

Abstract
Treatment assessment and patient outcome for sepsis depend predominantly on the timely administration of appropriate antibiotics1,2,3. However, the clinical protocols used to stratify and select patient-specific optimal therapy are extremely slow4. In particular, the major hurdle in performing rapid antimicrobial susceptibility testing (AST) remains in the lengthy blood culture procedure, which has long been considered unavoidable due to the limited number of pathogens present in the patient’s blood. Here we describe an ultra-rapid AST method that bypasses the need for traditional blood culture, thereby demonstrating potential to reduce the turnaround time of reporting drug susceptibility profiles by more than 40–60 h compared with hospital AST workflows. Introducing a synthetic beta-2-glycoprotein I peptide, a broad range of microbial pathogens are selectively recovered from whole blood, subjected to species identification or instantly proliferated and phenotypically evaluated for various drug conditions using a low-inoculum AST chip. The platform was clinically evaluated by the enrolment of 190 hospitalized patients suspected of having infection, achieving 100% match in species identification. Among the eight positive cases, six clinical isolates were retrospectively tested for AST showing an overall categorical agreement of 94.90% with an average theoretical turnaround time of 13 ± 2.53 h starting from initial blood processing.
Similar content being viewed by others

Main
Infection caused by the invasion of microbial pathogens can trigger a dysregulated host response5,6,7. If not recognized and treated promptly, this condition can further progress to sepsis or septic shock, which is one of the leading causes of death8. Every year sepsis affects more than 40 million people globally, with a highly concerning mortality rate of 20–50% and socioeconomic burdens of around US $25 billion in the USA alone9,10. Although the disease can often be treatable, patient outcome relies heavily on timing of treatment. Delays in appropriate antibiotic delivery have been shown to significantly affect patient survival rates, particularly among those with septic shock1,2,3. As such, rapid initiation of targeted antimicrobial therapy has been recognized as a key intervention during initial care.
Nonetheless, current protocols used to select patient-specific optimal treatment through AST in patients with bacteraemia are extremely time consuming. AST consists of a collective series of complex diagnostic procedures to inform the presence of infection, identify the causative microbial species and assess susceptibility profiles in response to various drugs and their concentration. The entire process typically requires at least 2–3 days of turnaround time (TAT), which necessarily warrants and yet forces the clinician to initiate empirical, broad-spectrum antibiotic administration4. This implies that often inadequate prescriptions made during the initial treatment phase might jeopardize patient survival until a valid AST result is available. Among antimicrobial drugs prescribed to human patients, 14–78% were estimated to be unnecessary or ineffective11,12,13. Recent studies have shown that patient mortality risk, length of hospital stay and medical costs can potentially be improved by reduction in TAT14,15. Therefore, the development of new strategies to substantially reduce total TAT for AST is critical.
From conventional broth microdilution (BMD), disc diffusion and density-based measures16,17,18,19 in which pathogen growth or inhibition is directly examined in bulk, various AST methods have been under continuous development using microfluidics20,21,22, nanoarrays23, advanced optics24,25,26,27,28,29 and single-cell analysis30,31. Alternatively, strategies detecting cell viability surrogates including metabolites23,26,32, electrical impedance33, genomic contents34 and transcriptional changes35 have been proposed with a focus on reducing the time required for drug susceptibility determination. However, total TAT relies not only on the duration of susceptibility testing but also on the time required for two additional culture preparation steps, which, in fact, account for the major proportion of delay. The microbial load in patient blood is extremely low, ranging between 1 and 100 colony-forming units (CFU) ml−1 (ref. 36), and is vastly outnumbered by blood cells. Due to this disparity, prior steps—including blood culture (BC) to amplify the number of pathogens followed by pure culture to subculture purified colonies of isolates—have been essential for subsequent pathogen species identification (ID) and AST.
Several ‘rapid’ phenotypic AST techniques have recently been demonstrated that avoid the need for pure culture by combining strategies such as blood cell lysis20,27, centrifugation16,24,25,26,27,35 or filtration22,26, thereby performing AST directly from positive BC bottles. Nevertheless, the lengthy BC period remains indispensable and is unlikely to improve due to the inherent growth properties (doubling time) of invading pathogens. Although several genotypic AST37,38,39 and ID40,41 assays have been introduced for direct application to whole-blood specimens, these molecular-based approaches typically suffer from low sensitivity caused by limited cell counts, in addition to surrounding blood components and loss during downstream reactions. Furthermore, it has been observed that genetic resistance does not always correlate with phenotypic susceptibility and, more importantly, due to challenges in measuring the minimum inhibitory concentration (MIC), such assays are restricted to facilitating drug avoidance rather than guiding clinicians in selecting optimal therapeutics.
To this end we present a phenotype-based, ultra-rapid AST (uRAST) platform capable of performing drug susceptibility profiling directly from the patient’s whole blood, obviating the need for BC. In addition, we also introduce a highly sensitive, BC-free, pathogen ID assay that provides concurrent species information during its workflow, which is essential for precise AST interpretation. Compared with commercial, hospital-based AST methods, our approach theoretically reduces TAT by more than 40–60 h, enabling antibiotic prescriptions with fast TATs. Finally we design a clinical study and apply our technology to a total of 190 hospitalized, infection-suspected patients for validation.
Design and workflow of uRAST
Starting from the initial therapy treatment, broad-spectrum antibiotics are periodically administered to patients every 6–8 h (ref. 42). Although first-line delivery cannot be avoided, AST results should ideally be available as early as the second dose of antibiotic prescription. However, depending on the microorganism(s) involved, BC can take up to 24 h or more, which already exceeds such a critical time point (Fig. 1a). To reduce this growth period, pathogen proliferation must be accelerated and the required number of inocula for AST analysis minimized. uRAST aims to achieve both, first by isolating the microorganisms from whole blood, which are surrounded by host cells, antibiotics and growth inhibitors (Fig. 1b), performing species identification (Fig. 1c), expediating culture under pure media (Fig. 1d) and implementing a phenotypic imaging-based AST chip that conducts susceptibility testing at cell numbers orders of magnitude lower compared with standardized protocols (Fig. 1e), all of which contribute to significant reduction in total TAT (Fig. 1f and Supplementary Table 1).

a, Clinical diagnosis and treatment (Tx) timeline for infection-suspected patients based on conventional AST, rapid AST and uRAST. TAT required to obtain susceptibility profiles and time reduction for optimal antibiotic administration are compared. b, The uRAST procedure starts with isolation of blood-borne pathogens directly from the patient’s whole blood, which eliminates BC from the AST pipeline. c–e, Quick species identification (c) is performed using a subset of samples following isolation. In parallel, the remaining sample is subjected to rapid culture in fresh medium to expedite pathogen proliferation (d), followed by low-input AST through a custom 96-well-based AST chip (e). BF, bright field. f, Comparison between phenotypic AST technologies as a function of TAT and categorical agreement. Methods are classified into three categories based on AST starting point: direct from whole-blood samples, following a positive BC and following pure culture (PC). Methods are colour coded based on the means of detection, with circle size representing the number of drug–pathogen combinations included in each test. Numbers next to circles correspond to references. aAST studies without specific blood-preprocessing procedures or that were conducted on urine samples are categorized as ‘after PC(+)’.
Pathogen isolation from whole blood
Because the invading pathogens are usually unknown, enrichment must be carried out covering a wide spectrum of strains with the microorganisms being alive and intact for subsequent analysis42,43. To achieve this we utilized magnetic nanoparticles decorated with synthetic beta-2-glycoprotein I (sβ2GPI) peptides, which we previously introduced for viral isolation44. β2GPI is an acute-phase plasma protein known to be involved in human innate immune responses that naturally recognize common motifs of pathogen-associated molecular patterns43,45. To characterize pathogen enrichment performance (Fig. 2a), five of the most frequently observed infecting strain cell lines were spiked into 5 ml of PBS or blood with varying concentrations of bacterial load. The entire procedure was completed within 1 h following nanoparticle injection, pathogen binding, magnetic separation and resuspension into 1 ml of PBS or fresh cation-adjusted Mueller–Hinton broth (MHB) culture medium (Fig. 2b). With an optimized protocol (Extended Data Fig. 1), capture efficiency of 96.23 ± 3.31 and 91.54 ± 4.35% was confirmed for Escherichia coli and Staphylococcus aureus, respectively, with a spiking concentration of 1,000 CFU ml−1 (Extended Data Fig. 2a). To match scales relevant to the physiological range, efficiency was tested again with lower bacterial loads (1–10 CFU ml−1) using E. coli, Klebsiella pneumoniae, Pseudomonas aeruginosa (all Gram-negative) and S. aureus and Enterococcus faecalis (both Gram-positive). Although yield was slightly reduced, all strains were successfully captured (Fig. 2c,d and Extended Data Fig. 2b–d). In addition, blood cell depletion rate was above 99.90 and 99.99% for white blood cells (WBC) and red blood cells (RBC), respectively (Fig. 2e).

