

Abstract
Hybridization allows adaptations to be shared among lineages and may trigger the evolution of new species1,2. However, convincing examples of homoploid hybrid speciation remain rare because it is challenging to demonstrate that hybridization was crucial in generating reproductive isolation3. Here we combine population genomic analysis with quantitative trait locus mapping of species-specific traits to examine a case of hybrid speciation in Heliconius butterflies. We show that Heliconius elevatus is a hybrid species that is sympatric with both parents and has persisted as an independently evolving lineage for at least 180,000 years. This is despite pervasive and ongoing gene flow with one parent, Heliconius pardalinus, which homogenizes 99% of their genomes. The remaining 1% introgressed from the other parent, Heliconius melpomene, and is scattered widely across the H. elevatus genome in islands of divergence from H. pardalinus. These islands contain multiple traits that are under disruptive selection, including colour pattern, wing shape, host plant preference, sex pheromones and mate choice. Collectively, these traits place H. elevatus on its own adaptive peak and permit coexistence with both parents. Our results show that speciation was driven by introgression of ecological traits, and that speciation with gene flow is possible with a multilocus genetic architecture.
Similar content being viewed by others
Main
Biodiversity has long been depicted as a ‘tree of life’, but a wealth of genomic data has made clear that the branches and leaves of the tree often do not represent neatly defined units. Instead, they comprise a braided delta of evolutionary lineages linked by hybridization and introgression4. Although gene flow tends to homogenize populations5, it may also contribute to adaptation and even drive speciation if introgressed variants cause reproductive isolation1,2,6. Polyploid (chromosome-doubling) hybrid speciation is common in plants7,8, but compelling examples of homoploid hybrid speciation (without a change in chromosome number) remain scarce and contested, especially in animals3. This is because it is difficult to prove that hybridization had a pivotal role in creating reproductive isolation between the hybrid lineage and the parental species3. Here we present evidence for homoploid hybrid speciation in Heliconius butterflies. We show that introgression of key adaptive traits from H. melpomene caused H. elevatus to diverge from H. pardalinus, despite ongoing gene flow among sympatric H. elevatus and H. pardalinus, which homogenizes 99% of their genomes (Fig. 1a).

a, Evolutionary relationships and main introgression events described in this study. We test the hypothesis that introgression of major pre-mating and post-mating ecological isolating traits from H. melpomene led to the establishment of H. elevatus as a new stable hybrid species. Mya, million years ago. b, Geographical distributions of major clades. Locations at which both H. elevatus and H. pardalinus were sampled are numbered. c, Distance-based network using genome-wide independent SNPs. This concatenated tree shows the existence of two distinct clusters, Amazonian versus non-Amazonian, in both H. elevatus and H. pardalinus. d, Topology weighting analysis (TWISST) showing the percentage of the 11,509 non-overlapping genomic windows of 1,000 SNPs in which the majority of subtrees (that is, topology weighting ≥ 0.5) clusters H. elevatus (ele) with either H. pardalinus (par) (93.2%; top) or H. melpomene (mel) (0.52%; bottom). Note that although H. elevatus groups with H. pardalinus in 93.2% of windows, only 1.61% of those trees yield the two species as reciprocally monophyletic. By contrast, all three species are monophyletic in 81% of the windows for which H. elevatus groups with H. melpomene. Subscripts indicate geographical distributions for H. elevatus and H. pardalinus (Ama, Amazon; And, Andes; Gui, Guianas) and subspecies for H. melpomene (Agl, aglaope; Ams, amaryllis). e, A multispecies coalescent model with introgression supports a hybrid origin of H. elevatus, with the introgression time coinciding closely with the origin of the species (the 95% HPD intervals are given within parenthesis). Images of butterfly wings are copyright of the authors and Michel Cast.
Heliconius butterflies are chemically defended by cyanogenic glycosides, either sequestered from larval passion-vine host plants (Passifloraceae) or synthesized de novo9,10. Adults signal their toxicity to predators through their brightly coloured wing patterns. The cost of educating predators is shared with other defended butterflies and moths through mutualistic Müllerian mimicry11. Mimicry among species is not restricted to colour pattern, because co-mimics also converge in flight behaviour and wing shape12,13. Hybrids with intermediate phenotypes are selected against because predators do not recognize them14,15. Therefore, different mimetic phenotypes correspond to fitness peaks in an adaptive landscape maintained by disruptive selection. How populations transition to new fitness peaks remains an unanswered question, but adaptive introgression provides a possible route.
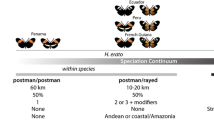
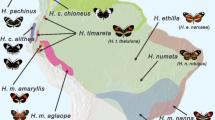
Heliconius elevatus, H. pardalinus and H. melpomene present an excellent system with which to elucidate the role of hybridization in speciation. All three species are sympatric across the Amazon basin16,17,18 (Fig. 1b). Heliconius elevatus and H. pardalinus are closely related19, but they have strikingly different colour patterns (Fig. 1a). Heliconius pardalinus has a ‘tiger’ mimetic colour pattern typical of its close relatives. By contrast, H. elevatus has a red, black and yellow pattern, mimicking the much more distantly related H. melpomene19. This phenotypic convergence results in part from introgression at the major colour patterning loci cortex and optix20. Because Heliconius elevatus uses colour pattern as a partial cue in mate choice17, these introgressed alleles are likely to promote pleiotropic reproductive isolation from H. pardalinus. This suggests that H. elevatus has a hybrid origin, and here we test that hypothesis.
Hybrid genome of H. elevatus
We analysed whole-genome sequences of 92 wild-caught individuals of these three species: 42 H. elevatus (12 locations), 33 H. pardalinus (7 locations) and 17 H. melpomene (4 locations). For H. elevatus and H. pardalinus, our sampling spanned their combined geographical range (Fig. 1b and Supplementary Table 1). A concatenated whole-genome phylogenetic network groups H. pardalinus with H. elevatus, whereas H. melpomene forms a much deeper lineage separated by several other species21 (Fig. 1c). This topology is echoed in 93.2% of the genealogies estimated in sliding windows of 1,000 single-nucleotide polymorphisms (SNPs) across the genome, whereas 0.52% of the genealogies cluster H. elevatus with H. melpomene (Fig. 1d). This is suggestive of introgression between these two species but could also be explained by retention of ancestral polymorphisms. Testing this hypothesis under the multispecies coalescent with introgression framework22 (Fig. 1e), we find that most of the H. elevatus genome is derived from H. pardalinus, with a 0.71% (95% high posterior density (HPD) 0.32–1.11%) contribution from H. melpomene. Heliconius elevatus arose as an independent lineage around 180,000 years ago (kya) (Fig. 1e; 95% HPD 137–216 kya; see also Extended Data Fig. 1). This divergence time coincides closely with the divergence from H. pardalinus (212 kya, 95% HPD 201–224 kya) and the timing of introgression from H. melpomene (193 kya, 95% HPD 142–247 kya). These coalescent-based estimates are therefore consistent with H. elevatus being a hybrid lineage that formed through admixture between H. pardalinus and H. melpomene.
Ongoing local gene flow with H. pardalinus
Notably, the whole-genome concatenated phylogenetic network suggests that sympatric populations of H. elevatus and H. pardalinus in the Amazon are more closely related to each other than to allopatric conspecifics from the Peruvian Andes and the Guianas (Fig. 1c). Only 1.92% (1.50% + 0.42%; Fig. 1d) of the 11,509 windows across the genome yield reciprocally monophyletic genealogies for H. pardalinus and H. elevatus; this is confirmed by multispecies coalescent-based trees across the genome (Extended Data Fig. 2). We therefore investigated whether this apparent double species paraphyly could be explained by extensive ongoing gene flow between H. elevatus and H. pardalinus in sympatry in the Amazon.
Putative natural hybrids have been reported occasionally between H. elevatus and H. pardalinus23, but the two very rarely mate in captivity17. However, F1 hybrids from forced matings are fully fertile17. We therefore examined the genomes of wild-caught individuals of the two species from sympatric populations across their range for evidence of gene flow. Focusing on SNPs diagnostic for H. elevatus and H. pardalinus, we find a few individuals with long tracts of heterozygous ancestry, in some cases spanning whole chromosomes, indicating recent gene flow (Extended Data Fig. 3). Four-population (f4) tests comparing within- and between-species gene flow support ongoing interspecific gene flow in sympatry (Fig. 2a). Estimated levels of effective gene flow between the species in sympatry are high (Nm > 1, where N is the effective population size, and m is the migration rate per generation), quite sufficient to homogenize neutral variation between species; indeed, gene flow approaches the rates that are found among nearby populations of the same species (Fig. 2b and Supplementary Table 2). Finally, we performed demographic modelling based on the site-frequency spectrum under different demographic scenarios and topologies. The best supported model retrieved a tree in which H. elevatus and H. pardalinus were reciprocally monophyletic, and confirmed that gene flow has been prevalent throughout their combined history (Fig. 2c and Extended Data Fig. 4). After their initial split, populations of H. pardalinus in the Amazon diverged from those in the Andes, and Amazonian populations of H. elevatus diverged from those in the Guianas (Fig. 2c and Supplementary Table 3). The two species then began to overlap broadly in the Amazon from around 28 kya (95% confidence interval 25.6–30.0 kya) until the present, with high levels of gene flow in sympatry. Nonetheless, sympatric populations of H. elevatus and H. pardalinus in the Amazon form mutually monophyletic genetic clusters (Fig. 1c); thus, the two species remain differentiated and can clearly coexist despite extensive ongoing gene flow, implying the existence of strong sexual and ecological isolation.

