

Abstract
Studying the dynamics of genome replication in mammalian cells has been historically challenging. To reveal the location of replication initiation and termination in the human genome, we developed Okazaki fragment sequencing (OK-seq), a quantitative approach based on the isolation and strand-specific sequencing of Okazaki fragments, the lagging strand replication intermediates. OK-seq quantitates the proportion of leftward- and rightward-oriented forks at every genomic locus and reveals the location and efficiency of replication initiation and termination events. Here we provide the detailed experimental procedures for performing OK-seq in unperturbed cultured human cells and budding yeast and the bioinformatics pipelines for data processing and computation of replication fork directionality. Furthermore, we present the analytical approach based on a hidden Markov model, which allows automated detection of ascending, descending and flat replication fork directionality segments revealing the zones of replication initiation, termination and unidirectional fork movement across the entire genome. These tools are essential for the accurate interpretation of human and yeast replication programs. The experiments and the data processing can be accomplished within six days. Besides revealing the genome replication program in fine detail, OK-seq has been instrumental in numerous studies unravelling mechanisms of genome stability, epigenome maintenance and genome evolution.



Similar content being viewed by others
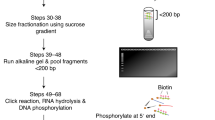
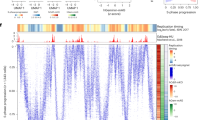
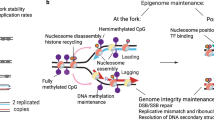
Data availability
Published available OK-seq raw and processed datasets analyzed in this work are available in SRA: SRP065949 (HeLa cells)27 and ENA: PRJEB36782 (S. cerevisiae)30.
Code availability
The bioinformatics tool and all example datasets underlying this paper are available at the following GitHub page: https://github.com/CL-CHEN-Lab/OK-Seq with DOI number: https://doi.org/10.5281/zenodo.7056979.
References
Huberman, J. A. & Riggs, A. D. On the mechanism of DNA replication in mammalian chromosomes. J. Mol. Biol. 32, 327–341 (1968).
Hamlin, J. L., Mesner, L. D. & Dijkwel, P. A. A winding road to origin discovery. Chromosome Res. 18, 45–61 (2010).
Hyrien, O. Peaks cloaked in the mist: the landscape of mammalian replication origins. J. Cell Biol. 208, 147–160 (2015).
Hulke, M. L., Massey, D. J. & Koren, A. Genomic methods for measuring DNA replication dynamics. Chromosome Res. 28, 49–67 (2020).
Lebofsky, R., Heilig, R., Sonnleitner, M., Weissenbach, J. & Bensimon, A. DNA replication origin interference increases the spacing between initiation events in human cells. Mol. Biol. Cell 17, 5337–5345 (2006).
Demczuk, A. et al. Regulation of DNA replication within the immunoglobulin heavy-chain locus during B cell commitment. PLoS Biol. 10, e1001360 (2012).
Anglana, M., Apiou, F., Bensimon, A. & Debatisse, M. Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell 114, 385–394 (2003).
Cadoret, J. C. et al. Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc. Natl Acad. Sci. USA 105, 15837–15842 (2008).
Besnard, E. et al. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 19, 837–844 (2012).
Karnani, N., Taylor, C. M., Malhotra, A. & Dutta, A. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol. Biol. Cell 21, 393–404 (2010).
Mukhopadhyay, R. et al. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet. 10, e1004319 (2014).
Langley, A. R., Gräf, S., Smith, J. C. & Krude, T. Genome-wide identification and characterisation of human DNA replication origins by initiation site sequencing (ini-seq. Nucleic Acids Res. 44, 10230–10247 (2016).
Mesner, L. D. et al. Bubble-chip analysis of human origin distributions demonstrates on a genomic scale significant clustering into zones and significant association with transcription. Genome Res. 21, 377–389 (2011).
Mesner, L. D. et al. Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res. 23, 1774–1788 (2013).
Hansen, R. S. et al. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl Acad. Sci. USA 107, 139–144 (2010).
Chen, C. L. et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 20, 447–457 (2010).
Zhao, P. A., Sasaki, T. & Gilbert, D. M. High-resolution Repli-Seq defines the temporal choreography of initiation, elongation and termination of replication in mammalian cells. Genome Biol. 21, 76 (2020).
Koren, A. et al. Genetic variation in human DNA replication timing. Cell 159, 1015–1026 (2014).
Lobry, J. R. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 13, 660–665 (1996).
Touchon, M. et al. Replication-associated strand asymmetries in mammalian genomes: toward detection of replication origins. Proc. Natl Acad. Sci. USA 102, 9836–9841 (2005).
