

Abstract
In vitro models of autoimmunity are constrained by an inability to culture affected epithelium alongside the complex tissue-resident immune microenvironment. Coeliac disease (CeD) is an autoimmune disease in which dietary gluten-derived peptides bind to the major histocompatibility complex (MHC) class II human leukocyte antigen molecules (HLA)-DQ2 or HLA-DQ8 to initiate immune-mediated duodenal mucosal injury1,2,3,4. Here, we generated air–liquid interface (ALI) duodenal organoids from intact fragments of endoscopic biopsies that preserve epithelium alongside native mesenchyme and tissue-resident immune cells as a unit without requiring reconstitution. The immune diversity of ALI organoids spanned T cells, B and plasma cells, natural killer (NK) cells and myeloid cells, with extensive T-cell and B-cell receptor repertoires. HLA-DQ2.5-restricted gluten peptides selectively instigated epithelial destruction in HLA-DQ2.5-expressing organoids derived from CeD patients, and this was antagonized by blocking MHC-II or NKG2C/D. Gluten epitopes stimulated a CeD organoid immune network response in lymphoid and myeloid subsets alongside anti-transglutaminase 2 (TG2) autoantibody production. Functional studies in CeD organoids revealed that interleukin-7 (IL-7) is a gluten-inducible pathogenic modulator that regulates CD8+ T-cell NKG2C/D expression and is necessary and sufficient for epithelial destruction. Furthermore, endogenous IL-7 was markedly upregulated in patient biopsies from active CeD compared with remission disease from gluten-free diets, predominantly in lamina propria mesenchyme. By preserving the epithelium alongside diverse immune populations, this human in vitro CeD model recapitulates gluten-dependent pathology, enables mechanistic investigation and establishes a proof of principle for the organoid modelling of autoimmunity.
Similar content being viewed by others


Main
Autoimmune diseases represent diverse and often intractable pathologies characterized by adaptive immunity against self antigens. CeD is an autoimmune disorder with a world-wide population prevalence of 1% that is caused by the ingestion of gluten, which is a storage protein of wheat, barley and rye1. In CeD, immune responses to dietary gluten promote the destruction of the duodenal mucosa in individuals carrying the MHC class II allotypes HLA-DQ2 or HLA-DQ8. After gluten ingestion, derivative gliadin peptides are deamidated by tissue TG2. This introduces negative charges into gliadin that enhance its binding to MHC class II, culminating in a gluten-specific CD4+ T-cell response with T-cell-mediated intestinal inflammation, mucosal injury and plasma cell anti-TG2 autoantibody production2,3. CeD pathology includes villous atrophy, crypt hyperplasia, inflammation of the lamina propria and intra-epithelial lymphocytosis4. How gluten-specific CD4+ T cells elicit cytotoxic T-cell responses is poorly understood but may involve CD8+ T-cell expression of NKG2C and NKG2D, which bind the MHC-I-related molecules HLA-E and MICA (MHC class I chain-related protein A) in the epithelium, targeting epithelial cells for destruction5,6,7.
The study of CeD, and autoimmunity in general, has notably lacked in vitro models that incorporate both targeted tissues and their endogenous resident immune populations as a unified entity8. Previous studies have used cell lines9, short-term slice cultures9,10 or mixtures of immune cells with antigen-presenting cells (APCs)11. Conventional intestinal organoids (enteroids) are composed solely of epithelium and so omit immune and mesenchymal populations12. Despite lacking fully human context, mouse CeD models have used human HLA-DQ8 or HLA-DQ2.5 transgenes13 or required the overexpression of IL-15 to elicit full pathology14.
Potential cytokine mediators of CeD include IL-2, which is rapidly induced in CeD patients after oral gluten challenge15,16, but this cytokine has not been shown to be essential for CeD pathology. Although IL-15 is upregulated in CeD clinical biopsies, and its transgenic overexpression in mouse CeD models promotes intestinal epithelial damage14,17, clinical IL-15 blockade has not yet been shown to prevent mucosal injury in CeD patients after gluten challenge18. IL-2 and IL-15 are members of a cytokine superfamily with receptors that have a common signal-transducing γ-chain19. Another member is IL-7, which is necessary for T-cell survival, proliferation and thymic development, memory-T-cell maintenance and B-cell development, survival and activation19. IL-7 has been implicated in the pathogenesis of autoimmune diseases such as multiple sclerosis, rheumatoid arthritis, type I diabetes and inflammatory bowel disease20,21,22,23,24,25. However, the IL-7 signalling axis has not previously been implicated in CeD pathology.
Here we establish an ALI organoid culture system for intact fragments of human small intestine in which syngeneic intestinal epithelial cells, mesenchymal cells and diverse native immune populations are co-preserved as a cohesive unit without exogenous reconstitution. From duodenal endoscopic biopsies of CeD patients, we created ALI organoids to recapitulate numerous aspects of gluten-induced pathology and to explore a previously unsuspected role for IL-7 in CeD pathology.
Small-intestine ALI organoids retain epithelial, mesenchymal and immune components
We used an ALI system to culture intact human small intestine endoscopic or surgical biopsy fragments as organoids embedded within a collagen matrix in a transwell that was directly exposed to air. Tissue culture media containing WNT3A, epidermal growth factor (EGF), Noggin and R-spondin1 (WENR) was contained in an outer dish with access to the inner transwell through a permeable membrane. Organoids formed after approximately 3 days of culture and expanded with epithelial proliferation consistently for more than a year (370 days was the longest time attempted) (Fig. 1a–g). Over the first several weeks, ALI small intestine organoids often displayed epithelial protrusions with interdigitating basally localized Ki67+ proliferative cells (Fig. 1d,e), after which cystic morphology predominated, presumably resulting from long-term exposure to WNT and R-spondin (Fig. 1f,g). Immunofluorescence revealed numerous stromal populations present endogenously with epithelium, including distinct PDGFRA+ and SMA+ fibroblasts, PGP9.5+ neural cells and CD31+ endothelium (Fig. 1h,i and Extended Data Fig. 1a–c). Crucially, CD45+ haematopoietic cells (Fig. 1j), spanning CD3+ T cells (Fig. 1k), CD14+ monocytes and macrophages (Fig. 1k and Extended Data Fig. 1d), CD19+ B cells and plasma cells (Fig. 1l) were also present. CD8+ T cells localized to epithelium, compared with CD4+ T cells, which localized in distinct sub-epithelial locations in ALI organoids (Extended Data Fig. 1e).

a, Schematic outlining the steps in creating ALI organoids. b, ALI small-intestine organoid with haematoxylin and eosin (H&E) staining at culture day 14 (representative of n = 25 biological replicates). c, ALI organoid growth time course, brightfield (representative of n = 3 biological replicates cultured for more than 300 days). d, Epithelial protrusions at day 14, with H&E staining (representative of n = 25 biological replicates). e, Immunofluorescence staining with Ki67 (green) and DAPI (blue) at day 14 (representative of n = 9 biological replicates). f, ALI organoids at day 370, stained with H&E (representative of n = 3 biological replicates cultured for more than 300 days). g, ALI organoids at day 370 stained with Ki67 (red), ECAD (green) and DAPI (blue) (representative of n = 3 biological replicates cultured for more than 300 days). h–j, ALI organoid whole-mount immunofluorescence staining using ECAD (white) and DAPI (blue) with PDGFRA (red) (h), PGP9.5 (red) (i) or CD45 (red) (j) at day 14 (representative of n = 3 biological replicates). k,l, Immunofluorescence whole-mount staining of ALI organoids using: ECAD (white), CD14 (red) and CD3 (green) (k); and ECAD (white), DAPI (blue) and CD19 (green) (l), at day 14 (representative of n = 3 biological replicates). m, An scRNA-seq UMAP plot of FACS-sorted CD45+ cells from ALI organoids at day 14, n = 4 donors. n, Organoid immune subset frequencies (n = 6 patients) compared with fresh tissue (n = 1 patient) using scRNA-seq at day 14. o, TCR (n = 6 patients) and BCR (n = 3 patients) repertoire and clonal expansion from ALI organoids at day 14 from scRNA-seq analysis. p, An scRNA-seq UMAP plot (from m) of T-cell CD103 mRNA (n = 4 biological replicates). q, Immunofluorescence staining of CD3+ IELs (red), ECAD (white) and DAPI (blue) on day 14 (representative of n = 3 biological replicates). All figure panels are for duodenal organoids apart from c, f and g, which are ileal. Duodenal and ileal organoids did not exhibit qualitative differences. All scale bars are 100 µm except for c, which is 5 mm.
Single-cell RNA sequencing (scRNA-seq) of multiple CeD patient samples revealed that ALI small intestine organoids consistently preserved endogenous CD4+ and CD8+ T cells, regulatory (Treg) and replicating T cells, B and plasma cells, NK cells and myeloid cells (Fig. 1m,n and Extended Data Figs. 1d and 2a,b). Both immune diversity and T-cell receptor (TCR) repertoires were highly conserved between fresh intestinal tissue and organoids (Fig. 1n,o and Extended Data Fig. 2c,d). Intraepithelial lymphocytes (IELs) expressing CD103 were identified by scRNA-seq (Fig. 1p) and some CD3+ T cells were indeed present within the epithelium (Fig. 1q).
Murine Ly49+CD8+ T cells have been shown to suppress autoimmunity in mouse models26, and CD8+ T cells expressing inhibitory killer cell immunoglobulin-like receptors (KIRs), human paralogues of Ly49, are elevated in CeD patient blood and efficiently kill CeD pathogenic gluten-specific CD4+ T cells in vitro27. KIR+ lymphocytes expressing KIR3DL1 or KIR2DL3 constituted a distinct subset of organoid CD8+ T cells (Extended Data Fig. 2e,f). NKG2C and NKG2D, which are members of the C-type lectin-like receptor superfamily, were also detectable in CD8+ T cells (Extended Data Fig. 2g). Although KIR+CD8+ T cells were not clonally expanded, organoid NKG2C+ and NKG2D+CD8+ T cells did contain expanded TCR clonotypes (Extended Data Fig. 2h).
In contrast to extended epithelial growth, CD45+ haematopoietic cells in ALI organoids progressively declined over about 45 days. However, medium supplementation with IL-2 and IL-7 improved maintenance of CD45+ haematopoietic cells to 9% of total live organoid cells, with CD4+ and CD8+ T-cell preservation at day 47 (Extended Data Fig. 3a). Intestinal ALI organoids could be cryopreserved in-gel and recovered with subsequent growth and maintenance of similar proportions of epithelium and immune compartments compared with never-frozen cultures (Extended Data Fig. 3b–d).
Gliadin induces enteropathy and epithelial proliferation in CeD organoids
We next applied the ALI culture system to study CeD in which tissue-resident immune cells mediate gluten-dependent epithelial destruction. Under an approved institutional review board protocol, duodenal endoscopic biopsies were collected from a total of 135 individuals, including 81 donors with a prior diagnosis of CeD and 54 non-CeD control donors. Of the 81 CeD donors, 59 had active CeD and 22 had remission CeD with a prior CeD diagnosis but had become asymptomatic on a gluten-free diet (Supplementary Table 1). For 119 of these individuals with available genomic DNA (74 CeD and 45 non-CeD), we performed HLA-DQA1 and HLA-DQB1 genotyping and parallel organoid generation. Of the 74 HLA-typed CeD patients, 82.4% expressed HLA-DQ2.5 in cis, 8.1% expressed DQ2.5 in trans28, 2.7% had HLA-DQ2.2 (without HLA-DQ2.5 expression) and 6.8% had HLA-DQ8 (Supplementary Table 1).
