

Abstract
In response to pathogen infection, gasdermin (GSDM) proteins form membrane pores that induce a host cell death process called pyroptosis1,2,3. Studies of human and mouse GSDM pores have revealed the functions and architectures of assemblies comprising 24 to 33 protomers4,5,6,7,8,9, but the mechanism and evolutionary origin of membrane targeting and GSDM pore formation remain unknown. Here we determine a structure of a bacterial GSDM (bGSDM) pore and define a conserved mechanism of pore assembly. Engineering a panel of bGSDMs for site-specific proteolytic activation, we demonstrate that diverse bGSDMs form distinct pore sizes that range from smaller mammalian-like assemblies to exceptionally large pores containing more than 50 protomers. We determine a cryo-electron microscopy structure of a Vitiosangium bGSDM in an active ‘slinky’-like oligomeric conformation and analyse bGSDM pores in a native lipid environment to create an atomic-level model of a full 52-mer bGSDM pore. Combining our structural analysis with molecular dynamics simulations and cellular assays, our results support a stepwise model of GSDM pore assembly and suggest that a covalently bound palmitoyl can leave a hydrophobic sheath and insert into the membrane before formation of the membrane-spanning β-strand regions. These results reveal the diversity of GSDM pores found in nature and explain the function of an ancient post-translational modification in enabling programmed host cell death.
Similar content being viewed by others

Main
In mammalian pyroptosis, active GSDMs bind the inner leaflet of the plasma membrane and assemble into pores that secrete cytokines and induce lytic cell death1,2,3,4,5. Human cells encode six GSDM proteins (GSDMA, GSDMB, GSDMC, GSDMD, GSDME and PJVK), with individual GSDMs capable of targeting not only the plasma membrane but also membranes of mitochondria10 and intracellular bacteria11. Structures of human and mouse GSDM pores demonstrate that mammalian GSDM proteins form oligomeric assemblies of differing architectures6,7,8,9 with inner diameters ranging from 150–215 Å. Recent identification of GSDM homologues encoded in invertebrates12, fungi13,14 and bacterial anti-phage defence systems15 further suggests that GSDMs may form a large diversity of pore structures to support distinct cellular and antiviral functions.
bGSDM pores are large and diverse
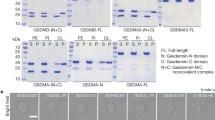
To define the mechanism and architectural diversity of GSDM pore formation beyond closely related mammalian proteins, we reconstituted pore formation in vitro using a panel of bGSDM homologues from diverse bacterial phyla (Fig. 1 and Extended Data Fig. 1a). Human and bacterial GSDM proteins use a shared mechanism of activation where proteolytic cleavage within an interdomain linker releases a pore-forming N-terminal domain (NTD) from a partnering inhibitory C-terminal domain (CTD). Most mammalian GSDM inhibitory CTDs are around 200 amino acids in length, but bacterial and fungal GSDMs achieve autoinhibition via a short 20-to-40-amino acid CTD14,15. GSDM activation in bacterial cells is controlled by a series of upstream proteins that sense unknown aspects of phage infection, limiting biochemical reconstitution of proteolytic activation and pore formation. We therefore sought to build on previous mammalian GSDM experiments4,6,7,11 to engineer bGSDMs for rational activation and enable analysis of pore formation across phylogenetically diverse systems. We first predicted bGSDM CTD cleavage sites based on our previous crystal structures of bGSDMs in inactive states15, and screened pairs of bGSDM full-length and ∆CTD constructs in vivo for toxicity in a cell growth assay (Extended Data Fig. 1a–d). For each bGSDM ∆CTD construct resulting in cellular toxicity, we next purified recombinant protein variants with a series of inserted sequence-specific protease motifs in the CTD linker and used a terbium-based liposome rupture assay to identify proteins capable of full membrane pore formation in vitro (Fig. 1b and Extended Data Fig. 1e,f). Using this approach, we successfully engineered controlled membrane pore formation for four bGSDM proteins from phylogenetically diverse bacteria (Fig. 1b and Extended Data Fig. 1f). In agreement with previous analyses of the mammalian GSDM lipid requirements for pore formation4,5, we observed that human GSDMD (hGSDMD) and mouse GSDMA3 (mGSDMA3) are able to form pores in liposomes prepared from Escherichia coli polar lipid extract but not phosphocholine lipids alone (Extended Data Fig. 2). By contrast, all of the engineered bGSDMs form pores in liposomes made of either simple phosphocholine lipids or E. coli polar lipid extract (Extended Data Fig. 2).

a, Schematic of bGSDM engineering and screen for functional pore formation. Predicted cleavage site loops were mutagenized to site-specific protease cleavage motifs and tested in liposome leakage assays monitoring release of terbium chloride (TbCl3). DPA, dipicolonic acid. b, Liposome leakage assay of engineered bGSDMs with matched site-specific proteases. The species of bGSDMs (and protease sites) are as follows: Unclassified Bacteroidetes (TEV site), Nostoc sp. Moss4 (WELQ site), Vitiosangium sp. GDMCC 1.1324 (thrombin) and Ideonella sp. 201-F6 (WELQ). Error bars represent the s.e.m. of n = 3 technical replicates. Data are representative of three independent experiments. RFU, relative fluorescence units. c, Size distributions of bGSDM pore inner diameters determined using negative-stain electron microscopy micrographs of pore–liposomes. The number of pores measured (n) for each species is as follows: Bacteroidetes (n = 117), Nostoc (n = 58), Runella (n = 189), Vitiosangium (n = 56) and Ideonella (n = 123). The black bar represents the average inner diameter of measured pores. The grey dashed lines represent the average inner diameters mammalian gasdermin pores, as previously measured6,7,11. d, Example negative-stain electron microscopy micrographs of bGSDM pores from five species. Scale bar, 50 nm.
We next reconstituted each successful pore-forming bGSDM variant into liposomes and visualized the membrane pores using negative-stain electron microscopy. Bacterial GSDM membrane pores are exceptionally large and diverse in architecture, spanning the range of known mammalian GSDM pores and other large pore-forming proteins16,17,18,19 (Fig. 1c,d and Extended Data Fig. 2). The Bacteroidetes bGSDM forms the smallest pores in our panel, with a mammalian-like inner diameter of around 180 Å. By contrast, Ideonella bGDSM pores are massive, spanning on average approximately 300 Å with some pores exceeding 400 Å and exhibiting dimensions substantially larger than the 260 Å pneumolysin cholesterol-dependent cytolysin pores produced by Streptococcus pneumoniae to kill target eukaryotic cells18,19. Notably, each bGSDM protein exhibits a distinct range of pore diameters, demonstrating that the variations in GSDM pore size are a programmed trait controlled by protein sequence specificity. Together, these results demonstrate that the size range of bGSDM pores extends substantially beyond that of characterized human GSDMs and that pore architecture is a diverse feature across GSDM protein evolution.
Mechanism of bGSDM oligomerization
To further compare the molecular basis of bacterial and human GSDM pore formation, we extracted bGSDM assemblies from pore–liposome samples and performed single-particle cryo-electron microscopy (cryo-EM) analysis. We focused on Bacteroidetes bGSDM pores, which form the smallest assemblies observed in our panel, and Vitiosangium bGSDM pores, which form some of the largest observed bGSDM assemblies. Cryo-EM and 2D classification analysis of extracted Bacteroidetes bGSDM pores reveals a rigid 30–31 protomer assembly that is remarkably similar to the hGSDMD 33-mer pore7 (Extended Data Fig. 3a,b). We confirmed the assembly of predominantly 31-mer Bacteroidetes bGSDM pores directly in the native lipid environment, demonstrating that some bGSDM proteins form pores with a similar overall size to that of GSDM proteins involved in human immunity (Extended Data Fig. 3a,b). In contrast to the smaller Bacteroidetes bGSDM pores, cryo-EM analysis of Vitiosangium bGSDM samples demonstrated giant assemblies formed by incorporation of 48–54 protomers (Fig. 2a and Extended Data Fig. 3c).

