

Abstract
Although the molecular composition and architecture of synapses have been widely explored, much less is known about what genetic programs directly activate synaptic gene expression and how they are modulated. Here, using Caenorhabditis elegans dopaminergic neurons, we reveal that EGL-43/MECOM and FOS-1/FOS control an activity-dependent synaptogenesis program. Loss of either factor severely reduces presynaptic protein expression. Both factors bind directly to promoters of synaptic genes and act together with CUT homeobox transcription factors to activate transcription. egl-43 and fos-1 mutually promote each other’s expression, and increasing the binding affinity of FOS-1 to the egl-43 locus results in increased presynaptic protein expression and synaptic function. EGL-43 regulates the expression of multiple transcription factors, including activity-regulated factors and developmental factors that define multiple aspects of dopaminergic identity. Together, we describe a robust genetic program underlying activity-regulated synapse formation during development.
Similar content being viewed by others


Main
The molecular composition and architecture of synapses have been extensively characterized and are highly conserved in metazoans. The majority of synapses are formed in small temporal windows during development1, suggesting the existence of a rapid and coordinated transcriptional program where cohorts of related genes are activated with precise control. These cohorts include pan-neuronally expressed genes, such as components of the synaptic vesicle cycle, and molecules that define specific neurotransmitter phenotypes. While neurotransmitter phenotype specification is known to be executed by terminal selector transcription factors (TFs)2, less is known about the genetic programs regulating pan-neuronally expressed genes. In a recent study in Caenorhabditis elegans, CUT homeobox family TFs were shown to function redundantly to activate pan-neuronal genes3, but how these and other genetic programs are regulated to ensure functional connectivity between neurons remains elusive.
One mechanism that neurons may use to modulate hardwired genetic programs during synaptogenesis is through neuronal activity. Although it remains unclear if CUT TFs are regulated by activity, numerous studies have demonstrated that both spontaneous and sensory-driven activity function in synapse formation, synaptic plasticity and synaptic strength4. A large body of work pioneered in ref. 5 demonstrated that visual input and spontaneous activity have essential roles in the synaptic organization of the visual cortex during a critical period. During this period, active neurons establish more synaptic connections, while inactive neurons have fewer synapses and smaller axonal arbors. It is thought that synaptic transmission in input neurons leads to depolarization and Ca2+ influx, which activates signal transduction pathways and ultimately results in activity-dependent transcriptional changes that impact synapse formation and plasticity6,7. The best-characterized TFs involved in this program are immediate-early genes (IEGs), such as Fos and Jun8. Fos has an important role in the development and function of the mammalian nervous system by acting as a strong transcriptional regulator9. Whether or how IEGs directly control synaptic gene expression is an important yet unanswered question.
Through a candidate approach using C. elegans dopaminergic neurons, we aimed to identify the mechanisms that drive activity-dependent synaptogenesis. Here we describe a new activity-regulated transcriptional program mediated by the conserved TFs EGL-43/MECOM and FOS-1/FOS. We demonstrate that both TFs bind directly to promoters of presynaptic genes, and loss of either factor reduces presynaptic gene expression. EGL-43 and FOS-1 mutually activate each other’s transcription, and enhancing the binding of FOS-1 to the egl-43 locus increases synaptic protein levels and subsequent synaptic transmission. In addition to activating presynaptic genes, EGL-43 modulates the expression of the terminal selector and CUT TFs, linking activity to hardwired genetic programs during development.
Results
Activity modulates synapse development in C. elegans
Although the role of activity in synapse development has been studied for decades, how activity regulates the expression of synaptic genes is unclear. To study these programs in vivo, we established the dopaminergic neurons of C. elegans as a model system. The posterior deirid (PDE) dopaminergic neurons possess bifurcating axons with numerous en passant synapses that can be visualized with endogenous, cell-type specific labeling of ELKS-1/ELKS, an active zone protein, using FLP/FRT-mediated recombination (FLP-on; Fig. 1a)10,11. We first tested if activity modulates presynaptic gene expression in PDE. To silence neurons, we expressed a histamine-gated chloride channel, HisCl1 (ref. 12), specifically in PDE. Silencing PDE during development led to a 22% decrease in ELKS-1 intensity along the length of the PDE axon (Fig. 1b–d). Animals lacking HisCl1 possess no defects in ELKS-1 intensity upon histamine treatment (Supplementary Fig. 1a,b). This finding contrasts with our previous work using overexpressed synaptic markers13, highlighting the importance of endogenous labeling to detect sensitive changes in protein abundance.

a, Schematic representation and line scan of the PDE axon and its synapses labeled using a combinatorial approach. Endogenous FLP-on (FRT) GFP::ELKS-1 labels active zones and endogenous FLP-on (FRT3) mScarlet::TBA-1 labels microtubules (for axon morphology) when combined with a transgene expressing a dopaminergic-specific flippase (dat-1p::FLP). b, Top: schematic representation of PDE-silencing experiments. Transgenic animals expressing dat-1p::HisCl1 were placed on media containing histamine at the L2 stage and imaged at the L4 stage. Bottom: schematic representation of PDE-excitation experiments. Transgenic animals expressing dat-1p::ChR2 were subjected to blue light at the L2 or L4 stage and imaged either 2 h or 48 h post-treatment. This schematic was created with BioRender.com. c, Line scans of ELKS-1 in the PDE axon of animals carrying wyEx8629(dat-1p::HisCl), treated with 0 mM or 10 mM histamine. d, Quantification of ELKS-1 in PDE of animals shown in c (n = 14 for both conditions). e, Line scans of ELKS-1 in the PDE axon of animals carrying wyEx10629(dat-1p::ChR2), treated with or without blue light. f, Quantification of ELKS-1 of animals shown in e (n = 19 for both conditions). g, Transgenic animals carrying wyEx8629(dat-1p::HisCl) were imaged for their endogenous FOS-1::GFP signal in PVD and PDE neuronal nuclei (white dashed circles) in the presence or absence of 10 mM histamine. h, Quantification of FOS-1::GFP in animals shown in f (n = 11 for both conditions). i, Normalized expression of PDE FOS-1::GFP presented in h, compared to each paired PVD’s (sister cell of PDE) expression (n = 11 for both conditions). j, Transgenic animals carrying wyEx10630(dat-1p::ChR2) were treated at L3 with or without blue light for ten cycles (15 s on and 60 s off) and allowed to recover for 2 h. FOS-1::GFP was imaged in PVD and PDE nuclei (white dashed circles). k, Quantification of FOS-1::GFP in animals shown in i (n = 15 for both conditions). l, Normalized expression of PDE FOS-1::GFP presented in k compared to each paired PVD’s expression (n = 15 for both conditions). For images with line scans, scale bar = 20 μm; for images of neuronal nuclei, scale bar = 10 μm. For all graphs, medians are represented in thick dashed lines and quartiles are represented in thin dashed lines. P values presented were calculated using two-tailed unpaired Student’s t tests. MGV, mean gray value.
We next asked if enhancing activity could increase presynaptic gene expression. We performed optogenetics using the blue light-gated sodium channel channelrhodopsin-2 (ChR2) to depolarize neurons14. We expressed ChR2 specifically in PDE and stimulated animals using blue light during axon outgrowth. Activation of ChR2 increased ELKS-1 intensity by 55% compared to controls (Fig. 1b,e,f and Supplementary Fig. 1c,d). Together, these results indicate that activity modulates synaptic gene expression in developing PDE neurons.
Given that transcription of Fos is highly induced by activity, we asked if C. elegans fos-1 is similarly dependent. We endogenously tagged fos-1, the sole Fos ortholog, and found that silencing PDE decreased FOS-1 intensity by 31% (Fig. 1g,h) and excitation of PDE increased FOS-1 intensity by 25% (Fig. 1j–l). Normalization of FOS-1 intensity in PDEs to their sister posterior ventral neuron D (PVDs), which do not express transgenes, revealed similar changes in FOS-1 intensity (Fig. 1i,l). Notably, animals lacking either transgene exhibited no changes in FOS-1 intensity upon histamine or blue light treatment (Supplementary Fig. 1e,f). These results demonstrate that FOS-1 expression is regulated by activity, in agreement with studies in other systems7. Thus, PDE neurons are a suitable system to study the mechanisms of activity-regulated synaptic gene expression in vivo.
EGL-43/MECOM is expressed in the nervous system
Because activity-dependent TFs are involved in various cellular processes, we postulated that specific activation of synaptic genes might require interactions between activity-dependent TFs and specialized TFs to achieve specificity during development (Fig. 2a). To identify candidates, we searched for TFs that interact with activity-dependent TFs and whose orthologs are expressed in C. elegans neurons. We identified MECOM, a conserved TF known to activate the expression of Fos, Jun and Egr1 and physically interacts with FOS, JUN and CREB15.

