
Abstract
The Changjiang Estuary and adjacent East China Sea are well-known hypoxic aquatic environments. Eutrophication-driven hypoxia frequently occurs in coastal areas, posing a major threat to the ecological environment, including altering community structure and metabolic processes of marine organisms, and enhancing diversion of energy shunt into microbial communities. However, the responses of microbial communities and their metabolic pathways to coastal hypoxia remain poorly understood. Here, we studied the microbial communities collected from spatiotemporal samplings using metagenomic sequencing in the Changjiang Estuary and adjacent East China Sea. This generated 1.31 Tbp of metagenomics data, distributed across 103 samples corresponding to 8 vertical profiles. We further reported 1,559 metagenome-assembled genomes (MAGs), of which 508 were high-quality MAGs (Completeness > 90% and Contamination < 10%). Phylogenomic analysis classified them into 181 archaeal and 1,378 bacterial MAGs. These results provided a valuable metagenomic dataset available for further investigation of the effects of hypoxia on marine microorganisms.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background & Summary
The Changjiang Estuary and adjacent East China Sea, as the largest marginal sea in the western Pacific1, are characterized by complex hydrological environment, where the Changjiang River freshwater, the Taiwan Warm Current, and the Yellow Sea coastal water mix2,3. Hypoxia, with dissolved oxygen (DO) levels < 2 mg/L, poses a severe problem in marine ecosystem4. Estuarine and coastal areas often experience hypoxia due to nutrients and organic matter enrichment5. Hypoxia creates profound ecological and economic consequences, including loss in biodiversity, decline in seagrass meadows, coral bleaching and mortality, fisheries collapse, and marine mammal mortalities6,7,8. Previous studies have linked the generation and development of hypoxia in estuaries and coastal waters to intensifying eutrophication caused by anthropogenic nutrient input9,10,11. In the East China Sea, hypoxia frequently occurs off the Changjiang Estuary, covering up to approximately 15,000 km2 areas12. The hypoxic area in the Changjiang Estuary and adjacent East China Sea is considered as one of the largest low-oxygen zones globally13,14. The recurring hypoxic occurrences in the Changjiang Estuary offer a natural laboratory to study how hypoxia influence microbial diversity and their function.
Marine microorganisms are highly abundant with a massive collective biomass, playing significant roles in biogeochemical cycles15. Hypoxia forces the energy flow to increasingly shifts from higher trophic levels of predators towards lower trophic levels of microbial community, influencing carbon and nutrient cycling16,17. Studies have shown that microbial life can thrive in low-oxygen environments18,19. Marine microorganisms often respond first to recover from ecological disturbances, serving as crucial bioindicators for assessing environmental shifts due to anthropogenic and natural factors20. Much of the current knowledge about microbial communities in low-oxygen regions focuses on oxygen minimum zones7,21,22,23. However, research on microbial community structure and metabolic functions in coastal hypoxic zones remains limited.
This study conducted a comprehensive space-time sampling expedition in the Changjiang Estuary and adjacent East China Sea from July to August 2022 (Fig. 1). Sample metadata was available in Table S1. Time-series sampling covering a 22-day period was conducted at S07 and S08. DO concentrations ranged from 2.73 to 4.54 mg/L in the bottom water, indicating the presence of low-oxygen environments. We collected size-fractionated bacteria (free-living: 0.2–3 μm; particle-associated: 3–20 μm) to distinguish different bacterial lifestyles at S07 and S08. Then we conducted sampling from the Changjiang Estuary to offshore, where DO concentrations ranged from 3.99 to 1.12 mg/L in the bottom water. Hypoxia was observed in the bottom layers of S03 and S09, with the DO concentration value of 1.12 and 1.98 mg/L, respectively. Free-living samples (0.2–3 μm) were collected at these sites. The metagenomes were sequenced, with each yielding approximately 10.2–19.6 Gbp of raw data. Illumina shotgun metagenome sequencing yielded a total of 1.31 Tbp, distributed across 103 samples corresponding to 8 vertical profiles. We further reconstructed 1,559 MAGs. Among them, 32.5% (n = 508) were high-quality (Completeness > 90% and Contamination < 10%), and 46.7% (n = 728) were medium-quality (Completeness: 70–90% and Contamination < 10%). Taxonomic classification, based on the Genome Taxonomy Database (GTDB), assigned these MAGs to 5 archaeal (181 MAGs) and 18 bacterial phyla (1,378 MAGs). Pseudomonadota (n = 658), Bacteroidota (n = 330), Actinomycetota (n = 251), Verrucomicrobiota (n = 37), and Planctomycetota (n = 30) were the dominant bacterial phyla (Fig. 2 and Table S2). The phylum Pseudomonadota was the most abundant, with MAGs assigned to Alphaproteobacteria (n = 319) and Gammaproteobacteria (n = 339). Archaeal phyla included Thermoplasmatota (n = 118), Thermoproteota (n = 52), Nanoarchaeota (n = 5), Asgardarchaeota (n = 4), and Huberarchaeota (n = 2) (Fig. 3 and Table S2). Detailed information of the MAGs was provided in Table S2. As far as we know, this is the first report of a multiple MAGs dataset being recovered from the coastal hypoxic areas. This collection of microbial genomes from deoxygenated seawater offers valuable insights into the species diversity, structure, and function of these communities, potentially revealing the ecological roles of specific taxa in estuarine-offshore environments.