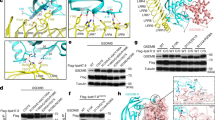
a, Cryo-EM micrograph and representative 2D class averages of HECAMEG detergent-extracted Vitiosangium bGSDM pores. The yellow arrowhead points to an example pore. Numbers in the top left corner of 2D classes represent the number of bGSDM protomers observed in that class. Scale bars, 50 nm. Micrographs are representative of more than 8,000 replicates. b, Cryo-EM micrograph and representative 2D class averages of DDMAB detergent-extracted Vitiosangium bGSDM slinkies. The yellow arrowhead points to an example slinky. Scale bars, 50 nm. Micrographs are representative of more than 8,000 replicates. c, Model of the structural relationship between bGSDM pores and slinky oligomers, with the pitch of a helical turn indicated. d, Cryo-EM map of an approximately 25-protomer turn of the Vitiosangium bGSDM slinky. In the bottom map, the dimension of the inner diameter is shown. e, bGSDM active model overlaid on a portion of the slinky map. Left, five-protomer model fit into the bGSDM slinky map. A single protomer is shown with darker magenta in the centre. Right, view along the oligomerization interface of the slinky onto a single protomer. The dimensions of the protomer are shown.
Previous X-ray structural analysis of the Vitiosangium bGSDM revealed the unexpected self-palmitoylation of an N-terminal cysteine and defined how bGSDMs are held in an inhibited conformation prior to proteolytic activation15. To determine the mechanism of how proteolysis and palmitoylation lead to the assembly of giant bGSDM pores, we focused on determining a high-resolution structure of Vitiosangium bGSDM in the active conformation. Owing to orientation bias of the exceptionally large Vitiosangium bGSDM pores, single-particle 3D reconstruction was not possible for these samples. While screening a series of detergents and extraction methods, we observed that in addition to single-ring pores, Vitiosangium bGSDM samples adopted double-ring assemblies and other open-ring assemblies, including cracked slit-shaped oligomers, as seen previously for human GSDM by cryo-EM, atomic force microscopy and molecular dynamics simulation experiments6,20,21. Unexpectedly, in N-dodecyl-N,N-(dimethylammonio)butyrate (DDMAB) detergent extractions we observed that open-ring assemblies of Vitiosangium bGSDM extend to form slinky-like oligomers with uniform radial dimensions closely mirroring the full pore (Fig. 2b,c and Extended Data Fig. 3c). Vitiosangium bGSDM slinkies are reminiscent of the helical oligomers of pneumolysin that form due to membrane-independent polymerization22. Additionally, mixtures of closed-ring pores and slinkies were observed in HECAMEG-extracted samples after prolonged storage, indicating that Vitiosangium bGSDM slinkies are likely to represent a stable non-native conformation derived from breaking closed-ring pores (Fig. 2c). Imaging Vitiosangium bGSDM slinkies by cryo-EM yielded 2D classes with excellent orientation distribution (Fig. 2b and Extended Data Fig. 4), and subsequent 3D classification and refinement of an approximately 25-protomer turn enabled us to determine a complete 3.3 Å structure of Vitiosangium bGSDM in the active oligomeric conformation (Fig. 2d,e and Extended Data Figs. 4b,c and 5).
Conservation of GSDM activation
The Vitiosangium bGSDM active-state cryo-EM structure reveals marked conformational rearrangements and an ancient mechanism of activation shared between bacterial and human GSDM proteins. Using DALI23 to compare the Vitiosangium bGSDM model against all structures in the Protein Data Bank (PDB), we observed active-state structures of hGSDMD and mGSDMA3 as the top hits, confirming that the Vitiosangium slinky-derived conformation corresponds to the bGSDM active oligomeric state (Fig. 3a). Notable structures more distantly related to the Vitiosangium bGSDM active state include the inactive conformation of mammalian GSDMs and members of the membrane attack complex perforin-like/cholesterol-dependent cytolysin (MACPF/CDC) family of pore-forming proteins.

a, Top, DALI z-scores from searching the active Vitiosangium bGSDM structure against the full PDB. PDB accessions with multiple similar chains were manually removed to show only unique hits. Below the graph are representative pore-forming protein structures from the MACPF/CDC family, in addition to active mammalian gasdermins. Pneumolysin and perforin structures have been reduced by 60% in size. b, Inactive-to-active-state transition of the Vitiosangium bGSDM structure. Left, transition from the inactive-state crystal structure (PDB ID 7N51) to the active-state cryo-EM structure (present study). Right, topology diagrams of the inactive and active structures labelling α-helices and β-sheets based on mammalian GSDM numbering6. The putative orientation of the palmitoyl modification is indicated in the active model. c, The active Vitiosangium bGSDM structure superimposed onto the mammalian GSDM structures from a and active hGSDMB (PDB ID 8ET2). Structures were superimposed by their globular domains and angles were measured using an axis at the base knuckle of the bGSDM to points at the fingertips of each structure. The inset shows the orientations of the α1 thumbs in the bGSDM and the mGSDMA3 structures. Selected positively charged residues are indicated, including R13 of mGSDMA3, which has been proposed to interact with the cardiolipin head group6. d, Views of Vitiosangium bGSDM slinky and pore models. Far right, the bGSDM pore is shown encompassing the hGSDMD pore model (PDB ID 6VFE). e, Top, a 6.5 Å resolution cryo-EM map of an elliptical closed-ring Vitiosangium bGSDM pore. The outlined area indicates a region where a pentamer of the bGSDM pore model was fit into the map (bottom).
Notably, despite sharing less than 15% sequence identity, the Vitiosangium bGDSM active-state model reveals strict conservation of the final overall ‘hand-shaped’ architecture of active mammalian GSDM proteins, including the α1 ‘thumb’, the globular ‘palm’, the β1–β2 ‘wrist’ loop, and the membrane-spanning ‘fingers’6,7 (Fig. 3b). Previous comparisons of the mammalian GSDM active-state structures revealed a key difference in the protein region between the globular domain and fingers region that controls the bend of the β-sheet fingers and directs the pitch of membrane insertion6,7. In contrast to the gentle bend of the ‘knuckles’ regions of the hGSDMD and mGDSMA3 active-state structures, the fingers and palm of the bGSDM active-state structure intersect at a nearly perpendicular angle and form a flat plateau from the inner palm to the knuckles (Fig. 3b). Superposition of the globular domains of the bGSDM and mammalian GSDM structures demonstrates that the bGSDM forms the sharpest angle with respect to the palm region, rotating 37° inwards compared with the hGSDMD conformation (Fig. 3c). This rotation compresses the bGSDM α1 thumb into the globular domain, where the N and C termini are held in unusually close proximity (Fig. 3c). Unlike the mammalian GSDM fingers, whose β-strands vary in length from 15 to 22 amino acids, the bGSDM fingers are nearly uniform at 17 or 18 amino acids each, and the fingertips, which resemble 2-to-7-amino acid β-hairpins in mammalian GSDMs, are expanded into 8-to-11-amino acid loops (Fig. 3b,c). Of note, multiple other bGSDM sequences, including the Lysobacter enzymogenes bGSDM involved in anti-phage defence, are each readily modelled using the Vitiosangium bGSDM active-state structure, supporting the idea that the conformational changes controlling bGSDM activation are likely to be conserved across diverse phyla (Extended Data Fig. 6a).
Comparison of the Vitiosangium bGSDM inactive-state crystal structure with the active-state cryo-EM model additionally explains bacteria-specific transitions that direct activation. Whereas the mammalian GSDM fingers form almost exclusively through the elongation of existing β-strands, the bGSDM fingers form via the local refolding of several alpha-helices (α2′′, α2′′′ and α4) and the β7′ strand, which are largely absent in mammalian GSDM structures (Fig. 3b and Extended Data Fig. 6b,c). In particular, the α2′′ helix contributes multiple residues to the hydrophobic groove housing the palmitoylated N-terminal cysteine residue that stabilizes the bGSDM autoinhibited state. Although no clear electron density could be observed for the cysteine in the active structure, reorientation of the α1 thumb is predicted to flip the covalently bound palmitoyl for insertion into the surrounding lipid membrane (Fig. 3b).
