
Abstract
Cytological and molecular studies have revealed that inversion chromosomal polymorphism is widespread across taxa and that inversions are among the most common structural changes fixed between species. Two major mechanisms have been proposed for the origin of inversions considering that breaks occur at either repetitive or non-homologous sequences. While inversions originating through the first mechanism might have a multiple origin, those originating through the latter mechanism would have a unique origin. Variation at regions flanking inversion breakpoints can be informative on the origin and history of inversions given the reduced recombination in heterokaryotypes. Here, we have analyzed nucleotide variation at a fragment flanking the most centromere-proximal shared breakpoint of several sequential overlapping inversions of the E chromosome of Drosophila subobscura —inversions E1, E2, E9 and E3. The molecular genealogy inferred from variation at this shared fragment does not exhibit the branching pattern expected according to the sequential origin of inversions. The detected discordance between the molecular and cytological genealogies has led us to consider a novel possibility for the origin of an inversion, and more specifically that one of these inversions originated on a heterokaryotype for chromosomal arrangements. Based on this premise, we propose three new models for inversions origin.
Similar content being viewed by others


Introduction
Chromosomal inversions are structural rearrangements with chromosomal segments in inverted orientation relative to non-inverted chromosomes. Their presence was first inferred in Drosophila almost a century ago from their genetic effect as suppressors of recombination when in heterozygosis1. They could be later observed at the cytological level in the larval salivary glands of Drosophila melanogaster as their cells present giant (polytene) chromosomes that exhibit somatic pairing2. Cytological methods propelled the study of inversion chromosomal polymorphism in natural populations of several Diptera species across the twentieth century3,4,5. The later molecular characterization of polymorphic inversions breakpoints allowed the identification of inversions by specific PCR amplification of their breakpoints6,7,8. Moreover, bioinformatics methods have been developed to identify inversions by comparing genomes of the same species (e. g.9) as well as of different species (e. g.10,11). The different methodological approaches have uncovered the pervasive existence of inversion chromosomal polymorphism across taxa (i.e., from bacteria to humans3,12,13,14,15,16,17,18). Moreover, comparisons between closely related species have revealed that at this level, inversions are the most common structural changes10,19,20,21 and that they play an important role in speciation22.
The extensive cytological studies of inversion polymorphism in Drosophila pseudoobscura and D. persimilis uncovered a series of chromosomal arrangements that differed by overlapping inversions, with similar studies in D. subobscura and other species also revealing such series in various chromosomes (as reviewed in3). These observations led to the establishment of chromosomal phylogenies under the generally accepted assumption that each inversion had a unique origin. However, the discovery of transposable elements raised the possibility that an inversion could originate repeatedly23. Inversions can originate through ectopic recombination between two transposable elements, or other repetitive sequences, present in opposite orientation on the same chromosome. Moreover, inversions can also result from erroneously joining the ends of two breaks on the same chromosome, simply through a cut-and-paste procedure but also from staggered breaks and their subsequent repair10,24. These mechanisms are often referred to, respectively, as NAHR —for non-allelic homologous recombination— and NHEJ —for non-homologous end joining. While a certain inversion resulting from NAHR could occur repeatedly23,25, the occurrence of any inversion resulting from NHEJ should be a unique event in the history of the species10,24. The molecular characterization of polymorphic inversions breakpoints in both D. melanogaster6,9,26 and D. subobscura27,28,29,30,31,32 has revealed that in these species the latter mechanism is prevalent, and therefore that most of their inversions have a unique origin. In contrast, the recent characterization of polymorphic inversions breakpoints in human populations has revealed that many of them are comparatively rather short and originated repeatedly by ectopic recombination between repetitive sequences33.
For inversions generated by the NHEJ mechanism, the extreme bottleneck due to their unique origin implies that they are originally devoid of variation. Variation in the inverted region accrues through time as a result of new mutations. An inversion can also acquire variation through recombination —by either double crossovers or gene conversion— with non-inverted chromosomes. The size of the inversion affects this exchange of variation as recombination is highly suppressed near the breakpoint regions of the inverted fragment and it increases with distance to the breakpoints34. Variation at regions closer to the breakpoints or flanking the breakpoints themselves might therefore better reflect the history of large inversions like those detected in different Drosophila species.