Huvet, M. et al. Human gene organization driven by the coordination of replication and transcription. Genome Res. 17, 1278–1285 (2007).
Chen, C. L. et al. Replication-associated mutational asymmetry in the human genome. Mol. Biol. Evol. 28, 2327–2337 (2011).
Audit, B. et al. Open chromatin encoded in DNA sequence is the signature of ‘master’ replication origins in human cells. Nucleic Acids Res. 37, 6064–6075 (2009).
Guilbaud, G. et al. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput. Biol. 7, e1002322 (2011).
Baker, A. et al. Replication fork polarity gradients revealed by megabase-sized U-shaped replication timing domains in human cell lines. PLoS Comput. Biol. 8, e1002443 (2012).
Green, P., Ewing, B., Miller, W., Thomas, P. J. & Green, E. D. Transcription-associated mutational asymmetry in mammalian evolution. Nat. Genet. 33, 514–517 (2003).
Petryk, N. et al. Replication landscape of the human genome. Nat. Commun. 7, 10208 (2016).
Smith, D. J. & Whitehouse, I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 483, 434–438 (2012).
McGuffee, S. R., Smith, D. J. & Whitehouse, I. Quantitative, genome-wide analysis of eukaryotic replication initiation and termination. Mol. Cell 50, 123–135 (2013).
Hennion, M. et al. FORK-seq: replication landscape of the Saccharomyces cerevisiae genome by nanopore sequencing. Genome Biol. 21, 125 (2020).
Liu, Y., Wu, X., D’aubenton-Carafa, Y., Thermes, C. & Chen, C.-L. OKseqHMM: a genome-wide replication fork directionality analysis toolkit. Nucleic Acids Res., https://doi.org/10.1093/nar/gkac1239 (2022).
Blin, M. et al. DNA molecular combing-based replication fork directionality profiling. Nucleic Acids Res. 49, e69 (2021).
Wang, W. et al. Genome-wide mapping of human DNA replication by optical replication mapping supports a stochastic model of eukaryotic replication. Mol. Cell 81, 2975–2988 (2021).
Wu, X. et al. Developmental and cancer-associated plasticity of DNA replication preferentially targets GC-poor, lowly expressed and late-replicating regions. Nucleic Acids Res. 46, 10157–10172 (2018).
Petryk, N. et al. MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 361, 1389–1392 (2018).
Chen, Y. H. et al. Transcription shapes DNA replication initiation and termination in human cells. Nat. Struct. Mol. Biol. 26, 67–77 (2019).
Li, Z. et al. DNA polymerase alpha interacts with H3-H4 and facilitates the transfer of parental histones to lagging strands. Sci. Adv. 6, eabb5820 (2020).
Tubbs, A. et al. Dual roles of poly(dA:dT) tracts in replication initiation and fork collapse. Cell 174, 1127–1142 (2018).
Kirstein, N. et al. Human ORC/MCM density is low in active genes and correlates with replication time but does not delimit initiation zones. eLife 10, e62161 (2021).
Hyrien, O., Maric, C. & Méchali, M. Transition in specification of embryonic metazoan DNA replication origins. Science 270, 994–997 (1995).
Dijkwel, P. A., Wang, S. & Hamlin, J. L. Initiation sites are distributed at frequent intervals in the Chinese hamster dihydrofolate reductase origin of replication but are used with very different efficiencies. Mol. Cell Biol. 22, 3053–3065 (2002).
Powell, S. K. et al. Dynamic loading and redistribution of the Mcm2-7 helicase complex through the cell cycle. EMBO J. 34, 531–543 (2015).
Gros, J. et al. Post-licensing specification of eukaryotic replication origins by facilitated Mcm2-7 sliding along DNA. Mol. Cell 60, 797–807 (2015).
Promonet, A. et al. Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites. Nat. Commun. 11, 3940 (2020).
Brison, O. et al. Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide. Nat. Commun. 10, 5693 (2019).
Letessier, A. et al. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470, 120–123 (2011).
Le Tallec, B. et al. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 4, 420–428 (2013).
Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T. & Cimprich, K. A. Transcription–replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 170, 774–786 (2017).
Manzo, S. G. et al. DNA topoisomerase I differentially modulates R-loops across the human genome. Genome Biol. 19, 100 (2018).
Park, K. et al. Aicardi–Goutières syndrome-associated gene SAMHD1 preserves genome integrity by preventing R-loop formation at transcription-replication conflict regions. PLoS Genet. 17, e1009523 (2021).
Bayona-Feliu, A., Barroso, S., Muñoz, S. & Aguilera, A. The SWI/SNF chromatin remodeling complex helps resolve R-loop-mediated transcription-replication conflicts. Nat. Genet. 53, 1050–1063 (2021).