a, A potential model for activity-dependent synaptogenesis. Upstream, specialized TFs bind cooperatively with activity-dependent IEGs to activate downstream presynaptic targets. This schematic was created with BioRender.com. b, Comparison of MECOM-related proteins. From top to bottom: C. elegans EGL-43, Drosophila melanogaster Hamlet, Homo sapiens MECOM and Mus musculus MECOM. Features—red boxes represent ZF domains, purple boxes represent SET domains, blue boxes represent nuclear localization signal, gold boxes represent CTBP-binding motif 1 and green boxes represent CTBP-binding motif 2. c, Endogenous GFP-tagged EGL-43 is expressed in various sensory neuron nuclei, including head sensory neurons, the PVD and PDE neurons, and tail sensory neurons at the L4 stage (n > 50 animals examined, repeated thrice). d, Line scans of L4 PDE axons from egl-43(wy1514) animals expressing FLP-on ELKS-1 (to label active zones) and FLP-on TBA-1 (to label microtubules). Animals were treated with either 0 mM or 4 mM auxin. Line scans marked with single asterisk are imaged from the same worm to demonstrate the expression of both labels in the same animal, with the final line scan of each set with both channels merged (denoted with two asterisks). White arrowheads denote ELKS-1 puncta detected in the distal axon. e, Quantification of line scan intensities of animals shown in d (n = 20 for both conditions). Medians are represented in thick dashed lines, and quartiles are represented in thin dashed lines; P = 1.6215 × 10−18. f, Line scans of L4 PDE axons from egl-43(wy1514) animals expressing FLP-on RAB-3 and FLP-on TBA-1. Animals were treated with either 0 mM or 4 mM auxin. Line scans marked with single asterisk are imaged from the same worm to demonstrate the expression of both labels in the same animal, with the final line scan of each set with both channels merged (denoted with two asterisks). White arrowheads denote weak RAB-3 puncta detected in the distal axon. g, Quantification of line scan intensities of animals shown in f (n = 13 (untreated) and n = 22 (treated)). Medians are represented in thick dashed lines and quartiles are represented in thin dashed lines; P = 6.33578 × 10−7. P values presented were calculated using two-tailed unpaired Student’s t tests. Scale bar = 10 μm for all images.
In mammals, the Mecom locus encodes for three distinct TFs (PRDM3, EVI1 and MDS1) that are implicated in diverse biological processes, including cell proliferation, invasion and differentiation16,17,18. Partial loss-of-function mutations of Mecom in mice lead to embryonic hypocellularity in the neuroectoderm and failure of peripheral nerve formation19. egl-43, the sole C. elegans ortholog, encodes a C2H2 zinc-finger (ZF) TF possessing six evolutionarily conserved ZF domains (Fig. 2b)20. EGL-43 has been primarily studied in non-neuronal tissues but has been implicated in the migration of various neurons20,21,22,23. Notably, fos-1 is a key target of EGL-43 in anchor cell invasion23,24.
To study the expression of EGL-43 in the nervous system, we generated an endogenous green fluorescent protein (GFP)-tagged allele of egl-43. EGL-43 is first detected during embryogenesis in sensory neuron precursor nuclei (Supplementary Fig. 2a) and remains nuclear localized in sensory neurons throughout and after development (Supplementary Fig. 2b,c). Notably, EGL-43 is expressed in all dopaminergic neurons, including PDE. To understand the temporal activation of EGL-43 in neuronal differentiation, we compared the expression of EGL-43 to the well-characterized pro-neuronal basic-helix–loop–helix TF, HLH-2/E/Daughterless, which is ubiquitously expressed in neuronal lineages25. We generated an endogenous mScarlet-tagged allele of hlh-2 and simultaneously imaged HLH-2 and EGL-43 in L2 to follow the V5.paa lineage, which ultimately gives rise to PVD and PDE (Supplementary Fig. 3). HLH-2 is first detected in the V5 seam cell (neuronal precursor) and is continuously expressed during subsequent cell divisions that generate PVD and PDE. Shortly after each cell has conferred neuronal fate, HLH-2 expression disappears. In contrast, strong expression of EGL-43 is only found in postmitotic neurons and is maintained until death (Supplementary Fig. 2b). These results suggest that EGL-43 is expressed in nascent postmitotic neurons, likely after commitment to become a neuron.
Loss of EGL-43 affects presynaptic protein expression
Because egl-43 is rapidly expressed during PDE development, we tested its requirement for synapse formation. Null alleles of egl-43 exhibit lethality and animals die before PDE is born22. To circumvent this, we used the auxin-inducible degron (AID) system where proteins of interest are tagged with an AID* tag, allowing for temporally precise degradation. In the presence of the synthetic hormone auxin, an exogenously expressed F-box protein, TIR1, recognizes AID*-tagged proteins, leading to poly-ubiquitination and subsequent degradation by the proteasome26. We used a strain carrying endogenous egl-43::TagRFP-T::AID* paired with a single-copy transgene expressing TIR1 ubiquitously26.
Shortly after PDE birth, but before axon outgrowth, L2s were moved onto media containing either 0 mM or 4 mM auxin and treated until L4. Degradation of EGL-43 led to a striking decrease of both endogenous ELKS-1 and RAB-3 along the axon of PDE (Fig. 2d–g). The reduction of synaptic markers was more pronounced in the distal axon than in the proximal axon, possibly due to proximal synapses forming first with less time for degradation of EGL-43 to occur. With this protocol, we did not detect any growth defects, exemplified by the presence of sparse synaptic markers in distal axons (arrowheads in Fig. 2d,f) and the presence of microtubules throughout the axon. This observation suggests that certain aspects of neurodevelopment are independent of EGL-43. Notably, auxin treatment of animals lacking egl-43::TagRFP-T::AID* did not possess defects in synaptic markers (Supplementary Fig. 4), indicating that our degradation protocol is specific.
EGL-43 binds a conserved DNA binding sequence
Having established that EGL-43 is critical for presynaptic protein expression in PDE, we next investigated how EGL-43 promotes synaptogenesis. We analyzed a whole-animal chromatin immunoprecipitation followed by sequencing (ChIP–seq) dataset of endogenous EGL-43 at L3 (ref. 24) and found a strong signal within 250 bp of transcription start sites (TSSs; Fig. 3a). EGL-43 binds numerous potential targets, but with minimal neuronal enrichment by Gene Ontology (GO) term analysis (Supplementary Fig. 5 and Supplementary Table 6). However, EGL-43 is expressed in many non-neuronal cells, which may contribute to the lack of neuronal terms we observed.

a, Aggregation plot of EGL-43 ChIP–seq signal across C. elegans genes. b, Aggregation plot of EGL-43 dopaminergic-specific NanoDam signal across C. elegans genes. For a and b, bold lines represent the mean signal, darker shading represents the estimated s.e. and lighter shading represents the 95% confidence interval. c, Venn diagram comparing EGL-43 ChIP–seq and dopaminergic-specific NanoDam datasets. Numbers represent peaks, either overlapping or unique to each dataset. d, GO analysis performed on the overlapping peaks between ChIP–seq and NanoDam datasets (PANTHER biological process). e, JASPAR TF binding profile for mouse (M. musculus) MECOM. f, Proportion of peaks within EGL-43 profiling datasets that contain a 5′-AAGATAA-3′ motif. g, Line scans of three PDE axons in animals carrying FLP-on ELKS-1 in the wild type and elks-1(wy1823), which possess a mutant EGL-43 motif. h, Quantification of PDE axon line scans of animals shown in f. P = 2.50832 × 10−6 and was calculated using a two-tailed unpaired Student’s t test. n = 15 for both genotypes. Medians are represented in thick dashed lines, and quartiles are represented in thin dashed lines. i, Representative micrographs of endogenous DAT-1 signal in animals carrying EGL-43::TagRFP-T::AID*. Animals were subjected to auxin treatment for 48 h. Arrowheads denote cell bodies of dopaminergic neurons (white, 0 mM auxin, and pink, 4 mM auxin). Numbers indicate the number of animals that resemble the phenotype shown in images. This experiment was repeated four times with similar penetrance. j, Representative micrographs of endogenous DAT-1 signal in wild-type animals and animals and dat-1(wy1824), which possess a mutant EGL-43 motif. White and yellow arrowheads indicate dopaminergic neuron cell bodies of respective genotypes. Numbers indicate the number of animals that resemble the phenotype shown in images. This experiment was repeated thrice with similar penetrance. Scale bar = 10 μm for all images.