a, Gene flow between species in sympatry is typically significantly greater (f4 > 0, filled points) than the within-species gene flow between populations across the Amazon basin. Numbers next to the points indicate the population pairs compared (see Fig. 1b). A significant positive correlation indicates that the within-species gene flow between populations X and Y declines with increasing geographical distance relative to between-species gene flow at populations X and/or Y. b, Estimates of effective migration rate (Nm) within and between species using G-PhoCS. In the Amazon, between-species Nm (parAma/eleAma) is similar to within-species Nm between locations (parAma/parAma and eleAma/eleAma). The estimates for parAma/eleAma are denoted as filled and open circles and correspond to within and between location comparisons, respectively. c, The best supported demographic model based on the site-frequency spectrum analysis supports H. elevatus and H. pardalinus as reciprocally monophyletic (Extended Data Fig. 4). They initially diverged with continuous gene flow (933 to 221 kya) and after splitting into Amazonian and non-Amazonian populations, they came into secondary contact and continued exchanging genes until the present (45 kya to the present). Numbers within the blocks are effective population sizes in thousands. Arrows between groups represent continuous gene flow; numbers above or below arrows indicate 2Nm values.
Lack of gene flow with H. melpomene
The genome of Heliconius elevatus is, on average, more distantly related to its other parental species H. melpomene than it is to H. pardalinus (Fig. 1d and Extended Data Fig. 5a). Yet gene flow from H. melpomene is plausible, because the latter is known to hybridize occasionally with other equally distant species in the wild24,25. None of the 31 H. elevatus or 17 H. melpomene individuals collected from areas of sympatry show any tracts of heterospecific ancestry (Extended Data Fig. 5b). Likewise, f4 tests do not detect any signals of gene flow (Supplementary Table 4). These data indicate that, in contrast to the extensive ongoing gene flow detected between H. elevatus and H. pardalinus, any recent gene flow between H. elevatus and H. melpomene is extremely rare. Because the H. elevatus and H. melpomene colour pattern phenotypes are essentially identical, this trait is probably not used to discriminate conspecifics26. Instead, their coexistence is likely to be due to strong assortative mating mediated by traits such as male sex pheromones and host plants (Extended Data Fig. 6), as well as female-limited hybrid sterility, which evolves rapidly27,28 and helps to isolate H. melpomene from other sympatric, co-mimetic species29.
Barriers inherited from H. melpomene
As a result of extensive ongoing gene flow, differentiation (FST) between sympatric populations of H. elevatus and H. pardalinus is approximately zero across around 99% of their genomes (Fig. 3). Only around 1% of the genome shows increased differentiation (FST ≥ 0.2) and retrieves both species as reciprocally monophyletic on the basis of topology weighting by iterative sampling of subtrees (TWISST) analysis, comprising 44 genomic islands of divergence. Notably, genealogies within genomic islands resolve all populations of both species, including the peripheral allopatric lineages, as reciprocally monophyletic (Fig. 3). Furthermore, introgression from H. melpomene is especially prevalent in these islands and is found in 32 of the 44 genomic islands of divergence (Fisher’s exact test P < 0.001; Fig. 3). Because these genomic islands resist homogenization despite gene flow in sympatry, we hypothesize that they contain the genetic basis for species differences.

Patterns of genomic divergence between sympatric H. elevatus and H. pardalinus in the Amazon together with locations of mapped traits. The black line and y axis show FST in 25-kb sliding windows across the genome. Coloured bars show significant QTLs for different traits, with the QTL peak indicated by the triangle and the Bayesian credible intervals by the length of the bar. For colour pattern and wing shape, only QTLs with non-overlapping credible intervals are shown. Most of the genome shows very low FST due to gene flow in the Amazon, causing the double paraphyly topology for H. elevatus and H. pardalinus in Fig. 1c. Genomic regions with rare phylogenetic topologies (bottom right) supporting introgression from H. melpomene (white circles, introgression tree) and resolving the pardalinus–elevatus species tree (grey circles, species tree) are shown above the plot. These topologies often coincide with one another and with FST peaks. O, outgroup (Heliconius ethilla). FW, forewing; HW, hindwing.
To understand the genetic architecture of traits that allow the coexistence of H. elevatus and H. pardalinus, we crossed sympatric Amazonian populations of these species. We identified quantitative trait loci (QTLs) for several species-specific traits, including colour pattern, male sex pheromones, male preference for female colour pattern, wing shape, flight and female host plant preference, in F2 and backcross offspring (Fig. 4). These traits contribute to reproductive isolation because they are under divergent selection and/or directly determine mate choice. For example, host preference is likely to be under divergent ecological selection and also confers non-random mating because Heliconius mate in the vicinity of their host plants30,31. In total, we identified 63 QTLs associated with species differences at these traits, which mapped to 14 of the 21 chromosomes (Fig. 3 and Supplementary Table 5).

a, Heliconius elevatus and H. pardalinus differ in host plant preference during egg-laying; female H. elevatus show a stronger preference for Passiflora venusta relative to Passiflora riparia. Heliconius melpomene has a very distinct host plant preference and lays eggs on neither of these plants (Extended Data Fig. 6). Point sizes here and in b,c are scaled by the log of sample size. Error bars are 95% confidence limits. b, The two species differ in flight dynamics; H. elevatus beats its wings significantly faster than H. pardalinus, and converges towards H. melpomene aglaope. c, Given a choice, male H. elevatus individuals preferentially court model female wings with their own colour pattern relative to H. pardalinus, whereas H. pardalinus males exhibit no preference. d, Principal component analysis (PCA) of forewing colour pattern in hybrid crosses with the parental species and H. melpomene aglaope rotated and projected into this space. Wings show the top 10% of pixels contributing to the variance in PC1. e, PCA of male sex pheromones in hybrid crosses, with parental species rotated and projected. Differences between the species are driven mainly by variance in alkanes. Selected loadings: (1) hexahydrofarnesylacetone; (2) (Z)-9-heneicosene; (3) (Z)-11-eicosenylacetate; (4) (Z)-9−tricosene; (5) 11-methylhexacosane; (6) 11-methylpentacosane; (7) heptacosane; (8) tricosane; (9) heneicosane (inset figure); and (10) homovanillyl alcohol. f, PCA of hindwing shape in hybrid crosses, with parental species rotated and projected. Inset wing shows changes in hindwing shape observed along PC2. Only landmarks along the margin of the wing are shown. Individual specimens are depicted as circles: H. elevatus, blue; H. pardalinus, red; F2s and backcrosses, grey; and H. melpomene, yellow.
QTLs for colour pattern mapped to chromosomes 1, 5, 10, 12, 13, 15 and 18, with those on chromosomes 10, 15 and 18 containing the known colour patterning genes WntA, cortex and optix (refs. 16,17,18). We identified a large effect locus on chromosome 20 that determined variation in hindwing shape (H. elevatus ancestry is associated with wider and shorter hindwings). Hybrid flight dynamics were quantified using high-frame-rate video footage. A single locus on chromosome 12 predicted wing beat frequency and explained 43% of the variance. Consistent with species differences (Fig. 4b), individuals with genotype EE (homozygous ancestry for H. elevatus) beat their wings faster (11.2 ± 0.1 Hz) than did PP (homozygous ancestry for H. pardalinus) individuals (10.9 ± 0.2 Hz), in which E is the H. elevatus allele and P is the H. pardalinus allele. In controlled insectary experiments, Heliconius elevatus females exhibited a strong preference for Passiflora venusta relative to Passiflora riparia (Fig. 4a), concordant with wild host plant records17. A single locus on chromosome 2 predicted the preference of female hybrids for different host plants (Fig. 3); the probability of ovipositing on P. venusta increased from 0.3 (s.e. 0.19–0.42) for genotype PP to 0.87 (s.e. 0.81–0.91) for genotype EE.
Mate choice among sympatric populations is further mediated by female preference for male sex pheromones secreted on wing androconia and male preference for colour pattern (attractiveness of females to males)13. We found large effect QTLs for male androconial volatiles on chromosomes 19 and 20. These genomic regions (see Supplementary Table 5) contain many genes encoding enzymes that are involved in fatty acid metabolism, such as reductases and Δ9-desaturases32—strong candidates for controlling differences between the saturated-fatty-acid-derived androconial volatiles of H. elevatus, and the unsaturated-fatty-acid-derived blend of H. pardalinus. For example, genotype EE at the chromosome 19 locus is associated with an approximately 100-fold increase in the concentration of heneicosane, relative to genotype PP, with the QTL explaining about a third of the variance.
The male colour pattern preference QTL with the highest LOD score (3.14) was tightly linked to QTLs for androconial volatiles and wing shape on chromosome 20. However, this QTL was not statistically significant. This might be explained if male preference is highly polygenic. In support of this, we found that the probability of a male courting the H. elevatus colour pattern is positively correlated with the total fraction of the male’s H. elevatus chromosomal ancestry (P < 0.05, generalized linear mixed model with binomial errors and individual-level random effect). For comparison, we applied the same test to host plant choice and found no such association, suggesting that host preference has a simpler genetic basis.
Consistent with hybridization driving speciation, QTLs underpinning species-specific traits are linked to genomic windows introgressed from H. melpomene far more often than when the position of these QTLs is randomized across the genome (mean recombination rate between QTLs and nearest introgression topology, c = 0.26; randomized mean c = 0.39; P < 0.001). QTL peaks that are tightly linked to H. melpomene introgression regions (c < 0.05) include those that determine colour pattern mimicry on chromosomes 10, 15 and 18, wing shape on chromosomes 19 and 20, male sex pheromones on chromosome 19 and 20, host plant preference on chromosome 2 and male preference on chromosome 20. Moreover, for colour pattern, wing shape, male sex pheromones and flight behaviour, H. elevatus exhibits trait values similar to those of H. melpomene (Fig. 4), providing a direct link between introgression, genotype and phenotype. Hence, these loci influencing ecological traits and derived from introgression represent key genomic regions that enabled hybrid speciation (Fig. 3).