Sampling sites in the Changjiang Estuary and adjacent East China Sea. The black dots represent stations where samples were collected from various depths. The inset map highlights time-series sampling conducted at stations S07 and S08 near Dongfushan Island. Detailed sample metadata can be found in Table S1.

The phylogenomic tree of 1,378 bacterial MAGs reconstructed from the Changjiang Estuary and adjacent East China Sea. The universally 120 conserved bacterial markers genes were used to build this maximum-likelihood phylogenomic tree. Detailed MAGs taxonomy assignment, associated with completeness and contamination information can be found in Table S2.

The phylogenomic tree of 181 archaeal MAGs reconstructed from the Changjiang Estuary and adjacent East China Sea. The universally 122 conserved archaeal markers genes were used to build this maximum-likelihood phylogenomic tree. Detailed MAGs taxonomy assignment, associated with completeness and contamination information can be found in Table S2.
Methods
Sample sites and sample collection
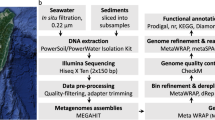
A total of 103 water samples were collected in July to August 2022 from the Changjiang Estuary and adjacent East China Sea. Environmental factors including depth, salinity, temperature, pH, Chlorophyll a concentration, and DO concentration were obtained through Conductivity-Temperature-Depth (CTD) sensors. About 5 L seawater were pre-filtered using a 20 μm pore size filter, and then filtered through 3 μm and 0.2 μm pore-size polycarbonate membranes (Millipore, USA) for each sample for metagenomic analysis. All filters were collected, quick-frozen in liquid nitrogen and then stored at −80 °C.
DNA extraction, metagenomic sequencing
Total microbial DNA was extracted from the filter membranes using DNeasy Powersoil Kit (MoBio, USA), and its quality and concentration was assessed by QuantusTM Fluorometer (Promega, USA) after 1% agarose gel electrophoresis. The genomic DNA was fragmented into about 500 bp segments using the Covaris M220 (Covaris, USA). Subsequently, libraries were constructed the manufacturer’s protocol with NovaSeq 6000 Reagent Kit (Illumina, USA). Metagenomic sequencing was conducted using Illumina PE150 chemistries on the NovaSeq 6000 at Hanyu Bio-Tech (Shanghai, China).
Metagenomic assembly, gene prediction and annotation, and genome binning
For each metagenomic data, trimmomatic v0.36 was used to quality control for removing adaptor and low-quality reads24. Clean reads were de novo assembled to generate contigs using MEGAHIT v1.1.3 with default parameters25. Genes from contigs were predicted using Prodigal v2.6.3 (-p meta)26. Predicted genes in all samples were combined, and CD-HIT v4.8.1 was used to remove redundant genes (-c 0.95, -aS 0.9) to construct non-redundant gene catelog27. Abundance of each gene in all samples was calculated using Salmon v1.10.128. For genome binning, MAGs are reconstructed from metagenome data as described in the previous study29. Bowtie2 v2.3.230 was used to align clean reads to the contigs with length exceeding 1,500 bp, and then bam file was sorted using SAMtools v.1.731. We used MetaBAT2 v.2.10.2 to calculate the coverage of contigs and carry out binging analysis, employing different parameters of maxP, minS, maxEdges (60, 60, 200; 95, 60, 200; 60, 95, 200; 60, 60, 500; 95, 60, 500; 60, 95, 500; 95, 95, 200; and 95, 95, 500)32. We then used DAS_Tool v.1.0 to dereplicate and aggregate these binning results to construct accurate bins33, and assessed the quality of these MAGs (including completeness, contamination, and strain heterogeneity) using CheckM2 v.1.0.134.