a, Schematic illustration of the native β2GPI protein and sβ2GPI peptide coated around a magnetic nanoparticle. Average nanoparticle diameter is 200 nm. b, Timeline and workflow of the pathogen isolation process using sβ2GPI nanoparticles. The resuspended pathogens are later subjected to ID and AST analysis. c,d, Total number of American Type Culture Collection (ATCC) standard E. coli (c; nPBS = 20, nblood = 20) and S. aureus (d; nPBS = 15, nblood = 20) cells captured using sβ2GPI nanoparticles with respect to the initial spike number of 1–10 CFU ml−1 in 5 ml of PBS (blue) and blood (red). Shaded regions represent 95% confidence intervals obtained from linear regressions. e, Blood cell counts remaining following bacterial enrichment from 5 ml of whole blood (purity, n = 4). Boxes and whiskers show interquartile range and maximum and minimum values of data; centre lines in boxes represent the median. f, Capture efficiency of sβ2GPI nanoparticles compared with various patient-derived clinical isolates in blood using a spike concentration of 5 CFU ml−1 (n = 10). Data shown as mean ± s.d. g, Repeatability test for yield and limit of detection (LOD) calculation (n = 20). c–g, Data points represent biologically independent experimental replicates.
For assessment of the coverage of sβ2GPI nanoparticles, varying species (n = 18) of clinical isolates were collected from patients and tested (Fig. 2f). Regardless of type, sβ2GPI nanoparticles efficiently captured a broad range of blood-borne pathogens. Despite both interaction and binding being slightly favourable to Gram-negative species over Gram-positive, in general accordance with previous hypotheses43, overall affinity was more strain dependent. Finally, for evaluation of the limit of detection, each bacterial strain was inoculated in blood at a concentration of either 1 or under, 2 or 4 CFU ml−1 (Fig. 2g). The limit of detection was determined when at least 18 (90%) of 20 specimens repeatedly tested showed a positive recovery. The collective limit of detection, combining all clinical strains, was constantly below 4 CFU ml−1, with over 60% (11 of 18) even below 1 CFU ml−1 (Extended Data Fig. 2e–g).
Quick bacterial species identification
For determination of susceptibility breakpoints for each drug–pathogen combination during AST, the ID of the invading species must be specified. To confirm the presence of and discriminate pathogenic type, we developed a quick-mapping ID assay (QmapID) using shape-encoded microdiscs (Fig. 3a). The assay consisted of a library of silica-coated microdiscs (diameter 50 µm), each immobilized with single-stranded DNA probes (5′-amine modified) on their carboxylated surface (Supplementary Table 2), which can hybridize genomic sequences of the corresponding pathogen. Unique hole patterns were assigned to each gene and encoded for one-pot multiplexing (more than 100-plex), with magnetic nanoparticles embedded to facilitate microdisc manipulation (Extended Data Fig. 3a). For a single reaction, 100 each of 27 different microdisc particle types with distinct DNA probe sets were mixed for use. From 1 ml of resuspended solution following enrichment, bacterial cells were lysed for genomic DNA (gDNA) extraction followed by amplification using two-step nested PCR and biotin labelling. Amplicons were then hybridized to target specific microdiscs and probed using fluorescent dyes for imaging (Extended Data Fig. 3b). By decoding hole patterns and analysis of fluorescent intensity distribution and mean, the presence of bacteria-specific genes was verified within 3 h (Fig. 3b).

a, Schematic workflow of multiplexed QmapID assay for species determination and analysis of resistance genes. Resuspended pathogens are lysed for DNA extraction, followed by amplification and biotin labelling. Amplicons are then mixed with microdiscs functionalized with DNA detection probes targeting bacteria-specific ID and resistance genes. Finally the resulting product is fluorescently labelled for imaging and subsequent analysis. b, Image-processing algorithm for microdisc detection, decoding and quantification. BF and fluorescent images are used for hole pattern recognition and positivity detection, respectively. A threshold of 800 arbitrary units (a.u.) was determined based on negative control experiments. The resulting images are representative of more than 100 independent experiments. Scale bars, 100 µm. c, Heatmap illustrating mean intensity signals acquired from QmapID assay using patient-derived clinical isolates. Staphylococcus lentus was not detectable using the Staph DNA probe due to a different nucleotide sequence at the targeted locus. See Supplementary Table 2 for definitions of abbreviations. d, LOD comparison using conventional blood-processing procedures for pathogen ID for E. coli (n = 3). Data are mean ± s.d. from biologically independent experimental replicates. Shaded area denotes the mean intensity range of blood samples (without pathogens) following sβ2GPI capture and QmapID, indicating the baseline interference of blood cells. e, Merged micrograph image of QmapID microdiscs and their corresponding fluorescent intensity profiles following pathogen enrichment from blood and QmapID analysis. The resulting images are representative of three independent experiments. Scale bars, 50 µm. E.C., E. coli; E.F., E. faecalis; K.P., K. pneumoniae; SA, S. aureus; MRSA, methicillin-resistant S. aureus; MSSA, methicillin-sensitive S. aureus; PA, P. aeruginosa; PAA, polyacrylic acid.
For evaluation of the assay, pre-extracted gDNA from 36 clinical isolates was prepared in PBS at a concentration of 100 fg per reaction (Fig. 3c). Based on the average weight of genomic content extracted and measured from a single bacterial cell (approximately 8–10 fg), this amount roughly corresponded to 10 CFU of pathogen. Overall, the bacterial ID from all clinical strains was successfully differentiated with no notable cross-reactions, except for Enterobacter cloacae. Nevertheless, using the combined outcomes from multiplexed detection we were able to negate such interference caused by non-specific binding. Furthermore, this multiplexed functionality allowed us to concurrently analyse resistance genes (Supplementary Fig. 1) using QmapID, simply by extending the gene panel beyond species ID (Supplementary Table 3). Next, combined with bacterial enrichment using sβ2GPI nanoparticles, the limit of detection of the QmapID assay for blood was re-examined (Fig. 3d). Several blood-preparation procedures, including simple dilution, RBC lysis and centrifugation, were simultaneously used to evaluate the depletion effect for each approach. Compared with the remainder, which required more than 5 × 103 CFU ml−1 E. coli for reliable detection, the limit of detection of our method was below 5 CFU ml−1, resulting in a 1,000-fold increase in gene detection sensitivity. Finally the test was repeated using six clinical isolates of different species with 3–5 CFU ml−1 inoculum in 5 ml of blood (Fig. 3e).
Rapid culture for reduction in growth time
In parallel with QmapID, blood-purified pathogens collected following the enrichment process were also used for AST (Fig. 4a). To do so, we proliferated these cells in 1 ml of fresh medium to help expedite their growth (‘rapid culture’). To test this hypothesis, 3–5 CFU ml−1 E. coli or S. aureus was spiked in whole blood and either rapidly cultured following β2GPI nanoparticle enrichment or, for comparison, injected in commercial BC bottles containing resins that can absorb antibiotics or microbial growth inhibitors. Following 4–10 h incubation, bacterial concentration was quantified for both cases (Fig. 4b,c). As expected, pathogen growth was greatly enhanced in pure medium in which blood components were efficiently removed compared with the standard BC practice, resulting in an increase of 9.76-fold and 19.01-fold in concentration following 10 h culture for E. coli and S. aureus, respectively.

