub, S. C. et al. Navigating the tip of the genomic iceberg: Next‐generation sequencing for plant systematics. American Journal of Botany 99, 349–364 (2012).
Rahn, K. A phylogenetic study of the Plantaginaceae. Botanical Journal of the Linnean Society 120, 145–198 (1996).
Dhar, M., Kaul, S., Sharma, P. & Gupta, M. Plantago ovata: cultivation, genomics, chemistry and therapeutic applications. Genetic resources, chromosome engineering and crop improvement (2011).
Samantaray, S., Dhagat, U. M. & Maiti, S. Evaluation of genetic relationships in Plantago species using Random Amplified Polymorphic DNA (RAPD) markers. Plant biotechnology 27, 297–303 (2010).
Kour, B., Kotwal, S., Dhar, M. K. & Kaul, S. Genetic diversity analysis in Plantago ovata and some of its wild allies using RAPD markers. Russian agricultural sciences 42, 37–41 (2016).
Dhar, M., Kaul, S., Sareen, S. & Koul, A. Plantago ovata: genetic diversity, cultivation, utilization and chemistry. Plant Genetic Resources 3, 252–263 (2005).
RØNSTED, N., Chase, M. W., Albach, D. C. & Bello, M. A. Phylogenetic relationships within Plantago (Plantaginaceae): evidence from nuclear ribosomal ITS and plastid trnL-F sequence data. Botanical Journal of the Linnean Society 139, 323–338 (2002).
Dhar, M. K., Friebe, B., Kaul, S. & Gill, B. S. Characterization and physical mapping of ribosomal RNA gene families in Plantago. Annals of botany 97, 541–548 (2006).
Singh, N., Lal, R. & Shasany, A. Phenotypic and RAPD diversity among 80 germplasm accessions of the medicinal plant isabgol (Plantago ovata, Plantaginaceae). Genet. Mol. Res 8, 1273–1284 (2009).
Zhu, A., Guo, W., Gupta, S., Fan, W. & Mower, J. P. Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytologist 209, 1747–1756 (2016).
Asaf, S. et al. Comparative analysis of complete plastid genomes from wild soybean (Glycine soja) and nine other Glycine species. PloS one 12, e0182281 (2017).
Wang, R. J. et al. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol Biol 8, 36, https://doi.org/10.1186/1471-2148-8-36 (2008).
Choi, K. S., Chung, M. G. & Park, S. The complete chloroplast genome sequences of three Veroniceae species (Plantaginaceae): comparative analysis and highly divergent regions. Frontiers in plant science 7, 355 (2016).
Asaf, S. et al. Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: Structures and comparative analysis. Scientific reports 7, 7556 (2017).
Sugita, M. & Sugiura, M. Regulation of gene expression in chloroplasts of higher plants. Plant molecular biology 32, 315–326 (1996).
Asaf, S. et al. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Frontiers in plant science 8, 304 (2017).
Xu, J. et al. The first intron of rice EPSP synthase enhances expression of foreign gene. Science in China Series C: Life Sciences 46, 561 (2003).
Mascarenhas, D., Mettler, I. J., Pierce, D. A. & Lowe, H. W. Intron-mediated enhancement of heterologous gene expression in maize. Plant Mol Biol 15, 913–920 (1990).
Luehrsen, K. R. & Walbot, V. Intron enhancement of gene expression and the splicing efficiency of introns in maize cells. Molecular & general genetics: MGG 225, 81–93 (1991).
Akua, T., Berezin, I. & Shaul, O. The leader intron of AtMHX can elicit, in the absence of splicing, low-level intron-mediated enhancement that depends on the internal intron sequence. BMC Plant Biol 10, 93, https://doi.org/10.1186/1471-2229-10-93 (2010).
Mascarenhas, J. P. Gene activity during pollen development. Annual review of plant biology 41, 317–338 (1990).
Clark, A. J. et al. Enhancing the efficiency of transgene expression. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 339, 225–232 (1993).