Taxonomic classification and genome tree construction
The taxonomic affiliation of the 1,559 MAGs was determined using GTDB-Tk v2.1.1 with the reference database GTDB r2.1.435,36. GTDB-Tk identified marker genes (120 bacterial and 122 archaeal conserved genes) to infer phylogenetic relationships37. Then the reference trees for 181 archaeal and 1,378 bacterial MAGs were generated using FastTree v2.1.1038. The trees were visualized and annotated using ChiPlot (https://www.chiplot.online/)39.
Data Records
The metagenome raw reads generated in this study haven been deposited and publicly available in the NCBI Sequence Read Archive (SRA) database with the BioProject number PRJNA108158340 and accession numbers SRP49221941. The reconstructed MAGs have been uploaded to NCBI GenBank database with the same project PRJNA108158340 and accession numbers JBERCU000000000-JBERRB000000000, JBERRC000000000-JBESFQ000000000, JBESFR000000000-JBESUD000000000, JBESUE000000000-JBETJF000000000, and JBETJG000000000-JBETKS000000000. MAGs sequences generated, MAGs trees, and non-redundant gene catalog in this study have been deposited at Figshare42.
Technical Validation
The quality of metagenomic datasets and contig assemblies was assessed using QUAST v5.2.043, and the quality of the MAGs (including completeness, contamination, and strain heterogeneity) was assessed using CheckM2 v.1.0.134. The taxonomic assignments of MAGs, such as Roseobacteraceae, Flavobacteriaceae, SAR11, and SAR86, were consistent with typical composition of marine microorganisms in coastal seawater44,45. The novelty of the MAGs was assessed through comparison with OceanDNA46 and Tara Oceans47 using dRep v3.4.248 at 95% average nucleotide identity.
Code availability
All versions and parameters for the software used in this study are described in the Methods section.
References
Xu, Z. L., Shen, X. M. & Gao, Q. in Ecological Continuum from the Changjiang (Yangtze River) Watersheds to the East China Sea Continental Margin (ed Jing Zhang) Ch.8 (Springer International Publishing, 2015).
Chen, J. et al. The processes of dynamic sedimentation in the Changjiang Estuary. J Sea Res 41, 129–140 (1999).
Zhang, W., Wu, H. & Zhu, Z. Transient Hypoxia Extent Off Changjiang River Estuary due to Mobile Changjiang River Plume. J. Geophys. Res. Oceans 123, 9196–9211 (2018).
Vaquer-Sunyer, R. & Duarte, C. M. Thresholds of hypoxia for marine biodiversity. Proc. Natl. Acad. Sci. USA 105, 15452–15457 (2008).
Breitburg, D. et al. Declining oxygen in the global ocean and coastal waters. Science 359, eaam7240 (2018).
Chai, F. et al. Persistent eutrophication and hypoxia in the coastal ocean. Cambridge Prisms: Coastal Futures 1, e19 (2023).
Diaz, R. J. & Rosenberg, R. Spreading dead zones and consequences for marine ecosystems. Science 321, 926–929 (2008).
Paulmier, A. & Ruiz-Pino, D. Oxygen minimum zones (OMZs) in the modern ocean. Prog. Oceanogr. 80, 113–128 (2009).
Rabalais, N. N. et al. Dynamics and distribution of natural and human-caused hypoxia. Biogeosciences 7, 585–619 (2010).
Fennel, K. & Testa, J. M. Biogeochemical Controls on Coastal Hypoxia. Ann Rev Mar Sci 11, 105–130 (2019).
Kemp, W. M., Testa, J. M., Conley, D. J., Gilbert, D. & Hagy, J. D. Temporal responses of coastal hypoxia to nutrient loading and physical controls. Biogeosciences 6, 2985–3008 (2009).
Zhu, Z. et al. Hypoxia off the Changjiang (Yangtze River) estuary and in the adjacent East China Sea: Quantitative approaches to estimating the tidal impact and nutrient regeneration. Mar Pollut Bull 125, 103–114 (2017).
Chen, C.-C., Gong, G.-C. & Shiah, F.-K. Hypoxia in the East China Sea: One of the largest coastal low-oxygen areas in the world. Mar Environ Res 64, 399–408 (2007).