Similar to members of the MACPF/CDC family16,17,18, GSDMs assemble into large membrane-spanning β-barrels, in which each protomer contributes four β-strands to the complete pore. Despite this similarity, a previous analysis concluded that GSDMs were unlikely to have arisen by divergent evolution from the MACPF/CDC proteins, in part, owing to differences in pore-forming mechanisms and the absence of conserved glycines at the knuckle region of the GSDM structures6. We confirmed that these glycines are largely absent from the bGSDMs and also did not detect notable homology in the structures of the bGSDM and pneumolysin active-state models outside of the central β-strand region (Extended Data Fig. 6b,c and Extended Data Fig. 7a). However, the above noted bGSDM-specific refolding of domains which form the central β3 and β5 strands suggests that substantial variations in the mechanism of pore formation of distant-related GSDMs are possible. We therefore hypothesized that features or shared homology between GSDMs and MACPF/CDC proteins might be hidden in more distantly related bGSDM homologues. To test this hypothesis, we used FoldSeek24 to query the AlphaFold database and uncovered an unexpected cluster of bGSDM-like proteins with cytolysin-like features (Extended Data Fig. 7b,c). These proteins are encoded by multiple Gammaproteobacteria including Pseudomonas and Lysobacter species (more than 200 homologues on the Integrated Microbial Genomes (IMG) database) and have predicted homology to the pore-forming domain of bGSDMs but not to MACPF/CDC proteins. Of note, unlike the bGSDMs studied so far, these proteins are not encoded by operons with proteases and are C-terminally fused to immunoglobulin-like β-sandwich domains with homology to the cholesterol-recognition domains of pneumolysin and other cytolysins19. The direct fusion of the bGSDM-like pore-forming domain with an immunoglobulin-like β-sandwich domain raises the possibility that bGSDMs share a common ancestor with a cytolysin-like pore-forming toxin. We therefore propose that GSDM self-intoxication arose by divergent evolution from ancient pore-forming toxins that target non-self membranes.
To visualize the Vitiosangium bGSDM in its natural closed-pore state, we assembled a 54-mer structure based on the radius of the slinky oligomeric conformation and then realigned protomers into a circular 52-mer geometric model corresponding to the dominant size observed in 2D classification of intact pores (Fig. 3d). The resulting model supports continuous hydrogen bonding around a giant pore formed by 208 β-strands. Notably, the Vitiosangium bGSDM pore has an inner diameter of 330 Å that is wide enough to completely encompass the largest known mammalian pore of hGSDMD (Fig. 3d). To verify that the model represents the conformation of the closed-ring pore, we optimized the extraction and vitrification of Vitiosangium bGSDM pores and identified a condition in which pores could be observed with minimal orientation bias (Extended Data Fig. 8a,b). A dataset collected on this sample revealed 3D classes of closed-ring structures of multiple sizes, a double-ring pore, and a slinky-like structure (Extended Data Fig. 8b,c). Refinement of the dominant class yielded a map of the flexible pore assembly at 6.5 Å resolution that is slightly oval in shape (Extended Data Fig. 8c). We docked an elliptical 52-mer of the slinky-derived pore structure and confirmed that all major features of the the model closely match the oligomeric conformation of the closed-ring pore (Fig. 3e and Extended Data Fig. 8d,e). We further demonstrated that most residue–residue contacts at the subunit interface are preserved between the slinky-like oligomer and the circular pore (Extended Data Fig. 9a).
We next introduced mutations to Vitiosangium bGSDM residues at key regions including along the Vitiosangium bGSDM oligomeric interface and used a cell death assay to analyse bGSDM-mediated pore formation in bacterial cells. We observed that all 17 Vitiosangium bGSDM single-substitution mutants tested retained the ability to induce cell death, and confirmed that 5 of these mutants can be purified as stable proteins and rupture liposomes in vitro (Extended Data Figs. 9b–d and 10a,b). Combining the single mutants with the strongest effects, we identified double mutants that ablated the ability of Vitiosangium bGSDM to induce cell death (Extended Data Fig. 9c,d). These results indicate that bGSDM pore assembly is remarkably stable and robust to minor perturbations across the protein length—a feature that is distinct from mammalian GSDM counterparts6,7.
Mechanism of bGSDM membrane assembly
The inability of single mutations in conserved regions and at the oligomeric interface to inhibit bGSDM-mediated cell death suggests that bGSDM pore formation is a robust and energetically favourable process. Previously, X-ray crystal structures of Vitiosangium and other bGSDMs revealed palmitoylation of a conserved N-terminal cysteine residue that is also required for bGSDM-mediated cell death15. We therefore hypothesized that palmitoylation may contribute to the mechanism of membrane pore formation. To determine the function of Vitiosangium bGSDM palmitoylation, we mutated the conserved cysteine and residues in the surrounding pocket of the inactive structure and tested the function of these bGSDM variants for the ability to induce bacterial cell death. We observed that most mutations of the palmitoyl-binding pocket did not compromise bGSDM-mediated cell death, including mutations of V89 and F111, which rearrange into the active-state β3–β5 fingertip loop and knuckles of the β5 strand, respectively (Fig. 4a,b and Extended Data Fig. 10). However, we found that alanine mutations of conserved L211 and F213 residues located between the β8 and β9 strands in the active state partially disrupted bGSDM function, and a C4A mutation to the modified cysteine residue itself resulted in bGSDM inactivation and near-complete rescue of bacterial growth (Fig. 4a,b). We next purified C4A mutants of Vitiosangium and Nostoc bGSDMs and analysed both proteins in vitro with liposome rupture assays. Similar to our previous analysis of the Runella bGSDM15, the Vitiosangium and Nostoc bGSDMs purified as stable proteins but each were significantly reduced in the ability to form membrane pores and cause liposome leakage (Fig. 4c). These results demonstrate that the N-terminal palmitoyl modification has a direct role in bGSDM pore formation (Fig. 4c).

a, Hydrophobic pocket surrounding the palmitoylated cysteine (C4) of Vitiosangium bGSDM in the inactive state (PDB ID 7N51). Select hydrophobic residues are indicated. b, Cysteine palmitoylation is critical for toxicity in bacteria, whereas surrounding hydrophobic residues are not. Growth on LB agar plates of E. coli (Extended Data Fig. 9) harbouring plasmids encoding full-length bGSDM (full), the N-terminal pore-forming domain alone (∆CTD), or single mutations of the ∆CTD. Data are mean ± s.e.m. of n = 3 biologically independent samples. Data are representative of three independent experiments. CFU, colony-forming units; WT, wild type. c, Cysteine palmitoylation stimulates pore formation in vitro. Liposome leakage assay of wild-type and cysteine-mutated (C4A) Vitiosangium and Nostoc bGSDMs. Engineered bGSDMs were paired with site-specific proteases. Data are mean ± s.e.m. of n = 3 technical replicates; data are representative of 3 independent experiments. d, Structural transitions during bGSDM activation. Schematic of the anchoring process starting from the inactive bGSDM (i), followed by molecular dynamics simulation snapshots of the inactive ∆CTD adhered to the membrane at 37 °C, with the palmitoyl in the hydrophobic sheath (ii), and at 97 °C, where the palmitoyl spontaneously inserts into the membrane (iii). e, Schematic of unresolved intermediate steps of oligomerization and membrane insertion including a snapshot of a membrane-adhered dimer of ∆CTDs after 1 µs of high-temperature molecular dynamics simulation (top) and an oligomer in pore conformation (bottom). f, Molecular dynamics simulation of the fully assembled 52-mer pore with and without palmitoylated C4. Snapshots of the pore top (left) and front (bottom right) views after 1 μs of simulation. Solvent and membrane lipid tails are omitted for clarity. r.m.s.d. fluctuations (top right) of palmitoylated and the non-palmitoylated pores with respect to the average structure of the last 900 ns of the simulation (transparent lines) and smoothed fluctuations (opaque lines; smoothed using a 30-ns window).
