

Abstract
Protein aggregation is the hallmark of neurodegeneration, but the molecular mechanisms underlying late-onset Alzheimer’s disease (AD) are unclear. Here we integrated transcriptomic, proteomic and epigenomic analyses of postmortem human brains to identify molecular pathways involved in AD. RNA sequencing analysis revealed upregulation of transcription- and chromatin-related genes, including the histone acetyltransferases for H3K27ac and H3K9ac. An unbiased proteomic screening singled out H3K27ac and H3K9ac as the main enrichments specific to AD. In turn, epigenomic profiling revealed gains in the histone H3 modifications H3K27ac and H3K9ac linked to transcription, chromatin and disease pathways in AD. Increasing genome-wide H3K27ac and H3K9ac in a fly model of AD exacerbated amyloid-β42-driven neurodegeneration. Together, these findings suggest that AD involves a reconfiguration of the epigenome, wherein H3K27ac and H3K9ac affect disease pathways by dysregulating transcription- and chromatin–gene feedback loops. The identification of this process highlights potential epigenetic strategies for early-stage disease treatment.





Similar content being viewed by others


Data availability
The epigenomic and transcriptomic data that support the findings of this study are available through the National Center for Biotechnology Information Gene Expression Omnibus repository under accession no. GSE153875. Part of the input libraries were previously generated18 and are available under accession no. GSE84618. The proteomic data are available through the repository Chorus under accession no. 1684.
Code availability
The code developed for the analyses performed in this study is available at https://github.com/yeminlan/ADEpigenetics.
Change history
08 October 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
World Alzheimer Report 2015 (Alzheimer’s Disease International, 2015); https://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf
Van Cauwenberghe, C., Van Broeckhoven, C. & Sleegers, K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 18, 421–430 (2016).
Cummings, J. Lessons learned from Alzheimer disease: clinical trials with negative outcomes. Clin. Transl. Sci. 11, 147–152 (2018).
Mehta, D., Jackson, R., Paul, G., Shi, J. & Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 26, 735–739 (2017).
Sen, P., Shah, P. P., Nativio, R. & Berger, S. L. Epigenetic mechanisms of longevity and aging. Cell 166, 822–839 (2016).
Benayoun, B. A., Pollina, E. A. & Brunet, A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 16, 593–610 (2015).
Berson, A., Nativio, R., Berger, S. L. & Bonini, N. M. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 41, 587–598 (2018).
Gräff, J. & Tsai, L.-H. Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 14, 97–111 (2013).
Gjoneska, E. et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518, 365–369 (2015).
Frost, B., Hemberg, M., Lewis, J. & Feany, M. B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 17, 357–366 (2014).
Gräff, J. et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226 (2012).
Xu, K., Dai, X.-L., Huang, H.-C. & Jiang, Z.-F. Targeting HDACs: a promising therapy for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2011, 143269 (2011).
Anand, R., Gill, K. D. & Mahdi, A. A. Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology 76, 27–50 (2014).
Lu, T. et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454 (2014).
Ben-Hattar, J. & Jiricny, J. Methylation of single CpG dinucleotides within a promoter element of the Herpes simplex virus tk gene reduces its transcription in vivo. Gene 65, 219–227 (1988).
De Jager, P. L. et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 17, 1156–1163 (2014).
Lunnon, K. et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 17, 1164–1170 (2014).
Nativio, R. et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 21, 497–505 (2018).
Marzi, S. J. et al. A histone acetylome-wide association study of Alzheimer’s disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat. Neurosci. 21, 1618–1627 (2018).
Klein, H.-U. et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 22, 37–46 (2019).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Perez Ortiz, J. M. & Swerdlow, R. H. Mitochondrial dysfunction in Alzheimer’s disease: role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 176, 3489–3507 (2019).
Swerdlow, R. H. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimers Dis. 62, 1403–1416 (2018).
Chien, H.-C. et al. Targeted disruption in mice of a neural stem cell-maintaining, KRAB-Zn finger-encoding gene that has rapidly evolved in the human lineage. PLoS ONE 7, e47481 (2012).
Vincent-Fabert, C. et al. PLZF mutation alters mouse hematopoietic stem cell function and cell cycle progression. Blood 127, 1881–1885 (2016).