Given the strong phenotype upon loss of EGL-43 in PDE, we investigated the targets of EGL-43 in dopaminergic neurons. We turned to tissue-specific genomics using NanoDam, a modified version of DamID27. Although NanoDam results in larger peaks and does not provide the same resolution as ChIP–seq, it enables the identification of targets on a tissue-specific basis27. Briefly, a Dam-methylase fused to a GFP-nanobody was driven using the dat-1 promoter and inserted into the genome as a single-copy transgene. This transgene was then crossed into worms carrying endogenous EGL-43::GFP. The Dam-methylase nanobody fusion is recruited to genomic regions where EGL-43 is bound, leading to proximity-based labeling of DNA. We examined an EGL-43 NanoDam dataset that we have recently generated28 and detected large peaks upstream of TSSs (Fig. 3b), complementing our observations by ChIP–seq. We observed a significant overlap between dopaminergic-specific NanoDam and whole-animal ChIP–seq (955/2,479 peaks, Fisher’s exact test: P = 2.8179 × 10−158; Fig. 3c and Supplementary Tables 2 and 3). GO term analysis revealed strong enrichment for neuronally associated terms in the overlapping genes (Fig. 3d and Supplementary Tables 7 and 8). Combining the findings from both datasets, EGL-43 was bound to genes encoding many presynaptic proteins, including synaptic vesicle-associated proteins, such as the Rab GTPase rab-3/RAB3, and active zone proteins, such as elks-1/Elks, cla-1/Bassoon/Piccolo, syd-2/Liprin-α and unc-10/Rim.
Because EGL-43 binds to many genes encoding presynaptic proteins and loss of EGL-43 reduces presynaptic protein intensity, we wondered if EGL-43 directly activates presynaptic gene expression. Although the consensus binding motif of EGL-43 has never been characterized, its ZF domains are highly homologous (~80%) to those of MECOM20. The binding motif of the first four ZF domains of mouse MECOM has been well studied, with a consensus sequence of 5′-GA(C/T)AAGA(T/C)AAGATAA-3′ (ref. 29; Fig. 3e). We hypothesized that EGL-43 binds a similar sequence and examined our datasets for the core motif of 5′-AAGATAA-3′. We found that 22% of ChIP–seq peaks and 36% of NanoDam peaks contained this putative motif (Fig. 3f). However, of the overlapping peaks between the datasets, the motif was present in 73% (700/955) of the peaks, supporting our hypothesis that EGL-43 binds a conserved sequence.
We scanned the combined EGL-43 NanoDam and ChIP–seq peaks within the promoters of multiple presynaptic genes, including elks-1 and the dopamine transporter dat-1, and found several putative motifs (5′-AAAAGATAG-3′ and 5′-AAAAGATAA-3′) that resemble the consensus sequence for MECOM. These motifs were present in regions of open chromatin, as identified through assay for transposase-accessible chromatin with sequencing (ATAC–seq) of neuronal nuclei, reinforcing the idea that EGL-43 might directly bind DNA to modulate gene expression (Supplementary Fig. 6a). To validate binding, we performed ChIP–quantitative polymerase chain reaction (qPCR) on endogenous EGL-43 using primer sets that span elks-1. We found increased binding of EGL-43 to an upstream region corresponding to a ChIP–seq peak containing a putative EGL-43 motif (Supplementary Fig. 6b).
We then tested if EGL-43 binding is necessary to modulate gene expression by mutating its putative binding sites. Using genome editing, we mutated the putative site (5′-AAAAGATAG-3′ to 5′-AACCCCCCC-3′) upstream of elks-1 in animals carrying endogenously FLP-on::GFP::elks-1. This permutation was shown in vitro to abolish the binding of EVI1, a TF encoded by Mecom, to the phosphatase and tensin homolog locus30. Mutation of this sequence reduced elks-1 transcript levels (Supplementary Fig. 7a), ultimately leading to reduced ELKS-1 intensity in PDE (Fig. 3g,h). Although this reduction in ELKS-1 is not as prominent as that observed upon degradation of EGL-43, it indicates that other factors, which may be controlled by EGL-43, contribute to elks-1 expression.
Having found a critical role for EGL-43 in the expression of elks-1, a core synaptic protein, we next considered if EGL-43 was more broadly regulating presynaptic proteins. The synaptic function of dopaminergic neurons is also critically dependent on dopamine transporters. We endogenously tagged dat-1, the plasma membrane dopamine transporter and the ortholog of human DAT, which is continuously expressed in all dopaminergic neurons (cephalic (CEP), anterior deirid (ADE) and PDE (Fig. 3i)). Auxin-induced degradation of EGL-43 at L2 resulted in a completely undetectable GFPnovo2::DAT-1 signal in PDE (Fig. 3i) by L4. We then tested if abolishing the binding of EGL-43 to dat-1 would similarly decrease DAT-1 expression. Using genome editing, we mutated the putative binding site (5′-AAAAGATAA-3′ to 5′-AACCCCCCC-3′) upstream of dat-1 and observed a striking reduction of DAT-1, to the extent where GFP was only detected in the cell body (Fig. 3j). We performed quantitative PCR with reverse transcription and confirmed that this mutation reduced dat-1 transcripts (Supplementary Fig. 7b), indicating that EGL-43 directly controls dat-1 expression. Although our degradation experiments did not yield as strong reduction of DAT-1 in other neurons compared to PDE (Fig. 3i), this may be due to limited exposure to auxin as those neurons are born and express DAT-1 embryonically. Together, these data provide evidence that EGL-43 functions as a transcriptional activator by binding directly to presynaptic genes to promote expression.
FOS-1 is critical for dopaminergic presynaptic gene expression
Increasing evidence suggests that synapse formation is regulated by activity, possibly through activity-triggered TF activation31. Therefore, we considered if EGL-43 links activity and synapse formation by activating IEGs to modulate synaptic gene expression. Consistent with mammalian studies, EGL-43 activates multiple IEGs, including fos-1/Fos during the development of non-neuronal cells23. FOS-1 has been implicated in activity-dependent axonal regeneration but has not been thoroughly investigated for its role in neuronal development32.
C. elegans FOS-1 is normally expressed constitutively in all cells, including PVD and PDE (Fig. 4a). To understand the relationship between FOS-1 and EGL-43, we generated animals carrying both endogenous FOS-1::GFP and EGL-43::TagRFP-T::AID*. Auxin-induced degradation of EGL-43 decreased FOS-1 intensity in both PVD and PDE (Fig. 4a,b). Auxin treatment did not affect the expression of FOS-1 in animals lacking EGL-43::TagRFP-T::AID* (Supplementary Fig. 8a). These results indicate that FOS-1 expression is partially dependent on EGL-43, further supported by the presence of EGL-43 ChIP–seq peaks at the fos-1 locus (Supplementary Table 1).

a, Images of L4 animals carrying endogenous FOS-1::GFP and EGL-43::TagRFP-T::AID* treated with 0 mM or 4 mM auxin for 48 h. PVD and PDE neuronal nuclei are outlined (white dashed circles). Auxin experiments were repeated independently with similar results (n = 3). b, Quantification of FOS-1::GFP intensity of nuclei (n = 15 for both conditions). c, Venn diagram displaying overlap of peaks between dopaminergic FOS-1 NanoDam and EGL-43 NanoDam. d, Left: GO term analysis of genes mapping to FOS-1 NanoDam peaks. Right: GO term analysis of genes mapping to the overlap between FOS-1 and EGL-43 NanoDam peaks. e, Line scans of FLP-on GFP ELKS-1 in PDE axons. The top two sets of line scans correspond to animals carrying endogenous FOS-1::TagRFP-T::AID* that were treated with either 0 mM or 4 mM auxin. Images were thresholded in the same fashion. The line scan denoted with single asterisk was thresholded to increase the visibility of synapses for illustrative purposes. Arrowheads point to small synapses that are nearly undetectable in the distal axon. The final set of PDE line scans corresponds to FLP-on ELKS-1 expression in animals with the AP-1 site deleted upstream of the TSS of elks-1. f, Quantification of line scans in e, where n = 14 for all conditions. For graph comparisons, P = 5.77664 × 10−21 for auxin (−) versus auxin (+) and P = 8.76057 × 10−21 for auxin (−) versus ΔAP-1 site. g, Line scans of FLP-on GFP RAB-3 in PDE axons of animals carrying FOS-1::TagRFP-T::AID*, treated with 0 mM or 4 mM auxin. h, Quantification of animals i (n = 15 for both conditions, P = 5.21856 × 10−8). For all graphs, medians are represented in thick dashed lines, and quartiles are represented in thin dashed lines. P values were calculated using two-tailed unpaired Student’s t tests. Scale bar = 10 μm for all images.
To assess if EGL-43 is acting specifically or broadly affecting the expression of nuclear proteins, we tested the requirement of EGL-43 on endogenous SWSN-4 expression. swsn-4/Brg1 encodes a subunit of the SWI/SNF chromatin remodeling complex and is expressed broadly, similar to fos-1. The swsn-4 locus lacks EGL-43 ChIP–seq peaks, unlike fos-1, and degradation of EGL-43 did not affect SWSN-4::GFP (Supplementary Fig. 8). Thus, it is likely that EGL-43 controls a discrete genetic program as opposed to causing global changes in transcription.