After 9–12 days of growth, organoids were treated with a 1:1 mixture of deamidated gliadin peptides representing immunodominant HLA-DQ2.5-glia-α1 (LQPFPQPELPYPGS) and HLA-DQ2.5-glia-α2 (APQPELPYPQPGS) gluten epitopes29,30,31. As a control, ALI organoids were treated with CLIP peptide derived from an MHC invariant chain, which is competent to be presented by all MHC class II molecules32. Because we used HLA-DQ2.5-resticted gliadin peptides, we employed corresponding HLA-DQ2.5+ organoids throughout this study. Gliadin or CLIP treatment was for 2 days unless stated otherwise (Extended Data Fig. 4a).
A hallmark of CeD pathophysiology is a gluten-induced increase in epithelial IL-15 production, which is postulated to facilitate trans-presentation of this cytokine to CD4+ and CD8+ IELs, promoting epithelial killing and villous atrophy5,6,7,33. Notably, gliadin, but not CLIP, induced IL-15 immunofluorescence in CeD organoids restricted to CK19+ epithelial cells; stimulation was not observed in control non-CeD organoids (Fig. 2a,b and Extended Data Fig. 4b). The induction of IL-15 in CeD organoids contrasts with mouse CeD models, which do not natively upregulate endogenous IL-15 and require constitutive transgenic IL15 expression to elicit full CeD pathology14.

a, Representative immunofluorescence staining of ALI organoids from control or active CeD patients after 2-day treatment with CLIP or gliadin, showing IL-15 (red), CK19 (green) and DAPI (blue). b, Quantification of a, where each point is an individual organoid from control (n = 3 biological replicates) or CeD (n = 4 biological replicates). **P = 0.0093; two-tailed Mann–Whitney test. c, Representative immunofluorescence staining after 2-day treatment with CLIP or gliadin, showing cleaved caspase-3 (green), ECAD (red) and DAPI (blue). d, Quantification of c, where each point is an individual organoid from control (n = 3 biological replicates) or CeD (n = 5 biological replicates). ***P = 0.0005 (CeD CLIP versus control CLIP); ***P < 0.0001 (CeD gliadin versus CeD CLIP); two-tailed Mann-Whitney test. e,f, Representative immunofluorescence staining showing SI (red) and DAPI (blue) (e) and APOA4 (red) and DAPI (blue) (f) for control or active CeD organoids treated with CLIP or gliadin for 2 days. g, Quantification of e, where each point is an individual organoid from control or CeD cultures (n = 3 biological replicates each). ***P = 0.0004; two-tailed Mann-Whitney test. h, Quantification of f, where each point is an individual organoid from control or CeD cultures (n = 3 biological replicates each). *P = 0.0339; two-tailed Mann-Whitney test. i,j, Representative flow cytometry of gliadin-induced EPCAM+ cell death by caspase-3/7 activity (i) in active CeD but not in control organoids (n = 3 biological replicates each); this is abrogated by anti-MHC-II (j). Scatter plots show the median as the centre line and whiskers represent minimum and maximum. **P ≤ 0.01, one-way analysis of variance (ANOVA). k. Gliadin (HLA-DQ2.5-restricted peptides) does not induce epithelial killing in HLA-DQ8+-derived CeD organoids (n = 5 patients). NS, not significant; P = 0.8125, two-tailed Wilcoxon test. All scale bars are 100 µm and all CeD organoids were HLA-DQ2.5+ (except in k). All box plots show the median as the centre line, the interquartile range as the box limits, and whiskers represent minimum and maximum.
Crucially, gliadin, but not CLIP, induced epithelial cell death in CeD organoids, with the appearance of apoptotic cleaved caspase-3+ epithelium. By comparison, gliadin did not significantly induce epithelial cleaved caspase-3 in control organoids (Fig. 2c,d). Gliadin-treated CeD organoids also exhibited pronounced reductions in the villus enterocyte markers sucrase–isomaltase (SI) and APOA4, compared with CLIP controls (Fig. 2e–h and Extended Data Fig. 4c,d). Downregulation of SI mRNA was also observed by quantitative PCR with reverse transcription (RT–qPCR) of organoid EPCAM+ cells purified by fluorescence-activated cell sorting (FACS) (Extended Data Fig. 4e). Similarly, in CeD organoids, gliadin, but not CLIP, reduced epithelial protrusions by around 25%, with increased spacing between remnants (Extended Data Fig. 4f,g).
Detailed HLA-DQ genotyping allowed correlation of CeD organoid responses to HLA-DQ2.5-restricted glia-α1 and glia-α2 gliadin peptides against the patient MHC-II allotypes. Although we focused on prevalent HLA-DQ2.5-expressing CeD organoids, we also evaluated a limited number of rarer HLA-DQ8+ CeD cultures. Apoptosis of EPCAM+ epithelium in active CeD organoids representing the two HLA haplotypes after challenge with HLA-DQ2.5-restricted glia-α1 and glia-α2 gliadin peptides was assayed in a quantitative fluorescence caspase-3/7 activity FACS assay (Fig. 2i). HLA-DQ2.5-restricted glia-α1 and glia-α2 gliadin peptides induced epithelial apoptosis in HLA-DQ2.5+ organoids, which was ablated by anti-MHC-II antibody (Fig. 2j). However, HLA-DQ8+ organoids were unresponsive to the HLA-DQ2.5-restricted gliadin peptides (Fig. 2k), demonstrating the specificity of the CeD ALI organoid system.
Another hallmark of CeD is crypt hypertrophy, which represents compensatory proliferation after epithelial damage1. Despite placing CeD and control organoids in culture medium lacking WNT3A and RSPO1 (EN medium) to induce epithelial quiescence, gliadin, but not CLIP, augmented epithelial Ki67+ proliferation (Extended Data Fig. 5a,b) and increased organoid size, as revealed by automated microscopy image analysis (Extended Data Fig. 5c,d). Under these conditions, gliadin increased transcripts for the stem cell or proliferative markers LGR5, PCNA and CCND1 in FACS-purified EPCAM+ epithelial cells from CeD but not from control organoids (Extended Data Fig. 5e–g). Overall, CeD organoids faithfully modelled both the epithelial destruction and the compensatory proliferation observed in clinical tissue biopsies.
CeD organoids contain gliadin-specific T cells
We next assessed the effects of gliadin on CeD organoid T-cell subsets after 2 days of treatment. Immunofluorescence microscopy of CeD organoids revealed that gliadin often led to an expansion of CD4+ and CD8+ T cells in clusters, which was not observed with CLIP (Fig. 3a). To confirm this observation, we used flow cytometry to analyse the abundance of T-cell subsets. Notably, in CeD organoids, 2-day treatment with gliadin, but not CLIP, increased the numbers of CD3+ and CD4+ T cells by 1.5- to 2.0-fold with a smaller but significant increase in CD8+ T cells. These elevated gliadin:CLIP ratios of CD3+, CD4+ and CD8+ abundance were highly specific for CeD compared with control organoids (Fig. 3b–d). Gluten-induced CeD pathology is initiated by rare tissue-resident CD4+ T cells that recognize gliadin and other gluten peptides only in the context of HLA-DQ2 or HLA-DQ8 (refs. 1,2,3). We assessed gluten-specific CD4+ T cells in remission CeD organoids using soluble tetramers comprising four HLA-DQ2.5 molecules loaded with DQ2.5-glia-α1 and DQ2.5-glia-α2 peptide epitopes as flow cytometry detection reagents. This revealed DQ2.5–gliadin tetramer-reactive gluten-specific CD4+ T cells in CeD organoids, which were moderately but not statistically significantly increased after a 6-day gliadin treatment compared with CLIP (Fig. 3e,f). The presence of gluten-specific CD4+ T cells in CeD organoids was further demonstrated by scRNA-seq-based TCR repertoire sequencing, revealing multiple CDR3 motifs in organoid-resident T cells that fully matched or exhibited GLIPH homology to known gliadin-binding TCRs from CeD patients34,35,36,37 (Extended Data Fig. 6).

a, Whole-mount immunofluorescence staining of CD4+ T cells (green), CD8+ T cells (red), ECAD (white) and DAPI (blue) in active CeD organoids treated with CLIP or gliadin for 2 days (n = 4 patients). Scale bar, 200 µm. b–d, Flow cytometry of T-cell abundance in control or active CeD organoids, expressed as a ratio of gliadin:CLIP treatment for each T-cell subset as a fraction of total live cells after 2 days of treatment. The graphs show: b, CD3+ T cells in organoids from control (n = 20) or CeD (n = 13) biological replicates; ***P < 0.0001, two-tailed Mann-Whitney test; c, CD4+ T cells in organoids from control (n = 20) or CeD (n = 13) biological replicates; ***P < 0.0001; two-tailed Mann-Whitney test; and d, CD8+ T cells in organoids from control (n = 18) or CeD (n = 12) biological replicates; ***P = 0.0006; two-tailed Mann-Whitney test. Scatter plots show the median as the centre line and whiskers represent minimum and maximum. e,f, Gluten-reactive CD4+ T cells in organoids from remission CeD patients (remission CeD organoids) treated with CLIP or gliadin for 6 days and HLA-DQ2–gliadin tetramer-reactive CD4+ T cells measured by FACS as a fraction of total organoid CD4+ cells. NS, P = 0.0547; two-tailed Wilcoxon test. Graphs show: e, HLA-DQ2-gliadin-PE (phycoerythrin) tetramer FACS analysis of organoids from a single representative CeD patient; and f, HLA-DQ2.5–gliadin tetramer FACS analysis of organoids from e; n = 9 CeD patients; two-tailed Wilcoxon test.
CeD organoids manifest gliadin-induced network immune activation
We then performed scRNA-seq analysis on FACS-sorted cells from 2-day gliadin- and CLIP-treated organoids from three different CeD patients. For CD45+ organoid cells, gliadin prominently induced several cytotoxic, chemokine, cytokine and other activation mRNAs in myeloid, plasma, B, Treg and CD8+ and CD4+ T cells compared with CLIP controls, indicating a complex immune network response (Fig. 4a and Extended Data Fig. 7). Organoids from these three CeD patients consistently exhibited a pronounced gliadin-induced upregulation of cytotoxic mRNAs in CD8+ T cells, including granzymes (GZMA/B/H/K/M) and PRF1. In multiple immune subsets, gliadin upregulated the mRNAs encoding the CCL3, CCL4, CCL5 and CCL20 chemokines, which recruit both innate (NK cells and monocytes) and adaptive (CD4+ and CD8+) T cells to sites of inflammation and consequent tissue damage38 and have been implicated in attracting gluten-specific CD4+ T cells to CeD patient gut lesions39. Gliadin also upregulated myeloid CSF1 and CSF2, encoding M-CSF and GM-CSF, which promote tissue damage, activate and expand innate immune cells during inflammation and induce MHC-II presentation40. IL-2 is induced in vivo after gluten challenge of CeD patients15; accordingly, gliadin increased mRNAs for IL2 and its receptors IL2RA and IL2RB in organoid CD4+ T cells (Fig. 4a and Extended Data Fig. 7). IFNG mRNA was also upregulated in CD8+ T cells, consistent with clinical CeD biopsy analyses indicating that cytotoxic CD8+ IELs are the predominant IFNG source after gluten challenge41 (Fig. 4a and Extended Data Fig. 7). Because MHC-II-restricted gliadin peptides specific for presentation to CD4+ T cells were used as the initiating primary stimulus, this observed widespread secondary network activation of diverse immune subsets was particularly notable.