Nagy, Z., Esiri, M. M. & Smith, A. D. The cell division cycle and the pathophysiology of Alzheimer’s disease. Neuroscience 87, 731–739 (1998).
Meikrantz, W. & Schlegel, R. Apoptosis and the cell cycle. J. Cell. Biochem. 58, 160–174 (1995).
Piu, F., Aronheim, A., Katz, S. & Karin, M. AP-1 repressor protein JDP-2: inhibition of UV-mediated apoptosis through p53 down-regulation. Mol. Cell. Biol. 21, 3012–3024 (2001).
Zhang, P. et al. Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep. 6, 1110–1121 (2014).
Wethkamp, N. & Klempnauer, K.-H. Daxx is a transcriptional repressor of CCAAT/enhancer-binding protein β. J. Biol. Chem. 284, 28783–28794 (2009).
Yang, X., Khosravi-Far, R., Chang, H. Y. & Baltimore, D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89, 1067–1076 (1997).
Tate, C. M., Lee, J.-H. & Skalnik, D. G. CXXC finger protein 1 restricts the Setd1A histone H3K4 methyltransferase complex to euchromatin. FEBS J. 277, 210–223 (2010).
Chang, B., Chen, Y., Zhao, Y. & Bruick, R. K. JMJD6 is a histone arginine demethylase. Science 318, 444–447 (2007).
Anderson, K. W. et al. Quantification of histone deacetylase isoforms in human frontal cortex, human retina, and mouse brain. PLoS ONE 10, e0126592 (2015).
Shen, X., Chen, J., Li, J., Kofler, J. & Herrup, K. Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. eNeuro 3, ENEURO.0124-15.2016 (2016).
Wu, Y. et al. Aberrant expression of histone deacetylases 4 in cognitive disorders: molecular mechanisms and a potential target. Front. Mol. Neurosci. 9, 114 (2016).
Lau, P. et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol. Med. 5, 1613–1634 (2013).
Szklarczyk, D. et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Petrij, F. et al. Rubinstein–Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 376, 348–351 (1995).
Roelfsema, J. H. et al. Genetic heterogeneity in Rubinstein–Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 76, 572–580 (2005).
Jin, Q. et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262 (2011).
Koutelou, E., Hirsch, C. L. & Dent, S. Y. R. Multiple faces of the SAGA complex. Curr. Opin. Cell Biol. 22, 374–382 (2010).
Spedale, G., Timmers, H. T. & Pijnappel, W. W. M. ATAC-king the complexity of SAGA during evolution. Genes Dev. 26, 527–541 (2012).
Allen, M. et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci. Data 3, 160089 (2016).
Wang, M. et al. The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci. Data 5, 180185 (2018).
Yuan, Z.-F. et al. EpiProfile 2.0: a computational platform for processing epi-proteomics mass spectrometry data. J. Proteome Res. 17, 2533–2541 (2018).
Wagner, E. J. & Carpenter, P. B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 13, 115–126 (2012).
Martin, C. & Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 6, 838–849 (2005).
Wang, Z. et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40, 897–903 (2008).
Kuo, A. J. et al. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier–Gorlin syndrome. Nature 484, 115–119 (2012).
Kaimori, J.-Y. et al. Histone H4 lysine 20 acetylation is associated with gene repression in human cells. Sci. Rep. 6, 24318 (2016).
Creyghton, M. P. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA 107, 21931–21936 (2010).
Calo, E. & Wysocka, J. Modification of enhancer chromatin: what, how, and why? Mol. Cell 49, 825–837 (2013).
Guillemette, B. et al. H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet. 7, e1001354 (2011).
Xhemalce, B. & Kouzarides, T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev. 24, 647–652 (2010).
Wu, R. S., Tsai, S. & Bonner, W. M. Patterns of histone variant synthesis can distinguish G0 from G1 cells. Cell 31, 367–374 (1982).
Wu, R. S. & Bonner, W. M. Separation of basal histone synthesis from S-phase histone synthesis in dividing cells. Cell 27, 321–330 (1981).
Gabrielli, F. et al. Histone complements of human tissues, carcinomas, and carcinoma-derived cell lines. Mol. Cell. Biochem. 65, 57–66 (1984).