Linkage or pleiotropy among traits are often thought to be necessary to circumvent the homogenizing effect of gene flow5,33. After removing overlapping loci in the colour pattern and wing shape phenotypic classes (Fig. 3), 28% of QTLs were tightly linked to at least one other species trait locus (recombination fraction c < 0.05), and only 11% were completely unlinked (c ≈ 0.5). The mean recombination fraction (c) between trait loci and their nearest neighbour was significantly lower than when positions of the loci were randomized across the genome (observed mean c = 0.26; randomized mean c = 0.37; P = 0.001). Thus, although QTLs for traits that underpin reproductive isolation are scattered across the genome, there is nonetheless significant clustering among traits. Inversions can be important for maintaining linkage disequilibria between traits that confer reproductive isolation as they suppress recombination34. However, with the exception of chromosome 15, in which a known inversion is associated with colour pattern differences between H. elevatus and H. pardalinus35, we found no candidate inversions overlapping QTL peaks (Extended Data Fig. 7).
Speciation was driven by introgression
The question of how new species originate and adapt to environments is fundamental to evolutionary biology. Hybridization might have a key role in establishing barriers to gene flow by creating new allelic combinations36,37. Many genomic studies have provided evidence of admixture among species4,38,39,40,41, but convincing cases of homoploid hybrid speciation remain scarce1,3,6. Here we show that H. elevatus is a hybrid species, the origin of which was triggered by introgression of traits from H. melpomene into a H. pardalinus-like ancestor (Fig. 1a). These traits place Heliconius elevatus on a separate adaptive peak and allow it to coexist in sympatry with both parental species, despite occasional but pervasive gene flow with H. pardalinus that distorts the evolutionary history of 99% of the genome away from the species tree. Furthermore, we estimate that H. elevatus has persisted in widespread sympatry as a distinct lineage for over 720,000 generations, suggesting that it is stable and not in the process of fusing with H. pardalinus. To our knowledge, this makes it the oldest reported case of homoploid hybrid speciation, and our study is among the few to fulfil the strict criteria for hybrid speciation that were laid out in a previous study3. Because H. elevatus overlaps broadly across its Amazonian distribution with both progenitors, it also differs from most other previously described putative hybrid species42,43,44, including Heliconius heurippa45, which overlap with only one or neither of the parental lineages. Consistent with models of sympatric speciation46, traits conferring mate choice and divergent selection are clustered within the genome. Nonetheless, there are multiple clusters of these species-specific QTLs across different chromosomes. Adaptive coupling among these unlinked loci therefore spreads the effects of selection across the genome, allowing multiple genomic regions to evolve as a coadapted unit47,48,49,50,51. The capacity of this multilocus genetic architecture to resist gene flow indicates that sympatric speciation can occur more readily than predicted by simple theory based on small numbers of traits or loci.
Methods
Data collection and whole-genome resequencing
Collections and library preparation
Adult butterflies were collected between 2009 and 2018 and stored at −20 °C in either salt-saturated dimethyl sulfoxide or 100% ethanol. RNA-free genomic DNA was extracted from the thorax of butterflies using Qiagen Blood and Tissue and E.Z.N.A Tissue DNA kits (Omega Bio-tek), and used to prepare 350-bp insert Illumina libraries for 33 individuals, which were sequenced using 100–150-bp paired-end sequencing on Illumina instruments. Collecting and export permit numbers are provided in the Acknowledgements. We complemented these samples with previously published sequences (see Supplementary Table 1 for sample details).
Read filtering, mapping and genotype calling
After demultiplexing, reads were filtered for Illumina adapters using cutadapt v.1.8.1 (ref. 52) and then mapped to the H. melpomene assembly v.2.5 (Hmel2.5, ref. 53)(ref. 54) using BWA-MEM v.0.7.15 (ref. 55) with default parameters and marking short split hits as secondary. Mapped reads were sorted and duplicate reads removed using sambamba v.0.6.8 (ref. 56) sort and markdup modules, respectively. Mapped reads were further realigned around indels using the Genome Analysis Toolkit (GATK) v.3.8 RealignerTargetCreator and IndelRealigner modules57,58, to reduce the number of indel miscalls. Read depth and other relevant read alignment quality control metrics were computed using QualiMap v.2.2.1 (ref. 59).
Genotype calling was performed using the bcftools v.1.5 (ref. 60) mpileup and call modules, requiring a minimum MQ (mapping quality) and QUAL (base quality) of 20. Genotyping was performed jointly for individuals belonging to the same population using the multiallelic and rare-variant calling option (-m) in bcftools call. Ploidy aware genotype calling was performed for the Z chromosome. Genotypes were filtered using the bcftools filter module. Both invariant and variant sites were required to have QUAL (quality of the variant call) ≥ 20 and MQ (root mean square mapping quality) ≥ 20, with DP (read depth) ≥ 8 for individual genotypes (DP ≥ 4 for females on the Z chromosome) and GQ (genotype quality) ≥ 20. All genotypes not fulfilling these criteria or within 5 bp of an indel (--SnpGap) were recoded as missing data.
Species relationships and demographic modelling of hybrid speciation
Relationships between H. elevatus, H. pardalinus, H. melpomene and other closely related species were investigated by building a phylogenetic network. The dataset was filtered to include only biallelic sites (excluding singletons) without missing data and at least 1 kb apart, using Plink v.1.9 (ref. 61). Pairwise absolute genetic distances between all pairs of samples were calculated using the disMat.py script (https://github.com/simonhmartin/genomics_general). The distance matrix was then used to construct a phylogenetic network using the NeighbourNet approach62, implemented in SplitsTree v.4.15.1 (ref. 63), with default parameters.
We also investigated species relationships by estimating a concatenated neighbour-joining tree. In this analysis, we included both variable and invariable sites, at least 1 kb apart and without missing data. The neighbour-joining tree was estimated from individuals’ pairwise distances using the R package ape v.5.7 (ref. 64) ‘read.dna’ and ‘nj’ functions. Trees were rooted using the ‘midpoint’ function from the R package phangorn v.2.11.1 (ref. 65). Bootstrap supports were obtained on the basis of 100 bootstrap replicates, using the ‘boot.phylo’ function in the R package ape v.5.7 (ref. 64).
Genealogical relationships along the genome between the three focal species (H. elevatus, H. pardalinus and H. melpomene) were further investigated using TWISST66 (https://github.com/simonhmartin/twisst), and using Heliconius nattereri as an outgroup species. Only SNPs fixed in the outgroup (H. nattereri), variable in the focal species and with a minimum allele frequency (MAF) of 0.05 were considered. Statistical phasing and imputation were performed using Beagle v.5.1 (ref. 67), with default settings. The phased filtered dataset was used to infer neighbour-joining phylogenies for non-overlapping windows of 1,000 SNPs (median size of around 23.6 kb), assuming the GTR substitution model, in PHYML (ref. 68). Exact weightings were computed for all phylogenies. Windows were classified into each of the following categories when weighting support was 0.5 or greater: (i) H. elevatus and H. pardalinus group together but are not reciprocally monophyletic; (ii) H. elevatus and H. pardalinus group together and are reciprocally monophyletic; (iii) H. elevatus and H. melpomene group together but are not reciprocally monophyletic; and (iv) H. elevatus and H. melpomene group together and are reciprocally monophyletic.
To infer the timing of introgression from H. melpomene into H. elevatus and its split from H. pardalinus, we used the multispecies coalescent-with-introgression (MSCi) model implemented in BPP v.4.6.2 (ref. 22) (A00 analysis). For each species of the three species, we selected four individuals to generate sequenced alignments. For H. melpomene, we used H. melpomene aglaope from Peru. Given the population structure between Amazonian and non-Amazonian population of both H. elevatus and H. pardalinus and evidence for gene flow between the two species in the Amazon, we first performed this analysis using the non-Amazonian populations (that is, H. elevatus bari and H. pardalinus sergestus). Loci were selected randomly from autosomes, requiring loci to be 2 kb long, a minimum distance of 20 kb to the next closest locus and 5 kb from the closest exon as annotated in H. melpomene assembly v.2.5. For each locus, individuals with more than 20% missing data and sites containing missing genotype calls were removed. Only loci containing all individuals and 800 bp passing filters were considered. Heterozygous sites were assigned IUPAC ambiguity codes. Demographic parameter estimation was performed using a fixed species tree, with introgression events (see Fig. 1e and Extended Data Fig. 1). An inverse gamma prior (invG) was applied both to the root age (τ0) and to all populations’ effective population sizes (θ) – invG(a = 3, b = 0.06) and invG(a = 3, b = 0.04), respectively. A beta prior was applied to the introgression probability (j) – Beta(a = 1, b = 1). The MCMC was run for 1,000,000 iterations after 50,000 iterations of burn-in, sampling every 10 iterations.