a, Dot plots from scRNA-seq of mean expression levels and corresponding percentage population expression for active CeD organoid CD4+ and CD8+ T cells, Treg, plasma B and myeloid cells after 2-day gliadin or CLIP treatment. Data are from organoids from a single CeD patient. Data from organoids from two other CeD patients are shown in Extended Data Fig. 7. b, Luminex protein analysis of organoid conditioned media from active CeD (n = 8 biological replicates) or control (n = 4 biological replicates) with fold-increases of different cytokines and growth factors as a ratio of gliadin:CLIP treatment after 2 days. Box plots show the median as the centre line, the interquartile range as the box limits and whiskers represent minimum and maximum. For SCD40L, IL-2, IL-4 and IL-8, *P = 0.028; for IL-1A, CCL2, MCSF and GROA, *P = 0.048; for IL-1RA and TGFA, **P = 0.008; for IL-15 and VEGF, *P = 0.016); for IL-25 and PDGFAA, **P = 0.004); two-tailed Mann-Whitney test. c, ELISA detection of anti-TG2 autoantibody production in conditioned media of ALI organoids from CeD (n = 7) or control (n = 4) biological replicates after 2-day gliadin or CLIP treatment; *P = 0.0156; two-tailed Wilcoxon test. All CeD organoids in this figure were HLA-DQ2.5+.
We further confirmed gliadin activation of multiple cytokines at the proteomic level. Cytokine production in conditioned medium from 2-day gliadin versus CLIP-treated CeD or control organoids was interrogated using a multiplex Luminex cytokine detection panel. This revealed significant elevation in numerous adaptive cytokines, including IL-2 and IL-15, as well as factors relevant to innate immunity, including IL-1A, M-CSF and CCL2. A protein signature relevant to epithelium, vasculature and stroma was also noted with significant induction of TGFA, VEGFA and PDGFAA as epithelial, endothelial and fibroblast growth factors, respectively (Fig. 4b).
Non-supervised integration of CeD organoid scRNA-seq data was performed to obtain interactomes based on the reciprocal expression of ligand and cell-surface receptors on distinct immune cell types. This defined signalling hubs centred on T cells, B cells and plasma B cells, NK cells and myeloid cells. The cell-type interactomes contained 331 cell-type-specific ligand–receptor pairs unique to gliadin stimulation, whereas only 78 pairs were unique to CLIP, implying a gliadin-induced acute switch in immune signalling pathways (Extended Data Fig. 8). Predicted B cell and plasma B cell interactomes exhibited particularly strong and specific gliadin induction, indicating possible B-cell and plasma B-cell HLA-E engagement with KLRK1 (NKG2D) or KLRC2 (NKG2C) on CD8+ T cells and NK cells (Extended Data Fig. 9a). Together, the CeD organoid scRNA-seq-based interactomes indicated extensive gliadin-dependent immune network crosstalk encompassing nearly all immune subsets (Extended Data Fig. 8 and Extended Data Fig. 9a).
Anti-TG2 autoantibody production in CeD organoids
In CeD, TG2 deamidates gliadin, enabling MHC binding and antigen presentation and recognition by gluten-specific CD4+ T cells. A hallmark of CeD is the plasma B-cell production of circulating anti-TG2 autoantibodies, the detection of which is widely used for CeD diagnosis42. We explored anti-TG2 autoantibody production in CeD organoids by scRNA-seq mapping of immunoglobulin heavy- and light-chain transcripts to individual plasma B cells, followed by single-cell B-cell receptor (BCR) repertoire analysis coupled to consensus sequence examination. This revealed immunoglobulin CDR3 motifs in plasma B cells from CeD organoids with complete or near-complete homology to published anti-TG2 BCR sequences43 (Extended Data Figs. 9b and 10).
Organoid production of anti-TG2 autoantibodies was quantified by clinical-grade enzyme-linked immunosorbent assay (ELISA) in conditioned media from control or CeD organoids after a 2-day gliadin or CLIP treatment. Strikingly, despite extensive changes of the medium, anti-TG2 autoantibodies were actively secreted and readily detected in the conditioned medium of CeD organoids compared with nearly undetectable levels in control organoids (Fig. 4c). Subsequent 2-day in vitro gliadin treatment of CeD organoids further augmented their already elevated baseline secretion of anti-TG2 autoantibodies, whereas gliadin did not induce anti-TG2 production in control organoids (Fig. 4c). Thus, CeD organoids recapitulate the pathological plasma B-cell anti-TG2 autoantibody production of CeD patients, which is further stimulated by MHC-II-restricted gliadin peptides.
IL-7 is essential and sufficient to kill CeD organoid epithelium
The events culminating in the histological changes of CeD are incompletely understood. One paradox is that gluten-specific TCRs have been found on only CD4+ T cells, not CD8+ T cells, indicating that epithelial killing by CD8+ IELs is secondary to CD4+ T-cell gliadin recognition. Proposed cytokine mediators include IL-2 and IL-15, which are induced by gluten in human CeD15,16, but which have not been functionally validated either experimentally or in clinical trials44. IL-7 is a member of the IL-2–IL-15 cytokine superfamily, which shares a common signal-transducing receptor γ-chain19. Because IL-7 regulates the function of memory T cells and memory B cells19, and is implicated in autoimmunity45, but had not been investigated in CeD, we examined potential IL-7 functionality in CeD organoids.
In CeD organoid scRNA-seq, the IL-7 receptor gene (IL7R) was highly expressed in a subset of CD8+ IELs expressing the tissue residency marker CD103 (ITGAE) (Fig. 5a–c). We then explored whether gluten-induced killing was regulated by IL-7. Strikingly, gliadin-induced epithelial cell death in CeD organoids was abrogated following IL-7 inhibition by adding the recombinant neutralizing, ligand-sequestering IL-7R extracellular domain (ECD) to the culture media (Fig. 5d). CD8+ IEL expression of the NK receptors NKG2C and NKG2D has been proposed to recognize and kill intestinal epithelial cells that have the counter-ligands HLA-E and MICA5,6,46. We therefore tested the potential operation of this pathway and its regulation by IL-7 in CeD organoids. NKG2C/D pharmacological inhibition with their cognate ECDs prevented gliadin-induced epithelial killing in CeD organoids (Fig. 5e). Furthermore, gliadin induced NKG2C and NKG2D expression on CeD organoid CD8+ T cells, which was notably reversed by IL-7 blockade using recombinant IL-7R ECD (Fig. 5f,g).

a,b, ScRNA-seq-derived violin plots of transcripts per cell type for IL7R (a) and CD103 (b). DN T, double negative T (CD4−CD8−CD3+). c, Scatter plot of CD103 and IL7R co-expression in CD8+ T cells from CeD organoids (n = 5 patients). d, Gliadin-induced CeD organoid EPCAM+ apoptosis is abrogated by blocking IL-7 using the IL-7RA ECD (n = 6 patients); **P = 0.0078 (gliadin versus CLIP); *P = 0.0129 (gliadin + IL7-RA ECD versus gliadin); one-way ANOVA. e, Gliadin-induced CeD organoid EPCAM+ apoptosis is abrogated by blocking NKG2C/D using cognate receptor ECDs (n = 4 patients). ***P < 0.0001 (gliadin versus CLIP); ***P < 0.0001 (gliadin + NKG2C/D ECD versus gliadin); one-way ANOVA. f,g, Flow cytometry results demonstrating that more CD8+ T cells expressed NKG2C (f) or NKG2D (g) after gliadin treatment, normalized to CLIP, with reversal by IL-7 inhibition. In f: *P = 0.0117 (gliadin versus CLIP, n = 4 patients); **P = 0.0093 (gliadin + IL7-RA ECD versus gliadin, n = 3 patients); in g: *P = 0.0113 (gliadin versus CLIP, n = 4 patients); **P = 0.0026 (gliadin + IL7-RA ECD versus gliadin, n = 4 patients). One-way ANOVA, bar graphs depict the median. h, IL-7 promotes EPCAM+ apoptosis with or without gliadin in CeD (n = 6 patients) but not in control organoids (n = 3 patients). ***P = 0.0006 (gliadin versus CLIP); ***P = 0.0007 (CLIP + IL-7 versus CLIP); **P = 0.0036 (gliadin + IL-7 versus CLIP); one-way ANOVA. i, Representative flow cytometry histograms of percentage (normalized to mode) NKG2C and NKG2D expression in CD8+ T cells and CD4+ T cells with or without IL-7 treatment. j, Flow cytometry quantification of CD8+ T cells depicting an increased percentage of cells expressing NKG2C (n = 5 biological replicates) or NKG2D (n = 8 biological replicates) after IL-7 treatment of control or CeD organoids. *P = 0.0312; **P = 0.0039; two-tailed Wilcoxon test. All CeD organoids were HLA-DQ2.5+ except for limited HLA-DQ8+ in j. All box plots show the median as the centre line and the interquartile range as box limits, with whiskers representing minimum and maximum.
Gain-of-function studies were then done to determine whether IL-7 was sufficient to induce epithelial cytotoxicity in CeD organoids. Notably, recombinant IL-7 strongly induced epithelial killing in active CeD organoids but not in organoids from non-CeD control patients. The degree of IL-7-stimulated apoptosis was equivalent to that of gliadin, and was not additive when combined with gliadin, indicating that they potentially operate in a single pathway (Fig. 5h). IL-7 induced NKG2C and NKG2D expression only in CeD organoid CD8+ T cells, not in CD4+ T cells, but this effect was seen in both CeD and non-CeD organoids (Fig. 5i,j). Thus, although IL-7 elicits the killing of CeD, but not non-CeD, organoid epithelium (Fig. 5h), IL-7 promotion of NKG2C/D expression in CD8+ T cells is not sufficient to confer this specificity (Fig. 5i,j).
IL-7 is induced by gliadin in CeD organoids and is overexpressed in active CeD
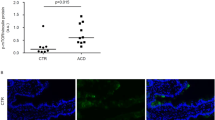
Finally, we examined whether gliadin could physiologically regulate IL-7 production in CeD. Whereas non-CeD organoids manifested little to undetectable IL-7 immunofluorescence staining, in vitro treatment with gliadin, but not with CLIP, strikingly increased IL-7 in active-CeD patient organoids. Gliadin induced IL-7 protein predominantly in the lamina propria-like compartment of the CeD organoids, with comparatively little epithelial expression present when treated with CLIP (Fig. 6a,b). Luminex analysis revealed a trend towards gluten induction of IL-7 secretion in the conditioned medium of active-CeD organoids that did not reach statistical significance (Extended Data Fig. 11a).