Lin, C.-P., Huang, J.-P., Wu, C.-S., Hsu, C.-Y. & Chaw, S.-M. Comparative chloroplast genomics reveals the evolution of Pinaceae genera and subfamilies. Genome biology and evolution 2, 504–517 (2010).
Khan, A. L., Asaf, S., Lee, I.-J., Al-Harrasi, A. & Al-Rawahi, A. First chloroplast genomics study of Phoenix dactylifera (var. Naghal and Khanezi): A comparative analysis. PLOS ONE 13, e0200104, https://doi.org/10.1371/journal.pone.0200104 (2018).
Khan, A. L., Asaf, S., Lee, I.-J., Al-Harrasi, A. & Al-Rawahi, A. First reported chloroplast genome sequence of Punica granatum (cultivar Helow) from Jabal Al-Akhdar, Oman: phylogenetic comparative assortment with Lagerstroemia. Genetica, 1–14 (2018).
Asaf, S. et al. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Frontiers in plant science 7, 843 (2016).
Khakhlova, O. & Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. The Plant Journal 46, 85–94 (2006).
Chen, J. et al. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Frontiers in plant science 6, 447 (2015).
Menezes, A. P. A. et al. Chloroplast genomes of Byrsonima species (Malpighiaceae): comparative analysis and screening of high divergence sequences. Scientific Reports 8, 2210, https://doi.org/10.1038/s41598-018-20189-4 (2018).
Iwanycki Ahlstrand, N. et al. Ancestral range reconstruction of remote oceanic island species of Plantago (Plantaginaceae) reveals differing scales and modes of dispersal. Journal of biogeography 46, 706–722 (2019).
Xu, W. Q. et al. Comparative genomics of figworts (Scrophularia, Scrophulariaceae), with implications for the evolution of Scrophularia and Lamiales. Journal of systematics and evolution 57, 55–65 (2019).
Magee, A. M. et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Research (2010).
Kode, V., Mudd, E. A., Iamtham, S. & Day, A. The tobacco plastid accD gene is essential and is required for leaf development. The plant journal 44, 237–244 (2005).
Haberle, R. C., Fourcade, M. L., Boore, J. L. & Jansen, R. K. Complete chloroplast genome of Trachelium caeruleum: extensive rearrangements are associated with repeats and tRNAs (2008).
Goremykin, V. V., Holland, B., Hirsch-Ernst, K. I. & Hellwig, F. H. Analysis of Acorus calamus chloroplast genome and its phylogenetic implications. Molecular biology and evolution 22, 1813–1822 (2005).
Martínez-Alberola, F. et al. Balanced gene losses, duplications and intensive rearrangements led to an unusual regularly sized genome in Arbutus unedo chloroplasts. PLoS One 8, e79685 (2013).
Zhao, Y. et al. The complete chloroplast genome provides insight into the evolution and polymorphism of Panax ginseng. Frontiers in plant science 5, 696 (2015).
Qian, J. et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PloS one 8, e57607 (2013).
Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E. & Tabata, S. Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA research 6, 283–290 (1999).
Zhang, Y.-J., Ma, P.-F. & Li, D.-Z. High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PloS one 6, e20596 (2011).
Yi, X., Gao, L., Wang, B., Su, Y.-J. & Wang, T. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome biology and evolution 5, 688–698 (2013).
Powell, W., Morgante, M., McDevitt, R., Vendramin, G. & Rafalski, J. Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proceedings of the National Academy of Sciences 92, 7759–7763 (1995).
Pauwels, M. et al. Nuclear and chloroplast DNA phylogeography reveals vicariance among European populations of the model species for the study of metal tolerance, Arabidopsis halleri (Brassicaceae). New Phytologist 193, 916–928 (2012).
Edh, K., Widén, B. & Ceplitis, A. Nuclear and chloroplast microsatellites reveal extreme population differentiation and limited gene flow in the Aegean endemic Brassica cretica (Brassicaceae). Molecular Ecology 16, 4972–4983 (2007).
Gong, Y.-Q. & Gong, X. Pollen-mediated gene flow promotes low nuclear genetic differentiation among populations of Cycas debaoensis (Cycadaceae). Tree genetics & genomes 12, 93 (2016).