Next, we wondered if FOS-1 modulates the expression of similar genes as EGL-43. FOS-1 NanoDam peaks were associated with a functionally diverse set of targets with no apparent neuronal enrichment (Fig. 4c,d and Supplementary Table 9). However, when we compared FOS-1 NanoDam to EGL-43 NanoDam, we found significant overlap (871/2,719 FOS-1 peaks; Fisher’s exact test, P = 3.2653 × 10−298), with enrichment of neuronally associated GO terms (Fig. 4d and Supplementary Tables 5 and 10). These results suggest that the expression of many neuronal genes might require binding of both EGL-43 and FOS-1.
To test this idea, we generated animals expressing endogenous FOS-1::TagRFP-T::AID* and visualized synaptic markers upon FOS-1 degradation. Degradation of FOS-1 caused a 99% reduction in ELKS-1 intensity, with synapses becoming detectable only with increased thresholding (Fig. 4e,f). Similarly, but to a lesser extent, we observed a 55% reduction in RAB-3 intensity (Fig. 4g,h). To test if the direct binding of FOS-1 to synaptic genes is critical for their expression, we deleted the AP-1/FOS-1 site upstream of elks-1. Loss of this site dramatically reduced ELKS-1 intensity, phenocopying loss of FOS-1 (Fig. 4e,f). Together, these data demonstrate that both FOS-1 and EGL-43 directly regulate presynaptic gene expression in PDE.
Because FOS-1 is expressed ubiquitously and EGL-43 is expressed in many neurons, including multiple head neurons (Fig. 2c and Supplementary Fig. 2b), we tested the requirement of EGL-43 and FOS-1 on presynaptic gene expression in the nerve ring, the C. elegans neuropil. The nerve ring is situated in the head of the animal and contains the majority of the worm’s synapses, as labeled by SYD-2/Liprin-α, a major structural component of active zones10 (Supplementary Fig. 9a). Auxin-induced degradation of either EGL-43 or FOS-1 for 48 h resulted in a 17% and 21% reduction of endogenous SYD-2, respectively (Supplementary Fig. 9b). Next, we tested the requirement of EGL-43 on rpm-1/Highwire/Pam, a RING E3 ubiquitin ligase that functions in parallel with SYD-2 to regulate synapse formation33. We did not detect any changes in endogenous RPM-1 expression upon EGL-43 degradation (Supplementary Fig. 9c). Thus, EGL-43 and FOS-1 control the expression of specific subsets of synaptic genes, as opposed to broadly affecting pan-neuronal gene expression.
EGL-43 participates in a positive feedback loop with FOS-1
Because both EGL-43 and FOS-1 bind and activate synaptic genes, we investigated the interplay between them. We degraded FOS-1 beginning in L2 and imaged EGL-43::GFP in L4. Loss of FOS-1 led to a slight but significant decrease of EGL-43 in PDE (Fig. 5a,b). This result, together with the data in Fig. 4a,b, suggests that egl-43 and fos-1 participate in a positive feedback loop where their expression is partially dependent on each other.

a, L4 animals carrying EGL-43::GFP and FOS-1::TagRFP-T::AID* were treated with 0 mM or 4 mM auxin for 48 h to degrade FOS-1. PVD and PDE nuclei are outlined (white dashed circles). b, Quantification of EGL-43::GFP intensity shown in a (n = 16 for both conditions). c, JASPAR TF binding profiles for C. elegans FOS-1 (top) and H. sapiens FOS (bottom). d, SNP effect matrix of H. sapiens FOS. The purple rectangle highlights the position of the consensus FOS motif that differs in wild-type C. elegans. e, EGL-43::GFP expression in either the wild type or in animals carrying a gain-of-function (denoted as egl-43(gof)) mutation in their FOS-1-binding site upstream of the egl-43. PVD and PDE nuclei are outlined (white dashed circles). This schematic was created with BioRender.com. f, Quantification of EGL-43::GFP intensity shown in e (n = 14 for both conditions). g, Line scans of FLP-on ELKS-1 in the PDE axon in the wild type and animals carrying egl-43(gof). h, Quantification of line scans of animals shown in g (n = 10 for both conditions, P = 5.45276 × 10−7). i, Line scans of FLP-on RAB-3 in the PDE axon in the wild type and animals carrying egl-43(gof). j, Quantification of line scans of animals shown in i (n = 10 for both conditions, P = 2.18919 × 10−5). For all graphs, medians are represented in thick dashed lines and quartiles are represented in thin dashed lines. P values displayed in this figure were calculated using two-tailed unpaired Student’s t tests. Scale bar for all images = 10 μm.
In C. elegans, the FOS-1/AP-1 conserved binding motif is a perfect palindrome of 5′-TGACTCA-3′ and is highly homologous to the binding motif of mammalian FOS (Fig. 5c). The FOS-1/AP-1 site upstream of egl-43 deviates from the consensus by a single nucleotide (5′-TTACTCA-3′), which is conserved across other Caenorhabditis species (Supplementary Fig. 10)22. Previous computational work on TF binding site mutations predicts that mutation of the FOS site at this position results in a 30% decrease in binding affinity (Fig. 5d)34. We wondered if changing this suboptimal FOS-1 site to the consensus sequence would affect the expression of egl-43. Using genome editing, we mutated a single nucleotide within the FOS-1 site from 5′-TTACTCA-3′ to 5′-TGACTCA-3′ in animals carrying endogenous EGL-43::GFP. This manipulation dramatically increased EGL-43 intensity in both PVD and PDE (denoted as gain-of-function (gof); Fig. 5e,f), suggesting that egl-43 activation is highly dependent on FOS-1 binding. Consequently, PDE axons of egl-43(gof) animals exhibited increased intensity of ELKS-1 and RAB-3 by 76% and 40%, respectively (Fig. 5g–j). These results illustrate the intricate molecular regulation between egl-43 and fos-1 and demonstrate that EGL-43 levels are critical for maintaining optimal levels of presynaptic gene expression.
Manipulation of EGL-43/FOS-1 impacts a behavioral circuit
Because we observed large changes in synaptic protein intensity through our manipulations, we investigated whether there was a functional consequence to these changes in behavior. All C. elegans dopaminergic neurons are mechanosensitive and sense stimuli from their food, bacteria35. Upon contacting bacteria, the neurons activate and trigger the basal slowing response, an adaptive mechanism that attenuates locomotion to increase time spent in the presence of food35. Previous studies revealed that artificial activation of dopaminergic neurons using ChR2 induces backing behavior36,37.
In the presence of food, we expressed a dopaminergic-specific ChR2 transgene to activate dopaminergic neurons. Constant blue light stimulation elicited a backing response followed by a reversal in wild-type worms, consistent with the literature (Fig. 6a)37. We assayed each worm five times, with 15 s between each trial, and measured the time from the onset of light to backward locomotion. Wild-type animals performed backing in 1.85 ± 0.22 s (Fig. 6a). Animals reared in the absence of all-trans-retinal (ATR), the ChR2 cofactor, did not elicit any backing in response to light within 8 s (n > 50). Next, we assayed egl-43(gof) animals that possess increased levels of EGL-43. Animals responded to light in 1.40 ± 0.43 s, significantly faster than the wild type (Fig. 6b). Thus, increasing EGL-43 expression and increasing synaptic protein levels accelerate the activation of a behavioral circuit.

a, Schematic representation of the backing assay. ChR2 was specifically expressed in dopaminergic neurons using the dat-1 promoter. Healthy, fed worms move forward, foraging on a bacterial lawn. On constant excitation by blue light, dopaminergic neurons activate and induce a backing response, followed by a reversal. The time from the onset of the blue light to a backing response was recorded. Wild-type worms perform the backing behavior in under 2 s under our conditions. b, Animals carrying a mutation to enhance FOS-1 binding upstream of the egl-43 locus exhibit a faster backing response than the wild type (n = 20 for both conditions, P = 2.60044 × 10−6). c, Auxin-induced degradation of either EGL-43 or FOS-1 results in a significantly slower backing response. n = 14 for all conditions tested; P = 0.0002 (−auxin versus +auxin for EGL-43), P = 2.52982 × 10−8 (−auxin versus +auxin for FOS-1). For all graphs, medians are represented in thick dashed lines, and quartiles are represented in thin dashed lines. P values were calculated using two-tailed unpaired Student’s t tests.
Because the loss of EGL-43 or FOS-1 strongly reduces presynaptic gene expression, we wondered if their losses would similarly affect the ChR2-induced backing response. We degraded EGL-43 or FOS-1 at L2 and assayed animals at L4. Loss of EGL-43 or FOS-1 dramatically delayed response time, where time to backing was 3.3 ± 1.27 s and 4.3 ± 1.17 s, respectively (Fig. 6c). Collectively, our experiments demonstrate that EGL-43 and FOS-1 directly control presynaptic gene expression, ultimately influencing the strength of synaptic signaling in dopaminergic circuits.