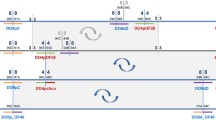
D. subobscura exhibits a rich inversion polymorphism that affects its five acrocentric chromosomes. Each of these chromosomes presents overlapping inversions that occur sequentially leading in some cases to chromosomal complexes formed by three or more extant chromosomal arrangements. The E chromosome (or Muller C element) stands out in this sense because it harbors a complex formed by the successive accumulation of inversions on the ancestral chromosome, hereafter referred as Est. Even if the E1+2+9 complex consists in eight different chromosomal arrangements35 formed through nine different inversions —E1, E2, E9, E3, E4, E5, E12, E15 and E18— only a subset of them coexist in any particular population. Here we have focused on the subset of these inversions—E1, E2, E9, E3 and E12— generating the most common derived chromosomal arrangements in the Mediterranean area —E1+2, E1+2+9, E1+2+9+3 and E1+2+9+12 (Fig. 1). At the cytological level, four of those inversions —E1, E2, E9 and E3— were considered to share their most centromere-proximal breakpoint36. Our molecular characterization of their breakpoints revealed that the proximal breakpoint regions of these inversions —i.e., regions AB, AG, AK, and AH2 in Fig. 1— share their A part28,32. It should be noted that the ~9-kb long fragment of the A part immediately flanking the breakpoint was duplicated when inversion E9 originated32. This duplicated fragment is therefore present in the here named GAL region of the E1+2+9 arrangement and its derivatives E1+2+9+3 and E1+2+9+12 (Figs 1 and 2). The A part is hereafter subdivided into two sections according to the extent of the duplication: Ap and Ad for the centromere-proximal and centromere-distal sections, respectively —corresponding the Ad section to the duplicated stretch. It should be, moreover, noted that arrangement E1+2+9+12 also shares region AK with arrangement E1+2+9, as the E12 inversion did not affect the E9 inversion breakpoints (Fig. 1).

Schematic representation of chromosomal arrangements Est, E1+2, E1+2+9, E1+2+9+3, and E1+2+9+12 of Drosophila subobscura. Inversions originating these arrangements are indicated by crossed lines. Breakpoint regions including the A fragment that is shared by various inversions are highlighted. The cytological location of these breakpoints on the Kunze-Mühl and Müller36 map is given. Ap and Ad refer, respectively, to the proximal and distal section of the A fragment (see text). The Ad section was duplicated when inversion E9 originated. Not at scale.

Functional annotation of breakpoint regions spanning fragment A. Annotation extracted from our previous work28,32. Breakpoint regions are named as in Fig. 1 except for the GAL region that is here presented in reverse orientation (and therefore named LAG) to facilitate its comparison with the other regions. Chromosomal arrangements presenting each breakpoint region are indicated on the rightmost part of the figure. Ap and Ad refer, respectively, to the proximal and distal sections of the A part (see text). Small black arrowheads indicate the location of amplification primers. The sequenced A fragment at each breakpoint is rose shadowed. Bp refers to the proximal fragment of the ancestral B part of the Est arrangement with the arrow indicating its orientation relative to the breakpoint. Colored arrowed boxes represent protein-coding regions. Green-striped boxes represent the presence of multiple snoRNA genes. SGM indicates an SGM element. α indicates an alpha element exhibiting some similarity to the SGM element.
In the present work, we have estimated the level of nucleotide variation in the ~2-kb long segment of the A part closest to the breakpoint shared by inversions E1, E2, E9 and E3 (hereafter named fragment A; Fig. 2) as variation in fragment A of chromosomal arrangements Est, E1+2, E1+2+9, E1+2+9+3 and E1+2+9+12 can be informative on the origin and history of these inversions.
Results and Discussion
Nucleotide variation at a fragment flanking multiple inversion breakpoints
Twenty-nine heterokaryotypic individuals from a wild population sampled on the outskirts of Barcelona in 20148 were used to separately obtain the nucleotide sequence of the A fragment from each homologous E chromosome whenever possible. Supplementary Table S1 shows these individuals karyotypes and also the breakpoint regions including fragment A —AB, AG, AK, AH2 and GAL (Fig. 1)— that were PCR amplified in each individual. Figure 2 presents the functional annotation of these breakpoint regions28,32 as well as the size of the amplified fragments spanning fragment A.
The 29 sampled individuals are expected to harbor 80 A fragments given the presence of the GAL region in E1+2+9, E1+2+9+3 and E1+2+9+12 chromosomes. Only 50 of these fragments were successfully amplified, sequenced and analyzed. Concerning amplification, the AG region —exclusive of the E1+2 arrangement— could only be independently amplified in Est/E1+2 heterokaryotypic individuals given that all other heterokaryotypes exhibit two copies corresponding to the AG and GAL regions (Supplementary Table S1). Moreover, some difficulties were encountered to completely sequence the A fragment of some amplified regions due to the presence of long thymidine (T) runs in the AG and GAL regions and the presence of inserted SGM (Subobscura Guanche Madeirensis) transposable elements37 in the AK region of various individuals. These characteristics and the presence of a series of small and similar snoRNA genes in fragment A increased the difficulty to perform multiple alignments of this fragment sequences, which required some manual curation. Supplementary Table S2 shows the nucleotide polymorphisms detected in the 50 sequenced A fragments.