Historic and recent gene flow
Species-diagnostic SNPs
To characterize instances of recent gene flow between Amazonian H. pardalinus and H. elevatus, we relied on ancestry-informative SNPs (allele frequency difference ≥ 0.8) between these two groups. Only ancestry-informative SNPs at least 10 kb apart were considered. For each SNP, an ancestry score of 0 and 1 was assigned for H. elevatus homozygous and H. pardalinus homozygous variants, respectively, and 0.5 for heterozygous. We then calculate each individual’s ancestry (average ancestry across SNPs) and heterozygosity, on the basis of the ancestry-informative SNPs passing the filters. A custom R script was used to visualize genotypes of species-diagnostic SNPs across the genomes of different individuals. The same approach was used to determine species-diagnostic SNPs between Amazonian H. elevatus and H. melpomene.
f 4 statistics
We calculated the f4 statistics in ADMIXTOOLS (ref. 69) to measure shared drift between pairs of populations of different species in the same location versus between pairs of populations of the same species in different locations. Shared drift between populations of different species in the same location is indicative of gene flow between species, and shared drift between populations of the same species in different locations is indicative of grouping by species. Only autosomal biallelic SNPs were considered in this analysis. Standard errors were estimated through a weighted block jackknife approach over 500-kb blocks. We also measured the Euclidean geographic distance between all possible pairs of locations and performed a Mantel test for its correlation with the f4 statistics.
Estimates of gene flow between population pairs
We used G-PhoCS (ref. 70) to estimate divergence times, effective population sizes and migration rates between pairs of populations of H. elevatus and H. pardalinus, both within and between species. In all analyses, we also include one individual from an outgroup species (Heliconius besckei) and estimate model parameters assuming possible bidirectional migration between the two ingroup species. G-PhoCS uses multiple independent neutrally evolving loci to infer demographic parameters. Therefore, we first defined regions of the genome within scaffolds larger than 1 Mb and at least 1 kb away from exons as annotated in H. melpomene assembly v.2.5. Within these regions we then selected 1-kb blocks that were at least 10 kb apart from the nearest block and produced sequence alignments, masking annotated repetitive elements and CpG islands identified with the software gCluster (ref. 71). Because previous studies have reported extensive introgression between both H. elevatus and H. pardalinus with other Heliconius species in large regions of the genome surrounding the three major colour pattern loci, we excluded blocks in chromosomes containing these loci (chromosomes 10, 15 and 18). We also excluded blocks in the Z chromosome owing to its different effective population size. For each alignment, we excluded individuals with more than 60% missing genotype calls, and only alignments with at least three individuals per population (or all individuals in the populations for those with fewer than three individuals) and a minimum of 100 bp for which no more than 25% of individuals had missing genotype calls were considered. We coded heterozygous genotype calls using IUPAC codes. A gamma prior with α = 2 and β = 100 was used for both the mutational-scaled effective population size (θ) and the divergence time (τ) between the two ingroup populations, whereas a gamma prior with α = 2 and β = 50 was used for the divergence time to the outgroup. For the mutation-scaled migration rates, we defined a gamma prior with α = 0.005 and β = 0.00001. The model was run three times, with a burn-in of 50,000 iterations (allowing for automatic fine-tuning of the parameters) followed by 200,000 iterations, sampling every 200 iterations. Convergence of the Markov chain and between the three different replicates was inspected using custom scripts. To convert the θ and τ estimates to absolute effective population size and divergence time, we assumed an average mutation rate (µ) of 2.9 × 10−9 substitutions per site per generation and an average generation time (g) of 0.25 years (ref. 72). We also obtain estimates of the effective migration rate (Nem) using the formula: NemAB = MAB × θB/4.
Simulations to infer robustness of G-PhoCS inferences
Whenever Nm > 1, estimates of Nm for the same population comparisons varied both in value and directionality between different replicate runs of G-PhoCS. To investigate the cause for these differences, we performed coalescent simulations using MSMS (ref. 73). We considered the same demographic scenario as for the G-PhoCS runs; that is, two sister populations (A and B) that diverged at TD1 and split from the outgroup (C) at TD2, and allowing either unidirectional or bidirectional migration between A and B. The split time between the two sister populations (TD1) was set to four million generations, and eight million generations for the split of the outgroup (TD2). An effective population size (Ne) of one million or five million was assumed for the two ingroup populations (400,000 for the outgroup), and varying levels of gene flow (Nm) were considered (0.01, 0.1, 1.0, 2.0 and 10.0). For each scenario, we simulated 100 trees in MSMS (ref. 73), from which we generated sequence alignments using Seq-Gen v.1.3.4 (ref. 74). Custom scripts were used to combine pairs of haploid sequences into diploid sequences, using IUPAC codes for heterozygous sites, and to convert the alignments to the G-PhoCS sequence format. Finally, we ran G-PhoCS for the simulated datasets using the same settings as described above. Whenever Nm > 1 in the simulated datasets, G-PhoCS showed a similar behaviour to what was seen in our analysis of the Heliconius data (Supplementary Table 6). We believe that this effect is due to the difficulty of estimating gene flow when the populations are nearly panmictic. Hence, for each population pairwise comparison, the highest Nm estimate among the three replicate runs is presented in Fig. 2b.
Species-tree inference
Phylogenetic relationships between the H. pardalinus and H. elevatus major groups were inferred using the multispecies coalescent (MSC) approach implemented in BPP v.4.6.2 (ref. 22), while accounting for incomplete lineage sorting. Three H. p. sergestus individuals (with the highest coverage) and three H. elevatus individuals from the Guianas (the individual with the highest coverage per location (French Guiana, Suriname and Venezuela)) were considered. For Amazonian H. pardalinus and H. elevatus, again, only the individual with the highest coverage from each of three locations—Ecuador, Bolivia and Brazil—was included. For this analysis, loci were selected by first defining regions of the genome within scaffolds larger than 1 Mb. To minimize the effect of linked selection, these regions also had to be at least 2 kb from exons as annotated in Heliconius melpomene v.2.5 (Hmel2.5, ref. 54). Because the analysis assumes no intra-locus recombination and independence between loci, we selected loci of 100–250 bp and at least 2 kb from neighbouring loci. Sequence alignments were produced for all loci, masking repetitive elements as annotated in the reference genome and CpG islands identified with the software gCluster (ref. 75). For each locus, individuals with more than 50% missing genotype calls were excluded from the alignment and only loci with at least two individuals per population were considered. Furthermore, sites with more than 20% of individuals with missing genotype calls were removed and loci with less than 50 bp passing filters were excluded. Loci were grouped into blocks of 100 loci, and those overlapping the inversion on chromosome 15 were grouped in a separate block. Species-tree estimation was then performed in BPP v.4.6.2 using the A01 analysis (species-tree inference assuming no gene flow). Inverse gamma priors (invGs) were applied both to the root age (τ0) and to effective population sizes (θ) – invG(3, 0.06) and invG(3, 0.04), respectively. Parameters were scaled assuming a mutation rate of 2.9 × 10−9 substitutions per site per generation and a generation time of 0.25 years (ref. 54). The MCMC was run for 1,000,000 iterations after 50,000 iterations of burn-in, sampling every 10 iterations. Three independent runs were performed for each block, using different starting species trees, and only blocks showing consistency among the three independent runs were considered. The most abundant estimated tree across the genome showed both species to be paraphyletic with respect to each other (Extended Data Fig. 2). We believe that this non-taxonomic arrangement is due to gene flow, which is not accounted for in the model.
Demographic modelling by analysis of site-frequency spectra
To understand the prevalence of gene flow at different stages of the speciation history of H. elevatus and H. pardalinus, we performed demographic modelling based on analysis of the site-frequency spectrum (SFS) using fastsimcoal2 v.2.7.0.2 (ref. 76). For this analysis, we considered all Amazonian and non-Amazonian populations of H. elevatus and H. pardalinus. Individuals with more than 50% missing data were excluded from the analysis and only sites genotyped in at least 80% of the individuals (including all four H. p. sergestus) were considered. Furthermore, only sites at least 2 kb apart and at least 10 kb from exons were considered, to mitigate the effects of linkage disequilibrium and linked selection, respectively. We further excluded sites within repetitive regions as annotated in the H. melpomene assembly Hmel2.5. The 209,115 sites that were retained after filtering were polarized by assessing the allele present in three outgroup species—H. besckei, Heliconius ismenius telchinia and Heliconius numata robigus. From each of the outgroup species, we chose one individual with the highest coverage and assigned the ancestral allele to each site if it was genotyped and monomorphic in the outgroup species. The unfolded multidimensional site-frequency spectrum (multiSFS) was generated using easySFS (https://github.com/isaacovercast/easySFS), using the recommended down projection approach (four individuals of H. p. sergestus; 10 northeastern group H. elevatus; and 20 H. pardalinus and 20 H. elevatus individuals from the Amazon) to maximize the number of segregating sites while accounting for missing data. For each demographic model, fitting of the simulated multidimensional site-frequency spectra to the empirical data was maximized using the composite-likelihood method implemented in fastsimcoal v.2.7 (ref. 77). For all model parameters, we used wide search ranges from which initial starting parameter values were randomly sampled. For each model, we performed 100 independent fastsimcoal2 runs. Parameter estimates optimization was performed for 40 expectation-maximization cycles and the expected SFS was estimated using 100,000 coalescent simulations. The best fitting model was identified by means of the Akaike information criterion, considering for each model the optimization run with the highest likelihood (using the script https://github.com/speciationgenomics/scripts/blob/master/calculateAIC.sh). To account for stochasticity in the likelihood approximation, we further compared likelihood distributions of the different models by performing 100 independent runs from parameter values estimated under the most likely replicate run for each model. Finally, for the best fit model, confidence intervals around the maximum likelihood parameter estimates were obtained by nonparametric block-bootstrapping. For this, the 209,115 sites were divided into 100 blocks and sampled with replacement.