a, Workflow of pathogen culture and AST chip loading using agarose. Micrograph image of the AST chip is shown on the right. b,c, Growth rate comparison in fresh medium and blood using E. coli (b) and S. aureus (c; n = 3). A standard BC bottle was used for whole-blood samples. d, Schematic illustration of the single-well unit of the AST chip. Within each well, varying types and concentrations of antibiotics (Abx) are preloaded and heat-dried. The entire chip is packaged under vacuum condition and stored at 4 °C before use. Scale bar, 1 mm. e,f, Effect of inoculum size with respect to the area of colony expansion of E. coli (e) and S. aureus (f) over time during culture (n = 3). Micrograph image of each bacterial colony following 12 h of culture is shown on the right. Experiments were carried out in control wells with no antibiotics. e,f, Scale bars 100 µm. g,h, Comparison between MIC values of E. coli (g) and S. aureus (h) using BMD and uRAST. Both species were cultured for 18 and 12 h for BMD and uRAST, respectively, before MIC determination. Shaded region indicates the drug’s essential agreement (EA) range (MIC within one degree of difference from BMD results). b,c,e,f, Data are mean ± s.d. from biologically independent experimental replicates.
Low-input phenotypic AST
Based on guidelines from either the Clinical and Laboratory Standards Institute (CLSI) or European Committee on Antimicrobial Susceptibility Testing, for testing of a single drug dose approximately 5 × 104 CFU of bacterial cells are needed during BMD46,47. Considering the number of drug–dose combinations recommended, minimal cell counts required to perform AST can reach up to 5 × 106 CFU. To further reduce TAT, apart from promotion of microbial growth, we designed a custom 96-well-based AST chip that accommodates a low number of cellular inputs for MIC determination and susceptibility analysis. The chip consists of an elliptically shaped pathogen-dispensing and pathogen-imaging area and a satellite reservoir in which varying types and concentrations of antibiotics are preloaded (Fig. 4d). For high-throughput AST, the rapidly cultured bacterial solution was mixed with 1% liquid agarose (1:1 ratio), immediately inoculated into each well (10 μl) and cooled for 10 min for gelation. Here the agarose matrix was used to immobilize motile pathogens to track and observe bacterial division over time. Next, 90 μl of MHB medium was supplied to provide nutrients and dissolve the preloaded antibiotics. Drug diffusion across the entire agarose gel occurred within 30 min (Supplementary Fig. 2). Finally the chip surface was partially sealed to prevent evaporation during incubation at 35 °C, and bacterial growth was monitored through time-sequential methods.
To acquire a comprehensive landscape of various antibiotic responses, two AST chips were used for each sample, which concurrently evaluate 179 and 160 drug–dose combinations for Gram-negative and Gram-positive species, respectively (Supplementary Tables 4 and 5). We set our target inoculum size to 500 CFU per well, which totals 5 × 104 cells for AST input. This corresponds to approximately 6 h of rapid culture considering a bacterial doubling time of 30–40 min. By decreasing the number of inocula, the colony area quickly expanded within the matrix due to a drop in local pressure caused by adjacent bacterial cells during culture, which aided observation and growth determination (Fig. 4e,f). Finally we evaluated the MIC of various antibiotics using E. coli and S. aureus to investigate AST accuracy (Fig. 4g,h). Compared with the BMD results, the MIC for all drugs was within one degree of difference except for penicillin, which showed an overall essential agreement of 100% (20 of 20) and 93.3% (14 of 15) for E. coli and S. aureus, respectively.
To avoid human error and reduce the time required for manual assessment, we developed an image-processing algorithm for automation of susceptible/intermediate/resistant (SIR) decisions (Extended Data Fig. 4a). Every hour, large scan images (×10 magnification) were acquired from each well, which were then aligned to the raw image taken at time zero and subtracted to remove background signals caused by dust particles or remaining blood cells. Next, the images were binarized followed by noise filtering, which underestimated signals intentionally to avoid false-positive growth detection. Finally, from the monochromic images, bacterial growth or inhibition was classified by a predefined threshold based on control experiments (Extended Data Fig. 4b,c). To optimize and establish the proper timing for SIR decisions, six different clinical isolates were spiked into 5 ml of whole blood and processed, starting from pathogen enrichment, 6 h of rapid culture and AST as described above. During 12 h of incubation and imaging we calculated both categorical agreement and errors (minor error, major error and very major error) over time, based on the BMD references (Extended Data Fig. 4d,e). The initial SIR decision was made for each AST chip when microbial growth was first detected from at least three of four control wells containing no antibiotics (‘dynamic decision’). The categorical agreement at this time point, from all strains combined, was 91.3% (84 of 92), showing an excellent match to BMD results with only a few errors. As incubation time was extended, in particular beyond 2 h from initial growth detection from the control wells (‘final decision’), AST accuracy was further improved, demonstrating an overall categorical agreement of 96.74% (89 of 92) and minor error of 3.26% (3 of 92), with no major error or very major error (Extended Data Fig. 4f and Supplementary Fig. 3).
Clinical validation
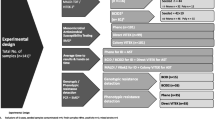
To validate our platform, a clinical study was designed to compare AST accuracy and TAT with respect to the diagnostic routine carried out in the hospital (Extended Data Fig. 5). A total of 190 hospitalized patients with haematologic malignancy and who were suspected of having an infection were enrolled in this study. For each patient, 5–10 ml of whole blood was sampled from four to five different sites, including several catheter lines and the peripheral body when an abnormal increase in temperature (pyrexia) was noticed. All samples were subjected to analysis based on hospital-established protocols following BC, Gram staining/chemical tests (catalase, coagulase and triple-sugar iron agar test), pure culture, ID using matrix-assisted laser desorption–ionization time-of-flight mass spectrometry (MALDI–TOF) and AST using either VITEK2 (bioMérieux, Gram-negative) or Phoenix M50 (Becton Dickinson, Gram-positive) (Supplementary Table 6). In addition, a conventional BMD test was performed from pure cultures to serve as the ground truth reference. During the initial blood draw, 5–10 ml was simultaneously sampled into EDTA tubes from the red-lumen Hickman catheter (H.Red) and tested using QmapID. Excluding 16 samples that did not satisfy the recruitment criteria due to hospital contamination, transportation delay and so on, 16 out of 174 patients (9.20%) tested positive for bacteraemia based on BC results (Supplementary Fig. 4). Considering samples only from the H.Red test, eight were positive among the 16 cases of infection (Supplementary Table 7). This exactly matched the results of the blind-tested QmapID and, more importantly, 100% concurrency was confirmed between ID information derived from our QmapID assay and MALDI–TOF (Fig. 5a).

a, Patient sample distribution based on infectivity. The patient’s infection status was determined based on combined BC results from all blood samples. b, Categorical agreement and error of uRAST results compared with BMD test. ME, major error; mE, minor error. c, MIC comparison between BMD and uRAST for essential agreement calculation. The shaded region signifies the drug’s essential agreement range (within one degree of difference from BMD results). Only data falling within the MIC test range (on-scale) are included. d, Time spent on each procedure for uRAST and hospital AST. TAT is calculated from the initiation of blood processing. Working time delay corresponds to the postponement resulting from limited operating hours for conducting routine AST in the hospital’s laboratory (lack of microbiology staff during night shifts); this is estimated based on the BC initiation time documented in the medical records for each patient. e, TAT comparison for hospital AST methods versus uRAST (n = 6 independent case scenarios). The total time reduction gained from using uRAST for drug susceptibility analysis is shown on the right. Boxes and whiskers show interquartile range and the maximum and minimum values of data; centre line in boxes represents the median. Statistical significance is calculated using a two-tailed, paired t-test (P < 0.0001). *P < 0.05, **P < 0.01, ***P < 0.001. f, Case study of patient P. 126 with sepsis caused by a multidrug-resistant organism (carbapenem-resistant K. pneumonia); diagnosis and treatment timelines are plotted based on the patient’s medical record. Inset represents AST results obtained from BMD, VITEK2 (hospital) and uRAST. The x axis abbreviations are defined in Supplementary Tables 4, 5. The susceptibility profile of colistin was later determined in a separate test from the hospital due to its absence in the VITEK2 drug panel. CRE, carbapenem-resistant Enterobacteriaceae; CRRT, continuous renal replacement therapy; Emp., empirical; ICU, intensive care unit; NA, not available.
