
Abstract
The Indo-Pacific humpback dolphin (Sousa chinensis), is a threatened marine mammal and belongs to the First Order of the National Key Protected Wild Aquatic Animals List in China. However, limited genomic information is available for studies of its population genetics and biological conservation. Here, we have assembled a genomic sequence of this species using a whole genome shotgun (WGS) sequencing strategy after a pilot low coverage genome survey. The total assembled genome size was 2.34 Gb: with a contig N50 of 67 kb and a scaffold N50 of 9 Mb (107.6-fold sequencing coverage). The S. chinensis genome contained 24,640 predicted protein-coding genes and had approximately 37% repeated sequences. The completeness of the genome assembly was evaluated by benchmarking universal single copy orthologous genes (BUSCOs): 94.3% of a total 4,104 expected mammalian genes were identified as complete, and 2.3% were identified as fragmented. This newly produced high-quality assembly and annotation of the genome will greatly promote the future studies of the genetic diversity, conservation and evolution.
Design Type(s) | sequence assembly objective • sequence annotation objective |
Measurement Type(s) | whole genome sequencing assay |
Technology Type(s) | DNA sequencing |
Factor Type(s) | |
Sample Characteristic(s) | Sousa chinensis • skin of body |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others
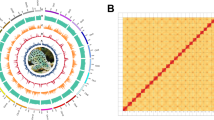

Background & Summary
The Indo-Pacific humpback dolphin (Sousa chinensis) normally appears in southeast Asia (in both the Indian and Pacific oceans), from at least the southeastern bay of Bengal east to central China, and then south to the Indo-Malay Archipelago1. The S. chinensis found in Chinese waters are locally known as Chinese white dolphins (the giant panda of the sea). Populations of S. chinensis in China have been known to be distributed from the Beibu Gulf near the border with Vietnam to the mouth of the Yangtze River2,3,4,5, the waters around Hainan island are also recently identified as one part of this species’ distribution6 (Fig. 1). At least four species are now indicated to make up the genus Sousa: the Atlantic humpback (Sousa teuszii), the Indian Ocean humpback (Sousa plumbea), the Australian humpback (Sousa sahulensis) and the Indo-Pacific humpback (S. chinensis) dolphins7. Further molecular evidence suggests that humpback dolphins in the bay of Bengal may comprise a fifth species7. However, as the classification and population genetics of genus Sousa was mainly based on the limited evidences from morphology, genetic markers and the mitochondrial sequences7,8,9, the newly produced genome of S. chinensis would greatly facilitate the classification and identification of Sousa genetic resources.

Geographical distribution and photograph of S. chinensis. (a) Distribution of S. chinensis reported in Chinese waters and the sampling site of this study. (b) S. chinensis photographed during the boat surveys in Guangxi Beibu Gulf, China.
S. chinensis are among the most threatened cetaceans for their coastal inhabitation, which are vulnerably impacted by human activities7. It has been listed in the First Order of the National Key Protected Wild Aquatic Animals List in China (refer to: List of Wildlife under Special State Protection, which was designated by the Chinese State Council in 1988) and in the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES). The species is currently categorized as Near Threatened by the International Union for Conservation of Nature (IUCN). The threats include entanglement in fishing nets (primarily gillnets), habitat destruction and degradation, vessel traffic and environmental pollutants, are all serious and fatal to S. chinensis1,10,11,12,13,14,15. As a result, much greater efforts are needed for conservation of this species to stop its apparent decline1. At present, most of the research has mainly focused on the morphology16, reproduction and growth15,17, population distribution1,18, biodiversity19 and toxicology studies of this species11,20,21. Genetic research of S. chinensis was mainly based on genetic markers9, specific genes22, mitochondrial DNA8,23 and transcriptome24. The genomic background and molecular mechanism of its evolution and conservation are still unknown. The high-quality whole genome sequences information would be a valuable resource for the biology, ecology, conservation and evolutionary studies.