EGL-43 activates activity-regulated and developmental TFs
Having demonstrated that EGL-43 can promote the expression of FOS-1 to modulate presynaptic gene expression, we explored if EGL-43 activates other activity-regulated TFs. We observed EGL-43 ChIP–seq peaks at the jun-1/Jun, crh-1/Creb, egrh-1/Egr1 and mef-2/Mef2 loci and used genome editing to fluorescently tag each TF. All four TFs are expressed broadly, similar to fos-1 (Fig. 7a–d). We performed auxin-induced degradation of EGL-43 beginning in L2 and examined TFs at L4. Remarkably, loss of EGL-43 increased JUN-1 expression, opposite to our observations with FOS-1, suggesting that EGL-43 might negatively regulate jun-1 (Fig. 7a). Different from jun-1, loss of EGL-43 decreased CRH-1 and EGRH-1 (Fig. 7b,c) but did not significantly affect MEF-2 expression (Fig. 7d). Because MEF2 is implicated in activity-dependent synapse elimination38, we wondered if MEF-2 functions in opposition to EGL-43 to control synaptogenesis. Using genome editing, we generated a full deletion of mef-2 but found no significant differences in synaptic markers in PDE (Supplementary Fig. 11a–d). Our findings suggest that EGL-43 controls a specific network of activity-regulated TFs, where CRH-1 and EGRH-1 might function together with FOS-1 to control presynaptic gene expression.

a–l, Left: images of L4 animals carrying endogenously tagged GFP or mNG TFs and EGL-43::TagRFP-T::AID* treated with 0 mM (A(−)) or 4 mM auxin (A(+)) for 48 h. PVD and PDE neuronal nuclei are outlined (white dashed circles). Right: quantification of TF intensity in the images shown on the left. Classification of the following factors tested with sample sizes: activity-dependent TFs JUN-1, n = 11 (A−) and n = 11 (A+) (a); CRH-1, n = 13 (A−) and n = 13 (A+) (b); EGRH-1, n = 10 (A−) and n = 10 (A+) (c); MEF-2, n = 10 (A−) and n = 11 (A+) (d). Classification of the following dopaminergic terminal selectors/associated factors: CEH-43, n = 14 (A−), n = 21 (A+), P = 8.6174 × 10−6 (e); AST-1, n = 15 (A−), n = 15 (A+) (f); CEH-20, n = 15 (A−), n = 15 (A+), P = 3.84033 × 10−11 (PDE) (g); UNC-62, n = 12 (A−), n = 14 (A+), P = 1.15877 × 10−12 (PVD), P = 2.78114 × 10−8 (PDE) (h). CUT homeobox TFs are as follows: CEH-38, n = 12 (A−), n = 10 (A+) (i); CEH-41, n = 12 (A−), n = 9 (A+), P = 4.09353 × 10−9 (PVD) (j); CEH-44 n = 8 (A−), n = 10 (A+) (k); CEH-48, n = 12 (A−), n = 10 (A+) (l). m, Line scans of PDE axons from animals expressing endogenous FLP-on RAB-3. Mutations were generated in the promoter of rab-3 corresponding to the CUT, EGL-43 or both CUT and EGL-43 motifs. n, Quantification of the line scans shown in m (n = 15 for all genotypes). For all graphs, medians are represented in thick dashed lines, and quartiles are represented in thin dashed lines. P values were calculated using ordinary one-way ANOVA except for e and f, which were calculated using two-tailed unpaired Student’s t tests. Scale bar = 5 μm for images of nuclei; scale bar = 10 μm for line scans of PDE axons.
Genetic screens in C. elegans have identified multiple terminal selectors that are critical for dopaminergic identity. Mutations in three genes, ceh-43/Dlx, ast-1/Etv1 and ceh-20/Pbx, all result in the loss of dopaminergic identity markers such as DAT-1 but do not affect the expression of pan-neuronal markers such as RAB-3 (refs. 39,40). Additionally, unc-62/Meis functions together with terminal selectors in PDE to specify dopaminergic identity41. Because degradation of EGL-43 results in the loss of both DAT-1 and RAB-3 expression, we asked if EGL-43 acts upstream of these TFs. We observed EGL-43 ChIP–seq peaks present at all four genes and used genome editing to fluorescently tag each TF. Degradation of EGL-43 reduced the expression of all four TFs in PDE, indicating that EGL-43 modulates terminal selector expression (Fig. 7e–h). Others have reported that the loss of individual terminal selectors results in only partial loss of DAT-1 due to compensation by other terminal selectors39,40. However, we found that degradation of EGL-43 resulted in a complete loss of DAT-1 in PDE, consistent with our data that EGL-43 functions upstream.
Unlike terminal selectors, CUT TFs are expressed throughout the nervous system and bind and regulate pan-neuronal genes, such as rab-3 (ref. 3). We observed EGL-43 ChIP–seq peaks near all CUT genes, and we tested the requirement of EGL-43 on four previously described tagged CUT alleles3. Degradation of EGL-43 significantly reduced CEH-41 and CEH-48 expression, but not CEH-38 and CEH-44 (Fig. 7i–l), indicating that EGL-43 controls specific CUTs in PDE. We then investigated the synergy between EGL-43 and CUTs to control RAB-3 expression. Using genome editing, we mutated the CUT, EGL-43 or both motifs upstream of rab-3 and imaged PDE axons at L4. These mutations resulted in similar decreases in RAB-3 intensity, suggesting that EGL-43 and CUTs might function in the same pathway to activate pan-neuronal genes.
Discussion
During development, synapses assemble in a rapid and organized fashion. To accomplish this, neurons use spontaneous and sensory-driven activity-dependent mechanisms to drive the transcription of specific genes. Using a model neuron, we discovered that EGL-43/MECOM and FOS-1/FOS function together to drive synapse formation. First, we found that both TFs bind promoters of many synaptic genes. Second, depletion of either TF reduces synaptic protein levels. Third, mutation of either TF’s binding sites in synaptic genes reduces synaptic protein levels. Finally, EGL-43 modulates the expression of genetic programs that constitute different aspects of neuronal identity. Notably, bidirectionally manipulating activity impacts FOS-1 expression and subsequent synaptic protein expression, suggesting that activity-dependent mechanisms for synaptogenesis use the EGL-43/FOS-1 program.
In addition to directly activating presynaptic genes, EGL-43 and FOS-1 function in a positive feedback loop. They each bind to the promoter of the other gene and are mutually required for their expression. Optimizing the FOS-1 site upstream of egl-43 profoundly increased EGL-43 levels, resulting in increased synaptic signaling and behavioral changes. Remarkably, the suboptimal site is conserved across millions of years of evolution22, suggesting positive selection pressure to maintain particular levels of EGL-43, and thus FOS-1, for survival.
Presynaptic assembly is a complex biological process that involves transcription of active zone and synaptic vesicle genes, selective expression of neurotransmitter phenotype-related genes and specification of presynaptic membrane by transsynaptic adhesion molecules. Recent work in C. elegans revealed that CUT TFs regulate pan-neuronal gene expression and function together with terminal selectors to define neuronal identity and function3. One important question is how these hardwired genetic programs are regulated to ensure functional connectivity between neurons.
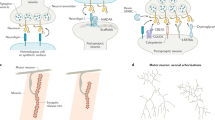
We propose that during development, pro-neuronal signaling activates EGL-43 to generate a robust activity-regulated gene expression program (Fig. 8). Here EGL-43 activates both terminal selectors and CUTs to execute expression of key neuronal genes. Simultaneously, EGL-43 and FOS-1 bind to presynaptic genes to modulate their expression. Notably, EGL-43 participates in a positive feedback loop with FOS-1, which is activated by neuronal activity. We hypothesize that during synaptogenesis, activity stimulates the EGL-43/FOS-1 loop, modulating presynaptic gene expression.

EGL-43 functions downstream of the initial pro-neuronal fate commitment and is a key organizer of synaptogenesis in dopaminergic neurons. Together with the activity-dependent TF FOS-1, EGL-43 functions in a positive feedback loop and binds upstream regulatory regions of synaptic targets to promote gene expression. Simultaneously, EGL-43 promotes the expression of hardwired genetic programs, such as terminal selector and pan-neuronal (CUT) TFs, that in turn also bind presynaptic loci and promote transcription through the same pathway. We hypothesize that during development, neuronal activity can modulate the strength of synaptic gene expression through EGL-43 and FOS-1 to ensure functional connectivity. This schematic was created with BioRender.com.
Strikingly, we found that EGL-43 is expressed predominantly in sensory neurons. Sensory neurons constantly monitor the environment, where their signaling leads to behavioral changes. For example, the thermosensory amphid finger (AFD) neuron uses short-term transcription-independent and long-term transcription and experience-dependent plasticity mechanisms to modulate behavior in response to environmental temperature42. Because the EGL-43/FOS-1 program can rapidly activate synaptic genes, it is plausible that sensory neurons use this program to enable plasticity and fine-tune behavior. Unlike sensory neurons, we did not detect EGL-43 in any motoneurons. The C. elegans motoneurons are a critical component of the locomotion circuit, which generates stereotyped forward and backward movement. Compared to the myriads of sensory modality, the wide range of stimulus intensity and rapid changes in response, the locomotion program is relatively simple and rigid, and thus may not require the plasticity mechanisms enabled by EGL-43.
