
Abstract
Eretmochelys imbricata, a critically endangered sea turtle inhabiting tropical oceans and protected across the world, had an unknown genome sequence until now. In this study, we used HiFi reads and Hi-C technology to assemble a high-quality, chromosome-level genome of E. imbricata. The genome size was 2,138.26 Mb, with contig N50 length of 123.49 Mb and scaffold N50 of 137.21 Mb. Approximately 97.52% of the genome sequence was anchored to 28 chromosomes. A total of 20,206 protein-coding genes were predicted. We also analyzed the evolutionary relationships, gene family expansions, and positive selection of E. imbricata. Our results revealed that E. imbricata diverged from Chelonia mydas 38 million years ago and had enriched olfactory receptors and aging-related genes. Our genome will be useful for studying E. imbricata and its conservation.
Similar content being viewed by others


Background & Summary
Sea turtles are a group with a long evolutionary history, having diverged for more than 100 million years1. Of the approximately 356 species of turtles worldwide2, only seven are sea turtles: the hawksbill turtle - Eretmochelys imbruaria, the green turtle - Chelonia mydas, the loggerhead turtle - Caretta Caretta, the olive ridley - Lepidochelys olivacea, the Kemp’s turtle - Lepidochelys kempii, the flatback turtle - Natator depressius, and the leatherback turtle - Dermochelys coriacea3. Sea turtles are widely distributed in global oceans and have highly migratory behavior, with migratory routes often spanning multiple seas and even oceans4. They are known for their remarkable survivability, reproductive capacity, and physiological diversity5. However, global sea turtle populations have been depleted in recent decades due to harvest for meat and eggs, commercial trade, fishery by-catch6, ecological degradation, and population gender disorders caused by global warming7,8. As a result, efforts to monitor, track, and protect sea turtles have increased in recent years.
Eretmochelys imbricata, commonly known as hawksbill turtle, is predominantly found in tropical and subtropical waters across the globe, and currently faces a very serious situation9. Among all globally distributed sea turtle species, it is the most endangered, and the IUCN has classified it as critically endangered (IUNC 2022). Despite concerted conservation efforts and interventions initiated since 1970, aimed at protecting and recovering E. imbricata populations, the species continues to face persistently low population levels10,11. Moreover, their significance in coral reef ecosystems cannot be understated, but the present global coral reef ecosystem faces severe degradation, further intensifying the threat to the survival of E. imbricata12,13. Conservation efforts for E. imbricata are particularly challenging due to their complex spatial structure and highly migratory nature14,15. The species needs may take decades to reach sexual maturity. Once mature, they return to their birthplace every few years to lay eggs, making it both difficult and costly to monitor their movements in the wild16. Most existing studies on E. imbricata primarily focus on counting nesting sites to assess their distribution17 and employing mitochondrial DNA haplotypes and microsatellite markers to examine their genetic structure18,19. Additionally, the development of Single Nucleotide Polymorphisms (SNPs) in E. imbricata has proved essential for evaluating their population structure20,21. However, despite these efforts, to date, there has been no reported genome assembly for E. imbricata.
In this study, we present the first high-quality, chromosome-level genome assembly of E. imbricata, achieved through PacBio HiFi and Hi-C sequencing technologies. The assembly resulted in a 2,138.26 Mb genome, with a contig N50 length of 123.49 Mb and a scaffold N50 of 137.21 Mb. Using Hi-C data, 97.52% of the assembled bases were successfully anchored to 28 chromosomes. This high-quality reference genome lays a robust groundwork for future population and conservation genetic studies of E. imbricata.
Methods
Sample collection and DNA extraction
An individual E. imbricata was obtained from the sea turtle rescue base on Naozhou Island, Zhanjiang City, Guangdong Province, China. A 10 mL blood sample was drawn from its jugular sinus and rapidly frozen for further analysis. Genomic DNA was extracted from the processed blood samples using the DNeasy Blood & Tissue Kit (Qiagen). The quality and quantity of the extracted DNA were assessed using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), a Qubit dsDNA HS assay kit on a Qubit 3.0 fluorometer (Life Technologies, Carlsbad, CA, USA), and 0.8% agarose gels.