Regions immediately flanking an inversion breakpoint are good markers of the inversion history as their variation in inverted chromosomes is mainly due to new mutations. Indeed, only gene conversion would contribute to the acquisition of variation from non-inverted chromosomes as double-crossover events are negligible in these regions. Variation at the A fragment of breakpoint regions AB, AG and AH2 will thus be considered to reflect variation at chromosomal arrangements Est, E1+2 and E1+2+9+3, respectively, whereas that of region AK would reflect variation at arrangements E1+2+9 and E1+2+9+12, and that of region GAL variation at arrangements E1+2+9, E1+2+9+3 and E1+2+9+12 (Figs 1 and 2). As the AK and GAL regions are present in both E1+2+9 and E1+2+9+12 chromosomal arrangements and even though the E12 inversion did not affect the E9 inversion breakpoints (Fig. 1), we tested for any putative differentiation of the AK and GAL sequenced regions between these chromosomal arrangements prior to analyzing variation in this fragment. The estimated FST value38 for each the AK and GAL regions —0.066 and 0.031, respectively— did not significantly differ from 0 as revealed by the corresponding permutation test (P = 0.18 and P = 0.31, respectively). Sequences from both arrangements (E1+2+9 and E1+2+9+12) were therefore grouped for subsequent analyses (hereafter referred to as E[1+2+9]).
Table 1 summarizes the analysis of nucleotide polymorphism and divergence —using the complete deletion option— at fragment A from each of the five different breakpoint regions considered (AB, AG, AK, GAL and AH2) and also when jointly considered. Nucleotide diversity at the A fragment varied between the different breakpoint regions, ranging from 0.006 at the AH2 region to 0.015 at the AB region. Similar values were obtained within arrangement when considering the pairwise deletion option (results not shown). The level of nucleotide diversity at fragment A in the different E chromosomal arrangements is of the same order than previously estimated in D. subobscura at regions affected by other autosomal and X-linked inversions39,40,41,42,43.
Variation at fragment A within the different chromosomal arrangements (i.e., at the AB, AG, AK and AH2 breakpoint regions) did not consistently increase with their relative age as inferred from the sequential occurrence of inversions E1, E2, E9 and E3 (Table 1 and Fig. 1). However, age is not the only aspect that can affect the level of variation at fragment A from the different arrangements. Indeed, its variation could also have been affected by i) the frequency attained by each arrangement, and ii) the putative recent fixation of an adaptive point or structural mutation in any of the arrangements.
Genetic differentiation between the A fragments of the different breakpoint regions
Table 2 summarizes the level of genetic differentiation at fragment A between the different breakpoint regions (AB, AG, AK, AH2 and GAL; Fig. 1) as measured by the FST statistic38. As expected from the relatively recent origin of the E1+2+9+3 chromosomal arrangement through the E3 inversion on an E1+2+9 chromosome (Fig. 1), the lowest FST estimate for the A fragment is that between the AK and AH2 breakpoint regions. However, in contrast to expectations from the sequential occurrence of inversion E9 on an E1+2 chromosome and of inversion E3 on an E1+2+9 chromosome (Fig. 1), the FST estimates for the A fragment were much lower between the AB breakpoint region and both the AK and AH2 breakpoint regions than between the AG breakpoint region and both the AK and AH2 breakpoint regions. This discordant result is clearly reflected in the genealogy inferred from variation at the A fragment, which is based on the 50 A fragment sequences of D. subobscura using the D. guanche sequence as outgroup (Fig. 3). Sequences from each particular breakpoint region of D. subobscura cluster together into differentiated clades. As expected from the cytological phylogeny44, sequences from regions AK and AH2 that correspond to the youngest arrangements —E[1+2+9] and E1+2+9+3— cluster together. Surprisingly, these sequences group together with the AB sequences that correspond to the oldest arrangement Est and not with the AG sequences that correspond to the E1+2 arrangement from which the E[1+2+9] and E1+2+9+3 arrangements are considered to be derived. In contrast, sequences from the GAL region corresponding to the E1+2+9 arrangement and its derivatives cluster together with the AG sequences, as expected from the sequential occurrence of inversions (Fig. 1).

Neighbor-joining tree of the A fragment sequences corresponding to the breakpoint regions of different E chromosomal arrangements. Bootstrap values >70% (based on 1000 replicates) are shown on the tree. Positions with over 5% alignment gaps, missing data, or ambiguous bases were not considered. D. guanche was used as outgroup.
The discordance between the molecular genealogy inferred from variation at the A fragment that flanks the proximal breakpoint of four sequentially originated inversions and their cytology-based phylogeny led us to check two possible sources of this being an artifactual result: i) sequence misalignment, and ii) putative bias in the sequenced sample. Concerning the first possible source, we checked again the multiple alignment of fragment A. No progress was made in this sense as the alignment had already been manually curated. Concerning the putative biased sampling of sequences from the natural population that might be associated to considering only heterokaryotypic individuals, comparison of the frequencies of the five chromosomal arrangements in the 29 sequenced heterokaryotypic individuals and in the complete sample from 20148 revealed no significant difference (G test = 5.195; d. f. = 4; P = 0.214). Moreover, the genealogy inferred from the A fragment sequences obtained from the homokaryotypic lines used to identify and characterize the different inversions breakpoint regions28,32 exhibits the same branching pattern than that inferred from the heterokaryotypic individuals (Supplementary Fig. S1).