uRAST was selectively performed retrospectively on infection-positive blood samples identified by QmapID. To mimic patient blood samples, six strains of patient-derived E. faecium (n = 2), E. coli (n = 1) and K. pneumoniae (n = 3), later obtained from the hospital, were individually spiked into whole blood (5 CFU ml−1) and processed. Correlating our results to BMD, we confirmed an overall categorical agreement of 94.90% (93 of 98) with 5.10% (5 of 98) minor error, and essential agreement of 94.44% (34 of 36) (Fig. 5b,c and Extended Data Fig. 6a). This performance coincided with US Food and Drug Administration requirements for AST platforms (categorical agreement 90% or above, minor error 10% or less, major error 3.0% or less, very major error 1.5% or less). Interestingly, the categorical agreement from uRAST was slightly higher than the results obtained from VITEK2 or Phoenix M50 (Extended Data Fig. 6b,c). Most importantly, TAT was significantly improved by applying uRAST compared with hospital AST, resulting in an average reduction of 47.99 ± 9.69 h (P < 0.0001) (Fig. 5d,e). Assuming full automation of all uRAST procedures and excluding working time delays caused by the laboratory’s limited AST practice hours (non-technical), the difference was even more pronounced at 62.99 ± 15.44 h (P = 0.0002) (Extended Data Fig. 7). Finally, TAT reduction on comparison of QmapID and hospital Gram staining/chemical tests was 13.11 ± 6.16 h (P = 0.0034) (Extended Data Fig. 8).
To underscore the urgency and importance of timely AST, we conducted a case study on one of the patients (P. 126) who died during our testing period (Fig. 5f). This patient was suffering from natural killer/T cell lymphoma and was under continuous treatment with chemotherapy (gemcitabine, dexamethasone and cisplatin). From the onset of fever, which occurred 12 days after starting chemotherapy, BC was performed following empirical treatment using meropenem. Without knowing the type of causative species, ambisome was consecutively administered considering the possibility of fungal infection. One day later, the disease advanced inducing acute kidney injury. From the second day following this, ID and AST results were reported, informing that the invading substance was the carbapenem-resistant Enterobacteriaceae, K. pneumoniae, and treatment was immediately switched to colistin and amikacin. However, with unstable blood pressure and exacerbating health conditions, the patient was transferred to the intensive care unit. Despite critical care the patient died on the following day. On the other hand, considering the TAT involved in applying uRAST, disease severity might have been reduced by delivery of optimal antibiotics before the occurrence of renal failure.
Discussion
Regardless of the extensive efforts being made towards rapid identification of pathogens and the provision of drug susceptibility information, prompt treatment of patients with sepsis continues to be a significant challenge. Over past decades, following the introduction of ‘rapid’ phenotypic AST concepts, most studies have competitively focused on reducing the time for SIR determination but with less or no attention given to the BC steps necessary for pathogen amplification, which account for more than 65–85% of TAT in these rapid approaches. To achieve marked acceleration of the initiation of targeted antimicrobial therapy that can completely change and affirmatively impact current treatment regimens and clinical outcomes, innovative strategies must now be comprehensively developed to potentially bypass or reduce the duration of traditional BC. The uRAST we describe herein addresses this root cause of delay by performing AST directly from whole blood, providing precise AST results even before bacteraemia is confirmed through traditional BC practice.
Using a sβ2GPI peptide to first collect and purify rare pathogens from the surrounding blood components, as well as residual antibiotics that are known to inhibit bacterial growth, we actively accelerate the proliferation process through rapid culture and further reduce the time, by orders of magnitude, required for this pathogen-expansion step by designing an AST chip that can perform SIR testing using microbial cells. Despite several blood-based bacterial isolation methods previously reported, the primary reason why barely any successful attempts have been made to apply these to the AST field is due not only to insufficient capability to isolate rare microorganisms from large volumes of complex patient specimens, but perhaps also from the limited breadth of pathogenic coverage. Unlike other diseases in which the physical properties or surface composition (types of protein, lipid or carbohydrate) of the targeted cells are predefined, in the case of bacteraemia the invading pathogens are typically unknown, making it difficult to probe them alive using classical antibodies, aptamers or nanobodies and thus rendering them impractical for clinical use. The sβ2GPI peptide we use, which was designed based on a protein molecule involved in human innate immunity, is particularly useful because it naturally recognizes common motifs shared by a broad range of pathogenic substances. This feature extends its capacity to encompass both singular or even polymicrobial infections (Extended Data Fig. 9). Ongoing discoveries and investigations towards such pathogen-binding biomolecules—and possibly the combination of various forms for synergistic enhancement of capture efficiency over a wide spectrum of microbial species—will enable alternative routes to surpass the detection limit, thereby overcoming the BC bottleneck in the current blood-based diagnostic routine.
The ability to specifically isolate various pathogens directly from whole blood using sβ2GPI peptides not only allows us to reduce the time for microbial expansion but also makes it feasible to monitor their growth in real time because the isolated pathogens are cultured in a purified medium. In the case of rapidly dividing species such as E. coli, a rapid culture time of 4 h has been found to be sufficient to reach a minimum required cell count for uRAST, which allowed us to record a complete AST result in under 8 h starting from whole blood. The integration of techniques that can continuously facilitate microbial quantification in our current workflow may facilitate optimization of growth duration based on the patient’s specific invading species, avoiding delays during culture and enabling precise control over the inoculum during AST. This will also allow the ready application of our present platform to various types of infection, including those caused by slow-growing microorganisms such as Candida spp. or carbapenem-resistant species (Extended Data Fig. 10).
As another means of reducing TAT, we developed the uRAST chip that enables accurate drug susceptibility determination using only a few microbial cells. By doing so, several doubling periods are instantly saved during rapid culture following the isolation of blood-borne pathogens. Comparing susceptibility results between uRAST and BMD, we also observe a high SIR concordance rate. However, for some drug–pathogen combinations, the MIC values from uRAST were slightly lower although most were within a single log2 dilution. Such a discrepancy may have arisen from a difference in the amount of inoculum, which was also observed from recent reports demonstrating rapid AST using low bacterial loads and often in species producing β-lactamase (inoculum effect)48,49. Currently, the parameters used to evaluate susceptibility profiles including breakpoints, which are annually announced and updated by both the CLSI and European Committee on Antimicrobial Susceptibility Testing, are generally based on an inoculum size of 5 × 105 CFU ml−1 for a BMD test. We speculate that this standardized level derives from the use of traditional BC along with the limited range of detection available for optical density-based measures (at least 107 CFU ml−1) used during sample preparation (dilution). However, now with options available to bypass BC, future studies are suggested to understand and calibrate the effect of using minimal inocula, in conjunction with the potential establishment of new regulations or standards, to support and guide the development of a low-input, ultra-rapid AST. Furthermore, attempts to distinguish between non-growing persisters and dead pathogens post antibiotic evaluation should be pursued, because the absence of growth does not always indicate susceptibility, a limitation present in all current phenotype-based AST diagnostic techniques.
Coinciding with the pressing need to implement rapid and accurate AST, we establish an approach that has the potential to circumvent the need for BC, thereby realizing pathogen identification over a wide range of species and delivering phenotypic drug susceptibility results on the day of patient blood draw. With the potential to guide clinicians towards timely administration of optimal therapy, thereby saving many lives, our platform can also help to de-escalate the spread of antimicrobial resistance and sustain the lifespan of existing antibiotics. Through future validation in a more diverse cohort of patients verifying its efficacy and clinical value, along with efforts to reduce costs through mass production and system automation to mitigate the substantial (non-technical yet dominant) delay caused by laboratory working hours, we anticipate that our method will completely transform the current standards of bacteraemia diagnostics and bring new focus on the development of next-generation BC-free AST platforms.