To obtain a high-quality genome sequence of S. chinensis, we first performed a pilot genome survey with low depth coverage sequencing (32.9X) (Table 1) by using Illumina Hiseq 4000 to estimate the genome size and heterozygosity of the species. The assembled genome size is about 2.29 Gb25 (contig N50 = 13 Kb and scaffold N50 = 163 Kb) and the completed BUSCO evaluated is just about 76% in genome survey26. The low depth sequencing estimated the genome size is about 2.7 Gb and generated an insufficient completeness genome26. Therefore, we constructed four additional insert size libraries (beside the previous 500 bp and 2 Kb in genome survey) and generated a total of 290.5 Gb (107.6X) clean data after filtering (Tables 1 and 2). The S. chinensis genome was finally assembled into scaffolds with a total size of 2.34 Gb27 (Tables 1 and 3). The contig and scaffold N50 of assembly results was 67 Kb and 9 Mb, the N50 number and N90 number of scaffolds was 78 and 283 respectively (Table 3). 94.3% of 4,104 conserved genes were completed identified by BUSCO28 (Table 4). The newly assembled genome quality was much better than the genome survey (Table 1). In total, 878.3 Mb (37.41%) of genomic regions consist of repeat sequences (Table 5). The gene annotation of the genome yielded 24,640 coding genes and 91.2% of the predicted genome were annotated from biological databases (Tables 6 and 7). Approximately 95% of the “total complete BUSCOs” were identified by BUSCO pipeline based on the annotation result (Table 8), which suggested a good quality genome annotation.
Methods
Sample collection, DNA extraction and sequencing
The same sample collection and DNA extraction methods have been reported in a previously published study26. In addition to the previously constructed 500 bp and 2 kb libraries, new 300 bp and 800 bp small insert and 5 kb and 10 kb mate pair libraries were constructed according to the manufacturer’s protocol (Illumina, San Diego, CA, USA). After library construction, we used Illumina HiSeq X Ten to sequence PE150 reads for 300 bp library. PE125 reads for 800 bp library, and PE50 reads for 5 Kb and 10 Kb libraries were sequenced by Illumina HiSeq 4000 platform. A total of approximately 370 Gb raw data was obtained. Then, we filtered the reads with stringent filtering criteria using SOAPnuke29 and 290.5 Gb of clean data was generated (107.6X genome coverage) (Table 2).
Genome assembly and evaluation
We used all the clean data to assemble the genome by Platanus30. First, the contigs were constructed based on the de Bruijn graphs from paired-end reads. Second, the order of the contigs was fixed using the paired end (mate-pair) information in the scaffold construction process. Third, in the Gap-closing step, each set of assembled reads were used to close the gaps, and each gap was covered with reads mapped on the scaffolds by the Platanus pipeline. After that, we filled the gaps with GapCloser31. Finally, scaffolds were extended by SSPACE32 using the mate-paired library data. The final total assembled genome length was 2.34 Gb with a contig N50 of 67 kb, and a scaffold N50 of 9 Mb (Table 3). The assembly and gene annotation qualities were assessed using BUSCO software28. The total number of mammal gene sets used in the evaluation was 4,104.
Genome annotation
The genome was searched for tandem repeats using Tandem Repeats Finder33. Interspersed repeats were mainly identified using homology-based approaches. The Repbase34 (known repeats) database and a de novo repeat library generated by RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html) were used. The database was mapped by using RepeatMasker (http://www.repeatmasker.org). The repeat content of this species is 37.4% (Table 5).
The coding genes in the S. chinensis genome were annotated based on evidence derived from known proteins and published RNA sequences. For protein homology-based prediction, proteins of B. taurus, T. truncatus, O. orca, and B. mysticetus were downloaded from NCBI and aligned to the S. chinensis genome using TBLASTN35 with an E-value ≦ 1E−5. Homologous genome sequences were aligned to the matched proteins to predict the gene models by Genewise36. We filtered the sequences for redundancy and retained the gene models with the highest scores. RNA-seq data provided a good supplement for gene prediction based on the homology-based method, as most of open reading frames (ORF) in the homology-based gene models were not intact. First, transcriptome data (total 4,305,634,920 nucleotides) of S. chinensis was downloaded from https://www.ebi.ac.uk/ena/data/search?query=ERP003522 which was sequenced by Illumina Hiseq2000 platform and published in 201324. These reads were aligned to the assembled genome sequence using hisat37. Subsequently, hisat mapping results were merged and sorted, and transcripts were assembled using stringtie with the default parameters38. Finally, the Genewise results were extended using the transcripts ORFs following the strategy of the Ensembl gene annotation system39. This method and strategy were used extensively in the genome research40,41,42,43,44. The 24,640 (Table 6) predicted genes were then functionally annotated by aligning to five databases: InterPro45, Gene ontology46, KEGG47, Swissprot48 and TrEMBL48, 91.2% of the predicted genes were annotated with function (Table 7).