Andrianova, M. A., Bazykin, G. A., Nikolaev, S. I. & Seplyarskiy, V. B. Human mismatch repair system balances mutation rates between strands by removing more mismatches from the lagging strand. Genome Res. 27, 1336–1343 (2017).
Jaksik, R., Wheeler, D. A. & Kimmel, M. Detection and characterization of replication origins defined by DNA polymerase epsilon. Preprint at https://doi.org/10.1101/2021.07.27.453931 (2021).
Shi, M. J. et al. APOBEC-mediated mutagenesis as a likely cause of FGFR3 S249C mutation over-representation in bladder cancer. Eur. Urol. 76, 9–13 (2019).
DeWeerd, R. A. et al. Prospectively defined patterns of APOBEC3A mutagenesis are prevalent in human cancers. Cell Rep. 38, 110555 (2022).
Flasch, D. A. et al. Genome-wide de novo L1 retrotransposition connects endonuclease activity with replication. Cell 177, 837–851 (2019).
Sultana, T. et al. The landscape of L1 retrotransposons in the human genome is shaped by pre-insertion sequence biases and post-insertion selection. Mol. Cell 74, 555–570 (2019).
Ming, X. et al. Kinetics and mechanisms of mitotic inheritance of DNA methylation and their roles in aging-associated methylome deterioration. Cell Res. 30, 980–996 (2020).
Reijns, M. A. et al. Lagging-strand replication shapes the mutational landscape of the genome. Nature 518, 502–506 (2015).
Daigaku, Y. et al. A global profile of replicative polymerase usage. Nat. Struct. Mol. Biol. 22, 192–198 (2015).
Clausen, A. R. et al. Tracking replication enzymology in vivo by genome-wide mapping of ribonucleotide incorporation. Nat. Struct. Mol. Biol. 22, 185–191 (2015).
Koh, K. D., Balachander, S., Hesselberth, J. R. & Storici, F. Ribose-seq: global mapping of ribonucleotides embedded in genomic DNA. Nat. Methods 12, 251–257 (2015).
Zhou, Z. X., Lujan, S. A., Burkholder, A. B., Garbacz, M. A. & Kunkel, T. A. Roles for DNA polymerase δ in initiating and terminating leading strand DNA replication. Nat. Commun. 10, 3992 (2019).
Koyanagi, E. et al. Global landscape of replicative DNA polymerase usage in the human genome. Nat. Commun. 13, 7221 (2022).
Pratto, F. et al. Meiotic recombination mirrors patterns of germline replication in mice and humans. Cell 184, 4251–4267 (2021).
Sriramachandran, A. M. et al. Genome-wide nucleotide-resolution mapping of DNA replication patterns, single-strand breaks, and lesions by GLOE-seq. Mol. Cell 78, 975–985 (2020).
Kara, N., Krueger, F., Rugg-Gunn, P. & Houseley, J., Genome-wide analysis of DNA replication and DNA double-strand breaks using TrAEL-seq. PLoS Biol. 19, e3000886 (2020).
Kit Leng Lui, S. et al. Monitoring genome-wide replication fork directionality by Okazaki fragment sequencing in mammalian cells. Nat. Protoc. 16, 1193–1218 (2021).
Audit, B. et al. Multiscale analysis of genome-wide replication timing profiles using a wavelet-based signal-processing algorithm. Nat. Protoc. 8, 98–110 (2013).
Muller, C. A. et al. Capturing the dynamics of genome replication on individual ultra-long nanopore sequence reads. Nat. Methods 16, 429–436 (2019).
Gansauge, M. T. et al. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res. 45, e79 (2017).
Salic, A. & Mitchison, T. J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl Acad. Sci. USA 105, 2415–2420 (2008).
Burgers, P. M. J. & Kunkel, T. A. Eukaryotic DNA replication fork. Annu. Rev. Biochem. 86, 417–438 (2017).
DePamphilis, M. L. Genome Duplication (Garland Science/Taylor & Francis Group, 2010).
Qu, D. et al. 5-Ethynyl-2′-deoxycytidine as a new agent for DNA labeling: detection of proliferating cells. Anal. Biochem. 417, 112–121 (2011).
Ligasova, A. et al. Dr Jekyll and Mr Hyde: a strange case of 5-ethynyl-2′-deoxyuridine and 5-ethynyl-2′-deoxycytidine. Open Biol. 6, 150172 (2016).
Manska, S., Octaviano, R. & Rossetto, C. C. 5-Ethynyl-2′-deoxycytidine and 5-ethynyl-2′-deoxyuridine are differentially incorporated in cells infected with HSV-1, HCMV, and KSHV viruses. J. Biol. Chem. 295, 5871–5890 (2020).