Methods
Bacterial strains
The five standard ATCC bacterial strains (E. coli ATCC 25922, K. pneumoniae ATCC 700603, P. aeruginosa ATCC 27853, S. aureus ATCC 33862 and E. faecalis ATCC 7080) were purchased from Liofilchem. All clinical isolates used in this study were derived from positive BC samples from patients and obtained from Seoul National University Hospital (SNUH) with informed consent under SNUH Institutional Review Board (IRB)-approved protocol (no. 1910-188-1076). Bacterial stocks were made at concentrations of 1.5 × 108 CFU ml−1 using 10% skim milk (Becton Dickinson) and stored at −80 °C. For testing, the stock solution was thawed and spread on a tryptic soy agar plate (Asan Pharm.), followed by overnight incubation at 35 °C and storage at 4 °C before use. Bacterial colonies were subsequently picked from these plates and inoculated into 5 ml of either MHB (Becton Dickinson) or tryptic soy broth (Becton Dickinson) and further expanded in a shaking incubator at 30 rpm and 35 °C for 20–24 h before experiments.
Blood specimen collection
Blood samples were drawn from infection-suspected patients with haematologic malignancies, as well as from over 50 healthy donors after obtaining informed consent under the same SNUH IRB-approved protocol. Participants in this study consented to publication with encoded personal data for confidentiality. The patient cohort was chosen due to their high vulnerability to infections and likelihood of benefiting from rapid diagnosis and appropriate antibiotic treatment. All specimens were collected in either BC bottles (Becton Dickinson BACTEC Plus Aerobic medium) or EDTA vacuum tubes (Becton Dickinson vacutainer) and were processed within 12 h.
Bacterial isolation
Bacterial strain solutions were prepared at a concentration of 1.5 × 108 CFU ml−1 (optical density of 0.5 McFarland standard) in MHB or tryptic soy broth medium using a DensiCHEK Plus spectrophotometer (bioMérieux), and serially diluted to a target cell concentration. Small volumes (100 μl) of each were then spiked into 5 ml of either PBS (Invitrogen) or whole blood to mimic the presence of bacteria in patient specimens. Concurrently, equal volumes of aliquot were spread directly onto tryptic soy agar or blood agar plates (n = 3) to backtrack the number of bacterial cells inoculated in the sample solution. However, taking into account that 10–40% of spiked bacterial cells were eliminated in healthy donor blood due to the blood effect, in our blood experiments the 100 μl aliquots were separately inoculated into the corresponding blood and incubated for under 1 h during the enrichment process before streaking on agar plates for quantification (n = 3). To minimize potential bias arising from various blood samples, the same blood was used no more than three times for each capture experiment characterizing a specific bacterial strain. The number of colonies from each plate was averaged and used as control to calculate capture efficiency.
For PBS experiments, 5 ml of PBS was additionally added to samples with 50 μl of sβ2GPI nanoparticle solution (concentration 1.9 × 1010 particles per microlitre) whereas for blood, 5 ml of binding buffer comprising Tris (below 3%) and Triton-X100 (below 0.4%) was mixed in a 1:1 ratio, to increase the electrostatic interaction between peptides and pathogens. Both samples were then placed in a thermomixer (KTM-100C, KBT) and incubated at 35 °C for 30 min under 800 rpm rotation for binding. Next, nanoparticle-bound bacterial cells were separated for 30 min using a MagListo magnetic rack (Bioneer) and the supernatant removed. Finally the remaining nanoparticle–cell complexes were resuspended in 100 μl of growth medium and streaked on tryptic soy agar plates to quantify the total number of cells captured. In addition, the number of blood cells was also estimated using a subset of the solution through a haemocytometer to evaluate overall purity following separation. The initial counts of WBC and RBC in 5 ml of whole blood were measured as 2.78 × 107 and 2.59 × 1010 cells, respectively (n = 4). For the remaining experiments, particle-bound bacteria were resuspended in 1 ml of PBS or MHB growth medium for the following QmapID and rapid culture/uRAST procedure, respectively.
Fabrication procedure for the shape-encoded microdisc library
The shape-encoded microdisc was fabricated using a protocol similar to that described in our previous report50. In brief, PAA-coated Fe3O4 nanoparticles were mixed with a photo-curable resin to form a homogenous prepolymer blend. The resin was made by combining ethoxylated trimethylolpropane triacrylate (molecular weight = 428, Sigma-Aldrich) and 3-(trimethoxysilyl)propyl methacrylate (92%, Sigma-Aldrich) at a 1:1 volumetric ratio. Here, the acrylate groups on both materials served as the polymer scaffold with the silanol group used as a silane-grafting matter for silica formation. In addition, 10 wt% 2-hydroxy-2-methylpropiophenone (97%, Sigma-Aldrich) was added as a photo-initiator. The prepared mixture was then exposed to ultraviolet light (200 W mercury-xenon lamp, Hamamatsu) for 0.25 s to create the microdisc with unique hole patterns using optofluidic maskless lithography through photopolymerization. Various patterns were generated by a combination of different hole counts and locations (angle or distance from the centre), including a parity element for error correction. For silica coating (modified Stöber method), the microdiscs were collected and immersed in a solution containing 3 ml of deionized water, 20 ml of 99% ethanol (Daejung) and 1 ml of 25–28% ammonium hydroxide (NH4OH, Daejung). Next, 0.1 ml of tetraethyl orthosilicate (98%, Sigma-Aldrich) was serially injected into the solution until the total volume of tetraethyl orthosilicate reached 1 ml. The microdiscs were finaly rinsed with ethanol.
To introduce amine groups on the surface, silica-coated microdiscs were treated with 5% vol./vol. 3-aminopropyltriethoxysilane (98%, Sigma-Aldrich) anhydrous ethanol solution for 12 h at 25 °C with agitation. After washing with ethanol, the microdiscs were baked at 110 °C for 1 h. Next, the microdiscs were added to a solution of N,N-dimethylformamide (99.8%, Sigma-Aldrich) solution containing 50 mM succinic anhydride (99%, Sigma-Aldrich) and 55 mM Tris-acetate-EDEA, shaken for 2 h at room temperature and washed with N,N-dimethylformamide and ethanol to form a carboxylic surface. For attachment of single-stranded DNA detection probes, the surface of the microdisc (approximately 3 × 104 count) was activated using 258 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (Alfa-Aesar) in MES buffered saline (pH 4.7, Thermo Fisher Scientific). Next, 5 µM 5′-amino-terminated DNA detection probes (Quantamatrix) were added to the microdisc solution and incubated for 1 h to carry out the coupling reaction with the carboxylic acids through amide bonds. Finally the microdiscs were rinsed with a washing buffer (no. RQID01-11, Quantamatrix) followed by 1× TE buffer (pH 8.0, Biosesang) to remove unbound excessive DNA probes and storage in 200 µl of TE buffer at 4 °C before use.
Bacterial identification and analysis of resistance genes
Following pathogen enrichment, resuspended bacterial samples were placed in a DynaMag-2 magnetic rack (Invitrogen) for 5 min and the supernatant removed. Next, samples were treated with 50 μl of 5% Chelex 100 chelating resin (no. 1421253, Bio-Rad) and incubated for 15 min at 95 °C to lyse bacterial cells for DNA extraction. The solution was then centrifugated at 14,000 rpm for 15 min at 4 °C to precipitate the remaining nanoparticles and cell debris. Finally, 8 μl of the purified supernatant containing bacterial DNA (PCR template) was collected and used for nested PCR. As an optional step, the weight of gDNA extraction was measured in between the use of Nanodrop 1 (Thermo Fisher Scientific). For DNA amplification, 20 μl of PCR master mix containing 10 μl of 2× PCR premix (no. RQID01-01, Quantamatrix), 2 μl of primer mix and 8 μl of PCR template was prepared. Here, different primer sets were used in separate amplification reactions for detection of pathogen ID and resistance genes. Following 55 cycles of nested PCR, 10 μl of 3× denaturation solution (no. RQID01-07, Quantamatrix) was added, with incubation for 5 min at room temperature, to obtain single-stranded PCR products of approximate length 200 base pairs.