Heintzman, N. D. et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318 (2007).
Pradeepa, M. M. et al. Histone H3 globular domain acetylation identifies a new class of enhancers. Nat. Genet. 48, 681–686 (2016).
Han, D. et al. A highly sensitive and robust method for genome-wide 5hmC profiling of rare cell populations. Mol. Cell 63, 711–719 (2016).
Sun, W., Zang, L., Shu, Q. & Li, X. From development to diseases: the role of 5hmC in brain. Genomics 104, 347–351 (2014).
Stroud, H., Feng, S., Morey Kinney, S., Pradhan, S. & Jacobsen, S. E. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 12, R54 (2011).
Tropberger, P. et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell 152, 859–872 (2013).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Wyss-Coray, T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Med. 12, 1005–1015 (2006).
De Ferrari, G. V. et al. Wnt/β-catenin signaling in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 13, 745–754 (2014).
Boonen, R. A. C. M., van Tijn, P. & Zivkovic, D. Wnt signaling in Alzheimer’s disease: up or down, that is the question. Ageing Res. Rev. 8, 71–82 (2009).
Banzhaf-Strathmann, J. et al. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 33, 1667–1680 (2014).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Satoh, J.-I., Kawana, N. & Yamamoto, Y. Pathway analysis of ChIP–seq-based NRF1 target genes suggests a logical hypothesis of their involvement in the pathogenesis of neurodegenerative diseases. Gene Regul. Syst. Bio. 7, 139–152 (2013).
Lee, C. S. et al. Loss of nuclear factor E2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc. Natl Acad. Sci. USA 108, 8408–8413 (2011).
Nativio, R. et al. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 5, e1000739 (2009).
Phillips, J. E. & Corces, V. G. CTCF: master weaver of the genome. Cell 137, 1194–1211 (2009).
Trimarchi, J. M. & Lees, J. A. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 3, 11–20 (2002).
Qin, X. Q., Livingston, D. M., Kaelin, W. G. Jr & Adams, P. D. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc. Natl Acad. Sci. USA 91, 10918–10922 (1994).
Shan, B. & Lee, W. H. Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol. Cell. Biol. 14, 8166–8173 (1994).
Mitxelena, J. et al. An E2F7-dependent transcriptional program modulates DNA damage repair and genomic stability. Nucleic Acids Res. 46, 4546–4559 (2018).
de Bruin, A. et al. Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem. 278, 42041–42049 (2003).
Liu, B., Shats, I., Angus, S. P., Gatza, M. L. & Nevins, J. R. Interaction of E2F7 transcription factor with E2F1 and C-terminal-binding protein (CtBP) provides a mechanism for E2F7-dependent transcription repression. J. Biol. Chem. 288, 24581–24589 (2013).
Zhang, S. & Cui, W. Sox2, a key factor in the regulation of pluripotency and neural differentiation. World J. Stem Cells 6, 305–311 (2014).
Vaeth, M. & Feske, S. NFAT control of immune function: new frontiers for an abiding trooper. F1000Res. 7, 260 (2018).
Lee, Y.-F. et al. Premature aging with impaired oxidative stress defense in mice lacking TR4. Am. J. Physiol. Endocrinol. Metab. 301, E91–E98 (2011).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Lee, P. H., O’Dushlaine, C., Thomas, B. & Purcell, S. M. INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics 28, 1797–1799 (2012).
Nica, A. C. & Dermitzakis, E. T. Expression quantitative trait loci: present and future. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 368, 20120362 (2013).
Zou, F. et al. Brain expression genome-wide association study (eGWAS) identifies human disease-associated variants. PLoS Genet. 8, e1002707 (2012).
Casas-Tinto, S. et al. The ER stress factor XBP1s prevents amyloid-β neurotoxicity. Hum. Mol. Genet. 20, 2144–2160 (2011).
Cutler, T. et al. Drosophila eye model to study neuroprotective role of CREB binding protein (CBP) in Alzheimer’s disease. PLoS ONE 10, e0137691 (2015).
Kouzarides, T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 19, 1176–1179 (2000).
Herz, H.-M. et al. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345, 1065–1070 (2014).
Tie, F. et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136, 3131–3141 (2009).