Zhu, Z. et al. Hypoxia off the Changjiang (Yangtze River) Estuary: Oxygen depletion and organic matter decomposition. Mar Chem 125, 108–116 (2011).
Karl, D. M. Microbiological oceanography - Hidden in a sea of microbes. Nature 415, 590–591 (2002).
Jessen, G. L. et al. Hypoxia causes preservation of labile organic matter and changes seafloor microbial community composition (Black Sea). Sci. Adv. 3, e1601897 (2017).
Wright, J. J., Konwar, K. M. & Hallam, S. J. Microbial ecology of expanding oxygen minimum zones. Nat Rev Microbiol 10, 381–394 (2012).
Han, H. et al. Adaptation of aerobic respiration to low O2 environments. Proc. Natl. Acad. Sci. USA 108, 14109–14114 (2011).
Beman, J. M. & Carolan, M. T. Deoxygenation alters bacterial diversity and community composition in the ocean’s largest oxygen minimum zone. Nat Commun 4, 2705 (2013).
Bunse, C. & Pinhassi, J. Marine Bacterioplankton Seasonal Succession Dynamics. Trends Microbiol 25, 494–505 (2017).
Campbell, L. G., Thrash, J. C., Rabalais, N. N. & Mason, O. U. Extent of the annual Gulf of Mexico hypoxic zone influences microbial community structure. PLoS ONE 14, e0209055 (2019).
Bertagnolli, A. D. & Stewart, F. J. Microbial niches in marine oxygen minimum zones. Nat Rev Microbiol 16, 723–729 (2018).
Mattes, T. E., Ingalls, A. E., Burke, S. & Morris, R. M. Metabolic flexibility of SUP05 under low DO growth conditions. Environ Microbiol 23, 2823–2833 (2021).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, D. et al. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11 (2016).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Liu, Y. et al. Expanded diversity of Asgard archaea and their relationships with eukaryotes. Nature 593, 553–557 (2021).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Sieber, C. M. K. et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843 (2018).
Chklovski, A. et al. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat Methods 20, 1203–1212 (2023).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2019).
Rinke, C. et al. A standardized archaeal taxonomy for the Genome Taxonomy Database. Nat. Microbiol 6, 946–959 (2021).
McCormack, J. E., Huang, H. & Knowles, L. L. Maximum Likelihood Estimates of Species Trees: How Accuracy of Phylogenetic Inference Depends upon the Divergence History and Sampling Design. Syst Biol 58, 501–508 (2009).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 5, e9490 (2010).
Xie, J. et al. Tree Visualization By One Table (tvBOT): a web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res 51, W587–W592 (2023).
NCBI BioProject https://identifiers.org/ncbi/bioproject:PRJNA1081583 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP492219 (2024).
Liu, S. Recovery of 1559 metagenome-assembled genomes from the East China Sea’s low-oxygen region. Figshare https://doi.org/10.6084/m9.figshare.25809334 (2024).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Gifford, S. M., Sharma, S., Booth, M. & Moran, M. A. Expression patterns reveal niche diversification in a marine microbial assemblage. ISME J 7, 281–298 (2012).
Wang, K. et al. Bacterial biogeography in the coastal waters of northern Zhejiang, East China Sea is highly controlled by spatially structured environmental gradients. Environ Microbiol 17, 3898–3913 (2015).
Nishimura, Y. & Yoshizawa, S. The OceanDNA MAG catalog contains over 50,000 prokaryotic genomes originated from various marine environments. Sci. Data 9, 305 (2022).
Paoli, L. et al. Biosynthetic potential of the global ocean microbiome. Nature 607, 111–118 (2022).
Olm, M. et al. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J 11, 2864–2868 (2017).
Acknowledgements
This study was supported by the National Key Research and Development Program of China (2020YFA0608300), and the National Natural Science Foundation of China project (92251306, 42188102, 42076160).
Author information
Authors and Affiliations
Contributions
S.L. and K.T. conceived this study. S.L., C.H., Q.R., C.D. and X.Q. conducted field sampling and DNA extraction. S.L. assembled the metagenomes, generated the MAGs and produced all figures under the supervision of K.T., S.L. interpreted the results and wrote the first draft. K.T. revised the draft. All authors reviewed and contributed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, S., Chen, Q., Hou, C. et al. Recovery of 1559 metagenome-assembled genomes from the East China Sea’s low-oxygen region. Sci Data 11, 994 (2024). https://doi.org/10.1038/s41597-024-03850-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03850-8
- Springer Nature Limited