Roy, P. S. et al. Nuclear and chloroplast DNA variation provides insights into population structure and multiple origin of native aromatic rices of Odisha, India. PloS one 11, e0162268 (2016).
Nie, X. et al. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PloS one 7, e36869 (2012).
Cavalier-Smith, T. Chloroplast evolution: secondary symbiogenesis and multiple losses. Current Biology 12, R62–R64 (2002).
Tangphatsornruang, S. et al. The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: structural organization and phylogenetic relationships. DNA research 17, 11–22 (2009).
Timme, R. E., Kuehl, J. V., Boore, J. L. & Jansen, R. K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: identification of divergent regions and categorization of shared repeats. American Journal of Botany 94, 302–312 (2007).
Gao, L., Yi, X., Yang, Y.-X., Su, Y.-J. & Wang, T. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern chloroplast genomes. BMC evolutionary biology 9, 130 (2009).
Raubeson, L. A. et al. Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC genomics 8, 174 (2007).
Yao, X. et al. The first complete chloroplast genome sequences in Actinidiaceae: genome structure and comparative analysis. PloS one 10, e0129347 (2015).
Lin, C.-P., Wu, C.-S., Huang, Y.-Y. & Chaw, S.-M. The Complete Chloroplast Genome of Ginkgo biloba Reveals the Mechanism of Inverted Repeat Contraction. Genome Biology and Evolution 4, 374–381, https://doi.org/10.1093/gbe/evs021 (2012).
Goulding, S. E., Olmstead, R. G., Morden, C. W. & Wolfe, K. H. Ebb and flow of the chloroplast inverted repeat. Molecular & general genetics: MGG 252, 195–206 (1996).
Frailey, D. C., Chaluvadi, S. R., Vaughn, J. N., Coatney, C. G. & Bennetzen, J. L. Gene loss and genome rearrangement in the plastids of five Hemiparasites in the family Orobanchaceae. BMC Plant Biology 18, 30, https://doi.org/10.1186/s12870-018-1249-x (2018).
Chumley, T. W. et al. The complete chloroplast genome sequence of Pelargonium× hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Molecular biology and evolution 23, 2175–2190 (2006).
Wu, C.-S., Lai, Y.-T., Lin, C.-P., Wang, Y.-N. & Chaw, S.-M. Evolution of reduced and compact chloroplast genomes (cpDNAs) in gnetophytes: selection toward a lower-cost strategy. Molecular Phylogenetics and Evolution 52, 115–124 (2009).
Kumar, M. et al. Phenotypic and molecular characterization of selected species of ‘Plantago’ with emphasis on ‘Plantago ovata’. Australian Journal of Crop Science 8, 1639 (2014).
Saeed, S., Munir, F., Naveed, I., Raja, G. K. & Mahmood, T. Phylogenetics of selected Plantago species on the basis of rps14 chloroplast gene. Journal of Medicinal Plants Research 5, 4888–4891 (2011).
Tay, M. L. Evolution of Australasian Plantago (Plantaginaceae) (2008).
Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L. & Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Scientific reports 5, 13957 (2015).
Zhang, Y. et al. The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Frontiers in plant science 7, 306 (2016).
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G. & Soltis, D. E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proceedings of the National Academy of Sciences, 200907801 (2010).
Goremykin, V. V., Hirsch-Ernst, K. I., Wölfl, S. & Hellwig, F. H. The chloroplast genome of Nymphaea alba: whole-genome analyses and the problem of identifying the most basal angiosperm. Molecular Biology and Evolution 21, 1445–1454 (2004).
Hohmann, N. et al. Taming the wild: resolving the gene pools of non-model Arabidopsis lineages. BMC evolutionary biology 14, 224 (2014).
Schäferhoff, B. et al. Towards resolving Lamiales relationships: insights from rapidly evolving chloroplast sequences. BMC Evolutionary Biology 10, 352 (2010).
Shi, C. et al. An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. Plos one 7, e31468 (2012).