Dai, J. et al. Probing nucleosome function: a highly versatile library of synthetic histone H3 and H4 mutants. Cell 134, 1066–1078 (2008).
Korzus, E., Rosenfeld, M. G. & Mayford, M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42, 961–972 (2004).
Caccamo, A., Maldonado, M. A., Bokov, A. F., Majumder, S. & Oddo, S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA 107, 22687–22692 (2010).
Zhang, M. et al. Role of CBP and SATB-1 in aging, dietary restriction, and insulin-like signaling. PLoS Biol. 7, e1000245 (2009).
Min, S.-W. et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966 (2010).
Weinert, B. T. et al. Time-resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell 174, 231–244.e12 (2018).
Toledo, J. B. et al. A platform for discovery: the University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement. 10, 477–484.e1 (2014).
Mirra, S. S. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: a commentary. Neurobiol. Aging 18, S91–S94 (1997).
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H. & Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404 (2006).
Risso, D., Ngai, J., Speed, T. P. & Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902 (2014).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Blalock, E. M., Buechel, H. M., Popovic, J., Geddes, J. W. & Landfield, P. W. Microarray analyses of laser-captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer’s disease. J. Chem. Neuroanat. 42, 118–126 (2011).
Sidoli, S., Bhanu, N. V., Karch, K. R., Wang, X. & Garcia, B. A. Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: from histone extraction to data analysis. J. Vis. Exp. 17, 54112 (2016).
Sidoli, S., Simithy, J., Karch, K. R., Kulej, K. & Garcia, B. A. Low resolution data-independent acquisition in an LTQ-Orbitrap allows for simplified and fully untargeted analysis of histone modifications. Anal. Chem. 87, 11448–11454 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Chen, X. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117 (2008).
Kang, H. J. et al. Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Moreno-Jiménez, E. P. et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 25, 554–560 (2019).
Acknowledgements
We thank A. Shilatifard for the histone H3.3WT and the histone mutants H3.3K27M and H3.3K9M. We thank members of the Berger laboratory for comments on the data analyses and P. Ortega for insightful scientific discussions. This work was supported by the Kleberg Foundation (S.L.B. and N.M.B.), a National Institutes of Health (NIH)/National Institute on Aging grant no. P01-AG031862 (S.L.B.) and an NIH/National Institute of Neurological Disorders and Stroke grant no. R35-NS097275 (N.M.B.). C.H. is supported by grant no. R01-HG006827. C.H. is a Howard Hughes Medical Institute Investigator. L.W. is supported by grant nos. U24-AG041689 (NIAGADS) and U54-AG052427 (Global Alliance for Chronic Diseases). A.A.-W. is supported by training grant no. T32-AG00255. B.A.G. is supported by NIH grant nos. R01-NS111997 and AI118891. J.Q.T. is supported by grant no. AG10124.
Author information
Authors and Affiliations
Contributions
R.N., N.M.B. and S.L.B. conceived the project. R.N. performed the ChIP–seq, RNA-seq and mass spectrometry experiments and supervised most of the analyses. Y.L. performed the ChIP–seq and RNA-seq analyses. G.D. performed the comparisons with published RNA-seq data and AD SNP enrichment analysis. S.S. performed the mass spectrometry and STRING analysis. A.B., A.R.S. and O.S. performed the fly experiments. R.N. extracted genomic DNA and J.N. performed 5hmC-Seal. X.C. processed the 5hmC data. A.A.-W. performed the AD eQTL enrichment analysis. C.H., L.W., B.A.G., J.Q.T., N.M.B. and S.L.B. contributed to the methodology and resources. R.N., N.M.B. and S.L.B. wrote the manuscript. All authors reviewed the manuscript and discussed the work.
Corresponding authors
Ethics declarations
Competing interests
C.H. holds a patent on the technology used (no. US8741567) and is a shareholder in Shanghai Epican Genetech.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 STRING network analysis for genes changing in AD.
a, Barplot showing the number of STRING (v11) interactions for genes with the top number of interactions in Fig. 1e. b, STRING interaction network for genes changing in AD vs Old (q < 0.05) that interact with EP300, CREBBP and TRAPP. Interactions that were identified in Fig. 1e are not shown in this network. The gene network was visualized with Cytoscape (v3.6)104. Size of nodes represents RNA expression values, the color represents gene expression changes (log2 fold-change) in the AD vs Old comparison (red for upregulated in AD; blue for downregulated in AD) and the thickness of the line is the confidence of the interaction calculated by STRING. Nodes circled in red or blue represent known transcription and chromatin genes.