For QmapID the shape-encoded microdiscs, coupled with various DNA detection probes, were washed to remove TE buffer, resuspended with 45 μl of hybridization buffer (no. RQID01-10, Quantamatrix) and transferred to a 96-well glass plate (no. QMPLATE01, Quantamatrix). Following loading of the 10 μl sample containing the PCR products to the well, the plate was incubated on a Grant Bio thermostatic shaker (Grant Instruments) for 30 min at 37 °C and 1,200 rpm. Next, the plate was left on a magnet for 30 s to settle and hold the microdiscs on the bottom surface of the well. The microdiscs were rinsed three times using 100 μl of washing buffer (no. RQID01-11, Quantamatrix). After discarding the washing buffer, 50 μl of detection solution (no. RQID01-12, Quantamatrix) containing streptavidin-conjugated R-phycoerythrin (PE) fluorescent dyes was injected. The plate was further incubated on a thermostatic shaker for 10 min at 37 °C and 1,200 rpm for microdisc labelling. Finally, the microdiscs were washed again three times to remove dyes that were non-specifically bound to the surface. With 100 μl of buffer remaining, the plate was loaded on an in-house plate analyser (no. QMAP 2.0, Quantamatrix) and image-processing software to count and measure the fluorescence intensity of each shape-encoded microdisc for analysis. On average, 50–60% of microdiscs per type were subjected to analysis based on the imaging field of view.
Design and fabrication process of the uRAST chip
The 96-well-based AST chip was designed using three-dimensional CAD software (SolidWorks 2018, Dassault Systèmes) and fabricated by injection moulding using a general-purpose polystyrene plastic (L.G. Chem.). Focus marks were imprinted on the bottom surface for automated chip alignment during time-lapse image acquisition. All antimicrobial agents were purchased from Sigma-Aldrich except for daptomycin (Tokyo Chemical Industry), clavulanic acid (Chong Kun Dang Pharm.), avibactam and varborbactam (MedChemExpress). Stock solutions were prepared following the manufacturer’s instructions and CLSI guidelines and stored at −80 °C before use. Varying types and concentrations of antimicrobial solution were diluted and dispensed in the satellite reservoir of each AST chip well following treatment of the surface with O2 plasma (Femto Science), and dried in a vacuum oven (Jeio Tech) for 50 min at 50 °C. The drug-loaded AST chip was packaged under nitrogen with silica gel and stored at 4 °C before experiments.
BMD
The BMD method was used as the gold standard to validate accuracy across various AST methods. Antimicrobial stock solutions were diluted with MHB medium to the appropriate testing conditions and inoculated into a 96-well microplate (SPL Life Sciences) with bacterial cells (5 × 105 CFU ml−1) at a final volume of 100 μl. Following 16–20 h incubation at 35 °C, the MIC of each drug was evaluated. Resistance was determined when a reduction in bacterial growth of at least 80% was observed through unaided visual inspection compared with controls. All tests were performed in triplicate.
Software
All figures were created using SolidWorks (v.2018) and Adobe illustrator (v.2021). Data and statistical analyses and plotting were carried out using Microsoft Excel (v.16.59), Matlab (v.R2022b) and R (v.4.1.0). All image-processing algorithms were developed using Python (v.3.9.12).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data from this study are available within the article. Additional data that support the findings of this study are available from the corresponding authors on reasonable request.
Code availability
All codes used to reproduce figures are available at GitHub (https://github.com/phisoart/uRAST). No additional custom code was generated in this study.
References
Heuverswyn, J. V. et al. Association between time to appropriate antimicrobial treatment and 30-day mortality in patients with bloodstream infections: a retrospective cohort study. Clin. Infect. Dis. 76, 469–478 (2022).
Lee, C.-C., Lee, C.-H., Hong, M.-Y., Tang, H.-J. & Ko, W.-C. Timing of appropriate empirical antimicrobial administration and outcome of adults with community-onset bacteremia. Crit. Care 21, 119 (2017).
Liu, V. X. et al. The timing of early antibiotics and hospital mortality in sepsis. Am. J. Respir. Crit. Care Med. 196, 856–863 (2017).
Tabak, Y. P. et al. Blood culture turnaround time in U.S. acute care hospitals and implications for laboratory process optimization. J. Clin. Microbiol. 56, e00500–e00518 (2018).
Singer, M. et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315, 801–810 (2016).
Hotchkiss, R. S. & Karl, I. E. The pathophysiology and treatment of sepsis. N. Engl. J Med. 348, 138–150 (2003).
Cohen, J. The immunopathogenesis of sepsis. Nature 420, 885–891 (2002).
Fleischmann, C. et al. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am. J. Resp .Crit. Care Med. 193, 259–272 (2016).
Rudd, K. E. et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet 395, 200–211 (2020).
Gaieski, D. F., Edwards, J. M., Kallan, M. J. & Carr, B. G. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit. Care Med. 41, 1167–1174 (2013).
Marquet, K., Liesenborgs, A., Bergs, J., Vleugels, A. & Claes, N. Incidence and outcome of inappropriate in-hospital empiric antibiotics for severe infection: a systematic review and meta-analysis. Crit. Care 19, 63 (2015).
Kadri, S. S. et al. Inappropriate empirical antibiotic therapy for bloodstream infections based on discordant in-vitro susceptibilities: a retrospective cohort analysis of prevalence, predictors, and mortality risk in US hospitals. Lancet Infect. Dis. 21, 241–251 (2021).
Vazquez-Guillamet, C. et al. Using the number needed to treat to assess appropriate antimicrobial therapy as a determinant of outcome in severe sepsis and septic shock. Crit. Care Med. 42, 2342–2349 (2014).
Battle, S. E. et al. Association between inappropriate empirical antimicrobial therapy and hospital length of stay in Gram-negative bloodstream infections: stratification by prognosis. J. Antimicrob. Chemother. 72, 299–304 (2016).
Shorr, A. F. et al. Inappropriate antibiotic therapy in Gram-negative sepsis increases hospital length of stay. Crit. Care Med. 39, 46–51 (2011).
Ling, T. K. W., Liu, Z. K. & Cheng, A. F. B. Evaluation of the VITEK 2 system for rapid direct identification and susceptibility testing of Gram-negative bacilli from positive blood cultures. J. Clin. Microbiol. 41, 4705–4707 (2003).
Ligozzi, M. et al. Evaluation of the VITEK 2 system for identification and antimicrobial susceptibility testing of medically relevant Gram-positive cocci. J. Clin. Microbiol. 40, 1681–1686 (2002).
Carroll, K. C. et al. Evaluation of the BD Phoenix automated microbiology system for identification and antimicrobial susceptibility testing of Enterobacteriaceae. J. Clin. Microbiol. 44, 3506–3509 (2006).
Baker, C. N., Stocker, S. A., Rhoden, D. L. & Thornsberry, C. Evaluation of the MicroScan antimicrobial susceptibility system with the autoSCAN-4 automated reader. J. Clin. Microbiol. 23, 143–148 (1986).
Li, H. et al. Adaptable microfluidic system for single-cell pathogen classification and antimicrobial susceptibility testing. Proc. Natl Acad. Sci. USA 116, 201819569 (2019).
Baltekin, Ö., Boucharin, A., Tano, E., Andersson, D. I. & Elf, J. Antibiotic susceptibility testing in less than 30 min using direct single-cell imaging. Proc. Natl Acad. Sci. USA 114, 9170–9175 (2017).
Malmberg, C. et al. Evaluation of the speed, accuracy and precision of the QuickMIC rapid antibiotic susceptibility testing assay with Gram-negative bacteria in a clinical setting. Front. Cell. Infect. Microbiol. 12, 758262 (2022).
Avesar, J. et al. Rapid phenotypic antimicrobial susceptibility testing using nanoliter arrays. Proc. Natl Acad. Sci. USA 114, E5787–E5795 (2017).
Fredborg, M. et al. Rapid antimicrobial susceptibility testing of clinical isolates by digital time-lapse microscopy. Eur. J. Clin. Microbiol. Infect. Dis. 34, 2385–2394 (2015).
Silva-Dias, A. et al. Evaluation of FASTinov ultrarapid flow cytometry antimicrobial susceptibility testing directly from positive blood cultures. J. Clin. Microbiol. 59, e00544-21 (2021).