Technical Validation
Evaluation the completeness of the genome assembly and annotation
To evaluate the completeness of the genome assembly and annotation, BUSCO pipeline28 was used to investigate the presence of highly conserved orthologous genes in the genome assembly and annotation result we obtained. BUSCO was run over the mammalian set, which includes total of 4,104 orthologue groups. 94.3% and 95.1% of the “total complete BUSCOs” were identified by BUSCO pipeline based on the genome assembly and annotation result respectively (Tables 4 and 8), which evidenced a good quality of the genome assembly and gene sets annotation.
To further evaluate the accuracy of genome, the paired-end short insert size library reads were aligned to the assembled genome by the BWA-mem (v0.7.15)50 with default parameters. After sorting mapped reads according to mapping coordinates in Picard (ver. 1.118) (http://broadinstitute.github.io/picard/), the mapping rate is 99.92% and the unique mapping rate is 75.81%. A total of 98.27% assembled genome was covered by the reads and the mapping coverage with at least 4X, 10X, 20X is respectively 98.16%, 97.97% and 97.32%.
Comparison with other cetacean genomes
A total of approximately 370 Gb raw data was generated using the Illumina HiSeq X Ten and 4000 platform for the S. chinensis genome with 6 different kinds of insert size libraries: 300 bp, 500 bp, 800 bp, 2 Kb, 5 Kb and 10 Kb49. After a data filtering process, approximately 290.5 Gb of clean data, representing approximately 107.6-fold genome coverage, was obtained for genome assembly (Table 1). After being assembled by the software Platanus, the total assembled genome length was approximately 2.34 Gb with a contig N50 of 67 kb, and a scaffold N50 of 9 Mb27 (Table 3), which was better than the published B. acutorostrata, L. vexillifer and B. mysticetus genomes (Table 9). We predicted 24,640 coding genes in the S. chinensis genome (Table 6) by using a homolog and RNA-seq supplemented approach which was used extensively in the genome research40,41,42,43,44. There were 27,924 genes predicted in O. orca and approximately 20,000–23,000 genes predicted in the B. mysticetus, L. vexillifer and B. acutorostrata (Table 9).
Here, we reported the updated high-quality genome sequence of the threatened Indo-Pacific humpback dolphin. The genome resource would greatly enhance the further studies of the gene function and conservation biology of S. chinensis. Our study is an important step towards comprehensive understanding of the genetic background of S. chinensis at the genomic level. The data will be also valuable for facilitating studies of cetacean evolution, as well as population genetic and ecology.
Code Availability
Several tools have been implemented in the data analyses, whose versions, settings and parameters are described below.
(1) SOAPnuke: version 1.5.3, parameters used were -n 0.1 -l 20 -q 0.4 -d -M 1 -Q 2 -i -G–seqType 1; (2) Platanus: version 1.2.4, parameters used were: contig step: -k 32 -u 0.1 -d 0.5 -c 2 -t 30 -s 10 -m 300G; scaffold step: -t 30 –u 0.1; gapclose step: default parameters; (3) GapCloser: version 1.12, parameters used were –l 150 –p 25 –t 30; (4) SSPACE: version 1.1, default parameters; (5) BUSCO: version 3.0.2; (6) TRF: version 4.07b, default parameters; (7) Repbase: version 21.01; (8) RepeatModeler: version 1.0.4, default parameters; (9) RepeatMasker: open-4-0-6, default parameters; (10) Blast: version 2.2.26, parameters used were -F F -m 8 -p tblastn -e 1e-05 -a 5; (11) Genewise: version 2.4.1, default parameters; (12) Hisat: version 2-2.0.1-beta, parameters used were -p 4–max-intronlen 50000–sensitive–dta–dta-cufflinks–phred64–no-discordant–no-mixed; (13) Stringtie: version 1.2.2, default parameters; (14) InterPro: version 5.16–55.0; (15) GO: version 20141201; (16) KEGG: version 84.0; (17) Swissprot: version release-2017-09; (18) TrEMBL: version release-2017-09; (19) BWA-mem: version 0.7.15, default parameters; (20) Picard: version 1.118, default parameters.
References
Jefferson, T. A. & Smith, B. D. In Adv Mar Biol Vol. 73 (eds Thomas, A. Jefferson & Barbara E., Curry) 1–26 (Academic Press, 2016).
Chen, B. et al. Conservation Status of the Indo-Pacific Humpback Dolphin (Sousa chinensis) in the Northern Beibu Gulf, China. Adv Mar Biol 73, 119–139 (2016).