A large body of evidence supports the notion that activity shapes synaptic connectivity during development4. Activity-regulated TFs, such as CREB, FOS and JUN, have important roles in this process. Here we demonstrate that FOS-1 directly activates presynaptic genes, providing a potential mechanism that underlies activity-regulated modification of synaptic circuits. In vertebrates, the majority of AP-1/FOS sites reside in enhancer regions43. Conversely, due to its compact genome size, C. elegans FOS-1 sites are found near TSSs44. While future experiments are needed to determine whether FOS can directly control synaptic programs in vertebrates, our work identifies a crucial link between activity and genetic programs to modulate synaptic gene expression.
Methods
C. elegans culture and maintenance
Nematodes were cultured under normal conditions on nematode growth media (NGM) plates seeded with Escherichia coli OP50 at 20 °C. To make histamine plates, histamine dihydrochloride (Sigma-Aldrich) was dissolved in water to generate a 1 M stock solution. Fresh histamine stock solution was added to molten NGM agar at approximately 65 °C to a final concentration of 10 mM. The worm strains used in this study are listed in Supplementary Table 13. All experiments used hermaphrodite worms, and the stages used for each experiment are outlined in each figure legend.
Auxin depletion
NGM plates were supplemented to a final concentration of 4 mM K-NAA (auxin salt; PhytoTech Labs) from a 1 M stock solution. Auxin-containing plates were seeded using OP50 bacteria and stored at 4 °C for up to 2 weeks before use. Well-fed hermaphrodite worms were moved onto seeded auxin plates at the L2 stage and imaged at the L4 stage, unless otherwise noted.
Molecular biology, cloning and microinjection
For genotyping, worms were lysed in worm lysis buffer as previously described. Worm lysates were amplified using either SuperFi II MasterMix (Thermo Fisher Scientific) or CloneAmp (Takara). To generate pCY37 (dat-1p::ChR2), a fragment containing the dat-1 promoter was digested from pCM558 (dat-1p::Cre) using SphI and AscI and purified using a Qiagen Gel Extraction Kit. pTLs30 (ser-2prom3::ChR2::unc-54 3′UTR) was digested with SphI and AscI, and the vector backbone containing ChR2 was similarly purified. The resulting two fragments were ligated together overnight using T4 ligase (Thermo Fisher Scientific). Transgenes were generated using standard gonad transformation by injection.
Genome editing by CRISPR–Cas9
For fluorophore insertions, primers providing at least 50 nucleotides of homology to the genomic locus were used to amplify PCR repair templates. Plasmids pWZ203 (TagRFP-T::AID*), pCM552 (GFPnovo2), pJW2098 (mScarlet::AID*) and pJW2171 (mNeonGreen::AID*) were used as templates for PCR repair products. CloneAmp PCR reactions with a volume of 4 × 50 μl were performed to generate repair products, which were subsequently pooled and purified together using a Qiagen PCR Purification Kit. PCR products were eluted in 10 μl of water and quantified using a NanoDrop instrument (Thermo Fisher Scientific). PCR templates were melted immediately before use, as previously described. For binding site mutations, long primers (<100 nucleotides (nt); Thermo Fisher Scientific) and ultramers/gBlocks (>100 nt; Integrated DNA Technologies (IDT)) were resuspended in water to a concentration of 100 μM.
cRNAs (IDT) were resuspended to a stock concentration of 100 μM in duplex buffer (IDT). In total, 0.5 μl of 100 μM cRNA was added to a tube containing 3 μl of 10 μM tracrRNA and heated in a PCR machine at 95 °C for 5 min followed by 25 °C for 5 min. The cRNA:tracrRNA reaction was transferred to a 1.5 ml tube containing 0.5 μl of ALT-R Cas9 enzyme and heated in a 37 °C water bath for 15 min. In total, 5 μl of freshly melted PCR product was added to the tube (final concentration = ~300 ng μl−1), with 1 μl of co-injection plasmid (pRF4, final concentration = 50 ng μl−1) to bring the volume of the injection mixture to 10 μl. Injection mixes were spun at 13,000 rpm for 15 min and subsequently kept on ice during microinjection. Details of individual CRISPR edits (guides, oligos and locations) are found in Supplementary Table 11. In many cases, it was necessary to mutate the PAM site to prevent Cas9 from editing the repair template, where NGG was mutated to NGT.
Neuronal silencing and activation
For neuronal silencing experiments, wyEx8629 (dat-1p::HisCl1) worms were placed on plates containing 0 or 10 mM histamine seeded with OP50. At 20 °C, hermaphrodite worms were placed on histamine plates at the L2 stage and observed at the L4 stage.
For neuronal activation experiments, all worms were crossed into a lite-1(ce314) background and were grown at 20 °C on plates with OP50 mixed with or without ATR. ATR (Sigma-Aldrich) was dissolved in pure ethanol to create a 100 mM stock concentration, which was stored at −20 °C. In total, 5 μl of the 100 mM ATR stock was added to 1 ml of OP50 culture, and 100 μl was added to NGM plates. Plates were kept in the dark and used the following day. Worms carrying wyEx10629 and wyEx10630 were grown in the presence of ATR and stimulated using blue light emitted from a SOLA Light Engine (Lumencor) pulsed for ten cycles of 15 s (on) and 60 s (off). For FOS-1::GFP imaging experiments, worms were treated with blue light at L2 and allowed to recover for 2 h before imaging. For imaging FLP-on::GFP::ELKS-1, worms were treated with blue light at L2 and imaged at L4. For the neuronal activation backing/reversal assays, free, forward-moving worms were stimulated using constant blue light using a SOLA Light Engine. A timer was used to measure the time required from the onset of blue light until a backing response was invoked. Each animal was assayed five times, with 15 s rest between trials.
Microscopy and imaging
Worms were mounted on 3% agarose pads made by melting agarose in M9 buffer. Worms were immobilized using 10 mM levamisole, which was diluted in M9 buffer. Imaging was performed either on a Zeiss Axio Observer Z1 microscope paired with a Yokogawa CSU-X1 spinning-disk unit and a Hamamatsu EM-CCD digital camera controlled by Metamorph (v.7.8.12.0) and using either a Plan-Apochromat ×100 (1.4 numerical aperture (NA)) objective or a ×63 (1.4 NA) objective or on an inverted Zeiss Axio Observer Z1 microscope paired with a Yokogawa CSU-W1 spinning-disk unit and a Prime 95B Scientific CMOS camera with a C-Apochromat ×40 (0.9 NA) objective with ×1 or ×2 enhanced digital magnification controlled by 3i Slidebook (v6) software. Image settings (laser power and exposure time) were identical for all genotypes or treatments across experiments. Images were taken typically with 1 um step sizes in z stacks of 20–30 μm range.
Raw image files were processed using Fiji (ImageJ) software. For synapse analyses, image stacks were z projected using maximum projection, and axons were straightened using the plugin ‘straighten to line’ to generate line scans. Images were thresholded and analyzed identically using the ‘analyze particles’ function, where axon length and integrated synapse intensity were measured as previously described13. The integrated intensity for each synapse (particle) was then summed and finally divided by PDE axon length to obtain an intensity per μm value. For normalization, all intensity per μm values corresponding to the control group were first averaged to generate a mean intensity per μm value. Each individual value for both control groups and mutant/treated groups was then divided by the control mean intensity per μm value and plotted. For all other measurements, including nerve ring, and nuclei intensities, images were z-projected using ‘sum’ stack. Regions of interest were manually drawn and quantified using the ‘measure’ function.
RNA extraction and qPCR
Worms were washed thrice in 1.5-ml tubes with M9 buffer and aspirated to remove as much buffer as possible. Worm pellets were flash frozen in liquid nitrogen and thawed thrice to facilitate cracking of the cuticle. Total RNA was extracted using Trizol (Thermo Fisher Scientific) according to manufacturer protocols. cDNA libraries were generated using SuperScript III or SuperScript VILO (Thermo Fisher Scientific), and qPCR was performed on a CFX90 thermocycler (Bio-Rad) using SsoFast EvaGreen Master Mix (Bio-Rad). qPCR results were analyzed using the 2−ΔΔCT method.