Zhang, M. et al. Rapid determination of antimicrobial susceptibility by stimulated Raman scattering imaging of D2O metabolic incorporation in a single bacterium. Adv. Sci. 7, 2001452 (2020).
Flentie, K. et al. Microplate-based surface area assay for rapid phenotypic antibiotic susceptibility testing. Sci. Rep. 9, 237 (2019).
Rosselin, M., Prod’hom, G., Greub, G. & Croxatto, A. Performance evaluation of the Quantamatrix QMAC-dRAST system for rapid antibiotic susceptibility testing directly from blood cultures. Microorganisms 10, 1212 (2022).
Truong, T. T., Mongkolrattanothai, K., Flores, I. I. & Bard, J. D. Evaluation of the performance and clinical impact of a rapid phenotypic susceptibility testing method directly from positive blood culture at a pediatric hospital. J. Clin. Microbiol. 60, e0012222 (2022).
Choi, J. et al. A rapid antimicrobial susceptibility test based on single-cell morphological analysis. Sci. Transl. Med. 6, 267ra174 (2014).
Zhang, F. et al. Direct antimicrobial susceptibility testing on clinical urine samples by optical tracking of single cell division events. Small 16, 2004148 (2020).
Tibbetts, R. et al. Performance of the Reveal rapid antibiotic susceptibility testing system on Gram-negative blood cultures at a large urban hospital. J. Clin. Microbiol. 60, e00098-22 (2022).
Spencer, D. C. et al. A fast impedance-based antimicrobial susceptibility test. Nat. Commun. 11, 5328 (2020).
Schoepp, N. G. et al. Rapid pathogen-specific phenotypic antibiotic susceptibility testing using digital LAMP quantification in clinical samples. Sci. Transl. Med. 9, eaal3693 (2017).
Bhattacharyya, R. P. et al. Simultaneous detection of genotype and phenotype enables rapid and accurate antibiotic susceptibility determination. Nat. Med. 25, 1858–1864 (2019).
Lamy, B., Dargère, S., Arendrup, M. C., Parienti, J.-J. & Tattevin, P. How to optimize the use of blood cultures for the diagnosis of bloodstream infections? A state-of-the art. Front. Microbiol. 7, 697 (2016).
Ganguli, A. et al. A culture-free biphasic approach for sensitive and rapid detection of pathogens in dried whole-blood matrix. Proc. Natl Acad. Sci. USA 119, e2209607119 (2022).
Abram, T. J. et al. Rapid bacterial detection and antibiotic susceptibility testing in whole blood using one-step, high throughput blood digital PCR. Lab Chip 20, 477–489 (2019).
Huang, T., Tzeng, Y. & Dickson, R. M. FAST: rapid determinations of antibiotic susceptibility phenotypes using label‐free cytometry. Cytometry A 93, 639–648 (2018).
Vincent, J.-L. et al. Rapid diagnosis of infection in the critically ill, a multicenter study of molecular detection in bloodstream infections, pneumonia, and sterile site infections. Crit. Care Med. 43, 2283–2291 (2015).
Ho, C.-S. et al. Rapid identification of pathogenic bacteria using Raman spectroscopy and deep learning. Nat. Commun. 10, 4927 (2019).
Tjandra, K. C. et al. Diagnosis of bloodstream infections: an evolution of technologies towards accurate and rapid identification and antibiotic susceptibility testing. Antibiotics 11, 511 (2022).
Sorgenfrei, M., Hürlimann, L. M., Remy, M. M., Keller, P. M. & Seeger, M. A. Biomolecules capturing live bacteria from clinical samples. Trends Biochem. Sci. 47, 673–688 (2022).
Kang, J. et al. Accurate diagnosis of COVID-19 from self-collectable biospecimens using synthetic apolipoprotein H peptide-coated nanoparticle assay. Anal. Chem. 94, 17186–17194 (2022).
Vutukuru, M. R. et al. A rapid, highly sensitive and culture-free detection of pathogens from blood by positive enrichment. J. Microbiol. Meth. 131, 105–109 (2016).
CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Ninth Edition (Clinical and Laboratory Standards Institute, 2012).
ESCMID. Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by agar dilution. Clin. Microbiol. Infec. 6, 509–515 (2000).
Smith, K. P. & Kirby, J. E. The inoculum effect in the era of multidrug resistance: minor differences in inoculum have ramatic effect on MIC determination. Antimicrob. Agents Chemother. 62, e00433-18 (2018).
Wiegand, I., Hilpert, K. & Hancock, R. E. W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175 (2008).
Kim, L. N. et al. Shape-encoded silica microparticles for multiplexed bioassays. Chem. Commun. 51, 12130–12133 (2015).
Acknowledgements
This work was supported by the Ministry of Science and ICT of the Republic of Korea and the National Research Foundation of Korea (nos. NRF-2020R1A3B3079653 and NRF-2020R1A2C1007242), the BK21 FOUR programme of the Education and Research Program for Future ICT Pioneers (Seoul National University, 2022), Bio-MAX Institute (Seoul National University), K-BIO KIURI Centre (no. 2020M3H1A1073304), QuantaMatrix Inc., Korea Disease Control and Prevention Agency (no. 2022-ER2207-00) and the SNUH Research Fund (grant no. 03-2020-0440).
Author information
Authors and Affiliations
Contributions
T.H.K., J.K., H. Jang, I.K., W.B.P. and S.K. conceived and designed the study. T.H.K., J.K., H. Jang, H. Joo, G.Y.L. and H.K. conducted pathogen-capture experiments using sβ2GPI nanoparticles. U.C., H.B. and J.J. designed PCR primers and optimized the protocol for bacterial gene amplification. U.C., H.B. and J.J. fabricated the shape-encoded microdiscs and developed the DNA detection probes used in the QmapID assay. T.H.K., J.K., H. Jang, H. Joo, U.C. and H.B. optimized the protocol and carried out the QmapID assay. J.J. and G.Y.L. designed and manufactured the 96-well-based uRAST chip. H.K. modelled and ran the computation simulation for drug diffusion. T.H.K., J.K., H. Jang, G.Y.L. and S.H. performed uRAST experiments. T.H.K., H. Jang and D.Y.K. developed the automated image-processing algorithm for SIR determination and analysed the data. C.M.L., C.K.K., P.G.C., N.J.K., M.O., T.S.K., I.K. and W.B.P. collected patient-derived bacterial strains and prepared patient samples used for the clinical study. T.H.K. and J.J. analysed the results from the clinical study. I.K. and W.B.P. provided general advice on clinical data and validated the results. T.H.K., J.K., H. Joo, I.K., W.B.P. and S.K. cowrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
P.G.C., T.S.K. and W.B.P. serve as consultants for QuantaMatrix, Inc. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature thanks Jason Biemond, Stephanie Popping, W. Wiersinga and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Optimization of pathogen isolation protocol.
To maximize enrichment yield, varying amount of sβ2GPI nanoparticles as well as incubation time for mixing and microbial binding is examined over 5 mL of PBS or whole blood, spiked with 500 cfu of S. aureus. Capture efficiency based on the volume of nanoparticle solution used for isolation on (a) PBS and (b) blood samples (n = 3). Each mixture is incubated at 35 °C for 30 min under gentle rotation prior to magnetic separation. Capture efficiency as a function of incubation time on (c) PBS and (d) blood specimens (n = 3). Here, 50 μL of nanoparticle solution is used. To ensure sufficient binding and separation for both PBS and blood samples, a minimum of > 50 μL of nanoparticle solution and > 20 min of incubation period is required. (e) Effect of binding buffer during magnetic separation. For blood, a pre-made binding buffer is added and mixed in a 1:1 ratio, to increase the electrostatic interaction between the synthetic peptides and pathogens. (f) Capture efficiency based on different E. coli serotype. Samples are prepared by spiking each cell serotype into 5 mL of PBS at a concentration of 5 cfu/mL (n = 5). Total number of (g) WBCs and (h) RBCs remaining after enrichment, based on different mixtures of particle solution to blood volume ratio (n = 4). Data points in a-d and f-h represent biologically independent experimental replicates. All data in a-d and f are represented as the mean ± SD. The boxes and whiskers in g and h display the interquartile range as well as the maximum and minimum values of each data. The line through the boxes shows the median.