Library construction and sequencing
The DNA extracted from the blood was used for sequencing library construction using the PacBio SEQUEL Platform. For 20 kb template library preparation, ten micrograms (μg) of E. imbricata genomic DNA were utilized, following the manufacturer’s protocol with the BluePippin Size Selection system (Sage Science, Beverly, MA, USA). The PacBio single molecule real-time (SMRT) library was prepared using the SMRT bell express template prep Kit 2.0 (Pacific Biosciences, Menlo Park, CA, USA) and sequenced on the PacBio Sequel II platform in CCS mode. The raw data was converted into high-precision HiFi reads using the CCS workflow13 (v. 6.3.0, https://github.com/pacificbiosciences/unanimity) (parameters: - minPasses 3). A total of 30.11 Gb of HiFi reads with 27.26x coverage was generated, and the N50 value was 14,598 bp (Table 1).
For Hi-C library preparation, the previously reported method22 was followed. Blood tissue was fixed with 2% formaldehyde, and the cross-linked DNA was digested with MboI enzyme. Biotin-labeled adapters were attached to the sticky ends of fragmented DNA. After reverse crosslinks by proteinase K (Thermo, Shanghai, China), DNA purification was performed using the QIAamp DNA Mini Kit (Qiagen) following the manufacturer’s instructions. The purified DNA was then sheared to a length of 300–500 bp to construct Hi-C libraries. A total of 186.13 G raw reads, which obtained from the MGI-SEQ. 2000 sequencing platform in paired-end 150 bp mode, were trimmed for sequencing adaptors and low-quality fragments using Trimmomatic (v0.39, parameters: LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:15). Finally, 181.16 Gb of high-quality Hi-C data were used to construct the chromosome-level genome. (Table 2).
For transcriptome sequencing, RNA was extracted from blood tissues using TRIzol reagent (Invitrogen, Waltham, MA, USA) following the manufacturer’s instructions. mRNA was then purified from the total RNA using poly-T oligo-attached magnetic beads. Sequencing libraries were generated from the purified mRNA using the V AHTS Universal V6 RNA-seq Library Kit for MGI (V azyme, Nanjing, China) with unique index codes following the manufacturer’s recommendations. The library quantification and size were assessed using Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Subsequently, sequencing was performed on the MGI-SEQ 2000 platform by Frasergen Bioinformatics Co., Ltd. (Wuhan, China).
Genome survey and assembly
To estimate the genome size, heterozygosity, and repeat rate of E. imbricata, we employed the k-mer frequency method. The raw reads obtained from the DNBSEQ-T7 platform were quality-filtered using SOAPnuke (v2.1.0)23 (main parameters: -lowQual = 20, -nRate = 0.005, -qualRate = 0.5, other parameters default). Subsequently, the quality-filtered reads were utilized to calculate the K-mer frequency with k = 17, using Jellyfish (v. 2.2.10)24 and GCE (https://github.com/fanagislab/GCE). Our estimation resulted in a genome size of 2138.26 Mb, with a peak 17-mer depth of 81. The heterozygosity and repeat rate were found to be 0.33% and 53.52%, respectively (Fig. 1). For the initial genome assembly, we used 30.11 Gb HiFi reads utilizing HiFiasm (v0.16.1)25 with default parameters. This preliminary assembly yielded a genome size of 2.30 Gb, with a contig N50 of 123.49 Mb (Table 3).

K-mer distribution of E. imbricata. Horizontal dotted line indicates heterozygosity rate, vertical dotted line represents a k-mer depth.
The paired-end reads obtained from the Hi-C library were mapped to the assembled genome using BWA (v 2.2.1) (parameters: -SP5M) to get the unique mapped paired-end reads, which were used to construct the Hi-C association scaffold26. The 3D-DNA pipeline was employed to cluster, sequence, and orient the contigs to construct a genome-wide interaction matrix27. Additionally, Juciebox (v1.11.08)28 was used for manual error correction, resulting in the final assembly of 28 chromosomes. The quality of the genome assembly was validated by a heatmap of the Hi-C assembly interaction bins, demonstrating excellent results (Fig. 2). The length of the final assembled genome was 2,296,181,705 bp, with a contig N50 of 123,485,570 bp and scaffold N50 of 137,212,766 bp (Table 3). Approximately 2,239,151,156 bp (97.52%) of the assembled result were anchored to 28 pseudochromosomes (Chr) (Table 4).

Hi-C interaction heatmap. The genome features of E. imbricata: genome-wide Hi-C heatmap of chromatin interaction counts. The color bar indicates contact density from red (high) to white (low).