Genomic islands of divergence and introgression
Summary statistics
We calculated between-population differentiation (FST) for Amazonian and non-Amazonian populations of both H. elevatus and H. pardalinus groups, in sliding windows of 25 kb (5 kb step size) along the genome using the script popgenWindows.py (https://github.com/simonhmartin/genomics_general). The script implements a version of Hudson’s KST (ref. 78), modified to avoid weighting nucleotide diversity in each population by sample size. Individuals with more than 50% missing data were removed. Only sites with a maximum of two alleles, and with at least three individuals with genotype calls per population (or the total number of individuals in populations with fewer than three individuals) were considered. Only windows with at least 10% of sites passing filters were considered in the analysis.
Topology weighting
To determine genomic regions in which H. elevatus and H. pardalinus are reciprocally monophyletic (that is, genomic regions that are potentially involved in species barriers), genealogical relationships between Amazonian and non-Amazonian populations along the genome were quantified using TWISST66 (https://github.com/simonhmartin/twisst). The same dataset as for FST was used, but also adding five individuals of representative outgroup species (H. besckei, H. ismenius, H. numata, H. nattereri and H. ethilla). Statistical phasing and imputation were performed using Beagle 5.1 (ref. 67), with default settings. Only SNPs fixed in all outgroup individuals and variable in the ingroup population with an MAF of 0.05 were considered. The phased filtered dataset was used to infer neighbour-joining phylogenies for windows of 100 SNPs (slide every 25 SNPs), assuming the GTR substitution model, in PHYML (ref. 68). Exact weightings were computed for all phylogenies. To estimate the proportion of trees supporting a grouping of individuals by species versus grouping by geography, we considered five groups: (i) H. elevatus from the Guianas (Venezuela, Suriname and French Guiana); (ii) H. elevatus from the Amazon; (iii) H. pardalinus from the Amazon; (iv) H. p. sergestus (Andes); and (v) an outgroup, H. nattereri. Because we hypothesize that introgression from H. melpomene into H. elevatus could be involved in speciation of the latter and H. pardalinus, the same analysis was performed including only Amazonian H. elevatus, Amazonian H. pardalinus and two H. melpomene populations (H. m. amaryllis and H. m. aglaope). By including H. ethilla (a sister species to H. elevatus and H. pardalinus) as a fifth population, we were able to polarize the genealogies, allowing determination of the direction of introgression.
Association between H. melpomene introgression and genomic islands of divergence
To test whether H. melpomene introgression in the genome of H. elevatus is associated with genomic islands of divergence between sympatric H. elevatus and H. pardalinus, we performed a Fisher’s exact test. First, we defined genomic islands of divergence as regions with FST ≥ 0.2 and in which TWISST recovered both H. pardalinus and H. elevatus as reciprocally monophyletic (with weight ≥ 0.8). Second, we defined as introgressed, genomic regions in which TWISST grouped H. elevatus with H. melpomene with a weight ≥ 0.8. We then performed a Fisher’s exact test, as implemented in bedtools v.2.30.0 (ref. 79), to test whether the two sets of genomic intervals overlap more than expected given the size of the reference genome.
Genetic mapping of traits involved in reproductive isolation
Captive populations of Amazonian H. elevatus pseudocupidineus, H. pardalinus butleri and H. m. agalope were established in outdoor insectaries in Tarapoto, Peru and in heated indoor insectaries in York, UK, as previously described17. Crosses for QTL mapping were generated by mating H. elevatus with H. pardalinus to produce F1 broods, and then by either crossing these amongst themselves to generate F2 broods or backcrossing to parental taxa.
Colour pattern phenotyping
Dorsal surfaces of wings from 12 H. elevatus, 19 H. pardalinus, 14 H. m. aglaope, 348 F2 and 50 backcross hybrids were photographed in a light box against a white background using a Canon EOS D1000 together with an X-rite ColorChecker Mini (Supplementary Table 7). From each image, we selected a single forewing and hindwing for analysis, clipped the image to the wing outlines and flipped wings when necessary to ensure that all were similarly orientated (resulting in two files; one forewing and one hindwing). To align the wings so that pixels represent homologous units among individuals, we used image registration80, a regression-based method that aligns two sets of wings (a source and a reference) according to intensity-based similarity. We chose the reference set of wings using the PCA of wing shape (see below). For forewing (36 PCs) and hindwing (26 PCs) we found the mean value for each PC across all F2 and backcross individuals. We assigned the reference individual as the individual that had the minimum deviation from these mean values (summed across all PCs). We then checked all alignments by eye. To allow for minor misalignment or damage to wings, we included pixels in which up to 5% of individuals had missing RGB values.
Wing shape
Wing shape was quantified in 31 H. elevatus, 26 H. pardalinus, 10 H. m. aglaope and 308 F2 and 36 backcross hybrids using landmark-based geometric morphometrics analyses (Supplementary Table 7). The ventral side of the butterfly wings was scanned using a flatbed scanner at 300 dpi and landmarks were placed at specific vein intersections81 on the forewing (20 landmarks) and hindwing (15 landmarks) using tpsDig282. Landmark coordinates were adjusted for size and orientation using a Procrustes analysis from the package geomorph83. Forewings and hindwings were analysed separately.
Flight
H. elevatus (n = 12), H. pardalinus (n = 13), H. m. aglaope (n = 5) and F2s (n = 40) were filmed flying freely in a large flight cage (5 × 2.5 × 2 m) using a GoPro HERO 4 Black camera at 239.7 frames per second at a resolution of 720p (Supplementary Table 7). Videos were studied in slow motion using GoPro Studio v.2.5.9.2658. Flight sequences in which an individual was flying straight and level for at least five wing beats were selected to measure wing beat frequency (WBF). WBF was measured by counting the number of complete wing beats and the number of video frames. Five WBF measurements were taken per individual from separate flight sequences and used to calculate the individuals’ mean WBF by dividing the total number of wing beats across all flight sequences by the total flight time estimated from the number of video frames.
Female host plant preference
Host plant preference assays for QTL mapping were performed by introducing single H. elevatus, H. pardalinus and F2 females (n = 24, 32 and 31, respectively) into cages measuring 1 m (W) × 2 m (L) × 1.7 m (H), with two approximately equally sized shoots of the host plants (P. riparia and P. venusta) placed in the back corners. At the end of each day, the number of eggs laid on each plant species was recorded and the eggs were removed (Supplementary Table 7). To compare the oviposition preference of Peruvian H. elevatus, H. pardalinus and H. melpomene, groups of females (wild-caught and/or reared) of a given taxon were released into a large cage (2.5 m (W) × 5 m (L) × 2 m (H)) containing single representatives of 21 species of Passiflora that are commonly found near Tarapoto, Peru and which represent potential host plants17. The number of eggs laid on each host plant was recorded at the end of each day. A total of 126 females were tested, resulting in a total of 889 eggs (176 from 35 H. elevatus females, 288 from 24 H. melpomene and 425 from 51 H. pardalinus).
Male sexual preference
To assay male preference for female colour pattern, we presented H. elevatus, H. pardalinus and F2 males (n = 46, 66 and 106, respectively) with a pair of model female wings (one H. elevatus and one H. pardalinus), and recorded courtship events (full details of the experimental set-up are provided in ref. 17). Males were tested individually and placed in the experimental cage one day earlier to allow acclimatization. Trials lasted 15–30 min. The number of courtships (defined as sustained flight 5–15 cm over a model) by the males directed towards each of the model wings was recorded (Supplementary Table 7).
Phenotyping of androconial volatiles
Male Heliconius produce complex chemical blends of volatile compounds from their hindwing androconia. These blends have been shown to function as sex pheromones in several other Heliconius species and in butterflies in general84,85. Androconial regions were excised from 13 H. elevatus, 10 H. pardalinus, 7 H. melpomene malleti individuals and 122 F2 and 17 backcross hybrids 21 days after eclosion, and suspended in dichloromethane. The extracts were analysed by gas chromatography–mass spectrometry (GC–MS), as reported previously16,86 (Supplementary Table 7) on a 7890A GC-System coupled with an MSD 5975C mass analyser (Agilent Technologies) instrument fitted with an HP-5MS column (50 m, 0.25 mm internal diameter, 0.25 µm film thickness). The ionization method was electron impact with a collision energy of 70 eV. Conditions were as follows: inlet pressure 9.79 psi, He 20 ml min−1, injection volume 1 µl. The GC was programmed as follows: start at 50 °C, increase at 5 °C min−1 to 320 °C and hold that temperature for 5 min. The carrier gas was He at 1.2 ml min−1. For all identified compounds, the concentration was calculated from the peak’s area, as reported by AMDIS software87. Each compound’s chromatogram was interpreted by AMDIS through the NIST databases and the additional databases compiled at the Institute of Organic Chemistry of Technische Universität Braunschweig. All identifiable compounds running between undecane and nonacosanal were scored. Potential contaminants or extraneous compounds were excluded, together with compounds that appeared fewer than 10 times across the entire dataset.
DNA extraction and RAD library preparation for QTL analysis
RNA-free genomic DNA was extracted from thoracic tissue using a Qiagen DNeasy Blood and Tissue Kit following manufacturer’s standard protocol. Restriction-site-associated DNA (RAD) libraries were prepared using a protocol modified from (ref. 88, using a PstI restriction enzyme, sixteen 6-bp P1 barcodes and eight indexes. DNA was Covaris sheared to 300–700 bp and gel size selected. A total of 128 individuals were sequenced per lane, with 125-bp paired-end reads, on an Illumina HiSeq 2500 (Supplementary Table 8).