We hypothesized that the palmitoyl group stimulates intermediate steps during bGSDM pore assembly. To test this hypothesis, we conducted atomistic molecular dynamics simulations of the early and late steps of pore assembly (Fig. 4d–f). Starting from the full-length inactive Vitiosangium bGSDM crystal structure (Fig. 4d(i)), we removed the 29-amino acid CTD and placed the inactive ∆CTD structure on a model bacterial membrane (Fig. 4d(ii)). At 37 °C, this structure was remarkably stable in 3× 2-µs long simulations, with a Cα root mean squared distance (r.m.s.d.) of approximately 2 Å from the initial structure and the palmitoyl remaining sheathed in the hydrophobic binding pocket. The ∆CTD monomers and dimers remained stable also at 97 °C (∆CTD r.m.s.d. of approximately 5 Å), including the proto-finger region and palmitoyl-binding pocket. Nevertheless, the palmitoyl spontaneously left the hydrophobic sheath to integrate into the bulk membrane within 1 µs in 12 out of 24 monomer simulations and in 9 out of 24 dimer simulations (Fig. 4d(iii)), subsequently being replaced in some simulations by a lipid acyl chain. In simulations of a single active-state protomer and of dimeric or trimeric bGSDMs in the membrane, the bGSDMs formed stable coiled assemblies. For the trimer, narrow membrane-spanning and water-filled channels formed, similar to those observed for hGSDMD21 (Fig. 4e and Extended Data Fig. 11). C4 palmitoylation did not noticeably affect the stability or shape of these small, possibly sublytic, pores. Finally, we simulated a complete 52-mer pore within a membrane. The pore was remarkably stable (r.m.s.d. of approximately 3 Å) with or without palmitoylation (Fig. 4f and Supplementary Video 1). Together, our simulations suggest that palmitoyl membrane insertion probably precedes the structural reorganization of the bGSDM proto-finger region. Integration of the covalently attached fatty acid tail may ensure stable membrane binding of the vulnerable, partially unfolded state and drive the system towards pore formation. Once the β-sheet is formed and membrane is inserted, the palmitoyl appears to have a subordinate role. The structural similarity of the hydrophobic cavity holding the palmitoyl in different bGSDMs15 suggests that a similar mechanism is shared in other species. To what extent the Vitiosangium bGSDM oligomerizes before it inserts into the membrane and whether it can form circular pre-pores, as proposed for eukaryotic GSDMs7,25, awaits further investigation.
Our study defines an ancient structural mechanism of GSDM pore formation. Comparison of the cryo-EM structure of the Vitiosangium bGSDM active state with a previously determined crystal structure15 enabled us to model how bacteria-specific features of the inactive state transform into membrane-spanning β-sheets to form huge pores, approximately 1.3 MDa in size. Reconstitution of pore formation using five diverse bGSDMs demonstrates that pore size is heterogeneous in nature and is intrinsically determined by protein sequence. Mammalian GSDM pores have well-defined roles in secretion of interleukin signals7. Although interleukins are not conserved in bacteria, the divergent sizes observed for bGSDM pores suggest that other molecules may be released to provide an additional layer of complexity to bGSDM-mediated anti-phage defence26. Whereas mammalian GSDMs and pore-forming toxins recognize specific lipids in target membranes4,5,27, a remarkable feature of bGSDMs is their ability to form pores on membranes with simple lipid compositions15. This lack of specificity may reflect the absence of selective pressures on bGSDMs to distinguish the plasma membrane from the organelle membranes present in eukaryotes, or self from non-self membranes required for pore-forming toxins. Consistent with this observation, the α1 thumb, which in mGSDMA3 mediates interactions with the cardiolipin head group via R136, is reoriented in the active-state bGSDM and the analogous residue of the Vitiosangium bGSDM is not required for GSDM-mediated cell death or liposome rupture (Fig. 3c and Extended Data Figs. 9 and 10). This finding suggests that cardiolipin-specific interactions are not conserved features of GSDM pores, despite the prevalence of cardiolipin in bacterial membranes. Nevertheless, future studies should investigate whether phylogenetically diverse bGSDMs are customized to the complex protein and lipid features of native bacterial membranes28.
Besides extrinsic membrane features, bGSDMs appear to broadly utilize intrinsic properties such as palmitoylation to strengthen membrane interactions and induce membrane disorder to lower the energy barrier for pore formation. Our experiments and molecular dynamics simulations support a model in which palmitoylation stimulates initial membrane interactions and stabilizes intermediate bGSDM assemblies prior to full pore formation. Analysis of bGSDM proteins naturally lacking the modified N-terminal cysteine will provide an opportunity to further explore the mechanistic basis of GSDM pore formation. Building on the identification of bGSDM palmitoylation15, two recent studies describe a requirement for enzyme-dependent palmitoylation of human and mouse GSDMD for pore formation in cells29,30. Although the palmitoylated cysteine (C191 in humans) is located in the fingertip region instead of on the thumb in bGSDMs, this finding raises the possibility that palmitoylation might provide a conserved function for gasdermins across the tree of life.
Methods
Bacterial growth assays
pBAD plasmids encoding full-length and ∆CTD GSDMs were constructed using gene synthesis (IDT) or PCR amplification from existing plasmids followed by Gibson assembly. Constructs expressing single amino acid mutations were generated by ‘around the horn’ (ATH) mutagenesis or gene synthesis (IDT). The following GSDM sequences were used in this study: hGSDMD (UniProt ID P57764), Runella bGSDM (IMG ID 2525253496), Unclassified Bacteroidetes metagenomic isolate bGSDM (simplified to ‘Bacteroidetes bGSDM’, IMG ID 2806880301), Nostoc bGSDM (IMG ID 2631173059), Vitiosangium bGSDM (IMG ID 2831770670), and Ideonella bGSDM (IMG ID 2684147428). C-terminal truncations for the ∆CTD GSDMs were based on experimentally demonstrated or predicted cleavage sites. For cleavage site predictions, proteolysis was assumed to occur within exposed loops of X-ray crystal or AlphaFold predicted structures, and before a small P1′ residue (Gly, Ala or Ser). The ∆CTD GSDMs comprised the following sequence lengths: 1–275 (hGSDMD), 1–247 (Runella bGSDM), 1–247 (Bacteroidetes bGSDM), 1–246 (Nostoc bGSDM), 1–237 (Vitiosangium bGSDM) and 1–244 (Ideonella bGSDM). Sequence-verified plasmids were transformed into E. coli TOP10 cells, the E. coli triple cardiolipin synthase knockout strain BKT1231, or the parental E. coli strain W3310 used to make BKT12. Transformations were plated on LB agar plates with 100 µg ml−1 ampicillin and 1% glucose. Single colonies were used in duplicate or triplicate to inoculate 5 ml liquid cultures in LB with 100 µg ml−1 ampicillin and 1% glucose and were incubated with shaking overnight at 37 °C. The next morning, cultures were 10-fold serially diluted in 1× phosphate-buffered saline (PBS) from 100 to 10−7 and 5 µl of each dilution was spotted onto LB agar plates with 100 µg ml−1 ampicillin and 1% glucose (to repress expression) or 0.2% arabinose (to induce expression from the arabinose promoter). Spots were allowed to dry at room temperature and plates were then grown overnight at 37 °C. The plates were imaged using a Chemidoc system (Bio-Rad) and the number of CFU was determined by counting colonies at appropriate dilutions. For conditions in which no colonies appeared in the 10−1 and the 100 dilution was uncountable, it was assumed that 1 colony was present in 100 and therefore 200 CFU ml−1 in total.