Repeat annotation
To identify tandem repeats and interspersed repeats (transposon elements), we employed a combination of two methods: homology-based and de novo prediction. For the homology-based analysis, RepeatMasker (v4.1.2, -nolow -no_is -norna -parallel 2) and RepeatProteinMask (v1.36, -engine ncbi -noLowSimple -pvalue 0.0001) (http://www.repeatmasker.org) were used to predict TEs within the E. imbricata genome based on the known TE protein database and RepBase library (v21.12)29. For de novo prediction, we constructed an ab initio repeat sequence library of the E. imbricata genome using RepeatModeler (v2.0.2a) and LTR_FINDER (v1.0.5)30. RepeatMasker was then used to search and classify the repeat regions against this newly constructed repeat library. Tandem Repeat Finder (TRF)31 was utilized to identify tandem repeats, while RepeatMasker was employed to identify non-dispersed repeat sequences. Genome annotation revealed that transposable elements make up approximately 55.51% of the E. imbricata genome (Table 5).
Gene prediction
Three strategies were used for E. imbricata gene structure annotation: ab initio annotation, homology prediction, and RNA-sequencing-assisted prediction. For homology prediction, we aligned protein sequences from closely related species (Chelonia mydas, Dermochelys coriacea, Trachemys scripta elegans, Chrysemys picta and Gopherus evgoodei) with E. imbricata genome sequence to define gene models using Exonerate (v2.2.0)32. Ab initio prediction was generated using Augustus (v3.3)33 and Genescan (v1.0)34. In addition, RNA-seq data from E. imbricata was assembled and aligned to the repeat-masked genome to identify splice sites and exonic regions. All data were then integrated using MAKER (v3.00)35. PASA36 was used to further refine the gene structure based on transcriptome data. The final comprehensive gene set comprised 20,206 genes (Table 6).
Gene function annotation
To perform functional annotation of the integrated gene set, we aligned the genes to several databases, including SwissProt37, KEGG38, TrEMBL39, GO Ontology (GO)40, and NR (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz), using Blastp (parameters: -e 1e-5). PfamScan and the InterProScan (v5.35–74.0) were used to search protein structural domains based on the PFAM and InterPro41 protein database, respectively. As a result, 99.48% of the predicted protein-coding genes were functionally annotated (Table 7).
Gene family evolution and phylogenetic relationships
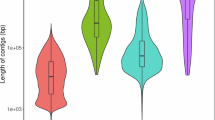
To identify orthologous gene groups, we conducted a comparative analysis of the protein sequences of E. imbricata with those of ten additional species, namely C. mydas (NCBI: GCA_015237465.2)42, D. coriacea (NCBI: GCA_009764565.4), T. scripta (NCBI: GCA_013100865.1)43, G. Evgoodei (NCBI: GCA_007399415.1), C. picta (NCBI: GCA_000241765.5), Gavialis gangeticus (NCBI: GCA_001723915.1), Thamnophis elegans (NCBI: GCA_009769535.1), Crocodylus porosus (NCBI: GCA_001723895.1)44, Gallus gallus (NCBI: GCA_016699485.1)45, and Homo sapiens (NCBI: GCA_000001405.29). The OrthoFinder2 (v2.5.4)46 tool was employed to cluster the genes from the 11 species into gene families using default parameters. After analysis of the gene family, a total of 94.2% (19039) of the 20206 protein-coding genes were clustered into 15,829 orthologous groups in E. imbricata (Fig. 3). The average ortholog group contained 1.20 genes per group, and we identified 62 gene families, comprising 320 genes, were found to be unique to E. imbricata (Table 8). Additionally, we identified 6,507 single-copy genes based on orthologous genes from the 11 species.

Gene family clustering status classification statistics. Distribution of gene, CDS, exon and intron length for protein-coding genes in Eretmochelys imbricata and other turtle genomes.