ChIP–qPCR
Worms were bleach-synchronized and grown at 25 °C until the L3 stage (24 h after placing L1s on food), washed thrice in PBS supplemented with Complete Mini protease inhibitor (Roche) and flash frozen in liquid nitrogen to make worm popcorn. Worm popcorn was stored at −80 °C until three biological replicates were obtained. Worm popcorn was ground in a mortar in liquid nitrogen, and powder was added to ten volumes of PBS containing 1.1% formaldehyde, Complete Mini protease inhibitor (Roche) and phosphatase inhibitor cocktail (Calbiochem) and incubated for 10 min at room temperature with rocking. Formaldehyde was quenched by adding glycine to a final concentration of 125 mM, and the solution was pelleted and washed in cold PBS supplemented with 1 mM phenylmethylsulfonyl fluoride. The resulting pellet was resuspended in FA buffer (50 mM HEPES/KOH (pH 7.5), 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate and 150 mM NaCl) supplemented with 0.1% sarkosyl and the aforementioned protease and phosphatase inhibitors. The resuspended pellet was sonicated using an EpiShear probe sonicator (Active Motif) for 2× 13-min intervals at 80% power (30 s on and 30 s off). Following sonication, immunoprecipitation (IP) was performed using GFP-Trap Agarose (ChromoTek) according to manufacturer protocols, with unconjugated agarose beads used as a control. In total, 25 μl was reserved as input DNA. IP and input DNA were purified using ChIP elution buffer (250 mM NaCl, 1% SDS, 10 mM Tris–Cl (pH 8) and 1 mM EDTA) and incubated with proteinase K (Thermo Fisher Scientific) overnight at 65 °C. DNA was purified using a PCR Purification Kit (Qiagen). DNA was quantified using a Qubit dsDNA High Sensitivity Kit and a Qubit 4.0 fluorometer (Thermo Fisher Scientific). qPCR using primers targeting elks-1 was performed on the resulting purified input and IP DNA using SsoFast EvaGreen SuperMix (Bio-Rad) on a Bio-Rad CFX96 thermocycler. Primers used for these experiments are listed in Supplementary Table 12.
NanoDam profiling
Nematodes carrying a NanoDam driver or both a NanoDam driver and an endogenous GFP-tagged TF were bleached and grown on NGM plates seeded with E. coli dam−/−dcm−/− lawns. Worms were cultured for at least three generations and then bleach-synchronized and grown at 25 °C for 24 h to obtain synchronized L3 animals. Worms were washed thoroughly in M9 buffer and flash frozen in liquid nitrogen. Worm pellets were stored at −80 °C until needed.
Three biological replicates were obtained for each condition (NanoDam control and NanoDam + GFP allele), and all pellets were processed together to avoid batch effects. gDNA was extracted using a Qiagen DNeasy spin column kit (Qiagen) and resuspended in water. gDNA was digested using DpnI (New England Biolabs), and double-strand adaptors were ligated using T4 DNA Ligase (Thermo Fisher Scientific). Ligated DNA was next amplified using HiDi DNA Polymerase (myPols Biotec) and run on a 1% gel to check for amplification. PCR was performed with four initial long cycles, followed by 20 regular PCR cycles. Amplicons were purified using AMPure magnetic beads (Beckman Coulter) and quantified using a Qubit 4.0 with a Qubit dsDNA High Sensitivity Kit (Thermo Fisher Scientific). Nanopore sequencing libraries were prepared according to manufacturer protocols using a Ligation Sequencing Kit (SQK-LSK-109 (ONT)) and Native Barcoding Expansion Kit (EXP-NBD104 and EXP-NBD114). Nanopore sequencing of pooled libraries was performed on a Nanopore MinION Mk1C device using Spot-ON Flow Cells (v.R9; FLO-MIN106D). Base calling and demultiplexing of fast5 files were performed onboard the MinION MK1C using fast basecalling and a minimum Q quality score of 8. Fastq files were mapped to the C. elegans ce10 genome using minimap2 and converted to bam using samtools (v.1.14) damidseq_pipeline (v.1.5.3) and find_peaks (v.1.0.1) were used to subsequently call NanoDam peaks.
Flow cytometry and ATAC–seq
Mixed-stage worms carrying a pan-neuronal nuclear GFP (otIs381) transgene were grown at 20 °C and washed thrice with M9 buffer. Nuclei were collected as previously described using a stainless-steel tissue grinder (Wheaton). Suspended nuclei were filtered using 5-μm cell strainers (Corning) and kept on ice. Nuclei were labeled with DAPI (Thermo Fisher Scientific) according to manufacturer instructions and sorted using a BD FACSAria Fusion with a 70 µm nozzle equipped with a blue laser for GFP detection (488 nm, 525/50 filter with B525 detector) and a UV laser for DAPI detection (355 nm, 515/30 filter with U515 detector). The gating strategy is illustrated in Supplementary Fig. 12. Initial FSC and SSC gates were drawn to select against debris, which was verified by imaging on a compound microscope at the Stanford FACS core. Negative GFP gates were generated using N2 animals that did not carry the pan-neuronal marker. Approximately 100,000 positive events were collected for three biological replicates. Samples were spun down and immediately subjected to transposition using Nextera reagents (25 μl tagmentation DNA buffer), 2.5 μl TDE1 and 22.5 μl H2O; Illumina). The transposition reaction was incubated at 37 °C for 30 min, followed by purification using a PCR purification kit (Qiagen). Transposed DNA was next amplified using NEBNext High-Fidelity 2× PCR Master Mix and standard ATAC primers, with an initial qPCR step to determine the cycles required for amplification. PCR products were purified using AMPure magnetic beads (Beckman Coulter) and assayed on an Agilent Bioanalyzer 2100 for quality and success of the ATAC reactions (presence of histone ladder). The resultant Nextera libraries were sequenced on the HiSeq 2000 platform (2 × 101 bp; Illumina) at the Stanford Genome Sequencing Service Center. Fastq reads were trimmed, filtered for quality using FastQC (v.0.11.9), mapped to the C. elegans ce10 genome using Bowtie 2 (v.2.5.1) and processed using the standardized ENCODE consortium ATAC–seq pipeline to obtain peaks (atac-seq-pipeline (v.1.14)).
TF peak analysis and Gene Ontology
Because ChIP–seq reads generated many small peaks that are not reflective of the occurrence of GATC fragments used by DamID, we merged processed peaks obtained from the averaged ChIP–seq and NanoDam signals that were within 1,500 bp using bedtools (v.2.28) merge. ChIP–seq and NanoDam peaks for EGL-43 and FOS-1 were processed using HOMER(annotatePeaks, v.4.11) to associate peaks with nearby genes. The resulting gene lists from ChIP–seq and NanoDam experiments were analyzed using the PANTHER overrepresentation test (release: 7 October 2022) with the GO database (release: 1 July 2022) and compared against the C. elegans reference list for GO biological process complete terms using Fisher’s exact test.
Statistics, data visualization and reproducibility
All data were analyzed either in Microsoft Excel or GraphPad Prism 9. IGV (v.2.15.2) was used to visualize the ATAC–seq and ChIP–seq signals. SeqPlots (v.3.0.12) was used to generate z-score aggregation plots for ChIP–seq and NanoDam signals. The protein structure of MECOM-related proteins shown in Fig. 2a was drawn using R Studio (v.4.1.1) and the drawProteins package (v.3.16). For statistical tests, single pairwise comparisons of genotypes or treatments were analyzed using two-tailed unpaired Student’s t tests. Multiple comparisons were performed using one-way analysis of variance (ANOVA). Data distribution was assumed to be normal, but this was not formally tested. Data collection and analysis were not performed blind to the conditions of the experiments. No data exclusion was performed. No randomization was necessary due to the experimental conditions. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications3,13. All imaging data were replicated in at least three independent imaging sessions. All experiments showing representative images were repeated with similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequencing data generated in this study are publicly available in the Gene Expression Omnibus (GEO) GSE260637. The accession for the previously reported EGL-43 ChIP–seq data is GEO GSE144292. We have the rights to publish BioRender figures, and Figs. 1b, 2a, 5e and 8 and Supplementary Fig. 10 were generated using BioRender. All raw data and reagents are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Code availability
All packages and software, described in Methods, were used according to standard protocols and without modifications. Custom code was not generated in this study.
References
Deguchi, Y., Donato, F., Galimberti, I., Cabuy, E. & Caroni, P. Temporally matched subpopulations of selectively interconnected principal neurons in the hippocampus. Nat. Neurosci. 14, 495–504 (2011).
Hobert, O. & Kratsios, P. Neuronal identity control by terminal selectors in worms, flies, and chordates. Curr. Opin. Neurobiol. 56, 97–105 (2019).
Leyva-Diaz, E. & Hobert, O. Robust regulatory architecture of pan-neuronal gene expression. Curr. Biol. 32, 1715–1727 (2022).
Andreae, L. C. & Burrone, J. The role of neuronal activity and transmitter release on synapse formation. Curr. Opin. Neurobiol. 27, 47–52 (2014).
Wiesel, T. N. & Hubel, D. H. Single-cell responses in striate cortex of kittens deprived of vision in one eye. J. Neurophysiol. 26, 1003–1017 (1963).
Greenberg, M. E., Ziff, E. B. & Greene, L. A. Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Science 234, 80–83 (1986).