Extended Data Fig. 2 Histone posttranslational modifications in Younger, Old and AD.
a, Amino acid sequence of canonical histone H3 (H3.1 and H3.2) tail and globular domain, and its H3.3 variant. The residue that differs between canonical H3 and H3.3 is highlighted in red. b, Amino acid sequence of histone H4 tail and globular domain. Bars below the amino acid sequence in panels a-b represent peptides generated in the trypsinization process that were identified on the mass spectrometer (LC-MS/MS). Grey bars represent peptides not reliably detected and therefore excluded from the analysis. c–e, Stacked bar plots showing relative abundance of histone modifications (methylation and acetylation) on histones H3, H3.3 and H4 in (c) Younger, (d) Old and (e) AD. The lysine residues (K) analyzed are listed below the stacked bar plots.
Extended Data Fig. 3 Histone acetyl marks are enriched at both TSS and enhancers.
Metaplots showing peak enrichment of H3K27ac, H3K9ac and H3K122ac and corresponding 5hmC and H3K4me1 enrichments for peaks at transcriptional start sites (TSSs) (≤ 1 Kb from TSS) and enhancer (Enh) sites (> 1Kb from TSS) in (a–f) Younger, (g-l) Old and (m-r) AD brains. Histone acetyl-peaks are enriched at both TSSs and enhancers, while 5hmC and H3K4me1 mark enhancer sites.
Extended Data Fig. 4 H3K27ac, H3K9ac and H3K122ac peak distribution in Younger, Old and AD.
a, Histogram of peak density for H3K27ac (light green), H3K9ac (light blue) and H3K122ac (light red), based on their distance from the transcriptional start site (TSS) for peaks detected in Younger, Old and AD. Grey vertical lines demark (from left to right): 5, 25, 50 and 100 Kb distance from TSS. b–d, Venn Diagram showing the overlap between H3K27ac, H3K9ac and H3K122ac peaks for (b) All peaks, (c) TSS peaks (≤ 1Kb from TSS) and (d) enhancer (Enh) peaks (> 1Kb from TSS) detected in Younger, Old and AD.
Extended Data Fig. 5 Correlation between ChIP-seq and RNA-seq data.
a-c, Scatterplot of (a) H3K27ac, (b) H3K9ac and (c) H3K122ac peak enrichment vs gene expression for genes expressed in Old. d-f, Scatterplot of (d) H3K27ac, (e) H3K9ac and (f) H3K122ac peak enrichment vs gene expression for genes expressed in AD. For graphical representation in a-b, 3000 randomly chosen points are shown in each panel. g-i, Scatterplot of (g) H3K27ac, (h) H3K9ac and (i) H3K122ac absolute peak fold-change vs absolute gene expression change for significantly (q < 0.05) differentially expressed genes in AD vs Old. j,k, Scatterplot of total acetyl-peak enrichment (H3K-total-ac; sum of H3K27ac, H3K9ac and H3K122ac peak enrichment at the same site) vs gene expression for genes expressed in (j) Old and (k) AD. l, Scatterplot of H3K-total-ac absolute peak fold-change vs absolute gene expression change for significantly (q < 0.05) differentially expressed genes in AD vs Old. The closest peak to the TSS was chosen for these analyses. Linear regression trendlines, Pearson’s correlation coefficients and p-values (test for association using Pearson’s product moment correlation coefficient implemented by R stats package, two-sided) are indicated in each panel (a-l).
Extended Data Fig. 6 Comparison between histone marks enrichments at sites with disease-specific changes.
a–c, Boxplots showing H3K27ac, H3K9ac, H3K122ac and H3K4me1 peak enrichment at sites with (a) H3K27ac, (b) H3K9ac, (c) H3K122ac (highlighted in blue) disease-specific gains. d–f, Boxplots showing H3K9ac, H3K122ac and H3K4me1 peak enrichment at sites with (d) H3K27ac, (e) H3K9ac and (f) H3K122ac (highlighted in blue) disease-specific losses. Asterisks in (a-f) denote level of significance comparing peak enrichment across Younger (N = 11-12), Old (N = 10) and AD (N = 9–11) (* P < 0.05; ** P < 0.01, 1-way ANOVA) (Supplementary Table 2). Boxplots show minimum, first quartile, median (center line), third quartile and maximum.