Recombinant protein expression and purification
bGSDM sequences were engineered for controlled activation by inserting site-specific protease cleavage motifs into predicted or verified cleavage sites using the target cleavage sequences for Tobacco Etch Virus (TEV) protease (ENLYFQ/G), the human rhinovirus (HRV) 3C protease (LEVLFQ/GP), the thrombin protease (LVPR/GS), or the WELQut protease (WELQ/G) where the backslash indicates the cleaved bond. Mammalian GSDMs were engineered for controlled activation by HRV 3C as described6,7, by inserting the cleavage site immediately after E262 in mGSDMA3 or replacing residues 259–275 of hGSDMD with the cleavage site. GSDM-encoding sequences were codon-optimized for E. coli and constructed by DNA synthesis (IDT). Gene fragments were cloned by Gibson assembly into a custom pET vector for expression fused to a 6×His-tagged SUMO2 tag. The N-terminal methionine residue was removed from each sequence and replaced by a single serine residue left as a scar following SUMO2 tag removal by the human SENP2 protease. For select constructs, alanine mutants of N-terminal cysteines were prepared by ATH mutagenesis or DNA synthesis (IDT). Forty total engineered bGSDM constructs were screened, and the best identified constructs were for the Bacteroidetes bGSDM with the TEV site replacing residues E242–G248, the Nostoc bGSDM with the WELQut site replacing residues V243–G247, the Vitiosangium bGSDM with the thrombin site replacing residues P234–M239, and the Ideonella bGSDM with the WELQut site replacing residues A241–G245.
Plasmids were transformed into BL21(DE3)-RIL E. coli and plated on MDG (0.5% glucose, 25 mM Na2HPO4, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 0.25% aspartic acid, 100 mg ml−1 ampicillin, 34 mg ml−1 chloramphenicol, and trace metals) agar plates. Three to five of the resulting colonies were used to inoculate starter cultures of MDG medium for overnight growth at 37 °C. MDG starter cultures were used to inoculate 2× 1 l of M9ZB medium (0.5% glycerol, 1% Cas-amino acids, 47.8 mM Na2HPO4, 22 mM KH2PO4, 18.7 mM NH4Cl, 85.6 mM NaCl, 2 mM MgSO4, 100 mg ml−1 ampicillin, 34 mg ml−1 chloramphenicol, and trace metals) and grown at 37 °C with shaking at 230 rpm until the culture reached an OD600 of ~2.5. Cultures were then chilled on ice for 20 min and induced for expression with 0.5 mM IPTG before shaking overnight at 16 °C and 230 rpm. The next morning, expression cultures were pelleted by centrifugation, washed with PBS, re-pelleted, and pellets were flash frozen on liquid nitrogen and stored at −80 °C until purification.
Protein purification was performed at 4 °C with all buffers containing 20 mM HEPES-KOH (pH 7.5) and other components as described below. Cell pellets were thawed and lysed by sonication in buffer containing 400 mM NaCl, 30 mM imidazole, and 1 mM DTT. Lysate was clarified by centrifugation and passing through glass wool, bound to NiNTA agarose beads (QIAGEN), which were subsequently washed with buffer containing 1 M NaCl, 30 mM imidazole, and 1 mM DTT. Protein was eluted with buffer containing 400 mM NaCl, 300 mM imidazole, and 1 mM DTT. Human SENP2 protease was added to the eluate to cleave off the SUMO2 tag before dialyzing overnight into buffer containing 125–250 mM KCl and 1 mM DTT. For initial screening of candidates, the dialysed protein was directly purified by size-exclusion chromatography (SEC) using a 16/600 Superdex 75 column equilibrated with 250 mM KCl and 1 mM TCEP. For select candidates, SEC was preceded by passing dialysed protein through a 5 ml HiTrap Q ion exchange column (Cytiva) in 125 mM KCl and 1 mM DTT. Sized proteins were brought to concentrations >20 mg ml−1, using 10 kDa molecular weight cut-off concentrators (Millipore), flash frozen on liquid nitrogen, and stored at −80 °C until use.
Liposome assays
Liposomes were prepared from 8 mg lipid total from powder or chloroform dissolved stocks (Avanti) of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), a 3:1 w/w mixture of DOPC or POPC and cardiolipin, or E. coli polar lipid extract (E. coli liposomes). Lipids were dried by transferring to a glass vial and evaporating chloroform under a stream of nitrogen gas and held under vacuum overnight in the dark. Liposomes for electron microscopy were prepared by resuspending the dried lipids with 20 mM HEPES-KOH (pH 7.5) and 150 mM NaCl. To prepare liposomes for terbium-based liposome rupture assays, and the resuspension buffer was supplemented with 15 ml TbCl3 and 50 mM sodium citrate. Liposomes were resuspended in a 800 µl volume (10 mg ml−1) by vortexing for 2 min, transferring to a 1.7 ml microfuge tube, and freeze thawing 3× with liquid nitrogen and a warm water bath. The crude liposome suspension was transferred to a glass tube, pulled into a glass syringe (Hamilton), and then extruded 21× with a mini-extruder (Avanti) using 19 mm nucleopore membranes with 0.2 µM pores (Whatman) and 10 mm filter supports (Avanti). Extruded liposomes were purified by SEC with a 10/300 Superdex 200 column equilibrated with 20 mM HEPES-KOH (pH 7.5) and 150 mM NaCl. Fractions containing liposomes were combined and stored at 4 °C until use.
Liposome leakage assays were carried out with purified DOPC or E. coli liposomes containing TbCl3. 40 µl liposome reactions were assembled in a clear well 96-well PCR plate (Bio-Rad) from 24 µl liposomes and 16 µl DPA dilution buffer at a final concentration of 20 µM DPA. For the data reported in Figs. 1 and 4 and Extended Data Fig. 1, cleavage reactions were assembled in triplicate for the addition of 10 µl per well with final concentrations of 50 µM bGSDM with or without paired proteases in a buffer containing 20 mM HEPES-KOH (pH 7.5) and 150 mM NaCl. Site-specific proteases were used at the following final concentrations: 5 µM homemade TEV protease, 0.1 µM HRV 3 C protease, 0.25 units ml−1 thrombin protease (MP Bioscience), or 0.1 units per µl WELQut protease (ThermoFisher). For the data reported in Extended Data Figs. 2 and 10, cleavage reactions were assembled in duplicate in the same buffer with the addition of 10 µl per well with final concentrations of 5 µM bGSDM with or without paired proteases used at the following final concentrations: 1 µM recombinant TEV protease, 0.05 µM human rhinovirus (HRV) 3 C protease, 0.1 units ml−1 thrombin protease (MP Bioscience), or 0.05 units per µl WELQut protease (ThermoFisher). After adding proteases, cleavage reactions were immediately added to wells containing liposomes and plate contents were spun to the bottom of wells by brief centrifugation. The plate was sealed and transferred to a Synergy HI microplate reader (BioTek) to monitor fluorescence resulting from Tb3+–DPA complex formation for 6 h with measurements every 2 min by excitation/emission at 276/545 nm.
Detergent screens and extraction of bGSDM pores
Four-millilitre bGSDM cleavage reactions were prepared in glass tubes with a buffer containing 20 mM HEPES-KOH (pH 7.5) and 150 mM NaCl. Reactions contained final concentration of liposomes at ~0.8 mg ml−1, bGSDMs at 20 µM, and proteases as described above. Screens were initially performed using liposome compositions found to be most ideal by negative-stain electron microscopy analysis. Reactions were carried out at room temperature overnight. The next day, liposomes were collected by centrifuging 150 µl portions at 60,000g at 4 °C for 30 min with a TLA100 rotor. The supernatant containing uncleaved bGSDM and protease was removed, and pellets were resuspended using 1/5 or 1/10 dilutions of detergents stocks from the JBScreen (Jena Bioscience) or Detergent Screen (Hampton). Detergent resuspensions were incubated for 30 min at room temperature, centrifuged at 60,000g at 4 °C for 30 min with a TLA100 rotor, and the supernatants were analysed by SDS–PAGE. Select candidate extractions were analysed by negative-stain electron microscopy and cryo-EM. All extractions were stored at 4 °C until use. Of the detergents screened, the Bacteroidetes bGSDM pores were found to extract in 0.34 mM n-dodecyl-d-maltoside (DDM), the Vitiosangium bGSDM pores were found to extract in 15.6 mM HECAMEG, and the Vitiosangium bGSDM slinky-like oligomers were found to extract in 5.16 mM DDMAB. HECAMEG-based extraction of the Vitiosangium bGSDM pore was optimized by titrating a range of detergent concentration against the Vitiosangium bGSDM incorporated into E. coli liposomes.