Inversion E9 originated in an inversion heterokaryotype
The overlapping character of inversions E1, E2, E9 and E3 implies their sequential occurrence, which is reflected in their cytology-based phylogeny (Fig. 1). The molecular genealogy inferred from variation at the shared A fragment of regions AB, AG, AK and AH2 would be expected to exhibit the same branching pattern than the cytology-based phylogeny given that this fragment immediately flanks the most centromere-proximal breakpoint of the corresponding inversions. Nevertheless, the molecular genealogy does not conform to afore mentioned expectations. The detected discordance —i.e., the clustering of arrangements E[1+2+9] and E1+2+9+3 with Est instead of with E1+2, as inferred from the A fragment sequences (Fig. 3 and Supplementary Fig. S1)— would place the focus on the origin of inversion E9.
A clue to understand the detected discordance between the molecular genealogy and the cytology-based phylogeny stems from the comparison of the extended AB, AG, AK and AH2 sequences (Fig. 2) from the homokaryotypic lines28,32. This comparison revealed that the AK and AH2 sequences share an ~500-nt long fragment adjacent to the distal end of section Ad (hereafter named fragment Bp; Fig. 2). In E1+2 chromosomes, fragment Bp is absent from their AG region and present in the B part of their BF region (Fig. 1). Moreover, fragment Bp is present in the B part of the AB region of Est chromosomes even if at a different position (Fig. 2). In order to ascertain whether fragment Bp was a repetitive element, it was used as query for both a RepeatMasker (http:// www.repeatmasker.org/) search, and a BLAST search against the D. guanche genome45. The negative result of the first search and the reduced number of very partial hits returned by the second search would not yield any support for it being a transposable element or any other repetitive sequence that could have been replaced since the E9 inversion originated. These observations clearly indicate that inversion E9 would have captured its Bp fragment from an Est chromosome (Fig. 2) when originating. Taking into account that the B part of Est chromosomes (Fig. 1) suffered several structural changes prior and after the E1+2 arrangement originated28, it can be inferred that the Bp fragment was ancestrally at a proximal position relative to the A fragment of Est. These results and the detected discordance between the molecular genealogy and cytology-based phylogeny of the studied arrangements have led us to consider that inversion E9 occurred in an individual heterokaryotypic for arrangements Est and E1+2. The newly formed E1+2+9 chromosome could have, thus, acquired some features of the Est A fragment during the E9 inversion process.
New models to explain the origin of inversion E9
The presence in inverted orientation of the ~9-kb long fragment named Ad at both inversion E9 breakpoints had been considered a clear signal that this fragment was duplicated when inversion E9 originated32. Two previous NHEJ models had been proposed to explain the presence at both breakpoints of inverted chromosomes of a duplicated fragment relative to the single copy present in only one of the breakpoints of non-inverted chromosomes10. These models are: i) the isochromatid model that considers two staggered breaks in a single chromatid occurring during premeiotic mitosis and ii) the chromatid model that considers two breaks in each of two sister chromatids, occurring during meiotic prophase. Neither of these previously proposed models can account for the detected discordance between the molecular genealogy and cytology-based phylogeny of the studied arrangements because they are both chromatid models. Here, we propose three new chromosome models that would explain the detected discordance under the assumption that inversion E9 originated in an individual heterokaryotypic for arrangements Est and E1+2.
The first model proposed —named NHEJ-4-chromosome model— considers that inversion E9 originated through the NHEJ mechanism and resulted from four breaks occurring on both homologous chromosomes of a heterokaryotypic individual (Fig. 4). According to this model, both homologous chromosomes —Est and E1+2— would have been simultaneously broken at two different sites (staggered break) in the proximal region and at the same site in the distal region. The proximal break on the Est arrangement would have occurred past the Bp fragment and that on the E1+2 arrangement at the limit between the Ap and Ad sections. The repair of these chromosomal breaks would have been resolved by the NHEJ mechanism so that the excised central part of the E1+2 chromosome would have been rejoined in inverted orientation to the external Est fragments, giving rise to inversion E9. The rest of chromosomal fragments could have been joined in different ways or even not have been joined at all, with their putative product/s not having survived to present.