To investigate the evolutionary relationships between E. imbricata and other sea turtle species, we performed protein sequence alignments for each species’ single-copy orthologues using MUSCLE (v3.8.31)47. These alignments were then translated into corresponding coding DNA sequences (CDS). The evolutionary tree was constructed using the maximum likelihood method in RAxML (v8.2.12, parameters: -f a -x 12345 -# 100 -m PROTGAMMAAUTO)48. Calibration times were obtained by integrating the constructed evolutionary trees with data from the TimeTree website49. Divergence times were estimated using R8s (v1.81, -b)50 and the MCMCTree program with default parameters in the PAML (v4.10.0)51 packages. The phylogenetic tree reveals the evolutionary relationships between E. imbricata and other sea turtle: D. coriacea diverged approximately 53.0 million years ago (mya) from a common ancestor with C. mydas and E. imbricata. In addition, C. mydas was the closest sequenced relative to E. imbricata, having diverged from their common ancestor around 36.7 to 40.3 mya. (Fig. 4).

Comparison of orthologous genes between Eretmochelys imbricata and 10 other species. Horizontal coordinates represent the species and vertical coordinates represent the number of genes. The dark blue blocks represent single-copy homologues orthologs; the light blue blocks represent multiple-copy orthologs; the red blocks represent unique paralogues; the orange blocks represent other orthologs and the green blocks represent unclustered genes.
Contraction and expansion of gene families
The time-calibrated phylogenetic tree was utilized to estimate gene family contractions and expansions through CAFÉ (v4.2.1)52. In comparison to 10 closely related species, the investigation revealed 292 expanded gene families and 895 contracted gene families in the E. imbricata genome (Fig. 5). Further functional annotation of the expanded gene families through GO and KEGG enrichment analyses highlighted their significant involvement in pathways related to olfactory transduction - olfactory receptor, the immune response - pathways for intestinal immune network for IgA production, and detoxification - cytochrome P450.

Phylogenetic tree of E. imbricata and other species. The maximum likelihood phylogenetic tree based on 6507 concatenated single-copy orthologous genes. The bootstrap value of all nodes is supported at 100/100. Numbers below the branches represent the number of expanded (green) and contracted (red) gene families. The scale at the bottom represents divergence time. The pie chart represents gene families (black, expanded; red, extracted; blue, others).
Positively selected genes
To gain insights into the selection pressure on the single-copy orthologous genes, the rate ratio (ω) of nonsynonymous (Ka) to synonymous (Ks) nucleotide substitutions was estimated based on the phylogenetic tree using the PAML (v4.10.0)53 package. Employing the branch-site model of Codeml54 within the PAML package, the rate ratio of the foreground branch of E. imbricata and all other branches was determined within the likelihood framework. As a result, a total of 1,487 positively selected genes were identified with a likelihood ratio test (LRT) significance level of ≤0.05 and false discovery rate (FDR) of ≤ 0.05 in the E. imbricata genome. The GO enrichment analysis demonstrated significant enrichment in the terms “binding,” “olfactory receptor,” as well as “ECM-receptor” and “Focal adhesion” in the KEGG pathway enrichment analysis.
In summary, we obtained the high-quality chromosome-level genome of E. imbricata. The newly generated reference genome will significantly contribute to our understanding of the genetic diversity of sea turtles and facilitate future comparative evolutionary studies and the conservation efforts for this endangered species.
Data Records
The E. imbricata genome project was deposited at NCBI under BioProject No. PRJNA872952. The Illumina sequencing data were deposited under NCBI Accession No. SRR2131239155; the PacBio sequencing data were deposited under NCBI Accession No. SRR2131191256; the Hi-C sequencing data were deposited under NCBI Accession No. SRR2131230057; the RNA-seq data were deposited under NCBI Accession No. SRR2131191358; the assembled genome sequence was deposited into NCBI under accession number JARRBA00000000059; the genome annotation files are available in Figshare60; the phylogenetic and molecular evolution analyses data are available in Figshare61.
Technical Validation
Genome assembly and gene prediction quality assessment
The completeness of the E. imbricata genome was assessed using BUSCO with the tetrapoda_odb10 (parameters: -m genome -l tetrapoda_odb10)62. The assembled genome exhibited approximately 97.4% complete BUSCO genes, with 96.8% being complete and single copy, 0.6% being complete and duplicated, 0.7% being fragmented, and 1.9% being missed (Table 9). Minimap2 (v2.12, parameters: -ax map-pb)63 aligned the assembly results with HiFi data to obtain the depth of coverage for each locus on the genome, which showed mapping and coverage rate were estimated to be 100% and 99.85%, respectively (Table 10). Subsequently, employing 1000 bp non-overlapping sliding windows along the chromosomes, we calculated the GC content and the average depth of reads (Fig. 6). Collectively, all of the above results indicate that we have obtained a high-quality genome of E. imbricata.