Al-Dous, E. K. et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nature biotechnology 29, 521 (2011).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Wyman, S. K., Jansen, R. K. & Boore, J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255 (2004).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic acids research 33, W686–W689 (2005).
Lohse, M., Drechsel, O. & Bock, R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 52, 267–274, https://doi.org/10.1007/s00294-007-0161-y (2007).
Kumar, S., Nei, M., Dudley, J. & Tamura, K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform 9, 299–306, https://doi.org/10.1093/bib/bbn017 (2008).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: computational tools for comparative genomics. Nucleic acids research 32, W273–W279 (2004).
Kurtz, S. et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic acids research 29, 4633–4642 (2001).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research 27, 573 (1999).
Mayer, C. Phobos Version 3.3. 12. A tandem repeat search program 20 (2010).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution 30, 772–780 (2013).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16, 111–120 (1980).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452, https://doi.org/10.1093/bioinformatics/btp187 (2009).
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Muller, K. F. & Quandt, D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol 76, 273–297, https://doi.org/10.1007/s11103-011-9762-4 (2011).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Wu, Z., Tembrock, L. R. & Ge, S. Are Differences in Genomic Data Sets due to True Biological Variants or Errors in Genome Assembly: An Example from Two Chloroplast Genomes. PLOS ONE 10, e0118019, https://doi.org/10.1371/journal.pone.0118019 (2015).
Posada, D. jModelTest: phylogenetic model averaging. Molecular biology and evolution 25, 1253–1256 (2008).
Gascuel, O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Molecular biology and evolution 14, 685–695 (1997).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evolution 4, vey016 (2018).
Lohmann, L. G., Bell, C. D., Calió, M. F. & Winkworth, R. C. Pattern and timing of biogeographical history in the Neotropical tribe Bignonieae (Bignoniaceae). Botanical Journal of the Linnean Society 171, 154–170 (2013).
Pigg, K. & Wehr, W. Tertiary flowers, fruits, and seeds of Washington State and adjacent areas-Part III. Washington Geology 30, 3–16 (2002).
Perret, M., Chautems, A., De Araujo, A. O. & Salamin, N. Temporal and spatial origin of Gesneriaceae in the New World inferred from plastid DNA sequences. Botanical Journal of the Linnean Society 171, 61–79 (2013).
Wesley, C. W. & Hopkins, D. Q. The Eocene orchards and gardens of Republic, Washington. Washington Geology 22, 27–34 (1994).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic biology 67, 901–904 (2018).
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2017R1D1A1B04035601) and The Research Council Oman through Research Grant (BFP/RGP/EBR/18/005).
Author information
Authors and Affiliations
Contributions
S.A., A.L.K. and A.K. (Adil Khan) planned, designed, and performed the research and documented the findings; Lubna., A.K. (Arif Khan). and I.J.L. edited the manuscript and arranged the experimental resources; A.H. and G.K. analysed the data and edited the language of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Asaf, S., Khan, A.L., Lubna et al. Expanded inverted repeat region with large scale inversion in the first complete plastid genome sequence of Plantago ovata. Sci Rep 10, 3881 (2020). https://doi.org/10.1038/s41598-020-60803-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60803-y
- Springer Nature Limited
This article is cited by
-
The complete plastome sequences of invasive weed Parthenium hysterophorus: genome organization, evolutionary significance, structural features, and comparative analysis
Scientific Reports (2024)
-
Insight into chloroplast genome structural variation of the Mongolian endemic species Adonis mongolica (Ranunculaceae) in the Adonideae tribe
Scientific Reports (2023)
-
Molecular marker studies on Balanites aegyptiaca and allied species for species delimitation, identification, and authentication
Tree Genetics & Genomes (2023)
-
Newly reported chloroplast genome of Sinosenecio albonervius Y. Liu & Q. E. Yang and comparative analyses with other Sinosenecio species
BMC Genomics (2022)
-
Uncovering the first complete plastome genomics, comparative analyses, and phylogenetic dispositions of endemic medicinal plant Ziziphus hajarensis (Rhamnaceae)
BMC Genomics (2022)