Schematic representation of the NHEJ-4-chromosome model for the origin of inversion E9. The sequential steps of how arrangement E1+2+9 could have originated from an Est/E1+2 heterokaryotypic individual through inversion E9 are graphically represented in the central part of the figure. Fragments flanking the different breakpoint regions are labeled as in Fig. 1. Initial state: pairing of the E homologous chromosomes of an Est/E1+2 heterokaryotypic individual with discontinuous arrows indicating the location of future breaks. Parts flanking future breaks are labeled in Est and E1+2 homologues by ordinal numbers and roman numerals, respectively. Upper left corner inset, image of an Est/E1+2 polytene chromosome preparation. First step: a total of four breaks considering both homologous chromosomes, with the two breaks in the proximal region occurring at different sites in both homologues —between sections Bp and Bd of the Est homologue and between sections Ap and Ad of the E1+2 homologue—, and those in the distal region (KL) occurring at the same site. Discontinuous lines indicate the location of breaks. Second step: inversion of the central fragment of the E1+2 homologue and resolution of the double-strand breaks. Insets on both sides of the central scheme highlight the resolution phase. Final state: result of the inversion process with the generation of the E1+2+9 arrangement. Also shown within a grey-shaded box are the chromosomal fragments that might have resulted —highlighted by a question mark,?— in an evolutionary unsuccessful arrangement.
The second model proposed —named NHEJ-3-chromosome model— also considers that inversion E9 originated through the NHEJ mechanism, but that it resulted from only three breaks (Fig. 5). According to this model, a staggered break similar to that of the NHEJ-4-chromosome model would have occurred at the proximal region. In contrast, only the E1+2 chromosome would have suffered an additional break at the distal region. The repair of these chromosomal breaks would have been resolved by the NHEJ mechanism so that the excised central part of the E1+2 chromosome would have been rejoined in inverted orientation to the proximal Est and distal E1+2 fragments, respectively, giving rise to inversion E9. This model is similar to that proposed by Sharakhov et al.24 as it also implies three breaks on both homologous chromosomes but it differs from that model in two fundamental aspects: i) the breaks would have been repaired by the NHEJ and not by the NAHR mechanism, and ii) the inversion would have originated in a heterokaryotypic individual.

Schematic representation of the NHEJ-3-chromosome model for the origin of inversion E9. The sequential steps of how arrangement E1+2+9 could have originated from an Est/E1+2 heterokaryotypic individual through inversion E9 are graphically represented in the central part of the figure. Fragments flanking the different breakpoint regions are labeled as in Fig. 1. Initial state: pairing of the E homologous chromosomes of an Est/E1+2 heterokaryotypic individual with discontinuous arrows indicating the location of future breaks. Parts flanking breakpoints are labeled as in Fig. 4. First step: a total of three breaks considering both homologous chromosomes, with the two breaks in the proximal region occurring at different sites in both homologues —between sections Bp and Bd of the Est homologue and between sections Ap and Ad of the E1+2 homologue—, and that in the distal region (KL) occurring at the E1+2 homologue. Discontinuous lines indicate the location of breaks. Second step: inversion of the central fragment of the E1+2 homologue and resolution of the double-strand breaks. Insets on both sides of the central scheme highlight the resolution phase. Final state: result of the inversion process with the generation of the E1+2+9 arrangement. Also shown within a grey-shaded box are the chromosomal fragments that might have resulted —highlighted by a question mark,?— in an Est chromosome lacking sections Ad and Bp.
The third model proposed —named BIR-NHEJ-chromosome model— considers that inversion E9 originated through two breaks on a single sister chromatid of the E1+2 homologous chromosome of an Est/E1+2 heterokaryotypic individual (Fig. 6). The proximal break would have been repaired through the Break-Induced Replication (BIR) pathway (i.e., through the resection and subsequent invasion and copying of the Est homologous chromosome46). According to this model, the proximal break would have also occurred at the limit between the Ap and Ad sections. Upon inversion of the central fragment of the E1+2 chromosome, both breaks would have been repaired and generated inversion E9. Repair of the Ap section would have, however, taken place through the BIR pathway and using the Est chromosome as template. Repair of the proximal break would have thus resulted in a copy of the Ad section of the Est homologue, which would explain the similarity observed between the Ad fragments present in the AB, AK and AH2 regions. In contrast, the distal break would have been repaired through the NHEJ pathway. In this case, the presence of the E1+2 Ad section in the distal break would explain the similarity observed between the Ad section present in the AG and GAL regions.

Schematic representation of the BIR-NHEJ-chromosome model for the origin of inversion E9. The sequential steps of how arrangement E1+2+9 could have originated from an Est/E1+2 heterokaryotypic individual through inversion E9 are graphically represented in the central part of the figure. Fragments flanking the different breakpoint regions are labeled as in Fig. 1. Initial state: pairing of the E homologous chromosomes of an Est/E1+2 heterokaryotypic individual with discontinuous arrows indicating the location of future breaks. Parts flanking breakpoints are labeled as in Fig. 4. First step: two breaks in the E1+2 homologue, with that in the proximal region occurring between sections Ap and Ad, and that in the distal region occurring between the K and L parts of the KL breakpoint region. Discontinuous lines indicate the location of breaks. Second step: inversion of the central fragment of the E1+2 homologue and resolution of the double-strand break of the proximal region through the BIR pathway and that of the distal region through the NHEJ mechanism. Insets on both sides of the central scheme highlight the different steps of the BIR and NHEJ pathways, respectively. Final state: result of the inversion process with the generation of the E1+2+9 arrangement. Also shown is the Est chromosome that did not undergo any break.