a, Representative immunofluorescence staining of IL-7 (red), CK19 (green) and DAPI (blue) in control or active CeD organoids treated with CLIP or gliadin for 2 days. Scale bar, 100 µm. b, Quantification of a, in which each data point represents an individual organoid from control (n = 3) or CeD (n = 3) biological replicates. ***P = 0.0002; two-tailed Mann-Whitney test. All CeD organoids were HLA-DQ2.5+. c, Immunofluorescence staining of IL-7 (red) in lamina propria, with CK19 (green) and DAPI (blue) in representative duodenal biopsy tissues from patients in remission or with active CeD. Further histology from n = 14 remission and n = 14 active CeD patients is shown in Extended Data Fig. 11b,c. Scale bar, 100 µm. d, Quantification of c and Extended Data Fig. 11b,c. Each data point corresponds to the median of ≥3 high-power fields from a different CeD patient in remission on a GFD (n = 15) or active CeD (n = 15). ***P < 0.0001; two-tailed Mann-Whitney test. e, Immunofluorescence staining of IL-7 (red), TAGLN (green) and DAPI (blue) in representative duodenal biopsy tissue from a patient with active CeD. Scale bar, 50 µm. f, Quantification of e, in which each data point is tissue IL-7 immunofluorescence from a patient with active CeD (n = 12). g, IL7 mRNA in situ hybridization (blue) in representative duodenal biopsy tissue from two CeD patients in remission on a GFD (left) and two patients with active CeD (right). Scale bars, 50 μm. h, IL7 mRNA quantification in lamina propria from g in which each data point corresponds to a different CeD patient in remission (n = 6) or with active CeD (n = 6). *P = 0.0152; two-tailed Mann-Whitney test. All box plots show the median as the centre line and the interquartile range as the box limits, with whiskers representing minimum and maximum.
We also investigated whether IL-7 was upregulated endogenously during CeD disease progression. IL-7 immunofluorescence staining was done on duodenal biopsies from 15 patients with active CeD and 15 patients with CeD in remission and on a gluten-free diet (GFD). IL-7 immunofluorescence was strongly augmented in biopsies from patients with active CeD compared with those in remission, again predominantly localized to the lamina propria with rare epithelial expression (Fig. 6c,d and Extended Data Fig. 11b,c). The specificity of IL-7 detection in biopsies from active CeD but not those in remission was confirmed with a second anti-IL-7 antibody (Extended Data Fig. 11d). Lamina propria IL-7 predominantly co-localized with mesenchyme expressing transgelin/smooth muscle 22α (TAGLN/SM22α) in active CeD biopsies (Fig. 6e,f). As with immunofluorescence staining, in situ hybridization confirmed strong induction of IL7 mRNA in active CeD compared with remission CeD biopsies (Fig. 6g,h). Taken together, the concordant results from organoids and biopsies (Fig. 6), combined with gain- and loss-of-function analyses (Fig. 5), strongly implicated IL-7 as a gluten-induced, functionally relevant mediator of CeD pathology.
Discussion
In vitro studies of human autoimmunity have been impeded by a lack of systems enabling the culture of affected tissues together with a full diversity of endogenous resident immune populations. Previous in vitro CeD models have used cancer cell lines9, gliadin-responsive monocytes and dendritic cells11 or establishment of T-cell lines from CeD biopsies47. Tissue explants9,10 progressively deteriorate over a few days, and conventional enteroids contain epithelium but lack immune components12. Here, we established a proof of principle for organoid modelling of autoimmunity, specifically CeD, by the holistic ALI culture of intestinal biopsies, enabling epithelial growth alongside mesenchyme and tissue-resident immune cells without exogenous cell addition. The ALI organoids preserved diverse infiltrating immune populations, including T, B, plasma B, NK and myeloid cell subsets, as highly relevant to tissue-resident immunity in CeD and in marked contrast to previous culture methods. Furthermore, the KIR+ suppressor CD8+ T cells present in CeD patients27 were also represented in CeD organoids. This system is an improvement on our previous ALI organoid cultures, which were restricted to neonatal mouse intestine or tumour tissue48,49.
Importantly, in vitro gliadin treatment of ALI CeD organoids is sufficient to robustly recapitulate pathognomonic histological hallmarks of CeD, including epithelial apoptosis and compensatory proliferation. The CeD organoid system manifests pronounced specificity whereby HLA-DQ2.5-restricted gliadin peptides presented to CD4+ T cells induce epithelial death only in organoids expressing cis- or trans-encoded HLA-DQ2.5, whereas HLA-DQ8 organoids are non-responsive and gluten responses are abrogated by MHC-II antibody blockade. This HLA selectivity and dependency further indicates that adaptive immunity is sufficient to induce the seminal characteristics of CeD in the organoid system, although further contributions from innate immunity cannot be excluded. Accordingly, HLA-DQ2.5-restricted gliadin peptides were fully competent to induce epithelial IL-15 expression, which has previously been ascribed to innate stimulatory effects of gluten during CeD50. CeD organoids endogenously upregulate IL-15 in response to gliadin, contrasting with mouse CeD models in which constitutive transgenic IL15 overexpression is not required to elicit mild disease but is necessary for full CeD-like pathology14. CeD ALI organoids also allow rapid determination of epithelial destruction within days, as opposed to weeks of gluten consumption in CeD patients or transgenic mouse models14.
The immune diversity of the ALI CeD organoid system is strongly reflected by gluten induction of a complex immune network response with cytotoxic T-cell, B-cell and myeloid activation and predicted autocrine and paracrine crosstalk, both among immune subsets and with epithelium, and plasma B-cell anti-TG2 autoantibody production. Such organoid recapitulation of gliadin-induced pathology may be facilitated by accurate preservation of TCR and BCR clonotypes representing known gluten-binding TCRs and TG2-binding BCRs.
The mechanism by which gliadin presentation to CD4+ T cells elicits a CD8+ IEL cytotoxic response has been an enduring ambiguity of CeD pathology. Numerous cytokines, including IL-2 and IL-15, are induced by gluten and upregulated in CeD-patient blood or tissue biopsies, respectively15,16,33. However, a functional role for IL-2 has not been formally demonstrated in CeD, and a clinical trial of IL-15 inhibition did not meet its primary endpoint18. The related cytokine IL-7 shares a common signal-transducing receptor γ-chain with IL-2 and IL-15 (ref. 19), and IL-7 was both necessary for gluten-induced cytotoxicity and sufficient to elicit epithelial cell death without gluten, in CeD organoid gain- and loss-of-function studies. In CeD, CD8+ IELs bearing IL-15-induced NKG2D have been proposed to destroy epithelium that has the NKG2D receptor MICA5,6,7. In this case, IL-7 was required for gluten promotion of NKG2C and NKG2D expression on CD8+, but not CD4+, T cells, and gliadin-induced epithelial apoptosis required NKG2C/D, further establishing IL-7 as an important modulator of intestinal inflammation51,52. However, because IL-7 stimulated NKG2C/D expression in both normal and CeD organoids, the specificity of IL-7-promoted tissue destruction for CeD epithelium likely requires additional molecular determinants, perhaps through the regulation of MICA or HLA-E receptors.
Our study implicates IL-7 as a previously unsuspected gluten-regulated target during active CeD. HLA-DQ2.5-restricted gliadin peptides were fully sufficient to stimulate IL-7 production in CeD organoids. Crucially, IL-7 protein and mRNA were strongly and concurrently upregulated in duodenal biopsies from patients with active, but not remission, CeD. IL-7 induction occurred predominantly in lamina propria mesenchyme expressing TAGLN, which is broadly expressed in intestinal fibroblasts and smooth muscle53, thus implicating this cellular compartment in CeD pathogenesis. A minor component of epithelial IL-7 expression was observed, as previously described54. Although increased circulating IL-7 has not been reported from human acute gluten challenge15,16, further IL-7 tissue and blood characterization, and exploration of IL-7 therapeutic manipulation during CeD, are warranted.
Our ALI organoid model, containing diverse epithelial, stromal and immune cell types, strongly indicates that the tissue-resident intestinal microenvironment is sufficient to initiate gluten-induced pathology in established CeD. However, our results by no means exclude potential contributions from secondary lymphoid organs, the inclusion of which could enhance the mild expansion of gliadin-reactive T cells in CeD organoids, which exhibit quiescence in CeD biopsies55. We also cannot exclude the potential activation of injury-sensing pathways during tissue processing for organoid culture, but these are not reflected in our data because the effects of gliadin were always compared with CLIP and/or other experimental conditions. The current study could be expanded to earlier and later temporal windows, enabling time-scale evaluation that would otherwise require sequential human biopsies. Numerous potential translational applications of CeD organoids include therapeutics screening or in vitro CeD diagnosis, particularly given the specificity of gliadin responses for ALI organoids from CeD patients versus non-CeD controls. Finally, these studies establish a general paradigm for the organoid modelling of autoimmunity, enabling mechanistic investigations, therapeutics testing and precision medicine.
Methods
Human specimens
The small-intestine tissue specimens from surgical resections that we used for ALI organoid characterization were obtained through the Stanford University Hospital Tissue Procurement Shared Resource facility. All experiments using human tissue were approved by the Stanford University institutional review board. For characterization of CLIP versus gliadin challenge experiments, ALI organoids were generated from 135 individuals, representing 54 non-coeliac controls and 81 CeD patient biopsies collected by endoscopy from the distal duodenum, under IRB-20362 (Studying the Molecular Factors Involved in Celiac Disease Pathogenesis) approved by the Research Compliance Office of Stanford University. The initial diagnostic criteria for active CeD were being positive for circulating TG2 IgA autoantibodies and enteropathy (villus atrophy) at the time of acquisition56. Remission CeD was determined for individuals on a GFD for more than a year with no circulating TG2 IgA antibodies and without enteropathy. Non-CeD controls were donors who underwent a clinically indicated endoscopy for gastrointestinal symptoms, including dysphagia, GERD and dyspepsia, but did not have CeD. Gender, age and diagnoses of donors are presented in Supplementary Table 1.
HLA-DQ genotyping
Of the 135 individual organoid donors, genomic DNA was available for 119 subjects (74 CeD and 45 non-CeD) and used for HLA-DQ genotyping. Patient and control genomic DNA samples were extracted either from peripheral blood specimens using an automated QIAsymphony SP platform (Qiagen) or from formalin-fixed paraffin embedded tissue specimens with a QIAmp DNA formalin-fixed, paraffin-embedded (FFPE) advanced kit (Qiagen, 56604). The HLA-DQ typing was then performed using a multiplex sequence-specific oligonucleotide DNA typing test with the One Lambda LABType assay (Thermo Fisher Scientific) on a Luminex FLEXMAP 3D analyser (Luminex).
Establishment of organoid ALI cultures
Inserts with a membranous bottom (Millipore, PICM03050) were placed into wells of a six-well plate. Collagen mixtures were prepared by mixing Cultrex Rat Collagen I (R&D System, 3443-100-01), 10× concentrated Ham’s F-12 (Gibco, 21700075) and reconstitution buffer (260 mM NaHCO3, 200 mM HEPES, pH 11) on ice at a ratio of 8:1:1, respectively, until use. Next, 1 ml reconstituted collagen mixture was added to each insert, which served as a bottom layer without tissue. This bottom layer was left to solidify for 10 min in an incubator at 37 °C, after which eight small-intestinal biopsy bites of 2.8 mm diameter were minced finely with iris scissors on a petri dish placed on ice and then added to the remaining collagen mixture. Then, 1 ml homogenized collagen-and-tissue mixture was layered on top of the pre-solidified 1 ml collagen gel. The six-well plate with the collagen double-layered inserts was left to solidify again for 10 min at 37 °C in an incubator. Next, 1 ml WENR media (50% ADMEM/F12 (Gibco, 12634028) with 50% WNT3A-, RSPO1- and Noggin-conditioned media (L-WRN, CRL-3276, from ATCC) and HEPES (1 mM, Gibco, 15630080), Glutamax (1X, Gibco, 35050061), nicotinamide (10 nM, Sigma, N0636-500G), N-acetylcysteine (1 mM, Sigma, A9165-100G), B-27 without vitamin A (1X, Gibco, 125870-01), A83-01 (0.5 µM, Tocris, 2939), pen–strep glutamine (1X, Gibco, 10378016), gastrin (10 µM, Sigma, G9145), SB-202190 (10 µM, Biogems, 1523072), EGF (50 ng ml−1, PeproTech, AF-100-15)), and supplemented with Normocin (Invivogen, ant-nr-2), 5% FBS (Biotechne, S11550), 10 μM Y-27632 dihydrochloride (Biogems, 1293823), 10 μM CHIR 99021 (Biogems, 2520691) added to each well of the six-well plate outside the inserts, generating an air–liquid interface. Organoid establishment efficiency, based on visible growth, was around 95%. The medium was changed after every 2–3 days of culture.