Negative-stain electron microscopy
Select bGSDMs were cleaved with paired proteases in the presence of liposomes in a volume of 50 µl in a buffer containing 20 mM HEPES-KOH (pH 7.5) and 150 mM NaCl. Reactions contained liposomes at concentrations of ~4 mg ml−1 and all bGSDMs at 25 µM with proteases concentrations as described above. Samples were incubated at room temperature overnight in glass tubes. To confirm cleavage, samples made in parallel without liposomes were analysed by 15% SDS–PAGE and stained by Coomassie blue (Extended Data Fig. 1). To identify optimal liposomes for imaging with each bGSDM pore sample, reactions were prepared using liposomes composed of DOPC or POPC lipids with or without cardiolipin. Reactions were diluted 1/10 in 20 mM HEPES-KOH (pH 7.5) and 150 mM NaCl buffer and 5 µl was applied to glow-discharged 400 mesh copper grids coated with formvar/carbon film (Electron Microscopy Sciences). Grids were blotted after 1 min, stained with 2% uranyl formate for 2 s, blotted immediately, and air dried. Imaging was performed at 80 kV using the JEOL JEM 1400plus using AMT Image Capture Engine Software version 7.0.0.255. Select samples were imaged at 40,000× magnification to acquire enough micrographs for measuring the inner diameters of >50 pores. For Fig. 1 and Extended Data Fig. 1, Bacteroidetes, Nostoc, and Ideonella bGSDM pores were all imaged in DOPC liposomes, while the Vitiosangium bGSDM pores were imaged from POPC–cardiolipin liposomes. For Extended Data Fig. 2, Bacteroidetes and Vitiosangium bGSDM pores were imaged from E. coli liposomes. Pore dimensions were measured manually using ImageJ version 2.9.0. Select detergent-extracted bGSDM pore samples were applied to grids and imaged and measured in the same manner as liposomes.
Cryo-EM sample preparation and data collection
For all bGSDM samples, 3 μl of sample was vitrified on grids using a Mark IV Vitrobot (ThermoFisher). Thirty minutes prior to sample vitrification, grids were glow-discharged using an easiGlow (Pelco). Grid type and blotting time was optimized for each sample using a double-sided blot with a constant force of 0, in a 100% relative humidity chamber at 4 °C, and a 10 s wait prior to plunging in liquid ethane before storing in liquid nitrogen. For the Bacteroidetes pores embedded in liposomes, the 1.2/1.3 Carbon Quantifoil grids were used with a blotting time of 10 s. For the DDM-extracted Bacteroidetes bGSDM pores, lacey carbon grids with carbon thin film support were used with a blotting time of 9 s. For the Vitiosangium bGSDM pores and pore–slinky mixtures extracted in HECAMEG detergent, lacey carbon grids with an additional 2 nm layer of carbon thin film were used with a blotting time of 9 s. The pore–slinky mixture represents the same sample as the pore-only sample, after leaving it for one week at 4 °C. For the Vitiosangium bGSDM oligomeric slinkies extracted in DDMAB detergent, 2/1 Carbon Quantifoil grids with an additional 2 nm layer of carbon thin film were used with a blotting time of 9 s. For the HECAMEG-extracted Vitiosangium bGSDM pore–slinky mixture that yielded side views, 1.2/1.3 UltrAuFoil grids were used with a blotting time of 7 s.
Cryo-EM data were collected either using a Talos Arctica (ThermoFisher) operating at 200 kV or a Titan Krios (ThermoFisher) microscope operating at 300 kV. Both microscopes were equipped with a K3 direct electron detector (Gatan). SerialEM software version 3.8.6 was used for all collections. Data collection on grids of the Bacteroidetes pores embedded in liposomes was performed using a Titan Krios microscope and a total of 4,340 movies were acquired at a pixel size of 0.83 Å, a total dose of 54.5 e− Å−2, dose per frame of 1.1e− Å−2 at a defocus range of −1 to −2 µm. Grids of the DDM-extracted Bacteroidetes bGSDM pores were imaged using a Talos Arctica microscope and a total of 5,364 movies were acquired at a pixel size of 1.1 Å, a total dose of 51.7 e− Å−2, dose per frame of 1.1 e− Å−2 at a defocus range of −1.0 to −2.0 µm. Data collection on grids of the Vitiosangium bGSDM pores extracted from POPC–cardiolipin liposomes in HECAMEG detergent was performed using a Titan Krios and a total of 8,930 movies were acquired at a pixel size of 0.83 Å, a total dose of 48.35 e− Å−2, dose per frame of 1.1 e− Å−2 at a defocus range of −1.0 to −2.0 µm. Data collection on grids of the Vitiosangium bGSDM slinky-like oligomers extracted from POPC–cardiolipin liposomes in DDMAB detergent was performed using a Titan Krios microscope and a total of 8,156 movies were acquired at a pixel size of 1.06 Å, a total dose of 51.8 e− Å−2, dose per frame of 1.04 e− Å−2 at a defocus range of −0.7 to −2.0 µm. Data collection on grids of the Vitiosangium bGSDM pore–slinky mixture extracted from POPC–cardiolipin liposomes in HECAMEG was performed using a Titan Krios microscope and a total of 4,818 movies were acquired at a pixel size of 0.83 Å, a total dose of 52.68 e− Å−2, dose per frame of 1.1 e− Å−2 at a defocus range of −1.0 to −2.0 µm. Data collection on grids of the Vitiosangium bGSDM pore–slinky mixture extracted from E. coli liposomes in HECAMEG was performed using a Titan Krios microscope and a total of 33,411 movies were acquired at a pixel size of 1.3 Å, a total dose of 53 e− Å−2, dose per frame of 1.06 e− Å−2 at a defocus range of −1.0 to −2.8 µm. Micrographs shown in Fig. 2 and Extended Data Figs. 4, 6, and 10 were prepared using IMOD version 4.11.3 and ImageJ Version 2.9.0.
Cryo-EM image processing and model building
For the Bacteroidetes bGSDM pores and the Vitiosangium bGSDM pores and pore–slinky mixtures, movies frames were imported to cryoSPARC32 for patch-based motion correction, patch-based CTF estimation, and 2D classification. For the Vitiosangium bGSDM oligomeric slinkies, movies frames were pre-processed using the on-the-fly processing scheme of RELION (version 3.1)33. Motion correction was performed using MotionCor234 and the motion corrected movies were imported to cryoSPARC for patch-based CTF estimation, 2D and 3D particle classification, non-uniform refinement, and masked local refinement. The resulting cryo-EM density was post-processed using DeepEMhancer35 prior to model building. For the Vitiosangium bGSDM pore–slinky mixture yielding pore side views, movies frames were imported to cryoSPARC32 for patch-based motion correction, patch-based CTF estimation. Particles were picked using the Topaz36 wrapper inside cryoSPARC before extraction and 2D classification, 3D classification, and homogenous and masked local refinements (see Extended Data Fig. 8 for processing workflow). Local resolution was estimated using cryoSPARC and visualized in ChimeraX.