The three models proposed to explain the origin of inversion E9 from an Est/E1+2 heterokaryotype differ in the number and location of double-strand breaks at the inversion breakpoints as well as in the pathway/s used to repair these breaks. Based on the number of breaks, the BIR-NHEJ model would seem the most likely as it only involves two double-strand breaks in a single chromosome whereas the NHEJ-4 model would seem the least likely as it does not only require the highest number of double-strand breaks affecting both homologous chromosomes but also the distal break to have occurred between the same two nucleotides in both homologous chromosomes. Nevertheless, as models also differ in the repair pathways involved, further discrimination among models should await a better characterization of these pathways in the Drosophila genus.
In summary, our study revealed that the molecular genealogy inferred from variation at the A fragment differed from the cytology-based phylogeny of inversions E1, E2, E9 and E3 by the clustering of chromosomal arrangements E[1+2+9] and E1+2+9+3 with Est instead of with E1+2 as expected. To explain this discrepancy, we propose that inversion E9 originated in an Est/E1+2 heterokaryotypic individual, and develop three alternative models for the origin of E9 in such a heterokaryotype. This is, to our knowledge, the first documented case where the two homologous chromosomes of a heterokaryotypic individual are required to explain the origin of an inversion. Even though this situation may apply to other inversions, it should be noted that it is the characteristics of the complex here studied —i.e., a system with multiple arrangements resulting from the sequential accumulation of overlapping inversions that share a breakpoint at the molecular level— that have permitted its detection.
Materials and Methods
We used 29 individuals of D. subobscura sampled from a wild population at Observatori Fabra (Barcelona, Catalonia, Spain). These individuals had been previously identified as heterokaryotypic for any pair of the five E chromosome arrangements considered in the present work8. Their heterokaryotypic status allowed in many cases the independent PCR amplification of the A fragment of each of its two homologous chromosomes (Supplementary Table S1).
Regions spanning the breakpoints were PCR amplified using TaKaRa DNA polymerase (Takara Bio Inc) and newly designed oligonucleotide pairs (Supplementary Table S3). For each amplified region (ranging from 5.4 to 6.6 kb), an ~2-kb long stretch that spans fragment A was sequenced (Fig. 2). Sequence reactions were performed with the ABI PRISM version 3.2 cycle sequencing kit and the sequencing products separated on an ABI PRISM 3730 sequencer. All sequences were obtained on both strands and assembled using the DNASTAR package47. Sequences newly obtained have been deposited in the European Nucleotide Archive (ENA) under project number PRJEB33551. The A fragment sequence of D. guanche was retrieved from its complete genome sequence45 (https://denovo.cnag.cat/genomes/dgua/).
The MUSCLE program in the MEGA7 package48 was used for sequence alignment. Genetic differentiation between chromosomal arrangements was measured using the FST statistic38 and its statistical significance established using the mstatspop beta version (https://bioinformatics.cragenomica.es/numgenomics/people/sebas/software/software.html) with a total of 10000 random permutations. Summary statistics for nucleotide polymorphism and divergence were obtained using the DnaSP v6 program49. MEGA7 was also used to infer the Neighbor-Joining trees using the partial deletion option, in which nucleotide positions with less than 95% site coverage were eliminated before computing the corresponding evolutionary distances using the Jukes and Cantor correction50.
Data availability
The D. subobscura sequences newly obtained have been deposited in the European Nucleotide Archive (ENA) under project number PRJEB33551.
References
Sturtevant, A. H. A Case of rearrangement of genes in Drosophila. Proc. Natl. Acad. Sci. USA 7, 235–237 (1921).
Painter, T. S. A new method for the study of chromosome rearrangements and the plotting of chromosome maps. Science 78, 585–586 (1933).
Krimbas, C. B. & Powell, J. R. Drosophila inversion polymorphism. (CRC Press, 1992).
Coluzzi, M., Petrarca, V. & Di Deco, M. A. Chromosomal inversion intergradation and incipient speciation in Anopheles gambiae. Bolletino di Zool. 52, 45–63 (1985).
Gupta, J. P. & Kumar, A. Cytogenetics of Zaprionus indianus Gupta (Diptera: Drosophilidae): Nucleolar organizer regions, mitotic and polytene chromosomes and inversion polymorphism. Genetica 74, 19–25 (1987).
Andolfatto, P., Wall, J. D. & Kreitman, M. Unusual haplotype structure at the proximal breakpoint of In(2L)t in a natural population of Drosophila melanogaster. Genetics 153, 1297–1311 (1999).