CeD organoid culture and CLIP or gliadin treatment
Small-intestine tissue from endoscopies of the distal duodenum were used to compare control and CeD ALI organoids after CLIP or gliadin treatment. First, ALI organoids were established and allowed to grow for 9–12 days in WENR media. Then, control or CeD organoids were treated with either 10 μM CLIP (PVSKMRMATPLLMQA, GenScript) or 10 μM gliadin (5 μM DQ2.5-glia-α1; LQPFPQPELPYPGS) plus 5 μM DQ2.5-glia-α2 (APQPELPYPQPGS; GenScript). The peptides were reconstituted in PBS and added to WENR media without Y-27632 dihydrochloride and without CHIR 99021, except for the proliferation/sizing assays shown in Extended Data Fig. 5, in which the peptides were diluted in growth factor starvation (EN) media (WENR without WNT3A, RSPO1, Y-27632 dihydrochloride and CHIR 99021). Organoids were treated with CLIP or gliadin for 2 days, except for the 6-day treatment in gliadin-DQ2.5 tetramer staining FACS experiments shown in Fig. 3e,f. All CeD organoids were obtained from CeD patients with active disease (with inflamed duodenum while consuming gluten-containing foods) except for the gliadin-DQ2.5 tetramer experiments shown in Fig. 3e,f for which CeD organoids were obtained from CeD patients with disease in remission (without duodenal inflammation while on a GFD).
Immunomodulation treatments
All experiments modulating the ALI organoid immune component were done in WENR media without Y-27632 dihydrochloride and without CHIR 99021, including CLIP and gliadin treatments. To block MHC-II presentation, an anti-HLA-DR, DP, DQ antibody (BD, clone TU39, 55557) was used at 1 µg ml−1 with CLIP or gliadin for 48 h. To block IL-7, soluble human IL7RA ECD (Sino Biological, 10975-H08H, used at 1 µg ml−1) was added to the media 2 days before the CLIP or gliadin challenge, and continued during the challenge for another 2 days, representing a 4-day total treatment. Equivalent experiments used human NKG2C (R&D Systems, 138-NK-050) and NKG2D (R&D Systems, 1299-NK-050) blocking ECD-Fc chimeras in combination, each at 1 µg ml−1. Similarly, recombinant human IL-7 (PeproTech, 200-07) was added to the media at 10 ng ml−1 (1,170 IU ml−1) 2 days before CLIP or gliadin challenge, and continued during the challenge for another 2 days, for a 4-day total treatment. For long-term preservation of the immune component in the ALI organoids, non-CeD control ALI intestinal organoids (from the ileum) were grown in WENR media with the addition of human IL-2 (1 ng ml−1, 10 IU ml−1, PeproTech, AF-200-02) in combination with human IL-7 at (10 ng ml−1, 1,170 IU ml−1, PeproTech, 200-07) from culture day 3 to more than 50 days, with at least three data points (≥2 organoid-containing inserts per data point) collected every 2 weeks.
Tissue sectioning and histological analysis
For FFPE block sectioning, ALI organoids embedded in collagen matrix were fixed with 10% formalin phosphate for 1 h, washed in PBS and cut into strips into a cassette in 70% ethanol. The organoid-containing strips were paraffin-embedded and sectioned (5–10 µm) into slides. Sections were deparaffinized overnight at 60 °C. For histological analysis, sections were stained with H&E and images were acquired on a Keyence BZ-X700 or a Leica DMC4500 microscope. The number of epithelial protrusions per millimetre was quantified in H&E-stained sections and the circumference of the organoids was determined by delineating the contours of the organoid cells, excluding the lumen area, using ImageJ. Epithelial protrusions were defined as structures approximately 50 μm in length along the organoid epithelial lining and counted by a researcher blinded to donor CeD diagnostic status and to in vitro treatment. For frozen sectioning, 1–3 fresh endoscopy bites were placed in a mould and embedded in Tissue-Tek O.C.T. compound (Sakura, 4583) on dry ice until frozen and sectioned (5–10 µm) into slides. Sections were washed in PBS, fixed with 4% PFA for 1 h, washed again and PFA was quenched for 30 min in glycine solution.
Immunofluorescence
Unstained sections (FFPE or frozen; see above) were first permeabilized in 0.2% Triton X-100 in PBS (PBST) for 1 h and then steamed for 20 min in antigen unmasking solution (Vector laboratories, H-3301). Sections were then covered in blocking buffer (10% donkey serum (Jackson ImmunoResearch, 017-000-121) diluted in PBS) for 1 h at room temperature, followed by incubation with primary antibodies in staining diluent (Epredia, TA125ADQ) overnight at 4 °C. Sections were then washed in PBST and incubated with fluorescent secondary antibodies and DAPI at room temperature (RT) for 1 h. After more washes, sections were submerged in mounting medium (Vector Laboratories, H-5501) and mounted on chambered cover slips for imaging. All images were taken using a Keyence BZ-X700 microscope or a Zeiss LSM900 confocal microscope. For whole-mount immunofluorescence staining, the organoid-containing collagen matrix was cut into smaller pieces and washed with PBS for 5 min with rocking at RT, fixed with 4% paraformaldehyde (PFA) for 1 h at RT and washed with PBS. PFA was quenched with PBS-glycine (130 mM NaCl, 13.2 mM Na2HPO4, 3.5 mM NaH2PO4, 100 mM glycine in PBS at pH 7.4) for 30 min at RT with gentle rocking. After washing, the collagen pieces were incubated in blocking solution (10% donkey serum diluted in a permeabilizing solution (130 mM NaCl, 13.2 mM Na2HPO4, 3.5 mM NaH2PO4, 7.7 mM NaN3, 15 μM BSA, 2% Triton X-100, 0.5% TWEEN-20 in PBS at pH 7.4)) for 2 h at RT with gentle rocking. The collagen pieces were then stained with primary antibodies diluted in the blocking solution for 1–3 days at RT with gentle rocking. After several washes with permeabilizing solution, the collagen pieces were incubated with secondary antibodies diluted in blocking solution for 4 h at RT with gentle rocking. After several washes, the collagen pieces were transferred onto slides and mounted on chambered cover slips for imaging. Vacuum grease was used around the collagen pieces to avoid flattening of the organoids inside. All images were taken using a Zeiss LSM880 or a Zeiss LSM900 confocal microscope. Total fluorescence of cleaved caspase-3, SI and APOA4 signals was measured by delineating the outline of each organoid and calculating the integrated density using ImageJ. Quantification of Ki67+ cells was determined as the percentage of total organoid nuclei using the ImageJ particle analyser.
Antibodies used for immunofluorescence
Immunofluorescence staining of sections and whole-mount organoids used the following antibodies for recognizing human antigens (manufacturer, catalogue number, dilution): anti-SI (Santa Cruz, sc393424, 1:50), anti-APOA4 (Sigma, HPA001352, 1:250), anti-IL-15 (Abcam, ab55276, 1:250), anti-CK-19 (R&D systems, AF3506, 1:400), anti-CD3 (BioLegend, 300431, 1:250), anti-PDGFRA (R&D Systems, AF-307-NA, 1:250), anti-ECAD (BD, 610181, 1:400), anti-PGP9.5 (Genetex, GTX109637, 1:250), anti-CD45 (Cell Signaling, 13917, 1:250), anti-CD14 (Abcam, ab196169, 1:250), anti-CD19 (Antibodies Online, ABIN724595, 1:250), anti-CD3 (Abcam, ab5690, 1:250), anti-cleaved caspase-3 (Cell Signaling, 9664 S, 1:250), anti-Ki67 (Invitrogen, MA5-14520, 1:250), anti-CD4 (Abcam, ab133616, 1:250), anti-CD8 (BioLegend, 344702, 1:250), anti-SMA (Abcam, ab5694, 1:250), anti-MUC2 (Abcam, ab90007, 1:250), anti-lysozyme (Agilent, A0099, 1:250), anti-CD31 (Abcam, ab28364, 1:250), anti-chromogranin A (Abcam, ab15160, 1:250), rabbit anti-IL-7 (Abcam, ab175380, 1:400), mouse anti-IL-7 (Santa Cruz, sc-365306, 1:100) and anti-TAGLN (Abcam, ab14106, 1:250). All secondary antibodies and DAPI were used at 1:1,000 dilution.
Organoid cryopreservation
ALI ileal or duodenal organoids were generated as above and grown for 5 days. Each organoid-containing collagen gel was carefully removed from its insert in one piece and placed into a cryovial containing 1 ml of freezing media (WENR media, supplemented with 10% DMSO, 15% FBS and 10 μM Y-27632 dihydrochloride) and stored at −80 °C for at least 24 h. For cryorecovery, cryovials were quickly submerged in a 37 °C water bath and each organoid-containing collagen gel was transferred to a petri dish, spread out with forceps and washed three times with DMEM with 10% FBS. Each organoid-containing collagen gel was then placed and spread out with forceps on top of a new insert containing freshly solidified 1 ml of collagen mixture (corresponding to the bottom layer) in a well of a six-well plate. A further 200 µl of cold collagen-mixture solution was layered on top. After solidifying for 10 min in an incubator at 37 °C, 1 ml of media was added to each well as usual.
Organoid sizing
Brightfield images of organoid-containing inserts were acquired on a Keyence BZ-X700 microscope and used to manually outline the organoids and the separating spaces between them. This was used to train pixel-based machine-learning Ilastik software (https://www.ilastik.org) to automatically identify organoids by generating probability maps while eliminating the background57. The brightfield images and Ilastik probability maps were exported to CellProfiler58 (https://cellprofiler.org/) to calculate the surface area occupied by each CLIP- or gliadin-treated CeD organoid present in the insert surface areas.
Flow cytometry analysis and FACS isolation
Organoids were dissociated in collagenase IV (2,030 units ml−1; Worthington, LS004212) at 37 °C for 40 min, washed twice in PBS and digested in 400 µg ml−1 Liberase-TL (Roche, 5401020001) and 416 units ml−1 DNase I (Worthington, LS006328) at 37 °C for 30 min with intermittent shaking. Samples were washed twice in FACS buffer (0.6 mM EDTA (Invitrogen, AM9261), 5% FBS, in PBS) and stained in LIVE/DEAD kit (Invitrogen, L34957) diluted in FACS buffer (1:1,000) at RT for 30 min with apoptotic quantification using Nucview488 (Biotium, 30072, 1:500, fluorogenic substrate of caspase 3 and caspase 7 conjugated to AF488), followed by washes in FACS buffer. Surface marker staining was done using antibody cocktails that included anti-CD3-Pacific blue (BioLegend, 300431), anti-CD45-PE (BioLegend, 304008), anti-CD8-BV786 (BD, 563823) or anti-CD8-AlexaFluor 700 (BioLegend, 301028), anti-CD4-BV605 (BioLegend, 317438), anti-CD326(EPCAM)-PerCP-Cy5.5 (BioLegend, 324213), anti-CD19 (BioLegend, 302212), anti-NKG2C (R&D Systems, FAB138A) and anti-NKG2D (BioLegend, 320811), all diluted at 1:500 in FACS buffer. All staining was done on ice. After two washes in FACS buffer, the organoid cells were sorted in a BD FACSAria-II SORP machine after sequential gating for all cells, live cells and single cells. For the HLA-DQ2.5-gliadin tetramer staining experiments, biotinylated HLA-DQ2.5 monomers with covalently attached DQ2.5-glia-α1 and DQ2.5-glia-α2 epitopes were produced as previously described59, conjugated with PE streptavidin, and CD45+ cells were stained with anti-CD45-FITC instead (BioLegend, 304006). For epithelial caspase-3 and caspase-7 activity assays, single live cells were initially gated according to the LIVE/DEAD kit (Invitrogen, L34957). Subsequently, EPCAM+ epithelial cells were gated and NucView488 positivity was assessed. A representative gating strategy is shown in Supplementary Fig. 1.