Extended Data Fig. 2 Isolation efficiency of the sβ2GPI nanoparticles in blood.
(a) Isolation efficiency of the sβ2GPI nanoparticles in blood based on high (n = 3) and low (n = 20) bacteria load. Total number of ATCC standard, (b) K. pneumoniae (npbs = 20, nblood = 20), (c) P. aeruginosa (npbs = 15, nblood = 20), and (d) E. faecalis (npbs = 15, nblood = 20) cells captured using sβ2GPI nanoparticles with respect to the initial spike number of 1-10 cfu/mL in 5 mL of PBS (blue) or blood (red). The shaded regions, blue and red, signify the 95% confidence intervals obtained from the linear regressions. Data points in a-d represent biologically independent experimental replicates. (e) Total positivity rate derived from the repeatability test shown in Fig. 2g. Each data point represents 60 sets of experiments conducted on individual species. The box and whisker display the interquartile range as well as the maximum and minimum values. The line through the box shows the median. (f) Positivity rate based on spiking concentrations. At a spiking concentration of around 4 cfu/mL, a 100% positivity rate is achieved within all clinical strains examined. Each data point represents the results obtained from 20 repeated experiments conducted across 19 different species. (g) Positivity rate based on gram type. Each data point represents 60 experiments. All data in a and e-g are represented as the mean ± SD.
Extended Data Fig. 3 Schematic illustration of the QmapID protocol.
(a) Fabrication and functionalization procedure of the QmapID micro-discs. (b) Workflow of the QmapID assay.
Extended Data Fig. 4 Image processing algorithm and report time for SIR determination.
(a) Flow chart of the automated image processing and SIR decision algorithm. Prior to SIR determination, raw images taken from each AST well are aligned to the initial acquisition and processed through a background subtraction, binarization, and noise filtering algorithm to monitor changes in the bacteria’s kinetic growth patterns. Representative time-lapse image of (b) E. coli and (c) S. aureus during culture in the AST chip. Resistance and susceptible cases are shown at the top and bottom, respectively. The purple square indicates the time when pathogenic growth is first detected from the control wells (dynamic decision) and the red square depicts the time after an additional 2 h of incubation in which a final SIR decision is made. CA evaluated over time using six patient-derived (d) GN and (e) GP strains to optimize the SIR report time during AST. (f) Combined CA and errors calculated as a function of time starting at the moment when a positive growth is observed from the control wells. E.C., E. coli; K.P., K. pneumoniae; P.A., P. aeruginosa; E.F., E. faecalis; MSSA, Methicillin-sensitive S. aureus; MRSA, Methicillin-resistant S. aureus.
Extended Data Fig. 5 Design of the clinical study.
Design and workflow of the clinical study performed to validate the QmapID and uRAST platform. The accuracy of ID and AST as well as the overall TAT is compared to the hospital protocols and the conventional BMD tests. The approximate time for each procedure is stated below in parentheses. The total TAT calculated for each procedure does not account for non-technical delays (e.g., sample transport delay or work-hour delay).
Extended Data Fig. 6 Comparison between BMD, uRAST, and the hospital AST result.
(a) SIR results from the clinical study based on the BMD, hospital AST, and uRAST. (b) CA and error of the hospital AST and uRAST results compared to the BMD test. Since the drugs listed in each AST panel are different, only the antimicrobial agents mutually included in both testing methods are considered for comparison. (c) Combined CA and error calculated from the six samples using uRAST and hospital AST methods.
Extended Data Fig. 7 The impact of hospital laboratory working hours on the TAT in conducting uRAST.
(a) The average work-hour delay attributed to constraints in the hospital’s laboratory working hours for conducting AST is estimated to be 15 ± 7.01 h, which is considerably high compared to the actual run time of uRAST (n = 6 independent case scenarios). This signifies the importance of system automation for future clinical applications. Data is represented as the mean ± SD. (b) TAT comparison between the hospital AST methods versus uRAST without considering work-hour delays (n = 6 independent case scenarios). Starting from initial blood processing and incubation, the average theoretical TAT for uRAST is determined to be 13 ± 2.53 h. The total time reduction of using uRAST for drug susceptibility analysis is shown on the right. The boxes and whiskers display the interquartile range as well as the maximum and minimum values of each data. The line through the boxes shows the median. Statistical significance is calculated using a two-tailed, paired t-test (P = 0.0002). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Extended Data Fig. 8 Comparison between the QmapID assay and hospital gram staining and chemical tests.
(a) Time spent for each procedure during QmapID and gram staining/chemical tests over the six infection-positive patient samples. BC, blood culture; GS, gram stain. (b) TAT comparison between the hospital gram staining/chemical test method versus QmapID assay (n = 6 independent case scenarios). The total time reduction of using the QmapID for pathogen identification is shown on the right. The boxes and whiskers display the interquartile range as well as the maximum and minimum values of each data. The line through the boxes shows the median. Statistical significance is calculated using a two-tailed, paired t-test (P = 0.0034). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Extended Data Fig. 9 Isolation and identification from samples mimicking polymicrobial infections using sβ2GPI nanoparticles and QmapID assay.
Characterization of bacteria isolation using synthetic β2GPI peptide-coated nanoparticles for polymicrobial infections. To demonstrate the feasibility of detecting polymicrobial infections, 2-4 cfu/mL of S. aureus and S. epidermidis are both inoculated into blood, followed by sβ2GPI capture. The two species have been specifically chosen as they represent one of the most frequently observed combinations in polymicrobial infections. (a) Color difference between S. aureus and S. epidermidis colonies on a CHROMagar Staphylococcus plate. CHROMagar purchased from CHROMagar™ (France) and used to differentiate the two species for quantification purposes. Capture efficiency from (b) single-species samples of S. aureus and S. epidermidis (n = 10) or (c) mixed (n = 5). All data in b-c are represented as the mean ± SD from biologically independent experimental replicates. The independent capture efficiency observed in the mixed cases is comparable to that of the single-species samples. (d) Image of the mixed sample plated after capture. The images are taken after placing the same agar plate on top of a green (top) or blue surface (bottom) to enhance colony visibility. Differential detection of closely related bacterial species using QmapID. Heatmap illustrating outcomes from QmapID for species identification. Results are obtained from combinations of bacterial cell inputs: (e) S. aureus/S. epidermidis, (f) S. pneumoniae/S. mitis.
Extended Data Fig. 10 Effect of low inoculum on AST results for carbapenem-resistant bacterial strain.
Changes in MIC values based on the concentration of inoculum used for (a) BMD test and (b) uRAST (ndrug = 22). The clinical isolate acquired from P. 126, identified as CRE, has been employed for the evaluation. The thickness of the connecting lines represents the frequency of drugs manifesting a particular trend. Red solid lines indicate the mean (thick line) and SD (narrow line) of the MIC errors. Overall, the MIC values mostly remained stable, falling within a single log2-dilution difference, when using uRAST. However, in the BMD test, MICs from IP and MP, which are both classified under the β-lactam/carbapenem antibiotics, exhibited deviations from the essential agreement range as the inoculum size decreased. This suggests that AST outcomes from uRAST are more robust against varying inoculum concentrations compared to BMD, possibly due to the difference in drug testing conditions (liquid phase vs. agar embedded). We speculate that the bacterial cell immobilization within the agarose matrix have minimized the inoculum effect by localizing the antibiotic-inhibiting metabolites or proteins. (c) Corresponding SIR decisions based on the concentration of inoculum used for BMD test and uRAST. (d) log2-diluation MIC errors of MP as a function of inoculum size (n = 3). All data are represented as the mean ± SD from biologically independent experimental replicates.
Supplementary information
Supplementary Information
Supplementary Tables 1–7 and Figs. 1–4.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kim, T.H., Kang, J., Jang, H. et al. Blood culture-free ultra-rapid antimicrobial susceptibility testing. Nature 632, 893–902 (2024). https://doi.org/10.1038/s41586-024-07725-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07725-1
- Springer Nature Limited
This article is cited by
-
Testing fast with uRAST
Nature Reviews Microbiology (2024)