Karczmarski, L. et al. Humpback Dolphins in Hong Kong and the Pearl River Delta: Status, Threats and Conservation Challenges. Adv Mar Biol 73, 27–64 (2016).
Wang, J. et al. A framework for the assessment of the spatial and temporal patterns of threatened coastal delphinids. Sci Rep 6, 19883 (2016).
Wang, J. Y. et al. Biology and Conservation of the Taiwanese Humpback Dolphin, Sousa chinensis taiwanensis. Adv Mar Biol 73, 91–117 (2016).
Li, S. et al. First record of the Indo-Pacific humpback dolphins (Sousa chinensis) southwest of Hainan Island, China. Mar Biodivers Rec 9, 3 (2016).
Jefferson, T. A. & Curry, B. E. Humpback Dolphins: A Brief Introduction to the Genus Sousa. Adv Mar Biol 72, 1–16 (2015).
Chen, L., Caballero, S., Zhou, K. & Yang, G. Molecular phylogenetics and population structure of Sousa chinensis in Chinese waters inferred from mitochondrial control region sequences. Biochem Syst Ecol 38, 897–905 (2010).
Lin, W. et al. Differentiated or not? An assessment of current knowledge of genetic structure of Sousa chinensis in China. J Exp Mar Biol Ecol 416, 17–20 (2012).
Slooten, E. et al. Impacts of fisheries on the Critically Endangered humpback dolphin Sousa chinensis population in the eastern Taiwan Strait. Endanger Species Res 22, 99–114 (2013).
Gui, D. et al. Bioaccumulation and biomagnification of persistent organic pollutants in Indo-Pacific humpback dolphins (Sousa chinensis) from the Pearl River Estuary, China. Chemosphere 114, 106–113 (2014).
Hung, C. L. et al. A preliminary risk assessment of trace elements accumulated in fish to the Indo-Pacific Humpback dolphin (Sousa chinensis) in the northwestern waters of Hong Kong. Chemosphere 56, 643–651 (2004).
Ng, S. L. & Leung, S. Behavioral response of Indo-Pacific humpback dolphin (Sousa chinensis) to vessel traffic. Mar Environ Res 56, 555–567 (2003).
Jia, K. et al. In vitro assessment of environmental stress of persistent organic pollutants on the Indo-Pacific humpback dolphin. Toxicol In Vitro: an international journal published in association with BIBRA 30, 529–535 (2015).
Jefferson, T. A., Hung, S. K., Robertson, K. M. & Archer, F. I. Life history of the Indo-Pacific humpback dolphin in the Pearl River Estuary, southern China. Mar Mammal Sci 28, 84–104 (2012).
Song, Z., Zhang, Y., Berggren, P. & Wei, C. Reconstruction of the forehead acoustic properties in an Indo-Pacific humpback dolphin (Sousa chinensis), with investigation on the responses of soft tissue sound velocity to temperature. J. Acoust. Soc. Am. 141, 681 (2017).
Chang, W. L., Karczmarski, L., Huang, S. L., Gailey, G. & Chou, L. S. Reproductive parameters of the Taiwanese humpback dolphin (Sousa chinensis taiwanensis). Reg Stud Mar Sci 8, 459–465 (2016).
Jefferson, T. A. & Hung, S. K. A Review of the Status of the Indo-Pacific Humpback Dolphin (Sousa chinensis) in Chinese Waters. Aquat Mamm 30, 149–158 (2004).
Hayano, A., Yoshioka, M., Tanaka, M. & Amano, M. Population differentiation in the Pacific white-sided dolphin Lagenorhynchus obliquidens inferred from mitochondrial DNA and microsatellite analyses. Zool Sci 21, 989–999 (2004).
Yeung, L. W. et al. Total fluorine, extractable organic fluorine, perfluorooctane sulfonate and other related fluorochemicals in liver of Indo-Pacific humpback dolphins (Sousa chinensis) and finless porpoises (Neophocaena phocaenoides) from South China. Environ Pollut 157, 17–23 (2009).
Wu, Y. et al. Evaluation of organochlorine contamination in Indo-Pacific humpback dolphins (Sousa chinensis) from the Pearl River Estuary, China. Sci Total Environ 444, 423–429 (2013).
Zhang, X. et al. Low Major Histocompatibility Complex Class II Variation in the Endangered Indo-Pacific Humpback Dolphin (Sousa chinensis): Inferences About the Role of Balancing Selection. J Hered 107, 143–152 (2016).