RT–qPCR
Live single EPCAM+ cells were sorted using a PicoPure RNA isolation kit (Applied Biosystems, KIT0204) extraction buffer in different tubes. The tubes were heated at 42 °C for 30 min immediately after sorting. RNA from FACS-sorted organoid cells was extracted using a PicoPure RNA isolation kit (Applied Biosystems, KIT0204). All RNA samples were treated with RNase-free DNase (Qiagen, 79254) and cDNA synthesis used an iScript reverse transcriptase (Bio-Rad, 1708841). Undiluted cDNA, SsoAdvanced PreAmp Supermix (Bio-Rad, 1725160) and a custom-made Preamp assay pool (50 nM of each primer) were used for gene-specific pre-amplification. Power SYBR Green PCR Master Mix (Applied Biosystems, 4367659) was used for the 10 µl qPCR reactions. Reactions were run in 384-multiwell plates (Applied Biosystems, 4309849) on a Bio-Rad CFX384 instrument. At least 1,000 cells were collected for each population subset from more than 15 organoids per experiment. Primer sequences are shown in Supplementary Table 2.
Single-cell RNA-seq of organoid cultures and fresh tissues
CeD organoid cultures were dissociated (as described above) 2 days after gliadin or CLIP treatment. Fresh tissues were dissociated on the same day of receiving the sample. Single live CD45+ or EPCAM+ cells from dissociated organoids or fresh tissues were sorted by FACS into WENR media and subjected to droplet based scRNA-seq with the 10x Genomics Chromium single cell 5′ platform, with or without single-cell TCR/Ig V(D)J library, per the manufacturer’s protocol using the Chromium Next GEM Single Cell 5′ Kit v2 (PN-1000263), Library Construction Kit (PN-1000190), Chromium Single Cell Human TCR Amplification Kit (PN-1000252) and Chromium Single Cell Human BCR Amplification Kit (PN-1000253).
scRNA-seq analysis
Sequences from scRNA-seq were processed using Cell Ranger (v.3.0.2) software (10x Genomics) with demultiplexing, unique molecular identifier (UMI) collapsing and alignment to the GRCh38 human transcriptome. The scRNA-seq data from the CLIP versus gliadin treatment were loaded into Seurat (v.5.01) objects in R Studio (v.4.1.2) following the standard pipeline60. Data were filtered with nFeature RNA values set to more than 200 and less than 3,500, and percentage Mt values were set to less than 20. Per patient, the two data sets were then anchored together with 3,000 integration features and 30 dimensions to find integration anchors before being clustered in accordance with the standard Seurat pipeline. Immune lineage gene expressions were analysed to identify the phenotypic identity of the cell clusters. Differential gene expressions of cytotoxic markers, chemokines, cytokines and activation markers were visualized using DotPlot in Seurat, stratified for immune subsets and stimulation.
TCR repertoire analysis
TCRβ CDR3 sequences derived from the CeD organoids were pooled with CDR3 sequences of validated gluten-specific T-cell clones from the published literature. This dataset was subjected to GLIPH analysis (v.2.01), which identified TCRs that share CDR3 motifs and exhibited enrichment relative to their expected frequencies in a naive reference TCR dataset.
BCR repertoire analysis
CDR3 sequencing from all the samples were analysed using R studio (1.3.959), and graphs were generated using ggplot2 (3.3.3). CDR3 sequences between ALI organoids and previously published anti-TG2 human antibody sequences were compared and their similarity percentage was calculated using Stringdist (0.9.6.3), which is based on the Levenshtein distance. A match case was defined as when both the J and V genes of ALI organoids and those of known anti-TG2 human monoclonal antibodies were the same and their CDR3 sequence-similarity percentage was more than 85%. Consensus CDR3 motifs between ALI organoids and the published literature were generated using the Motif Comparison tool (5.3.0) on MEME SUITE2, and the P-value was calculated using the Pearson correlation.
Interactome analysis (CellPhoneDB)
Interactions between immune cells in the CeD organoids were identified using CellPhoneDB v.3.0 (ref. 61). The ligand–receptor interactions between two cell types were derived on the basis of the expression of a ligand in one cell type and a receptor in another. At first, the cluster labels of all cells were permuted 1,000 times and the mean of the average expression level of ligands and receptors in interacting clusters was calculated. To determine the significance of interactions, the proportion of the means that was the same as or was higher than the actual mean of all the interactions between two clusters was calculated and assigned a P-value on the basis of the likelihood of cell-type-specific interactions. Significant interactions were determined using the following criteria: first, ligands and receptors were expressed in more than 10% of the cells in the specific cluster; and second, the P-value for at least one cell-type-specific interaction was less than 0.05. Significant interactions were considered to be unique when observed only in either the CLIP or the gliadin treatment. In the absence of a significant interaction, the P-value was manually set to 1 and the mean was considered to be 0.
IgA anti-TG2 autoantibody detection (ELISA)
Organoids were grown for 9–12 days and changed into fresh media for CLIP or gliadin treatment for 2 days. The conditioned medium was immediately frozen at −80 °C before organoid processing. On the day of the assay, samples of medium were thawed at RT and the concentration of IgA anti-TG2 autoantibodies present in the medium was determined by an enzyme immunoassay in a 1:1 dilution according to the manufacturer’s instructions (Generic Assays, Anti-huTransG, 4033).
Luminex cytokine detection
After establishing organoids for 9–12 days, cultures were changed into fresh medium. Organoid medium was collected after 2 days of CLIP or gliadin treatment and immediately frozen at −80 °C before organoid processing. On the day of the assay, samples of medium were thawed at RT and added to a 96-well filter plate. Reading buffer was added to pre-wet the filter plate and was removed by filtration. Assay buffer and samples or antigen standards were then added. Antibody beads and AssayChex beads in the assay buffer were added to each well and incubated at RT for 2 h while being shaken at 500 rpm. The plate was then sealed and incubated at 4 °C overnight. After the plate was pre-warmed at RT, the solution was removed by filtration. After several washes, detection antibody was added to each well. The plate was then incubated at RT for 2 h while being shaken. The solution was then removed by filtration. After several washes, Streptavidin-PE was added to the plate and incubated at RT for 40 min while being shaken. Reading buffer was then added to each well and incubated at RT for 5 min while being shaken. The plate was sealed and read on Luminex instruments according to the kit’s specifications.
In situ hybridization
To detect IL7 mRNA in clinical FFPE samples, sections of duodenal biopsies from active CeD versus remission CeD patients on a GFD were sectioned to slides in an RNase-free environment. To detect human IL7 mRNA, a human IL7 probe (RNAscope Probe Hs-IL7, Homo sapiens interleukin 7 (IL7); 424251) was used with an RNAscope 2.5 HD duplex detection kit (322500), according to the manufacturer’s protocol (ACD Biotechne). Imaging was performed on an Olympus BX43 microscope using a SPOT Insight 5MPCMOS camera and SPOT software. For quantification, a model was trained using the IHC toolbox within ImageJ for colour detection and extraction of the IL7 signal. A positive threshold was set on the basis of an average of 12 images (six active CeD and six remission CeD). This model and threshold were applied to each of the biopsy images, and the total number of positive pixels was quantified in each image for four representative images from each active CeD (6 total samples and 24 images) and each remission CeD (6 total samples and 24 images) specimen. A second model was trained and the threshold was set in an analogous manner for the detection of haematoxylin signal and applied to the same image set to determine the number of nuclei per image. The total IL7 signal for an image was divided by the number of nuclei per image to yield the IL7 signal per nucleus for each image. This was repeated for the four images from a given specimen to determine the average of IL7 per nucleus value for each specimen.
Statistics and reproducibility
All data are representative of at least three different biological replicates. Biopsy samples were processed and seeded into eight inserts for organoid formation. Normally, four read-outs per sample were obtained by combining at least two inserts per experimental condition, after counting the number of organoids and ensuring an equal total number of organoids per condition. In the figures, box-plot horizontal lines represent median values and whiskers represent data-range minima or maxima. Non-parametric two-tailed Mann–Whitney tests were used to determine the statistical significance for two samples from different donors, and two-tailed Wilcoxon tests were used to determine the statistical significance for two culture conditions derived from the same donor. One-way ANOVA, comparing multiple parameters, was used to determine the statistical significance for three or more paired culture conditions from the same donor. P-values are denoted as: *P < 0.05, **P ≤ 0.01, ***P ≤ 0.001 and NS if P ≥ 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data sets for scRNA-seq have been deposited in Gene Expression Omnibus with the accession code GSE200075. Source data are provided with this paper.
References
Catassi, C., Verdu, E. F., Bai, J. C. & Lionetti, E. Coeliac disease. Lancet 399, 2413–2426 (2022).
Levescot, A., Malamut, G. & Cerf-Bensussan, N. Immunopathogenesis and environmental triggers in coeliac disease. Gut 71, 2337–2349 (2022).
Iversen, R. & Sollid, L. M. The immunobiology and pathogenesis of celiac disease. Annu. Rev. Pathol. 18, 47–70 (2023).
Marsh, M. N. Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity (‘celiac sprue’). Gastroenterology 102, 330–354 (1992).
Jabri, B. et al. Selective expansion of intraepithelial lymphocytes expressing the HLA-E-specific natural killer receptor CD94 in celiac disease. Gastroenterology 118, 867–879 (2000).
Hüe, S. et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 21, 367–377 (2004).
Meresse, B. et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 21, 357–366 (2004).
Pinto-Sanchez, M. I. et al. Society for the Study of Celiac Disease position statement on gaps and opportunities in coeliac disease. Nat. Rev. Gastroenterol. Hepatol. 18, 875–884 (2021).
Barone, M. V. et al. Gliadin-mediated proliferation and innate immune activation in celiac disease are due to alterations in vesicular trafficking. PLoS ONE 6, e17039 (2011).
Castellanos-Rubio, A. et al. Long-term and acute effects of gliadin on small intestine of patients on potentially pathogenic networks in celiac disease. Autoimmunity 43, 131–139 (2010).
Palova-Jelinkova, L. et al. Gliadin fragments induce phenotypic and functional maturation of human dendritic cells. J. Immunol. 175, 7038–7045 (2005).
Freire, R. et al. Human gut derived-organoids provide model to study gluten response and effects of microbiota-derived molecules in celiac disease. Sci. Rep. 9, 7029 (2019).
de Kauwe, A. L. et al. Resistance to celiac disease in humanized HLA-DR3-DQ2-transgenic mice expressing specific anti-gliadin CD4+ T cells. J. Immunol. 182, 7440–7450 (2009).