SNP calling
Fastq files from each RAD library were demultiplexed using process_radtags from Stacks89, and BWA-MEM90 was used with default parameters to map the reads to the H. melpomene assembly v.2.5 (ref. 91). BAM files were then sorted and indexed with Samtools (ref. 90), and Picard v.1.119 (https://github.com/broadinstitute/picard) was used to add read group data and mark PCR duplicates. To check for errors, confirm pedigrees and assign samples with unrecorded pedigree to families, we used Plink v.1.9 (ref. 61) to estimate the fraction of the genome that is identical by descent (IBD; \(\widehat{{\boldsymbol{\pi }}}\)) between all pairwise combinations of samples (siblings and parent-offspring comparisons should yield \(\widehat{{\boldsymbol{\pi }}}\) values close to 0.5). In addition, for specimens that were sequenced multiple times, we checked that samples derived from the same individual \((\widehat{{\boldsymbol{\pi }}}\approx 1)\). We then merged these samples, using the MergeSamFiles command from Picard Tools, and used Samtools mpileup command to call SNPs.
Linkage map construction
Linkage maps were built for hybrid and within-species crosses using Lep-MAP3 (ref. 92). Pedigrees are provided in Supplementary Table 8. SNPs were first converted to posterior genotype likelihoods for each of ten possible SNP genotypes. We used the ParentCall2 module to correct erroneous or missing parental genotypes and call sex-linked markers using a log-odds difference of >2 (ZLimit) and halfSibs = 1. We used Filtering2 to remove SNPs showing segregation distortion, specifying a P value limit of 0.01; that is, there is a 1:100 chance that a randomly segregating marker is discarded. We then separated markers into chromosomes using their Hmel2.5 scaffold. To obtain genetic distances between markers, we fixed the order of the markers to their order in Hmel2.5, and then evaluated this order, using all markers and specifying no recombination in females. We then used map2gentypes.awk to convert the Lep-MAP3 output to four-way fully informative genotypes with no missing data. To assign ancestry to phased haplotype blocks in the hybrid linkage map, we used biallelic sites with significantly different allele frequencies in the parental species (χ2 test applied to sequences for 26 H. elevatus and 47 H. pardalinus individuals from Peru and Ecuador).
QTL mapping
The colour pattern, androconial volatiles and wing shape datasets are multivariate and highly collinear. We therefore used PCA to reduce the phenotypic values for the hybrids to orthogonal vectors (PCs), which we then used as phenotypes in QTL mapping. For wing shape, we applied PCA to the Procrustes coordinates. For the androconial volatiles, we applied the PCA to the set of compounds that were significantly different between the two parental species (one-tailed paired t-test). For colour pattern, we performed a PCA on the concatenated RGB values from the aligned images and retained PCs that explained more than 1% of the variance.
For colour pattern, androconial volatiles, wing shape and WBF, we tested for associations between phenotype and genotype using linear models with normal errors. For wing shape, we included centroid size as a covariate to control for allometry. For female host plant choice and male preference for female colour pattern, we (i) logistically transformed the proportions and used linear models with normal errors; and (ii) used generalized linear mixed models with an individual-level random effect to account for overdispersion and binomial errors. The significance of QTL scans was assessed by permuting the phenotypes relative to the genotypes (1,000 permutations). For traits phenotyped in both males and females, a sex-specific significance threshold was used to avoid spurious sex linkage (see Supplementary Table 5).
We first analysed all data using F2s only, using R/qtl (ref. 93) to estimate genotype probabilities at 1-cM intervals, using the Haldane mapping function and an assumed genotyping error rate of 0.001. These genotype probabilities were then used as the dependent variable in models, and for traits phenotyped in both males and females we included sex and cross direction as covariates for markers on the sex chromosome. For traits for which backcrosses had been scored in addition to F2s, we performed an additional round of analyses combining F2s with backcrosses. In this case, we used the categorical genotypes (EE, EP and PP) inferred from linkage mapping as the dependent variables, and added random effects for cross type (three levels: F2, backcross to H. elevatus, backcross to H. pardalinus), sex or individual. Model structures and estimated coefficients are provided in Supplementary Table 5.
To test whether QTLs are significantly clustered (that is, genetically linked), for each QTL we estimated the recombination probability with its nearest neighbouring QTLs (using the position of the maximum LOD score), and took the mean of the resulting vector (low values indicate that most QTLs are linked to at least one other QTL; high values indicate that most QTLs are unlinked). We then randomized the position of the QTLs 10,000 times and compared the observed data to the randomized dataset using a two-tailed test (P = the proportion of randomized datasets that give a result more extreme than the observed data × 2). When multiple QTLs overlapped within the phenotypic classes forewing colour pattern, hindwing colour pattern, forewing shape and hindwing shape, we included only the best supported QTL (highest LOD score). To test whether species and introgression topologies are associated with QTLs, we applied the same test.
To identify putative structural rearrangements between H. elevatus and H. pardalinus, we compared recombination rates between F2s and within-species crosses (F2s, 441 individuals across 26 families; H. elevatus, 179 individuals across 9 families; H. pardalinus, 296 individuals across 15 families). Regions that are freely recombining within species but not in F2s represent candidate rearrangements that might facilitate divergence and speciation. The probability of the within-species recombination events observed within an F2 breakpoint can be given as pn, where p is the fraction of parental individuals in the mapping crosses and n is the observed number of recombination events. We estimated pn within each F2 breakpoint and considered breakpoints in which p < 0.01 to be candidate rearrangements.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Newly generated whole-genome sequencing data used in the population genomic analyses and RAD-sequencing data used in the cross analyses have been uploaded to the NCBI Sequence Read Archive (SRA) (PRJNA1074694). NCBI SRA accessions for individual samples are listed in Supplementary Tables 1 and 8. Phenotypic data are available in Supplementary Table 7 and at https://doi.org/10.5281/zenodo.10685466 (ref. 94) and https://doi.org/10.5281/zenodo.10689714 (ref. 95).
Code availability
Custom code used for the genomic analyses (https://github.com/FernandoSeixas/HeliconiusHybridSpeciation) and the QTL mapping (https://github.com/heliconius-maps/HeliconiusHybridSpeciation) is available from GitHub.
Change history
13 May 2024
In the version of the article initially published, an earlier version of the Reporting Summary was included (that listed the sample size as 83 rather than 92). This has now been updated in the HTML and PDF versions of the article.
References
Lamichhaney, S. et al. Rapid hybrid speciation in Darwin’s finches. Science 359, 224–228 (2018).
Abbott, R. et al. Hybridization and speciation. J. Evol. Biol. 26, 229–246 (2013).
Schumer, M., Rosenthal, G. G. & Andolfatto, P. How common is homoploid hybrid speciation? Evolution 68, 1553–1560 (2014).
Lamichhaney, S. et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518, 371–375 (2015).
Coyne, J. A. & Orr, H. A. Speciation (Sinauer Associates, 2004).
Olave, M., Nater, A., Kautt, A. F. & Meyer, A. Early stages of sympatric homoploid hybrid speciation in crater lake cichlid fishes. Nat. Commun. 13, 5893 (2022).
Barker, M. S., Arrigo, N., Baniaga, A. E., Li, Z. & Levin, D. A. On the relative abundance of autopolyploids and allopolyploids. New Phytol. 210, 391–398 (2016).
Mallet, J. Hybrid speciation. Nature 446, 279–283 (2007).
Engler-Chaouat, H. S. & Gilbert, L. E. De novo synthesis vs. sequestration: negatively correlated metabolic traits and the evolution of host plant specialization in cyanogenic butterflies. J. Chem. Ecol. 33, 25–42 (2007).
Engler, H. S., Spencer, K. C. & Gilbert, L. E. Preventing cyanide release from leaves. Nature 406, 144–145 (2000).
Joron, M. & Mallet, J. Diversity in mimicry: paradox or paradigm? Trends Ecol. Evol. 13, 461–466 (1998).
Page, E., Queste, L., Rosser, N., Mallet, J. & Dasmahapatra, K. K. Pervasive mimicry in flight behavior among aposematic butterflies. Proc. Natl Acad. Sci. USA 121, e2300886121 (2024).
Jones, R. T. et al. Wing shape variation associated with mimicry in butterflies. Evolution 67, 2323–2334 (2013).
Merrill, R. M. et al. Disruptive ecological selection on a mating cue. Proc. R. Soc. B 279, 4907–4913 (2012).
Arias, M. et al. Crossing fitness valleys: empirical estimation of a fitness landscape associated with polymorphic mimicry. Proc. R. Soc. B 283, 20160391 (2016).
Cama, B. et al. Exploitation of an ancestral pheromone biosynthetic pathway contributes to diversification in Heliconius butterflies. Proc. R. Soc. B 289, 20220474 (2022).
Rosser, N. et al. Geographic contrasts between pre- and postzygotic barriers are consistent with reinforcement in Heliconius butterflies. Evolution 73, 1821–1838 (2019).
Benson, W. W., Brown, K. S. & Gilbert, L. E. Coevolution of plants and herbivores: passion flower butterflies. Evolution 29, 659–680 (1975).
Kozak, K. M. et al. Multilocus species trees show the recent adaptive radiation of the mimetic Heliconius butterflies. Syst. Biol. 64, 505–524 (2015).
Heliconius Genome Consortium. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487, 94–98 (2012).
Thawornwattana, Y., Seixas, F. A., Yang, Z. & Mallet, J. Major patterns in the introgression history of Heliconius butterflies. eLife 12, RP90656 (2023).
Flouri, T., Jiao, X., Rannala, B. & Yang, Z. A Bayesian implementation of the multispecies coalescent model with introgression for phylogenomic analysis. Mol. Biol. Evol. 37, 1211–1223 (2020).
Brower, A. V. Z. Alternative facts: a reconsideration of putatively natural interspecific hybrid specimens in the genus Heliconius (Lepidoptera: Nymphalidae). Zootaxa 4499, 1–87 (2018).