GC content and sequencing depth distribution density map. The x-axis represents the GC content; the y-axis represents the average depth.
Usage Notes
All data analyses were performed according to the manual and protocols of the published bioinformatic tools. The version and parameters of software have been described in Methods section.
Code availability
No specific code or script was used in this work. Commands used for data processing were all executed according to the manuals and protocols of the corresponding software.
References
Naro-Maciel, E., Le, M., Fitzsimmons, N. N. & Amato, G. Evolutionary relationships of marine turtles: a molecular phylogeny based on nuclear and mitochondrial genes. Molecular Phylogenetics and Evolution. 49, 659–662 (2008).
Rhodin, A. G. K. J. Turtles of the world annotated checklist and atlas of taxonomy, synonymy, distribution, and conservation status (9th Ed.). Phyllomedusa. 20, 225–228 (2021).
Bowen, B. W. & Karl, S. A. Population genetics and phylogeography of sea turtles. Molecular Ecology. 16, 4886–4907 (2007).
Monzón-Argüello, C. et al. Príncipe islands hawksbills: genetic isolation of an eastern Atlantic stock. Journal of Experimental Marine Biology and Ecology. 407, 345–354 (2011).
Chow, J. C., Anderson, P. E. & Shedlock, A. M. Sea turtle population genomic discovery: global and locus-specific signatures of polymorphism, selection, and adaptive potential. Genome Biology and Evolution. 11, 2797–2806 (2019).
Mcclenachan, L., Jackson, J. B. & Newman, M. J. Conservation implications of historic sea turtle nesting beach loss. Frontiers in Ecology and the Environment. 4, 290–296 (2006).
Hawkes, L. A., Broderick, A. C., Godfrey, M. H. & Godley, B. J. Investigating the potential impacts of climate change on a marine turtle population. Global Change Biology. 13, 923–932 (2007).
Witt, M. J., Hawkes, L. A., Godfrey, M. H., Godley, B. J. & Broderick, A. C. Predicting the impacts of climate change on a globally distributed species: the case of the loggerhead turtle. Journal of Experimental Biology. 213, 901–911 (2010).
Da Silva, V. R. F. et al. Adaptive threat management framework: integrating people and turtles. Environment, Development and Sustainability. 18, 1541–1558 (2016).
Casale, P. & Ceriani, S. A. Satellite surveys: a novel approach for assessing sea turtle nesting activity and distribution. Marine Biology. 166, (2019).
Mortimer, J. A., Donnelly, M., Meylan, A. B. & Meylan, P. A. Critically endangered hawksbill turtles: molecular genetics and the broad view of recovery. Molecular Ecology. 16, 3516–3517 (2007).
Carpenter, K. E. et al. One-third of reef-building corals face elevated extinction risk from climate change and local impacts. Science. 321, 560–563 (2008).
Jackson, J. B. C. et al. Historical overfishing and the recent collapse of coastal ecosystems. Science. 293, 629–637 (2001).
Rees, A. F. et al. Are we working towards global research priorities for management and conservation of sea turtles? Endangered Species Research. 31, 337–382 (2016).
Wallace, B. P. et al. Regional management units for marine turtles: a novel framework for prioritizing conservation and research across multiple scales. Plos One. 5, e15465 (2010).
Gaos, A. R. et al. Hawksbill turtle terra incognita: conservation genetics of eastern Pacific rookeries. Ecology and Evolution. 6, 1251–1264 (2016).
Askari Hesni, M., Tabib, M. & Hadi Ramaki, A. Nesting ecology and reproductive biology of the hawksbill turtle, Eretmochelys imbricata, at Kish Island, Persian Gulf. Journal of the Marine Biological Association of the United Kingdom. 96, 1373–1378 (2016).
Miro-Herrans, A. T., Velez-Zuazo, X., Acevedo, J. P. & Mcmillan, W. O. Isolation and characterization of novel microsatellites from the critically endangered hawksbill sea turtle (Eretmochelys imbricata). Molecular Ecology Resources. 8, 1098–1101 (2008).
Nishizawa, H., Joseph, J. & Chong, Y. K. Spatio-temporal patterns of mitochondrial DNA variation in hawksbill turtles (Eretmochelys imbricata) in Southeast Asia. Journal of Experimental Marine Biology and Ecology. 474, 164–170 (2016).