Lobo, N. F. et al. Breakpoint structure of the Anopheles gambiae 2Rb chromosomal inversion. Malar. J. 9, 1–9 (2010).
Orengo, D. J., Puerma, E., Cereijo, U., Salguero, D. & Aguadé, M. An easy route to the massive karyotyping of complex chromosomal arrangements in Drosophila. Sci. Rep. 7, 12717 (2017).
Corbett-Detig, R. B., Cardeno, C. & Langley, C. H. Sequence-based detection and breakpoint assembly of polymorphic inversions. Genetics 192, 131–137 (2012).
Ranz, J. M. et al. Principles of genome evolution in the Drosophila melanogaster species group. PLoS Biol. 5, e152, https://doi.org/10.1371/journal.pbio.0050152 (2007).
von Grotthuss, M., Ashburner, M. & Ranz, J. M. Fragile regions and not functional constraints predominate in shaping gene organization in the genus Drosophila. Genome Res. 20, 1084–1096 (2010).
Hughes, D. Evaluating genome dynamics: the constraints on rearrangements within bacterial genomes. Genome Biol. 1, 1–8 (2000).
Stefansson, H. et al. A common inversion under selection in Europeans. Nat. Genet. 37, 129–137 (2005).
Lowry, D. B. & Willis, J. H. A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biol., 8 (2010).
Salm, M. P. A. et al. The origin, global distribution, and functional impact of the human 8p23 inversion polymorphism. Genome Res. 22, 1144–1153 (2012).
Martínez-Fundichely, A. et al. InvFEST, a database integrating information of polymorphic inversions in the human genome. Nucleic Acids Res. 42, 1027–1032 (2014).
Nagle, D. L., Kozak, C. A., Mano, H., Chapman, V. M. & Bućan, M. Physical mapping of the Tec and Gabrb1 loci reveals that the W sh mutation on mouse chromosome 5 is associated with an inversion. Hum. Mol. Genet. 4, 2073–2079 (1995).
Davis, K. M., Smith, S. A. & Greenbaum, I. F. Evolutionary implications of chromosomal polymorphisms in Peromyscus boyliifrom southwestern Mexico. Evolution (N. Y). 40, 645–649 (1985).
Sharakhov, I. V. et al. Inversions and gene order shuffling in Anopheles gambiae and A. funestus. Science 298, 182–185 (2002).
Ayala, F. J. & Coluzzi, M. Chromosome speciation: Humans, Drosophila, and mosquitoes. Proc. Natl. Acad. Sci. 102, 6535–6542 (2005).
Romanenko, S. A. et al. Multiple intrasyntenic rearrangements and rapid speciation in voles. Sci. Rep. 8, 1–9 (2018).
Fuller, Z. L., Leonard, C. J., Young, R. E., Schaeffer, S. W. & Phadnis, N. Ancestral polymorphisms explain the role of chromosomal inversions in speciation. PLoS Genet., 1–26, https://doi.org/10.1371/journal.pgen.1007526 (2018).
Engels, W. R. & Preston, C. R. Formation of chromosome rearrangements by P factors in Drosophila. Genetics 107, 657–678 (1984).
Sharakhov, I. V. et al. Breakpoint structure reveals the unique origin of an interspecific chromosomal inversion (2La) in the Anopheles gambiae complex. Proc. Natl. Acad. Sci. USA 103, 6258–6262 (2006).
Cáceres, M., Sullivan, R. T. & Thomas, J. W. A recurrent inversion on the eutherian X chromosome. Proc. Natl. Acad. Sci. 104, 18571–18576 (2007).
Matzkin, L. M., Merritt, T. J. S., Zhu, C.-T. & Eanes, W. F. The structure and population genetics of the breakpoints associated with the cosmopolitan chromosomal inversion In(3R)Payne in Drosophila melanogaster. Genetics 170, 1143–1152 (2005).
Papaceit, M., Segarra, C. & Aguadé, M. Structure and population genetics of the breakpoints of a polymorphic inversion in Drosophila subobscura. Evolution (N. Y). 67, 66–79 (2013).
Puerma, E. et al. Characterization of the breakpoints of a polymorphic inversion complex detects strict and broad breakpoint reuse at the molecular level. Mol. Biol. Evol. 31, 2331–2341 (2014).
Puerma, E., Orengo, D. J. & Aguadé, M. The origin of chromosomal inversions as a source of segmental duplications in the Sophophora subgenus of Drosophila. Sci. Rep. 6, 30715, https://doi.org/10.1038/srep30715 (2016).
Puerma, E., Orengo, D. J. & Aguadé, M. Multiple and diverse structural changes affect the breakpoint regions of polymorphic inversions across the Drosophila genus. Sci. Rep. 6, 36248, https://doi.org/10.1038/srep36248 (2016).
Puerma, E., Orengo, D. J. & Aguadé, M. Inversion evolutionary rates might limit the experimental identification of inversion breakpoints in non-model species. Sci. Rep. 7, 17281 (2017).