The globular domain of the inactive-state Vitiosangium bGSDM (7N51) was used as a starting model after docking it into a single protomer of the slinky-like oligomer using Coot37. A complete protomer model was manual built in Coot and iterative corrections were made based on Phenix real-space refine38. The protomer model was validated in Phenix with MolProbity39 (Extended Data Table 1). The 54-mer slinky model in Fig. 3d was constructed by extending from a docked protomer by continuously fitting new protomers within the cryo-EM map density. To make the 52-mer pore model in Fig. 3d, the protomer models were realigned using a custom script and a geometric model based on the radius of a low-quality 3D reconstruction of anisotropic cryo-EM data of the 52-mer pore. Protomers were realigned to preserve the inter-subunit hydrogen bonding pattern of the slinky-like oligomer. Structure figure panels were generated using UCSF ChimeraX40 and PyMOL version 2.4.0.
Molecular dynamics simulations
Membrane
We set up the systems for molecular dynamics simulations using Charmm-GUI41. As membrane template, we used 15.2 × 15.2 nm2 patch mimicking the bacterial inner membrane (1-palmitoyl 2-cis-vaccenic phosphatidylethanolamine (PVPE):1-palmitoyl 2-cis-vaccenic phosphatidylglycerol (PVPG):cardiolipin, 15:4:1) surrounded with 150 mM NaCl solution. We minimized the potential energy of this patch in 5,000 steepest descent steps and performed 250 ps of NVT and 1.625 ns of NPT equilibration while stepwise decreasing the initial positional restraints of the phosphate z positions and the lipid tail dihedral angles. We used this membrane to set up simulation systems of 1–3 subunits of the Vitiosangium bGSDM. For simulations of the full 52-mer pore we multiplied the membrane patch along the xy plane to create a 62.3 × 62.3 nm2 large membrane patch.
bGSDM
We set up 1, 2, 3 and 52-mer Vitiosangium bGSDM systems in pore conformation by extracting the respective oligomer structures from the full 52-mer pore, with and without palmitoylated C4, inserting them into the membranes and removing all lipids with atoms overlapping with any protein (or C4 palmitoyl) atom, using Charmm-GUI to create simulation topologies. We alleviated membrane asymmetries by removing lipids from the overfilled leaflet. Subsequently, we resolvated the system with TIP3P water42, removed water molecules in the membrane, and then added NaCl to a sodium concentration of 150 mM and chloride atoms to neutralize the charge of the system.
For simulations of the inactive conformation of Vitiosangium bGSDM, we removed the 29-amino acid CTD from the inactive crystal structure (PDB ID 7N5115) and directly placed the inactive ∆CTD on the membrane patch. We used this system as starting point for 3 replicate 2-µs room-temperature molecular dynamics simulations. One of these was then continued in 24 replicate 1-µs molecular dynamics simulations of 1 µs each at 97 °C, each with different inintial velocities. We also performed 24 1-µs simulations of ∆CTD dimers at 97 °C. As starting dimer model, we placed two subunits from the last frame of a room-temperature replicate next to each other as in the electron microscopy ring structure and resolvated the system. To alleviate pressure in the protein-bound leaflet at elevated temperature, we randomly removed 10 (20) PVPE lipids from the protein-bound leaflet in the monomer (dimer) simulations. The dimer 1-µs production runs were preceded by potential energy minimization, as described below, and equilibration for 50 ns with position restraints on the protein, as described in Supplementary Table 1.
All molecular dynamics simulations were performed using Gromacs version 2022.443 and the CHARMM36m force-field44. We used a standard integration timestep of 2 fs. Electrostatic interactions were computed using the particle-mesh Ewald (PME) algorithm45. Real-space electrostatic and van-der-Waals interactions were cut-off at 1.2 nm. We constrained the distance of bonds with hydrogen atoms using LINCS46. We maintained a constant temperature of 37 °C using the velocity-rescale algorithm47 with a time constant of 1 ps, controlling protein, membrane and solvent (water and ions) individually. During system equilibration, we used the Berendsen barostat48 for semi-isotropic pressure coupling (x and y dimensions coupled together) with a time constant of 5 ps, a reference pressure of 1 bar, and a compressibility of 4.5 × 10−5 bar−1. After the equilibration phase, we switched to the Parrinello–Rahman algorithm49 for pressure control, while keeping all other control parameters the same.
Each system was first energy minimized for 5,000 steepest descent steps. Then, equilibration simulations of 50 ns length each were performed. During these equilibration simulations, the initial positional restraints of the heavy atoms of the protein (and the phospholipids) were gradually lifted (Supplementary Table 1). No positional restraints were used during the production simulations. Simulations were run for times indicated in Supplementary Table 2 and in the case of replicate simulations were started with randomly drawn initial velocities according to the Maxwell–Boltzmann distribution. The three simulations of the membrane bound inactive structure were not started from the exact same system, but from two systems with initially slightly different conformations of the palmitoyl fatty acid tail.
To calculate the Cα r.m.s.d. of the pore, we first calculated the average root mean squared fluctuation (r.m.s.f.) of each residue’s Cα and excluded the highly flexible protein regions (termini and fingertip regions with r.m.s.f. > 3.5 Å) from the subsequent r.m.s.d. calculation. The r.m.s.d. was calculated with respect to the average structure of the last 900 ns of the production simulation.
Two residues on neighbouring subunits were considered to form a contact if at least one pair of heavy atoms was within 3.6 Å distance. We estimated the probability of interfacial contacts over the last 900 ns of the simulation of the palmitoylated 52-mer pore and the 52 distinct interfaces.
Visual analysis as well as image and movie rendering were performed using PyMOL, VMD50, and UCSF ChimeraX40. Quantitative analyses and system setup were implemented with Python 3.9 and use the MDAnalyis package (v2.4.2)51,52.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Coordinates and density maps of the active-state Vitiosangium bGSDM have been deposited with the PDB and the Electron Microscopy Data Bank (EMDB) under the accession codes 8SL0 and EMD-40570, respectively. Complete 52-mer circular and elliptical models are deposited at the PDB-Dev under the accession codes PDBDEV_00000369, PDBDEV_00000370, and PDBDEV_00000371. Low-resolution density maps of the Vitiosangium bGSDM pore and slinky have been deposited at the EMDB under the accession codes EMDB-43508, EMD-43509, EMD-43510, EMD-43511 and EMD-43513. Simulation parameter files, raw trajectories and code used for analysis are deposited in two Zenodo repositories (https://doi.org/10.5281/zenodo.782840353 and https://doi.org/10.5281/zenodo.827214354). All other data are available in the manuscript or the supplementary materials, including source data for Figs. 1, 3 and 4 and Extended Data Figs. 1–4 and 8–10. Source data are provided with this paper.
Code availability
Custom scripts used to make the Vitiosangium bGSDM 52-mer pore model are available in a Zenodo repository (https://doi.org/10.5281/zenodo.782840353).
References
Broz, P., Pelegrín, P. & Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 20, 143–157 (2020).
Lieberman, J., Wu, H. & Kagan, J. C. Gasdermin D activity in inflammation and host defense. Sci. Immunol. 4, eaav1447 (2019).
Shi, J., Gao, W. & Shao, F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci 42, 245–254 (2017).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016).
Ding, J. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016).
Ruan, J., Xia, S., Liu, X., Lieberman, J. & Wu, H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67 (2018).
Xia, S. et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593, 607–611 (2021).
Wang, C. et al. Structural basis for GSDMB pore formation and its targeting by IpaH7.8. Nature 616, 590–597 (2023).
Zhong, X. et al. Structural mechanisms for regulation of GSDMB pore-forming activity. Nature 616, 598–605 (2023).
Rogers, C. et al. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 10, 1–17 (2019).
Hansen, J. M. et al. Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions. Cell 184, 3178–3191.e18 (2021).
Jiang, S., Zhou, Z., Sun, Y., Zhang, T. & Sun, L. Coral gasdermin triggers pyroptosis. Sci. Immunol. 5, eabd2591 (2020).
Daskalov, A., Mitchell, P. S., Sandstrom, A., Vance, R. E. & Glass, N. L. Molecular characterization of a fungal gasdermin-like protein. Proc. Natl Acad. Sci. USA 117, 18600–18607 (2020).