Banerjee, S. M. et al. Single nucleotide polymorphism markers for genotyping hawksbill turtles (Eretmochelys imbricata). Conservation Genetics Resources. 12, 353–356 (2020).
Komoroske, L. M. et al. A versatile Rapture (RAD‐Capture) platform for genotyping marine turtles. Molecular Ecology Resources. 19, 497–511 (2019).
Belton, J. M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58, 268–276 (2012).
Chen, Y.-X. et al. SOAPnuke: A MapReduce acceleration supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience. 7, 1–6 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Cheng, H.-Y. et al. Haplotype-resolved de novo assembly using phased assembly graphs with HiFiasm. Nature Methods. 18, 170–175 (2021).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature Biotechnology. 31, 1119–1125 (2013).
Kajitani, R. et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Research. 24, 1384–1395 (2014).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems. 3, 95–98 (2016).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research. 110, 462–467 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research. 35, W265–W268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research. 27, 573–580 (1999).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. Bmc Bioinformatics. 6, 31 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research. 34, W435–W439 (2006).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. Journal of Molecular Biology. 268, 78–94 (1997).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. Bmc Bioinformatics. 12, 491 (2011).
Haas, B. J. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research. 31, 5654–5666 (2003).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research. 28, 45–48 (2000a).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research. 27, 29–34 (1999).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research. 28, 45–48 (2000b).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nature Genetics. 25, 25–29 (2000).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Research. 49, D344–D354 (2021).
Wang, Z. et al. The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nature Genetics. 45, 701–6 (2013).
Brian, S. et al. An annotated chromosome-level reference genome of the red-eared slider turtle (Trachemys Scripta Elegans). Genome Biology and Evolution. 12, 456–62 (2020).
Arnab, G. et al. A high-quality reference genome assembly of the saltwater crocodile, Crocodylus porosus, reveals patterns of selection in crocodylidae. Genome Biology and Evolution. 12, 3635–3646 (2020).
Wesley, C. W. et al. A new chicken genome assembly provides insight into avian genome structure. G3-Genes Genomes Genetics. 7, 109–117 (2017).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biology. 20, (2019).
Manuel, M. A new semi-subterranean diving beetle of the Hydroporus normandi-complex from south-eastern France, with notes on other taxa of the complex (Coleoptera: Dytiscidae). Zootaxa. 3652, 453–474 (2013).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30, 1312–1313 (2014).
Hedges, S. B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 22, 2971–2972 (2006).
Sanderson, M. J. R8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 19, 301–302 (2003).
Yang, Z.-H. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution. 24, 1586–1591 (2007).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 22, 1269–1271 (2006).
Yang, Z.-H. PAML: a program package for phylogenetic analysis by maximum likelihood. Computer Applications in the Biosciences Cabios. 13, 555–6 (1997).
Gao, F. et al. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecology and Evolution. 9, 3891–3898 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21312391 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21311912 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21312300 (2022).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRR21311913 (2022).
Guo, Y.-S., Tang, J. & Wang, Z.-D. The first high-quality chromosome-level genome of Eretmochelys imbricata using HiFi and Hi-C data, GenBank, https://identifiers.org/ncbi/insdc:JARRBA000000000 (2023).
Figshare https://doi.org/10.6084/m9.figshare.23805789 (2023).
Figshare https://doi.org/10.6084/m9.figshare.24011031 (2023).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods in molecular biology (Clifton, N.J.). 1962, 227–245 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Acknowledgements
Science and Technology Infrastructure Project of Department of Science and Technology of Guangdong Province, China (No. 2021B1212110005).
Author information
Authors and Affiliations
Contributions
Y.G. and Z.W. conceived the project. J.T., J.H., Z.F., J.S., M.L. and Z.D. collected the samples. Z.Z., C.H., Z.W., Y.F., M.L. and C.L. performed the genome assembly, gene annotation and other bioinformatics analysis. Y.G. and J.T. wrote and revised the manuscript. Y.G., Z.W. and M.L. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, Y., Tang, J., Zhuo, Z. et al. The first high-quality chromosome-level genome of Eretmochelys imbricata using HiFi and Hi-C data. Sci Data 10, 604 (2023). https://doi.org/10.1038/s41597-023-02522-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02522-3
- Springer Nature Limited