Orengo, D. J., Puerma, E., Papaceit, M., Segarra, C. & Aguadé, M. A molecular perspective on a complex polymorphic inversion system with cytological evidence of multiply reused breakpoints. Heredity (Edinb). 114, 610–618 (2015).
Aguado, C. et al. Validation and Genotyping of Multiple Human Polymorphic Inversions Mediated by Inverted Repeats Reveals a High Degree of Recurrence. PLoS Genet. 10, 14–22 (2014).
Navarro, A., Betrán, E., Barbadilla, A. & Ruiz, A. Recombination and gene flux caused by gene conversion and crossing over in inversion heterokaryotypes. Genetics 146, 695–709 (1997).
Krimbas, C. B. The inversion polymorphism of Drosophila subobscura. In Drosophila Inversion Polymorphism (eds Krimbas, C. B. & Powell, J. R.) 127–220 (CRC Press, 1992).
Kunze-Mühl, E. & Müller, E. Weitere Untersuchungen uber die chromosomale Struktur und die natürlichen Strukturtypen von Drosophila subobscura Coll. Chromosoma 9, 559–570 (1958).
Miller, W. J., Nagel, A., Bachmann, J. & Bachmann, L. Evolutionary dynamics of the SGM transposon family in the Drosophila obscura species group. Mol. Biol. Evol. 17, 1597–1609 (2000).
Hudson, R. R., Slatkin, M. & Maddison, W. P. Estimation of levels of gene flow from DNA sequence data. Genetics 132, 583–589 (1992).
Munté, A., Aguadé, M. & Segarra, C. Nucleotide Variation at the yellow Gene Region is not Reduced in Drosophila subobscura: A Study in Relation to Chromosomal Polymorphism. Mol Biol Evol 17, 1942–1955 (2000).
Munté, A., Rozas, J., Aguadé, M. & Segarra, C. Chromosomal inversion polymorphism leads to extensive genetic structure: A multilocus survey in Drosophila subobscura. Genetics 169, 1573–1581 (2005).
Llopart, A. & Aguadé, M. Nucleotide Polymorphism at the RpII215 Gene in Drosophila subobscura: Weak Selection on Synonymous Mutations. Genetics 155, 1245–1252 (2000).
Nóbrega, C., Khadem, M., Aguadé, M. & Segarra, C. Genetic exchange versus genetic differentiation in a medium-sized inversion of Drosophila: The A2/Ast arrangements of Drosophila subobscura. Mol. Biol. Evol. 25, 1534–1543 (2008).
Navarro-Sabaté, À., Aguadé, M. & Segarra, C. The relationship between allozyme and chromosomal polymorphism inferred from nucleotide variation at the Acph-1 gene region of Drosophila subobscura. Genetics 153, 871–889 (1999).
Krimbas, C. B. & Loukas, M. The inversion polymorphism of Drosophila subobscura. in Evolutionary Biology (eds Hecht, H., Steere, W. & Wallace, B.) 163–234 (Plenum Press, 1980).
Puerma, E. et al. The high-quality genome sequence of the oceanic island endemic species Drosophila guanche reveals signals of adaptive evolution in genes related to flight and genome stability. Genome Biol. Evol., 1956–1969, https://doi.org/10.1093/gbe/evy135 (2018).
Kramara, J., Osia, B. & Malkova, A. Break-Induced Replication: The Where, The Why, and The How. Trends Genet. 34, 518–531 (2018).
Burland, T. G. DNASTAR’s Lasergene sequence analysis software. Methods Mol. Biol. 132, 71–91 (2000).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302 (2017).
Jukes, T. H. & Cantor, C. R. Evolution of protein molecules. in Mamalian protein metabolism (ed. Munro, H. N.) III, 22–96 (Academic Press Inc, 1969).
Acknowledgements
We thank David Salguero for his excellent technical assistance and Carmen Segarra for critical comments. We also thank Servei de Genòmica, Serveis Cientifico-Tècnics, Universitat de Barcelona, for automated sequencing facilities. This work was supported by grants BFU2012-35168 and BFU2015-63732 from Ministerio de Economía y Competitividad, Spain, and 2014SGR-1055 and 2017SGR-1287 from Comissió Interdepartamental de Recerca i Innovació Tecnològica, Generalitat de Catalunya, Spain.
Author information
Authors and Affiliations
Contributions
D.J.O. and M.A. conceived the study; D.J.O., U. C. and E.P. conducted the experiments; D.J.O., E.P., U. C. and M.A. analyzed the results; D.J.O., E.P. and M.A. wrote the manuscript; all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Orengo, D.J., Puerma, E., Cereijo, U. et al. The molecular genealogy of sequential overlapping inversions implies both homologous chromosomes of a heterokaryotype in an inversion origin. Sci Rep 9, 17009 (2019). https://doi.org/10.1038/s41598-019-53582-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53582-8
- Springer Nature Limited