Extended Data Fig. 7 H3K9ac disease-specific gain at CREBBP but not EP300.
a–c, Boxplot showing (a) H3K9ac, (b) H3K27ac and (c) H3K122ac peak enrichment at the CREBBP gene in Younger, Old and AD. A H3K9ac disease-specific gain is observed at CREBBP (highlighted in blue in a). d–f, Boxplot showing (d) H3K9ac, (e) H3K27ac (f) H3K122ac peak enrichment at the EP300 gene in Younger, Old and AD showing no disease-specific changes. The closest peak to the gene was considered for this analysis. P-values comparing peak enrichment across Younger (N = 8-9), Old (N = 10) and AD (N = 9–11) (Supplementary Table 2) (1-way ANOVA) are reported in each panel. Boxplots show minimum, first quartile, median (center line), third quartile and maximum. Dots overlaid on boxplots represent individual data points.
Extended Data Fig. 8 Functional analysis of H3K27ac and H3K9ac disease-specific losses.
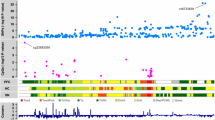
a,b, Barplot showing top GO terms (Biological Processes; GREAT, FDR < 5%, % by both the binomial and the hypergeometric tests) for (a) H3K27ac disease-specific losses and (b) H3K9ac disease-specific losses for terms with at least 20 genes. c,d, UCSC genome browser view showing an example of (c) H3K27ac disease-specific loss at the PCSK1 gene and (d) H3K9ac disease-specific loss at the SVOP gene. H3K27ac, H3K9ac, H3K122ac, H3K4me1 ChIP-seq and RNA-seq tracks are showed for Younger, Old and AD. e,f, Top DNA motifs (HOMER v4.6) for (e) H3K27ac disease-specific losses and (f) H3K9ac disease-specific losses in AD. Enrichment results are shown for known motifs (q < 0.05, Benjamini-Hochberg).
Extended Data Fig. 9 Functional analysis of disease-specific changes using DAVID.
a,b, Barplot showing top GO terms (Biological Processes, DAVID v6.7, FDR < 10%, Yekutieli) for genes targeted by (a) disease-specific gains (H3K27ac or H3K9ac) and (b) disease-specific losses (either H3K27 or H3K9ac or H3K122ac) for terms with at least 20 genes.
Extended Data Fig. 10 H3K27ac disease-specific gains are enriched with AD GWAS SNPs from Kunkle et al.
Bar plot showing the significance (-log10 p-value) of the association between each of the six classes of H3K27ac, H3K9ac and H3K122ac changes (age-regulated gains or losses, age-dysregulated gains or losses and disease-specific gains or losses) and AD SNP-regions from Kunkle et al.85 using INRICH. Red dashed horizontal line represents the threshold of significance (P < 0.05).
Supplementary information
Supplementary Information
Supplementary Notes 1–8 and Figs. 1–11
Supplementary Tables
Supplementary Tables 1–14
Rights and permissions
About this article

Cite this article
Nativio, R., Lan, Y., Donahue, G. et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat Genet 52, 1024–1035 (2020). https://doi.org/10.1038/s41588-020-0696-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-020-0696-0
- Springer Nature America, Inc.
This article is cited by
-
Promoting Alzheimer’s disease research and therapy with stem cell technology
Stem Cell Research & Therapy (2024)
-
Histone acetylation in an Alzheimer’s disease cell model promotes homeostatic amyloid-reducing pathways
Acta Neuropathologica Communications (2024)
-
Gut microbiota-host lipid crosstalk in Alzheimer’s disease: implications for disease progression and therapeutics
Molecular Neurodegeneration (2024)
-
Novel histone post-translational modifications in Alzheimer’s disease: current advances and implications
Clinical Epigenetics (2024)
-
The interaction between ageing and Alzheimer's disease: insights from the hallmarks of ageing
Translational Neurodegeneration (2024)