Abadie, V. et al. IL-15, gluten and HLA-DQ8 drive tissue destruction in coeliac disease. Nature 578, 600–604 (2020).
Goel, G. et al. Serum cytokines elevated during gluten-mediated cytokine release in coeliac disease. Clin. Exp. Immunol. 199, 68–78 (2020).
Tye-Din, J. A. et al. Patient factors influencing acute gluten reactions and cytokine release in treated coeliac disease. BMC Med. 18, 362 (2020).
Jabri, B. & Abadie, V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol. 15, 771–783 (2015).
Lähdeaho, M.-L. et al. Safety and efficacy of AMG 714 in adults with coeliac disease exposed to gluten challenge: a phase 2a, randomised, double-blind, placebo-controlled study. Lancet Gastroenterol. Hepatol. 4, 948–959 (2019).
Rochman, Y., Spolski, R. & Leonard, W. J. New insights into the regulation of T cells by γc family cytokines. Nat. Rev. Immunol. 9, 480–490 (2009).
Lei, X. et al. Down-regulation of interleukin 7 receptor (IL-7R) contributes to central nervous system demyelination. Oncotarget 8, 28395–28407 (2017).
Churchman, S. M. et al. Modulation of peripheral T-cell function by interleukin-7 in rheumatoid arthritis. Arthritis Res. Ther. 16, 511 (2014).
Penaranda, C. et al. IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc. Natl Acad. Sci. USA 109, 12668–12673 (2012).
Lee, L. -F. et al. Anti-IL-7 receptor-α reverses established type 1 diabetes in nonobese diabetic mice by modulating effector T-cell function. Proc. Natl Acad. Sci. USA 109, 12674–12679 (2012).
Belarif, L. et al. IL-7 receptor influences anti-TNF responsiveness and T cell gut homing in inflammatory bowel disease. J. Clin. Invest. 129, 1910–1925 (2019).
Watanabe, M. et al. Interleukin 7 transgenic mice develop chronic colitis with decreased interleukin 7 protein accumulation in the colonic mucosa. J. Exp. Med. 187, 389–402 (1998).
Saligrama, N. et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature 572, 481–487 (2019).
Li, J. et al. KIR+CD8+ T cells suppress pathogenic T cells and are active in autoimmune diseases and COVID-19. Science 376, eabi9591 (2022).
Sollid, L. M. The roles of MHC class II genes and post-translational modification in celiac disease. Immunogenetics 69, 605–616 (2017).
Anderson, R. P., Degano, P., Godkin, A. J., Jewell, D. P. & Hill, A. V. S. In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptide as the dominant A-gliadin T-cell epitope. Nat. Med. 6, 337–342 (2000).
Petersen, J. et al. T-cell receptor recognition of HLA-DQ2–gliadin complexes associated with celiac disease. Nat. Struct. Mol. Biol. 21, 480–488 (2014).
Sollid, L. M. et al. Update 2020: nomenclature and listing of celiac disease-relevant gluten epitopes recognized by CD4+ T cells. Immunogenetics 72, 85–88 (2020).
Sette, A., Southwood, S., Miller, J. & Appella, E. Binding of major histocompatibility complex class II to the invariant chain-derived peptide, CLIP, is regulated by allelic polymorphism in class II. J. Exp. Med. 181, 677–683 (1995).
Korneychuk, N. et al. Interleukin 15 and CD4+ T cells cooperate to promote small intestinal enteropathy in response to dietary antigen. Gastroenterology 146, 1017–1027 (2014).
Glanville, J. et al. Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98 (2017).
Broughton, S. E. et al. Biased T cell receptor usage directed against human leukocyte antigen DQ8-restricted gliadin peptides is associated with celiac disease. Immunity 37, 611–621 (2012).
Huang, H., Wang, C., Rubelt, F., Scriba, T. J. & Davis, M. M. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat. Biotechnol. 38, 1194–1202 (2020).
Dahal-Koirala, S. et al. Comprehensive analysis of CDR3 sequences in gluten-specific T-cell receptors reveals a dominant R-motif and several new minor motifs. Front. Immunol. 12, 639672 (2021).
Galeano Niño, J. L. et al. Cytotoxic T cells swarm by homotypic chemokine signalling. eLife 9, e56554 (2020).
Christophersen, A., Risnes, L. F., Dahal-Koirala, S. & Sollid, L. M. Therapeutic and diagnostic implications of T cell scarring in celiac disease and beyond. Trends Mol. Med. 25, 836–852 (2019).
Hamilton, J. A. GM-CSF-dependent inflammatory pathways. Front. Immunol. 10, 2055 (2019).
Olaussen, R. W. et al. Interferon-γ-secreting T cells localize to the epithelium in coeliac disease. Scand. J. Immunol. 56, 652–664 (2002).
Husby, S. et al. European Society Paediatric Gastroenterology, Hepatology and Nutrition guidelines for diagnosing coeliac disease 2020. J. Pediatr. Gastroenterol. Nutr. 70, 141–156 (2020).
Di Niro, R. et al. High abundance of plasma cells secreting transglutaminase 2-specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat. Med. 18, 441–445 (2012).
Kivelä, L. et al. Current and emerging therapies for coeliac disease. Nat. Rev. Gastroenterol. Hepatol. 18, 181–195 (2021).
Dooms, H. Interleukin-7: fuel for the autoimmune attack. J. Autoimmun. 45, 40–48 (2013).
Meresse, B. et al. Reprogramming of CTLs into natural killer-like cells in celiac disease. J. Exp. Med. 203, 1343–1355 (2006).
Lundin, K. E. et al. Gliadin-specific, HLA-DQ(α1*0501,β1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J. Exp. Med. 178, 187–196 (1993).
Ootani, A. et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 15, 701–706 (2009).
Neal, J. T. et al. Organoid modeling of the tumor immune microenvironment. Cell 175, 1972–1988 (2018).
Maiuri, L. et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 362, 30–37 (2003).
Yang, H., Spencer, A. U. & Teitelbaum, D. H. Interleukin-7 administration alters intestinal intraepithelial lymphocyte phenotype and function in vivo. Cytokine 31, 419–428 (2005).
Porter, B. O. & Malek, T. R. Thymic and intestinal intraepithelial T lymphocyte development are each regulated by the γc-dependent cytokines IL-2, IL-7, and IL-15. Semin. Immunol. 12, 465–474 (2000).
Dong, L.-H., Lv, P. & Han, M. Roles of SM22α in cellular plasticity and vascular diseases. Cardiovasc. Hematol. Disord. Drug Targets 12, 119–125 (2012).
Watanabe, M. et al. Interleukin 7 is produced by human intestinal epithelial cells and regulates the proliferation of intestinal mucosal lymphocytes. J. Clin. Invest. 95, 2945–2953 (1995).
Christophersen, A. et al. Distinct phenotype of CD4+ T cells driving celiac disease identified in multiple autoimmune conditions. Nat. Med. 25, 734–737 (2019).
Gandini, A., Gededzha, M. P., De Maayer, T., Barrow, P. & Mayne, E. Diagnosing coeliac disease: a literature review. Hum. Immunol. 82, 930–936 (2021).
Berg, S. et al. ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232 (2019).
Stirling, D. R. et al. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 22, 433 (2021).
Ráki, M. et al. Tetramer visualization of gut-homing gluten-specific T cells in the peripheral blood of celiac disease patients. Proc. Natl Acad. Sci. USA 104, 2831–2836 (2007).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 15, 1484–1506 (2020).
Acknowledgements
We thank members of the Kuo, Davis, Mellins and Sollid groups for discussions; the Stanford FACS, Functional Genomics, Human Histology (P. Chu), Cell Sciences Imaging and Human Immune Monitoring cores for technical expertise; the Stanford Tissue Bank for providing surgical samples; E. Sanjines, A. Adiao, D. Souki, G. Tan and G. Masarweh for collection and delivery of endoscopy samples; B. Simonsen for the HLA-DQ molecules used for tetramer assembly; and J. and R. Triebsch for support from the Stanford Celiac Translational Research Program. This work was also supported by funding from the Stanford Medicine Children’s Health Center for IBD and Celiac Disease. V.v.U. was supported by a Netherlands Organization for Scientific Research Rubicon grant (452181214). We acknowledge funding from the South-Eastern Norway Regional Health Authority (projects 2016113 and 2020027 to L.M.S.), a Stanford Maternal Child Health Research Institute seed grant (C.J.K.), NIH RM1-HG007735 (H.Y.C.), NIH U19AI057229 (M.M.D.), NIH U01DK085527, U19AI116484, R01CA251514, R01DK130414, R01DK115728 (C.J.K.), the NIDDK Intestinal Stem Cell Consortium and the NIAID Biomimetic U19 Consortium. We dedicate this work to the memory of Elizabeth D. Mellins, whose experimental design and guidance were crucial for this study.
Author information
Authors and Affiliations
Contributions
A.J.M.S. conceived, designed and performed experiments, analysed data and wrote the manuscript. V.v.U. conceived experiments and analysed scRNA-seq data. Z.L. designed experiments, did organoid cultures, confocal imaging and analysis, analysed scRNA-seq data and did RT–qPCR. S.M.C. collected tissue FFPE blocks and coordinated sectioning for haplotyping and IL7 in situ hybridization and analysed data. N.H. did organoid cultures and RT–qPCR. A. Batish did RT–qPCR. J.E.C. measured and analysed organoid sizes. J.C. did organoid cultures, cryopreservation and recovery, imaging and analysis. E.T.Z. sectioned frozen tissue blocks and helped with their staining and imaging. Q.M. did organoid cultures. A.G.-S. did confocal imaging and analysis. M.T. did RT–qPCR and data analysis. D.C. sectioned FFPE tissue blocks and did IL7 in situ hybridization. S.V. did IL7 in situ hybridization. S.S.C. did confocal imaging and schematic design. A.C. produced HLA-DQ monomers for tetramer assembly. A. Baghdasaryan provided resources. K.E.Y. prepared libraries for scRNA-seq. K.K. used Cell Ranger for scRNA-seq. A.H. assisted with 10x Genomics cell capture. J.L. did HLA genotyping. H.D. provided resources and supervision. Z.M.S. provided samples and guidance. H.Y.C. provided resources and supervision. J.C.Y.D. provided surgical samples. B.M.Z. did HLA genotyping and analysis. E.D.M. conceived and designed experiments and provided resources and supervision. L.M.S. provided resources, supervision and guidance. N.Q.F.-B. identified CeD patients and controls, coordinated and collected endoscopy samples and provided guidance. M.M.D. provided resources, supervision and guidance. C.J.K. conceived and designed experiments, analysed data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
C.J.K. and A.J.M.S. are inventors on patent WO 2020/247528 describing methods and uses of patient-derived celiac intestinal organoids. C.J.K. and M.M.D. are founders of Mozart Therapeutics and NextVivo, Inc. L.M.S has been a consultant during the last 3 years for BMS, GSK, Mozart Therapeutics, Ono Pharmaceutical, Precigen ActoBio, Sanofi-Aventis, SQZ Biotech, Takeda and Topas Therapeutics. All other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Nadine Cerf-Bensussan, Toshiro Sato and Detlef Schuppan for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Small intestine ALI organoids possess different mesenchymal and epithelial cell types.
a, IF whole-mount staining of small intestine organoids at day 14 showing SMA+ or PDGFRA+ fibroblasts, CD31+ endothelial cells and PGP9.5+ neurons (green), ECAD+ epithelium (white) and DAPI (blue) (representative images from n = 3 biological replicates). b, IF whole-mount staining of small intestine organoids at day 14 showing MUC2+ goblet cells, CHGA+ enteroendocrine cells, LYZ1+ Paneth cells (green), ECAD+ epithelium (white) and DAPI (blue) (representative images from n = 3 biological replicates). c, Enlargement of organoid cup-shaped MUC2+ goblet cells (representative image from n = 3 biological replicates). d, Violin plots of CD14 and CD68 mRNA expression from CeD organoid scRNA-seq and a scatter plot of CD14 and CD68 mRNA co-expression in the myeloid compartment. e, Whole-mount IF staining of small intestine ALI organoid CD4+ (green) and CD8+ (red) T cells, showing enrichment of CD8+ T cells within the EPCAM+ epithelial compartment (white). DAPI (blue). In contrast, CD4+ T cells localize to non-epithelial lamina propria-like areas (representative image from n = 3 biological replicates). All scale bars are 100 µm, except (c) in which the scale bar is 50 µm.