Dasmahapatra, K. K., Silva-Vásquez, A., Chung, J.-W. & Mallet, J. Genetic analysis of a wild-caught hybrid between non-sister Heliconius butterfly species. Biol. Lett. 3, 660–663 (2007).
Mallet, J., Beltrán, M., Neukirchen, W. & Linares, M. Natural hybridization in heliconiine butterflies: the species boundary as a continuum. BMC Evol. Biol. 7, 28 (2007).
González-Rojas, M. F. et al. Chemical signals act as the main reproductive barrier between sister and mimetic Heliconius butterflies. Proc. R. Soc. B 287, 20200587 (2020).
Rosser, N. et al. Complex basis of hybrid female sterility and Haldane’s rule in Heliconius butterflies: Z-linkage and epistasis. Mol. Ecol. 31, 959–977 (2022).
Jiggins, C. D. et al. Sex-linked hybrid sterility in a butterfly. Evolution 55, 1631–1638 (2001).
Sánchez, A. P. et al. An introgressed wing pattern acts as a mating cue. Evolution 69, 1619–1629 (2015).
Merrill, R. M., Naisbit, R. E., Mallet, J. & Jiggins, C. D. Ecological and genetic factors influencing the transition between host-use strategies in sympatric Heliconius butterflies. J. Evol. Biol. 26, 1959–1967 (2013).
Estrada, C. & Gilbert, L. E. Host plants and immatures as mate-searching cues in Heliconius butterflies. Anim. Behav. 80, 231–239 (2010).
Byers, K. J. R. P. et al. Clustering of loci controlling species differences in male chemical bouquets of sympatric Heliconius butterflies. Ecology and Evolution 11, 89–107 (2021).
Felsenstein, J. Skepticism towards Santa Rosalia, or why are there so few kinds of animals? Evolution 35, 124–138 (1981).
Rieseberg, L. H. Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16, 351–358 (2001).
Jay, P. et al. Supergene evolution triggered by the introgression of a chromosomal inversion. Curr. Biol. 28, 1839–1845 (2018).
Marques, D. A., Meier, J. I. & Seehausen, O. A Combinatorial view on speciation and adaptive radiation. Trends Ecol. Evol. 34, 531–544 (2019).
Hench, K., Helmkampf, M., McMillan, W. O. & Puebla, O. Rapid radiation in a highly diverse marine environment. Proc. Natl Acad. Sci. USA 119, e2020457119 (2022).
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010).
Palkopoulou, E. et al. A comprehensive genomic history of extinct and living elephants. Proc. Natl Acad. Sci. USA 115, E2566–E2574 (2018).
Li, G., Figueiró, H. V., Eizirik, E. & Murphy, W. J. Recombination-aware phylogenomics reveals the structured genomic landscape of hybridizing cat species. Mol. Biol. Evol. 36, 2111–2126 (2019).
Suvorov, A. et al. Widespread introgression across a phylogeny of 155 Drosophila genomes. Curr. Biol. 32, 111–123.e5 (2022).
Barrera-Guzmán, A. O., Aleixo, A., Shawkey, M. D. & Weir, J. T. Hybrid speciation leads to novel male secondary sexual ornamentation of an Amazonian bird. Proc. Natl Acad. Sci. USA 115, E218–E225 (2018).
Hermansen, J. S. et al. Hybrid speciation in sparrows I: phenotypic intermediacy, genetic admixture and barriers to gene flow. Mol. Ecol. 20, 3812–3822 (2011).
Nieto Feliner, G. et al. Is homoploid hybrid speciation that rare? An empiricist’s view. Heredity 118, 513–516 (2017).
Mavárez, J. et al. Speciation by hybridization in Heliconius butterflies. Nature 441, 868–871 (2006).
Gavrilets, S. Fitness Landscapes and the Origin of Species (Princeton Univ. Press, 2004).
Butlin, R. K. & Smadja, C. M. Coupling, reinforcement, and speciation. Am. Nat. 191, 155–172 (2018).
Barton, N. H. Multilocus clines. Evolution 37, 454–471 (1983).
Flaxman, S. M., Wacholder, A. C., Feder, J. L. & Nosil, P. Theoretical models of the influence of genomic architecture on the dynamics of speciation. Mol. Ecol. 23, 4074–4088 (2014).
Kautt, A. F. et al. Contrasting signatures of genomic divergence during sympatric speciation. Nature 588, 106–111 (2020).
Wessinger, C. A. et al. A few essential genetic loci distinguish Penstemon species with flowers adapted to pollination by bees or hummingbirds. PLoS Biol. 21, e3002294 (2023).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10 (2011).
Davey, J. W. et al. No evidence for maintenance of a sympatric Heliconius species barrier by chromosomal inversions. Evolution Letters 1, 138–154 (2017).
Edelman, N. B. et al. Genomic architecture and introgression shape a butterfly radiation. Science 366, 594–599 (2019).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at 10.48550/arXiv.1303.3997 (2013).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Bryant, D. Neighbor-Net: an agglomerative method for the construction of phylogenetic networks. Mol. Biol. Evol. 21, 255–265 (2003).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Schliep, K., Potts, A. J., Morrison, D. A. & Grimm, G. W. Intertwining phylogenetic trees and networks. Methods Ecol. Evol. 8, 1212–1220 (2017).
Martin, S. H. & Van Belleghem, S. M. Exploring evolutionary relationships across the genome using topology weighting. Genetics 206, 429–438 (2017).
Browning, S. R. & Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Gronau, I., Hubisz, M. J., Gulko, B., Danko, C. G. & Siepel, A. Bayesian inference of ancient human demography from individual genome sequences. Nat. Genet. 43, 1031–1034 (2011).
Gómez-Martín, C., Lebrón, R., Oliver, J. L. & Hackenberg, M. Prediction of CpG islands as an intrinsic clustering property found in many eukaryotic DNA sequences and its relation to DNA methylation. Methods Mol. Biol. 1766, 31–47 (2018).
Keightley, P. D. et al. Estimation of the spontaneous mutation rate in Heliconius melpomene. Mol. Biol. Evol. 32, 239–243 (2015).
Ewing, G. & Hermisson, J. MSMS: a coalescent simulation program including recombination, demographic structure and selection at a single locus. Bioinformatics 26, 2064–2065 (2010).
Rambaut, A. & Grass, N. C. Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Bioinformatics 13, 235–238 (1997).
Li, X., Chen, F. & Chen, Y. Gcluster: a simple-to-use tool for visualizing and comparing genome contexts for numerous genomes. Bioinformatics 36, 3871–3873 (2020).
Excoffier, L. et al. fastsimcoal2: demographic inference under complex evolutionary scenarios. Bioinformatics 37, 4882–4885 (2021).
Excoffier, L., Dupanloup, I., Huerta-Sánchez, E., Sousa, V. C. & Foll, M. Robust demographic inference from genomic and SNP data. PLoS Genet. 9, e1003905 (2013).
Hudson, R. R., Boos, D. D. & Kaplan, N. L. A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 9, 138–151 (1992).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Clayden, J., Modat, M., Presles, B., Anthopoulos, T. & Daga, P. RNiftyReg: Image registration using the ‘NiftyReg’ library. R version 2.8.1 https://cran.r-project.org/web/packages/RNiftyReg (2023).
Queste, L. M. The Evolution of Flight and Wing Shape in Heliconius Butterflies PhD thesis, Univ. York (2020).
Rohlf, F. J. tpsDig v.2.05 (State University of New York at Stony Brook, 2006). https://www.sbmorphometrics.org/soft-dataacq.html.
Adams, D. C., Collyer, M. & Kaliontzopoulou, A. Geomorph: Geometric morphometric analyses of 2D and 3D landmark data. R version 3.1.0 https://cran.r-project.org/web/packages/geomorph (2019).
Mérot, C., Frérot, B., Leppik, E. & Joron, M. Beyond magic traits: multimodal mating cues in Heliconius butterflies. Evolution 69, 2891–2904 (2015).
Darragh, K. et al. Male sex pheromone components in Heliconius butterflies released by the androconia affect female choice. PeerJ 5, e3953 (2017).
Ehlers, S., Blow, R., Szczerbowski, D., Jiggins, C. & Schulz, S. Variation of clasper scent gland composition of Heliconius butterflies from a biodiversity hotspot. ChemBioChem 24, e202300537 (2023).
Stein, S. E. An integrated method for spectrum extraction and compound identification from gas chromatography/mass spectrometry data. J. Am. Soc. Mass. Spectrom. 10, 770–781 (1999).
Etter, P. D., Preston, J. L., Bassham, S., Cresko, W. A., Johnson, E.A. Local de novo assembly of RAD paired-end contigs using short sequencing reads. PLoS ONE 6, e18561 (2011).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Davey, J. W. et al. Major improvements to the Heliconius melpomene genome assembly used to confirm 10 chromosome fusion events in 6 million years of butterfly evolution. G3 6, 695–708 (2016).
Rastas, P. Lep-MAP3: robust linkage mapping even for low-coverage whole genome sequencing data. Bioinformatics 33, 3726–3732 (2017).
Broman, K. W., Wu, H., Sen, Ś. & Churchill, G. A. R/qtl: QTL mapping in experimental crosses. Bioinformatics 19, 889–890 (2003).
Rosser, N. Image data for Rosser et al. 2024 Hybrid speciation driven by multilocus introgression of ecological traits. Zenodo 10.5281/zenodo.10685466 (2024).
Rosser, N. GCMS data for Rosser et al. 2024 Hybrid speciation driven by multilocus introgression of ecological traits. Zenodo 10.5281/zenodo.10689714 (2024).