Green, M. R. & Sambrook, J. Molecular Cloning: A Laboratory Manual. 4th edn (Cold Spring Harbor Laboratory Press, 2012).
Giacca, M., Pelizon, C. & Falaschi, A. Mapping replication origins by quantifying relative abundance of nascent DNA strands using competitive polymerase chain reaction. Methods 13, 301–312 (1997).
Tornøe, C. W.; Christensen, C.; Meldal, M. (2002).e, C. W., Christensen, C. & Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67, 3057–3064 (2002).
Rostovtsev, V. V., Green, L. G., Fokin, V. V. & Sharpless, K. B. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 41, 2596–2599 (2002).
Presolski, S. I., Hong, V. P. & Finn, M. G. Copper-catalyzed azide-alkyne click chemistry for bioconjugation. Curr. Protoc. Chem. Biol. 3, 153–162 (2011).
Kwok, C. K., Ding, Y., Sherlock, M. E., Assmann, S. M. & Bevilacqua, P. C. A hybridization-based approach for quantitative and low-bias single-stranded DNA ligation. Anal. Biochem. 435, 181–186 (2013).
Meyer, M. et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
R Core Team. R: a language and environment for statistical computing, https://www.r-project.org/ (R Foundation for Statistical Computing, 2020).
Himmelmann, L. HMM: HMM-Hidden Markov Models. R package version 1.0. (2016).
Morgan, M., Pages, H., Obenchain, V., and Hayden, N. Rsamtools: Binary alignment (BAM), FASTA, variant call (BCF), and tabix file import. R package version 1.30.0. (2017).
Lawrence, M. et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118 (2013).
RStudio Team. RStudio: Integrated Development for R, http://www.rstudio.com/ (RStudio, PBC, 2020).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
TrimGalore https://doi.org/10.5281/zenodo.5127899 (2021).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Picard Toolkit. Broad Institute, GitHub Repository. https://broadinstitute.github.io/picard/ (2019).
Acknowledgements
X.W. is supported by The Young Scientists Fund of the National Natural Science Foundation of China (grant no. 31900415). Y.L. thanks Agence Nationale pour la Recherche (ANR) for providing her PhD fellowship. C.T., Y.D.-C., C.-L.C., O.H. and N.P. thank the ANR grant BLAN2010–161501 (REFOPOL). Work in the O.H. laboratory is supported by the ANR grants 18-CE45-0002 (NanoPoRep) and 19-CE12-0028 (HUDROR). Work in the C.-L.C. laboratory is supported by the YPI program of I. Curie, the ATIP-Avenir program from Centre national de la recherche scientifique (CNRS) and Plan Cancer (grant number ATIP/AVENIR: N°18CT014-00); ANR grant 19-CE12-0016-02 (ReDeFINe) and 19-CE12-0020-02 (TELOCHROM); and Institut National du Cancer (INCa) grant PLBIO19-076. Work in the N.P. laboratory is supported by the ATIP-Avenir grant from CNRS and YPI funding from Institute Gustave Roussy; N.P. thanks the LabEx ‘Who Am I?’ ANR-11-LABX-0071; the Université de Paris IdEx ANR-18-IDEX-0001 and the ANR grant 19-CE12-0030-01 (INTEGER).
Author information
Authors and Affiliations
Contributions
O.H., C.-L.C. and N.P. conceived and supervised the project. N.P. developed the OK-seq method in mammalian cells; X.W. adapted the method for yeast cells. Y.L. Y.D.-C., C.T. and C.-L.C. developed the bioinformatics approach and built the analysis pipeline. X.W., Y.L., O.H., C.-L.C. and N.P. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks Kuhulika Bhalla, Bruce Stillman and Zhiguo Zhang for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Liu, Y. et al. Nucleic Acids Res., (2022): https://doi.org/10.1093/nar/gkac1239
Petryk, N. et al. Nat. Commun. 7, 10208 (2016): https://doi.org/10.1038/ncomms10208
Wu, X. et al. Nucleic Acids Res. 46, 10157–10172 (2018): https://doi.org/10.1093/nar/gky797
Hennion, M. et al. Genome Biol. 21, 125 (2020): https://doi.org/10.1186/s13059-020-02013-3
Supplementary information
Supplementary Information
Supplementary Protocol 1.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, X., Liu, Y., d’Aubenton-Carafa, Y. et al. Genome-wide measurement of DNA replication fork directionality and quantification of DNA replication initiation and termination with Okazaki fragment sequencing. Nat Protoc 18, 1260–1295 (2023). https://doi.org/10.1038/s41596-022-00793-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-022-00793-5
- Springer Nature Limited