Lin, W., Zhou, R., Porter, L., Chen, J. & Wu, Y. Evolution of Sousa chinensis: a scenario based on mitochondrial DNA study. Mol Phylogenet Evol 57, 907–911 (2010).
Gui, D. et al. De novo assembly of the Indo-Pacific humpback dolphin leucocyte transcriptome to identify putative genes involved in the aquatic adaptation and immune response. PLoS One 8, e72417 (2013).
Ming, Y., Jian, J., Yu, F., Yu, X., Wang, J. & Liu, W. Sousa chinensis isolate MY-2018, whole genome shotgun sequencing project. GenBank, http://identifiers.org/ncbi/insdc:QWLN00000000.1 (2018).
Ming, Y. et al. Molecular footprints of inshore aquatic adaptation in Indo-Pacific humpback dolphin (Sousa chinensis). Genomics, https://doi.org/10.1016/j.ygeno.2018.07.015 (2018).
Ming, Y., Jian, J., Yu, F., Yu, X., Wang, J. & Liu, W. Sousa chinensis isolate MY-2018, whole genome shotgun sequencing project. GenBank, http://identifiers.org/ncbi/insdc:QWLN00000000.2 (2019).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6 (2018).
Kajitani, R. et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res 24, 1384–1395 (2014).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res 14, 988–995 (2004).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295 (2015).
Curwen, V. et al. The Ensembl automatic gene annotation system. Genome Res 14, 942–950 (2004).
Buti, M. et al. The genome sequence and transcriptome of Potentilla micrantha and their comparison to Fragaria vesca (the woodland strawberry). Gigascience 7, 1–14 (2017).
Ni, G., Cavero, D., Fangmann, A., Erbe, M. & Simianer, H. Whole-genome sequence-based genomic prediction in laying chickens with different genomic relationship matrices to account for genetic architecture. Genet Sel Evol: GSE 49, 8 (2017).
Jiang, Y. et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 344, 1168–1173 (2014).
Brawand, D. et al. The genomic substrate for adaptive radiation in African cichlid fish. Nature 513, 375 (2014).
Sequencing, T. M. G. et al. The common marmoset genome provides insight into primate biology and evolution. Nat Genet 46, 850 (2014).
Mulder, N. & Apweiler, R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol Biol 396, 59–70 (2007).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28, 45–48 (2000).
NCBI Sequence Read Archive, http://identifiers.org/ncbi/insdc.sra:SRP157198 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Keane, M. et al. Insights into the evolution of longevity from the bowhead whale genome. Cell Rep 10, 112–122 (2015).
Yim, H. S. et al. Minke whale genome and aquatic adaptation in cetaceans. Nat Genet 46, 88–92 (2014).
Zhou, X. et al. Baiji genomes reveal low genetic variability and new insights into secondary aquatic adaptations. Nat Commun 4, 2708 (2013).
Foote, A. D. et al. Convergent evolution of the genomes of marine mammals. Nat Genet 47, 272–275 (2015).
Acknowledgements
This study was funded by the Ministry of Agriculture of China (Chinese White Dolphin Conservation Action), the China National Offshore Oil Corporation Foundation, the National Natural Science Foundation of China (Grant Nos 41676166 and 41776174). Funding was also provided by the Education Department of Guangxi Zhuang Autonomous Region Foundation (Grant Nos KY2016YB487 and KY2016YB476), the Foundation of Guangdong Provincial Key Laboratory of Marine Biotechnology (Grant No. GPKLMB201602) and the Guangxi Natural Science Foundation (Grant No. 2016GXNSFBA380142).
Author information
Authors and Affiliations
Contributions
Y.M. and W.H.L. conceived this study. X.Y.Y. and J.Z.W. collected and prepared the samples. Genome sequencing was performed by BGI-Shenzhen; Y.M. performed bioinformatics analyses and data statistics. Y.M., J.B.J., J.Z.W. and W.H.L. discussed and interpreted the results. Y.M. wrote the manuscript, J.B.J., J.Z.W., X.Y.Y. and W.H.L. revised the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
ISA-Tab metadata file
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Ming, Y., Jian, J., Yu, X. et al. The genome resources for conservation of Indo-Pacific humpback dolphin, Sousa chinensis. Sci Data 6, 68 (2019). https://doi.org/10.1038/s41597-019-0078-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0078-6
- Springer Nature Limited