Acknowledgements
This work was funded by the NERC grant NE/K012886/1 to K.K.D.; a National Geographic Waitt grant (W400-15) to N.R.; grant Schu 984/12-1 from the DFG to S.S.; and Harvard University. K.K. was supported by a fellowship from the Smithsonian Institution. A.V.L.F. acknowledges support from FAPESP (2021/03868-8), from the Brazilian Research Council–CNPq (304291/2020-0) and from the USAID–US National Academy of Sciences (NAS) (AID-OAA-A-11-00012). The University of York Viking cluster high-performance computing facility was used for some of the analyses. We thank SERFOR, the Peruvian Ministry of Agriculture and the Área de Conservación Regional Cordillera Escalera (0289-2014-MINAGRI-DGFFS/DGEFFS, 020-014/GRSM/PEHCBM/DMA/ACR-CE and 040–2015/GRSM/PEHCBM/DMA/ACR-CE) for collecting permits; the Ministerio del Ambiente and Museo Ecuatoriano de Ciencias Naturales in Ecuador (005-IC-FAU-DNBAPVS/MA) for collecting permits; the ICMBio for permits (52562-3 and 10438-1); and the Conselho Nacional de Desenvolvimento Científico e Tecnológico–CNPq for approving our scientific expedition (Expediente PR no. 01300.000477/2016-49, portaria no. 4.628). This study is registered at the Brazilian SISGEN (A752FC2). Field collections in Colombia were conducted under permit no. 530 issued by the Autoridad Nacional de Licencias Ambientales of Colombia (ANLA). We thank J. Caldwell, M. Chouteau, C. Córdova, N. Edelman, S. Galluser, C. López, M. McClure, C. Pérez, C. Segami and M. Tuanama in Peru, and R. Aldaz, A. Toporov and K. Willmott in Ecuador, for help and support with fieldwork; C. Thomas, W. Valencia-Montoya, A. Kautt and J. Coughlan for comments; and M. Cast for providing some of the butterfly images used in figures.
Author information
Authors and Affiliations
Contributions
N.R., F.S., J.M. and K.K.D designed the study. N.R. was responsible for fieldwork, including designing and conducting crosses, phenotyping of traits and performing QTL, linkage and recombination-rate analyses. F.S. performed the population genomic analyses (with D.K., N.R., J.M. and K.K.D.) and the demographic and coalescent analyses. L.M.Q. contributed to phenotyping host plant preference, colour pattern preference, wing shape, colour pattern (with B.C. and R.W.-H.) and flight (with M.G.). B.C. and S.S. led the analysis of androconial volatiles. R.M.-P. provided fieldwork assistance in Peru. M.N. constructed the RAD libraries. All other authors contributed to sample collection. N.R., F.S., J.M. and K.K.D wrote and finalized the paper with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Evaluating the hybrid speciation hypothesis under the MSCi model.
For each model, a schematic representation is depicted on top and the estimated values under the MSCi are presented in the table below. In the schematics, open circles denote internal nodes and arrows between internal nodes represent single migration pulses. Effective population sizes (Ne) are scaled to thousand individuals. The age (τ) of splits and nodes involved in hybridization events are given in kya. Introgression probabilities (φ) are depicted in blue and are given as a percentage. a, Model in which the two parental species, S (ancestral of H. melpomene) and T (ancestral of H. pardalinus), hybridize to originate the hybrid lineage H (ancestral of H. elevatus), with φ contribution from parent S and 1-φ from parent T. The nodes S and T may have distinct ages and are older than node H. b, Same as model a, but constraining introgression from S into H to occur after an initial split between H. elevatus and H. pardalinus (τH < τT). Introgression is instantaneous and occurs at time τH (which is the same as τS). c, Model allowing bidirectional migration between an H. melpomene ancestor (X) into the ancestral population of H. elevatus and H. pardalinus (Y), between an H. melpomene ancestor (S) and the lineage leading to H. elevatus (H), and between the lineage leading to H. elevatus (E) and the lineage leading to H. pardalinus (P). Note that in all models, the 95% HPD intervals of the age of gene flow from H. melpomene into H. elevatus and the split between H. elevatus with H. pardalinus overlap, in line with H. elevatus being a hybrid lineage. These are highlighted in red.
Extended Data Fig. 2 MSC analysis of H. elevatus and H. pardalinus.
Species-tree phylogeny along chromosomes calculated in blocks of 100 loci using BPP (ref. 22). Only the five major topologies are depicted (minor topologies are coloured in grey).
Extended Data Fig. 3 Species-diagnostic SNPs.
a, Number of species-diagnostic SNPs per chromosome. Species-diagnostic SNPs were defined as SNPs with an allelic difference of at least 0.8 between all Amazonian populations of H. elevatus and H. pardalinus. Chromosomes with at least 20 diagnostic SNPs are denoted with an asterisk (*) and shown in more detail in c. b, Triangular plot of hybrid index and observed heterozygosity, based on the 1,156 species-diagnostic SNPs, shows no evidence of early generation hybrids. c, Distribution of species-diagnostic SNPs along chromosomes in wild-caught H. elevatus and H. pardalinus. The physical location of SNPs along chromosomes (in Mb) are shown on top. Different blocks of SNPs within a chromosome, defined as groups of SNPs more than 500 kb apart, are denoted in alternating colours (black and grey). For visualization purposes, only chromosomes with at least 20 diagnostic SNPs are shown and SNP blocks were subsampled to show only one in every two SNPs. Long tracts of heterozygous genotypes (e.g. chromosome 19) suggest relatively recent hybridization followed by backcrossing.
Extended Data Fig. 4 Schematic of all demographic models tested with fastsimcoal2.
Two different tree topologies and 12 models per topology were tested. We considered the topology that retrieves both H. elevatus and H. pardalinus as monophyletic; that is, the species tree, (topology 1) and the most frequent topology across the genome (Extended Data Fig. 2), after excluding gene flow between H. elevatus and Amazonian H. pardalinus and in which H. p. sergestus is the first population to split (topology 2). The different demographic models are split into five main categories (depicted in different boxes): SI, strict isolation; AM, ancestral migration; SC, secondary contact; AM-SC, ancestral migration followed by secondary contact; IM, isolation with migration. Arrows between demes indicate gene flow (each direction being estimated as an independent parameter). Effective population sizes were allowed to change at split times. Note that for models under tree topology 1, the split times between H. elevatus populations and between H. pardalinus populations are different parameters and thus can assume different values.
Extended Data Fig. 5 Genetic evidence for current reproductive isolation between H. elevatus and H. melpomene.
a, Neighbour-joining tree based on autosomal sites sampled every 1 kb (166,989 sites). Values next to branches denote bootstrap values (based on 100 bootstrap iterations). Images of butterfly wings are copyright of the authors and Michel Cast. b, Distribution of species-diagnostic SNPs along chromosomes in wild-caught H. elevatus and H. melpomene. Species-diagnostic SNPs were defined as SNPs with an allelic difference of at least 0.8 between all Amazonian populations of H. elevatus and all H. melpomene populations. The physical location of SNPs along chromosomes (in Mb) are shown on top. For visualization purposes, SNP blocks were subsampled to show only 1 in every 20 SNPs. The lack of long tracts of heterozygous genotypes (or introgressed homozygous genotypes) suggests that there is no recent hybridization, followed by backcrossing, between these two species.
Extended Data Fig. 6 PCAs of male sex pheromones and host plant use show that H. elevatus, H. pardalinus and H. melpomene from the western Amazon form three distinct clusters in trait space.
a, PCA applied to concentrations of 30 male androconial volatiles. Loadings for selected compounds are annotated. b, PCA applied to oviposition preference of H. elevatus, H. pardalinus and H. melpomene for 21 species of Passiflora. Heliconius melpomene (24 females) laid 288 eggs and exhibited a strong preference for P. menispermifolia and P. triloba. Heliconius elevatus (35 females) laid 173 eggs and exhibited a preference for P. kaipiriensis. H. pardalinus butleri (51 females) laid 425 eggs and had a more generalized host plant use. To estimate the sample variance for each species, subsamples of 30 were drawn with replacement from the distribution of each species (1,000 replicates). PCA was then run on these bootstrapped replicates, polygons are minimum convex hulls encompassing all subsamples for each species. Images of butterfly wings are copyright of the authors and Michel Cast.
Extended Data Fig. 7 FST and genetic distances plotted against physical distance.
Physical distance is shown on the x axis; grey intervals are 1 Mb and black intervals are 5 Mb. Coloured bars show significant QTLs, with the QTL peak indicated by the triangle and the Bayesian credible intervals indicated by the length of the bar. Genetic distances are estimated using three crosses—within population (Heliconius elevatus; elev and Heliconius pardalinus; pard) and between population (F2). Candidate inversions (CIs; indicated by black arrows) are regions that recombine within species but not in hybrids (see Methods). The largest CI we identified was around 1.4 Mb long at the distal end of chromosome 16. However, we identified no CIs greater than 870 kb within the credible intervals of QTLs, and only one instance of a CI that was coincident with a QTL peak (on chromosome 15). Nonetheless, some CIs outside of QTLs present compelling targets for future investigation. Notably, at the proximal end of chromosome 19 and the distal end of chromosome 16, two large CIs overlap regions with elevated FST in which phylogenies resolve species boundaries (see Fig. 3).
Supplementary information
Supplementary Information
A full guide for Supplementary Tables 1–8 (Tables supplied separately).
Supplementary Tables
Supplementary Tables 1–8.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rosser, N., Seixas, F., Queste, L.M. et al. Hybrid speciation driven by multilocus introgression of ecological traits. Nature 628, 811–817 (2024). https://doi.org/10.1038/s41586-024-07263-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07263-w
- Springer Nature Limited