Lanahan, A. & Worley, P. Immediate-early genes and synaptic function. Neurobiol. Learn. Mem. 70, 37–43 (1998).
Herdegen, T. & Leah, J. D. Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res. Rev. 28, 370–490 (1998).
Sheng, M. & Greenberg, M. E. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron 4, 477–485 (1990).
McDonald, N. A., Fetter, R. D. & Shen, K. Assembly of synaptic active zones requires phase separation of scaffold molecules. Nature 588, 454–458 (2020).
Schwartz, M. L. & Jorgensen, E. M. SapTrap, a toolkit for high-throughput CRISPR/Cas9 gene modification in Caenorhabditis elegans. Genetics 202, 1277–1288 (2016).
Pokala, N., Liu, Q., Gordus, A. & Bargmann, C. I. Inducible and titratable silencing of Caenorhabditis elegans neurons in vivo with histamine-gated chloride channels. Proc. Natl Acad. Sci. USA 111, 2770–2775 (2014).
Maeder, C. I. et al. The THO complex coordinates transcripts for synapse development and dopamine neuron survival. Cell 174, 1436–1449 (2018).
Nagel, G. et al. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr. Biol. 15, 2279–2284 (2005).
Birdwell, C. et al. EVI1 dysregulation: impact on biology and therapy of myeloid malignancies. Blood Cancer J. 11, 64 (2021).
Chen, P. et al. MECOM promotes supporting cell proliferation and differentiation in cochlea. J. Otol. 17, 59–66 (2022).
Wang, Z. et al. EVI1 overexpression promotes ovarian cancer progression by regulating estrogen signaling. Mol. Cell Endocrinol. 534, 111367 (2021).
Leszczynski, P. et al. Deletiono of the Prdm3 gene causes a neuronal differentiation deficiency in P19 cells. Int. J. Mol. Sci. 21, 7192 (2020).
Kazama, H., Kodera, T., Shimizu, S., Mizoguchi, H. & Morishita, K. Ecotropic viral integration site-1 is activated during, and is sufficient for, neuroectodermal P19 cell differentiation. Cell Growth Differ. 10, 565–573 (1999).
Garriga, G., Guenther, C. & Horvitz, H. R. Migrations of the Caenorhabditis elegans HSNs are regulated by egl-43, a gene encoding two zinc finger proteins. Genes Dev. 7, 2097–2109 (1993).
Hwang, B. J., Meruelo, A. D. & Sternberg, P. W. C. elegans EVI1 proto-oncogene, EGL-43, is necessary for Notch-mediated cell fate specification and regulates cell invasion. Development 134, 669–679 (2007).
Rimann, I. & Hajnal, A. Regulation of anchor cell invasion and uterine cell fates by the egl-43 Evi-1 proto-oncogene in Caenorhabditis elegans. Dev. Biol. 308, 187–195 (2007).
Medwig-Kinney, T. N. et al. A developmental gene regulatory network for C. elegans anchor cell invasion. Development 147, dev185850 (2020).
Deng, T. et al. The Caenorhabditis elegans homolog of the Evi1 proto-oncogene, egl-43, coordinates G1 cell cycle arrest with pro-invasive gene expression during anchor cell invasion. PLoS Genet. 16, e1008470 (2020).
Grove, C. A. et al. A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell 138, 314–327 (2009).
Ashley, G. E. et al. An expanded auxin-inducible degron toolkit for Caenorhabditis elegans. Genetics 217, iyab006 (2021).
Tang, J. L. Y. et al. NanoDam identifies homeobrain (ARX) and scarecrow (NKX2.1) as conserved temporal factors in the Drosophila central brain and visual system. Dev. Cell 57, 1193–1207 e1197 (2022).
Yee, C. et al. A NanoDam toolkit for tissue-specific transcription factor profiling in C. elegans. Preprint at bioRxiv https://doi.org/10.1101/2023.05.31.543105 (2023).
Delwel, R., Funabiki, T., Kreider, B. L., Morishita, K. & Ihle, J. N. Four of the seven zinc fingers of the Evi-1 myeloid-transforming gene are required for sequence-specific binding to GA(C/T)AAGA(T/C)AAGATAA. Mol. Cell Biol. 13, 4291–4300 (1993).
Yoshimi, A. et al. Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood 117, 3617–3628 (2011).
West, A. E. & Greenberg, M. E. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 3, a005744 (2011).
Chung, S. H. et al. Novel DLK-independent neuronal regeneration in Caenorhabditis elegans shares links with activity-dependent ectopic outgrowth. Proc. Natl Acad. Sci. USA 113, E2852–E2860 (2016).
Opperman, K. J. & Grill, B. RPM-1 is localized to distinct subcellular compartments and regulates axon length in GABAergic motor neurons. Neural Dev. 9, 10 (2014).
Nishizaki, S. S. et al. Predicting the effects of SNPs on transcription factor binding affinity. Bioinformatics 36, 364–372 (2020).
Sawin, E. R., Ranganathan, R. & Horvitz, H. R. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26, 619–631 (2000).
Kindt, K. S. et al. Dopamine mediates context-dependent modulation of sensory plasticity in C. elegans. Neuron 55, 662–676 (2007).
Vidal-Gadea, A. et al. Caenorhabditis elegans selects distinct crawling and swimming gaits via dopamine and serotonin. Proc. Natl Acad. Sci. USA 108, 17504–17509 (2011).
Flavell, S. W. et al. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311, 1008–1012 (2006).
Doitsidou, M. et al. A combinatorial regulatory signature controls terminal differentiation of the dopaminergic nervous system in C. elegans. Genes Dev. 27, 1391–1405 (2013).
Flames, N. & Hobert, O. Gene regulatory logic of dopamine neuron differentiation. Nature 458, 885–889 (2009).
Jimeno-Martin, A. et al. Joint actions of diverse transcription factor families establish neuron-type identities and promote enhancer selectivity. Genome Res. 32, 459–473 (2022).
Goodman, M. B. & Sengupta, P. The extraordinary AFD thermosensor of C. elegans. Pflugers Arch. 470, 839–849 (2018).
Vierbuchen, T. et al. AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol. Cell 68, 1067–1082 (2017).
Kudron, M. M. et al. The ModERN resource: genome-wide binding profiles for hundreds of Drosophila and Caenorhabditis elegans transcription factors. Genetics 208, 937–949 (2018).
Acknowledgements
This work is dedicated to everyone who made it through these tough years. We thank members of the Shen Lab, L. Luo, K. Mizumoto, M. Killeen, P. Kurshan, O. Hobert, N. Flames and C. McLaughlin for the critical reading of this paper and for fun scientific discussions. We thank A. West and A. Narayanan for additional scientific discussion. Nuclei sorting for this project was performed on instruments in the Stanford Shared FACS Facility, primarily on PICI (purchased by the Parker Institute for Cancer Immunotherapy). Sequencing was performed at the Stanford Genomics Service Center on Illumina HiSeq 2000 and HiSeq 4000 instruments (National Institutes of Health (NIH) S10 Shared Instrumentation Grant (S10OD018220)). Some strains were provided by the Caenorhabditis Genetics Center (CGC; University of Minnesota), which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). We thank the Stanford Research Computing Center (SRCC) for providing computational resources (Sherlock cluster) that contributed to these research results. Some of the figures (Figs. 1b, 2a, 5e and 8 and Supplementary Fig. 10) were created with Biorender.com. C.Y. is supported by a Human Frontier Long Term Fellowship (LT000127/2016), T.N.M.-K. is supported by an NIH Predoctoral Fellowship (F31HD100091), W.A.H. is supported by an NIH Postdoctoral Fellowship (F32NS129942) and K.S. is a Howard Hughes Medical Institute Investigator. This work is supported in part by NIH (grants R37NS048392 to K.S., R01GM121597 to D.Q.M. and U24HG009293 to A.P.B.).
Author information
Authors and Affiliations
Contributions
C.Y., D.Q.M. and K.S. conceived and designed the study. Genome editing experiments were performed by C.Y., Y.X. and H.C. Genetics, genomics (ATAC–seq, ChIP and NanoDam), optogenetics and auxin treatment experiments were performed by C.Y. Microscopy was performed by C.Y. and A.R.R. C.Y. and A.P.B. performed computation analyses. B.X. and Y.K.X. provided technical support. T.N.M.-K., W.Z. and W.A.H. contributed to strain and reagent generation. C.Y. wrote the original draft and all authors edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Neuroscience thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–12.
Supplementary Tables
Supplementary Tables 1–13.
Supplementary Data
Supporting data for Supplementary Figs. 1, 4, 6–9 and 11.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yee, C., Xiao, Y., Chen, H. et al. An activity-regulated transcriptional program directly drives synaptogenesis. Nat Neurosci 27, 1695–1707 (2024). https://doi.org/10.1038/s41593-024-01728-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41593-024-01728-x
- Springer Nature America, Inc.