Extended Data Fig. 2 Duodenal ALI organoids contain diverse immune populations, related to Fig. 1.
a, Integrated UMAP plot of CD45+-sorted cells from scRNA-seq, revealing diverse immune populations in small intestine ALI organoids at day 14, n = 6 CeD patients. b, Violin plots showing expression of genes used to identify the immune populations shown in (a). c, UMAP plots of overlap between tissue and organoid CD45+ immune populations as in (a, b). d, scRNA-seq Jaccard index of TCR overlap between fresh small intestine tissue (n = 1 CeD patient) and ALI organoids (n = 4 CeD patients). e, Integrated UMAP from scRNA-seq of active CeD organoid T cells (n = 6 patients). Cells expressing KIR3DL1 or KIR2DL3 are rendered in red. f, Plot of CD8+ T cells from (e). g, Integrated UMAP from scRNA-seq of active CeD organoid T cells (top left) (n = 6 patients). Cells in red exhibit expression of KIR3DL1 or KIR2DL3 (top right), NKG2C (bottom left) and NKG2D (bottom right). h, Pie bar graph showing organoid-derived TCR counts in which each segment represents a unique clonotype, n = 5 patients. Expanded clonotypes (TCR counts ≥ 2) are indicated in red.
Extended Data Fig. 3 Cytokine supplementation and cryopreservation of intestinal ALI organoids.
a, FACS-based tSNE plots depicting time course abundance of EPCAM+ and CD45+ cells (top) and CD4+ and CD8+ T cells (bottom) as a percentage of total live single ileal organoid cells with or without addition of IL-2 and IL-7, representative experiment of n = 3 biological replicates. b, Organoids grown for 14 days (control) have similar percentages of epithelium and immune components as organoids grown for 5 days, frozen in-gel at −80 °C, cryorecovered, and replated for the indicated durations. c-d, ALI organoids demonstrate persistent growth after being frozen in-gel at −80 °C, cryorecovered and replated (c, arrows), with maintenance of epithelial protrusions by H&E (d). Numerous air bubbles in the collagen are present on initial plating post-cryorecovery and progressively disappear with culture. (b-d) depict representative experiments from n = 4 biological replicates. Scale bar is 5 mm for (c) and 100 µm for (d).
Extended Data Fig. 4 Gliadin induces loss of villus-like structures in CeD organoids.
a, Duodenal ALI organoids from celiac (CeD) or non-celiac control donors were established for 9–12 days followed by gliadin or CLIP treatment for 2 days before analysis, unless stated otherwise. The gliadin peptides were a 1:1 mixture of deamidated immunodominant, HLA-DQ2.5-restricted, glia-α1 (LQPFPQPELPYPGS) and glia-α2 (APQPELPYPQPGS) gluten epitopes. b-d, Confirmatory IF staining of sections of human duodenum tissue showing IL-15 (red) in (a), SI (red) in (b) and APOA4 (red) in (c); DAPI (blue) (representative images from n = 3 biological replicates). e, Quantification of SI mRNA in FACS-sorted organoid EPCAM+ cells from 2-day gliadin-treated control or active CeD organoids. RT-qPCR, expressed as a ratio of gliadin:CLIP treatment, from control (n = 4) or CeD (n = 5) biological replicates. Box plots show the median as the center line, the interquartile range as the box limits and the whiskers represent the min and max. *, P = 0.0381; two-tailed Mann-Whitney test. f, Representative H&E staining of different sections of control or active CeD organoids after 2-day gliadin or CLIP treatment. Arrows denote regions where epithelial protrusions are absent. g, Quantification of epithelial protrusions per organoid circumference from (f); control (N = 6 biological replicates), CeD (N = 7 biological replicates), each data point is from an individual organoid. Scatter plots show the median as the center line and the whiskers represent the min and max. ***=P < 0.0001; two-tailed Mann-Whitney test. All scale bars are 100 µm. All CeD organoids were DQ2.5+.
Extended Data Fig. 5 Gliadin induces epithelial proliferation in CeD organoids.
a, Representative IF staining of sections of active CeD organoids after 2-day gliadin or CLIP treatment in EN media showing proliferative KI67+ cells (green), ECAD (red) and DAPI (blue). Scale bar is 50 µm. b, Quantification of KI67 fluorescence from (a), control (n = 3 biological replicates), CeD (n = 5 biological replicates); each data point is from an individual organoid. ***, P < 0.0001; two-tailed Mann-Whitney test. c, Representative brightfield images of active CeD organoids before and after 2-day treatment with gliadin or CLIP peptides. Scale bar is 5 mm. d, Automated quantification of fold change in CeD organoid area from (c), 2 days after treatment with gliadin or CLIP. n = 10 CeD patients. **, P = 0.002; two-tailed Wilcoxon test. e, LGR5 RT-qPCR from FACS-sorted organoid EPCAM+ cells as ratio of gliadin:CLIP treatment for 2 days in organoids from control (n = 7 biological replicates) or active CeD (n = 8 biological replicates). **, P = 0.0012; two-tailed Mann-Whitney test. f, PCNA RT-qPCR from FACS-sorted organoid EPCAM+ cells as ratio of gliadin:CLIP treatment for 2 days in control or active CeD organoids, (n = 7 biological replicates each). **, P = 0.007; two-tailed Mann-Whitney test. g, CCND1 RT-qPCR from FACS-sorted organoid EPCAM+ cells as ratio of gliadin:CLIP treatment for 2 days in organoids from control (n = 8 biological replicates) or active CeD (n = 7 biological replicates). ***, P = 0.0003; two-tailed Mann-Whitney test. All box plots show the median as the center line, the interquartile range as the box limits and the whiskers represent the min and max. All CeD organoids were DQ2.5+.
Extended Data Fig. 6 TCR sequencing from CeD organoid scRNA-seq reveals known and suspected gliadin-specific TCR motifs.
a, scRNA-seq integrated UMAP plot highlighting TCR-expressing T cells in active CeD organoids at day 14, n = 5 CeD patients. b, GLIPH homology analysis showing conserved CDR3 motifs (red) found between active CeD organoids and gliadin-specific published sequences, n = 5 CeD patients. All CeD organoids in this figure were HLA-DQ2.5+.
Extended Data Fig. 7 Two additional biological replicates of scRNA-seq-derived dot plots.
Dot plots from organoid scRNA-seq from two active CeD patients (a, b); a third patient is shown in Fig. 4a. Depiction of mean expression levels and corresponding percent population expression amongst active CeD organoid CD4+ and CD8+ T cells, Treg, plasma B cells and myeloid cells after 2-day gliadin or CLIP treatment. The patient in (a) is HLA-DQ2.5, as is the patient in Fig. 4a. The patient in (b) is HLA-DQ2.2, which manifests low-affinity binding to the HLA-DQ2.5 gliadin peptides used in the study (Bodd et al, Gastroenterology, 2012 Mar;142(3):552-61).
Extended Data Fig. 8 ScRNA-seq-based interactome analysis of novel gliadin-induced immune interactions in CeD organoids.
a, Overview of unique CellPhoneDB immune interactions found in 2-day CLIP- or gliadin-treated active CeD organoids, stratified by immune cell type (CD4+ T, CD8+ T, myeloid, NK and Treg cells). Columns indicate sending:receiving cell type and rows indicate ligand-receptor pairs. P values are indicated by circle size. The mean (log2) average expression levels of interacting molecule 1 and interacting molecule 2 are indicated by the color gradient. b, Corresponding schematic showing potential interactions between immune cells in CeD. Integrated data from n = 4 CeD patients, 3 DQ2.5+ and 1 DQ2.2+.
Extended Data Fig. 9 Sc-RNAseq-based interactome of B cells and plasma cells, and BCR sequence consensus.
a, B cell- and plasma B cell-specific immune interactomes derived from scRNA-seq CellPhoneDB analysis showing 117 unique B and plasma-cell driven interactions in gliadin-treated organoids and absence of unique interactions in CLIP, integrated data from n = 4 CeD patients. b, BCR sequence consensus analysis from scRNA-seq of matched CDR3 sequences from active CeD organoids and published anti-TG2 CDR3 sequences categorized by length; n = 3 CeD patients.
Extended Data Fig. 10 BCR sequencing from CeD organoid scRNA-seq reveals extensive overlap with public anti-TG2 CeD-specific motifs.
a, scRNA-seq integrated UMAP plot highlighting BCR-expressing B and plasma cells in active CeD organoids, n = 3 CeD patients. b, Homology analysis showing conserved CDR3 sequences (red) found between active CeD organoids and anti-TG2 CeD-specific published sequences, n = 3 CeD patients.
Extended Data Fig. 11 IL-7 is upregulated in active celiac duodenal biopsy tissue.
a, Luminex protein analysis of organoid conditioned media from active CeD (N = 7 biological replicates) or control (N = 4 biological replicates) showing fold-increases of IL-7 as ratio of gliadin:CLIP treatment after 2 days. Box plots show the median as the center line, the interquartile range as the box limits and the whiskers represent the min and max. ns, P = 0.072; two-tailed Mann-Whitney test. b-c, Representative IF staining using a rabbit (Rb) anti-IL-7 antibody (red) in fresh duodenal biopsies from (b) 14 remission CeD patients (previously diagnosed with CeD but on gluten-free diet) versus (c) 14 CeD patients with active disease, showing increased IL-7 levels in the latter. Epithelium (CK19, green); DAPI (blue). Figure 6c shows staining for a 15th patient in remission and a 15th patient with active CeD, and quantitation is presented in Fig. 6d. (GFD, n = 15 donors) or active CeD (n = 15 donors). d, Representative IF staining using a mouse (Ms) anti-IL-7 antibody (red) in fresh duodenal biopsies from (b), 4 remission CeD patients versus 4 patients with active CeD. This confirmed elevated IL-7 expression in active CeD seen with a different antibody than in (b) and (c). Scale bars are 100 µm.
Supplementary information
Supplementary Information
This file contains sequential FACS gating strategy, donor DQ-typing and demographics, and primer sequences used for RT-qPCR.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Santos, A.J.M., van Unen, V., Lin, Z. et al. A human autoimmune organoid model reveals IL-7 function in coeliac disease. Nature 632, 401–410 (2024). https://doi.org/10.1038/s41586-024-07716-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07716-2
- Springer Nature Limited
This article is cited by
-
Organoids identify a role for IL-7 in coeliac disease
Nature Reviews Drug Discovery (2024)
-
Organoids identify a role for IL-7 in coeliac disease
Nature Reviews Immunology (2024)