Clavé, C. et al. Fungal gasdermin-like proteins are controlled by proteolytic cleavage. Proc. Natl Acad. Sci. USA 119, e2109418119 (2022).
Johnson, A. G. et al. Bacterial gasdermins reveal an ancient mechanism of cell death. Science 375, 221–225 (2022).
Pang, S. S. et al. The cryo-EM structure of the acid activatable pore-forming immune effector macrophage-expressed gene 1. Nat. Commun. 10, 4288 (2019).
Dudkina, N. V. et al. Structure of the poly-C9 component of the complement membrane attack complex. Nat. Commun. 7, 10588 (2016).
Tilley, S. J., Orlova, E. V., Gilbert, R. J. C., Andrew, P. W. & Saibil, H. R. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell 121, 247–256 (2005).
van Pee, K. et al. CryoEM structures of membrane pore and prepore complex reveal cytolytic mechanism of Pneumolysin. eLife 6, e23644 (2017).
Sborgi, L. et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35, 1766–1778 (2016).
Schaefer, S. L. & Hummer, G. Sublytic gasdermin-D pores captured in atomistic molecular simulations. eLife 11, e81432 (2022).
Gilbert, R. J. C. et al. Two structural transitions in membrane pore formation by pneumolysin, the pore-forming toxin of Streptococcus pneumoniae. Cell 97, 647–655 (1999).
Holm, L. Dali server: structural unification of protein families. Nucleic Acids Res. 50, W210–W215 (2022).
van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246 (2023).
Mari, S. A. et al. Gasdermin-A3 pore formation propagates along variable pathways. Nat. Commun. 13, 2609 (2022).
Johnson, A. G. & Kranzusch, P. J. What bacterial cell death teaches us about life. PLoS Pathog. 18, e1010879 (2022).
Peraro, M. D. & Van Der Goot, F. G. Pore-forming toxins: ancient, but never really out of fashion. Nat. Rev. Microbiol. 14, 77–92 (2016).
Strahl, H. & Errington, J. Bacterial membranes: structure, domains, and function. Annu. Rev. Microbiol. 71, 519–538 (2017).
Du, G. et al. ROS-dependent palmitoylation is an obligate licensing modification for GSDMD pore formation. Preprint at bioRxiv https://doi.org/10.1101/2023.03.07.531538 (2023).
Balasubramanian, A. et al. Palmitoylation of gasdermin D directs its membrane translocation and pore formation in pyroptosis. Preprint at bioRxiv https://doi.org/10.1101/2023.02.21.529402 (2023).
Tan, B. K. et al. Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proc. Natl Acad. Sci. USA 109, 16504–16509 (2012).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. CryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Abraham, M. J. et al. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). 27–8.
Gowers, R. et al. in Proc. 15th Python in Science Conference (eds Benthall, S. & Rostrup, S.) 98–105 (2016).
Michaud‐Agrawal, N., Denning, E. J., Woolf, T. B. & Beckstein, O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327 (2011).
Schaefer, S. L. & Hummer, G. Raw data for: Structure and assembly of a bacterial gasdermin pore. Zenodo https://doi.org/10.5281/zenodo.7828403 (2023).
Schaefer, S. L. & Hummer, G. Additional raw data for: Structure and assembly of a bacterial gasdermin pore. Zenodo https://doi.org/10.5281/zenodo.8272143 (2023).
Acknowledgements
The authors thank members of their laboratories for helpful discussions; S. Rawson, S. Sterling, R. Walsh, M. Yip and S. Shao for advice on cryo-EM; A. Lu for assistance with protein purification, and the Max Planck Computing and Data Facility for computational resources. Cryo-EM data were collected at the Harvard Cryo-EM Center for Structural Biology at Harvard Medical School and at PNCC supported by NIH grant U24GM129547. The work was funded by grants to P.J.K. from the Pew Biomedical Scholars programme, the Burroughs Wellcome Fund PATH programme, The Mathers Foundation, The Mark Foundation for Cancer Research, the Parker Institute for Cancer Immunotherapy, and the National Institutes of Health (1DP2GM146250-01). A.G.J. is supported through a Life Science Research Foundation postdoctoral fellowship of the Open Philanthropy Project. G.H. and S.L.S. are supported by the Max Planck Society and the Collaborative Research Center 1507 funded by the Deutsche Forschungsgemeinschaft (DFG project number 450648163).
Author information
Authors and Affiliations
Contributions
The study was designed and conceived by A.G.J. and P.J.K. All cell growth and biochemical assays were performed by A.G.J. Protein purification and detergent screens were performed by A.G.J. and N.K.M.-B. Samples for cryo-EM were prepared by A.G.J. and M.L.M. Electron microscopy data collection and processing was performed by A.G.J. and M.L.M. Model building and analysis was performed by A.G.J., S.L.S. and P.J.K. Molecular dynamics simulations were performed by S.L.S. and G.H. Figures were prepared by A.G.J., M.L.M. and S.L.S. The manuscript was written by A.G.J. and P.J.K. All authors contributed to editing the manuscript and support the conclusions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Kevin Corbett, James Whisstock and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Engineering diverse bGSDMs for controlled activation.
a, Crystal structure of an inactive bGSDM from a Vitiosangium species (PDB ID 7N51) and indication of the disordered loop that was targeted for cleavage site engineering. b, Colony forming units (CFU) per mL of E. coli derived from spot assays shown in panel (c). Error bars represent the SEM of n = 3 biologically independent samples. The data are representative of three independent experiments. c, TOP10 E. coli harboring plasmids encoding full-length GSDMs (full) or the N-terminal pore-forming domain alone (∆CTD) were grown on LB-agar plates in triplicate. LB-agar contained either 1% glucose or 0.2% arabinose to repress or induce expression, respectively. Cells were serially diluted and plated out from left (100) to right (10−7) with 5 µL per spot. Though the Ideonella ∆CTD construct does not drastically reduce the CFUs compared to the full construct, colonies grow more slowly and appear fainter in agreement with toxicity from pore formation. d, Parental (W3310) or the triple cardiolipin synthase knockout (BKT12) E. coli harboring plasmids for the ∆CTD GSDMs were grown and spotted out onto LB agar plates with 1% glucose or 0.2% arabinose in duplicate as in panel (c). e, Engineered bGSDMs were treated with or without paired site-specific proteases for 18 h at room temperature and analyzed by 15% SDS-PAGE and visualized by Coomassie staining. Cleaved bGSDM proteins are indicated with a yellow asterisk. f, Full time-course of liposome leakage assays related to Fig. 1b. Error bars represent the SEM of three technical replicates. The data are representative of three independent experiments. g, Negative-stain EM micrographs representing larger view fields micrographs shown in Fig. 1d or second example micrographs used to measure pore sizes for Fig. 1c. Scale bar = 50 nm. Micrographs are representative of >10 replicates with similar results.
Extended Data Fig. 2 Engineered bGSDMs form pores in liposomes with simple and complex lipid compositions.
a, Liposome leakage assay of engineered mammalian GSDMs and bacterial GSDMs with matched site-specific proteases. The left plot shows the results from an experiment performed with liposomes prepared from DOPC lipids (DOPC liposomes), while the right plot shows the result from an experiment using liposomes prepared from E. coli polar lipid extract (E. coli liposomes). The species and/or paralog of mammalian GSDMs or bGSDMs or mammalian GSDMs (and protease sites) are as follows: mGSDMA3 (HRV 3 C site), hGSDMD (HRV 3 C site), Unclassified Bacteroidetes (TEV site), Nostoc sp. Moss4 (WELQ site), Vitiosangium sp. GDMCC 1.1324 (thrombin), and Ideonella sp. 201-F6 (WELQ). In most cases, engineered bGSDMs exhibited robust pore formation in the presence of E. coli liposomes. The Nostoc bGSDM exhibited comparatively weak pore formation activity, possibly due to inefficient cleavage or exogenous amino acids remaining following WELQ protease cleavage compared to the ΔCTD variant tested in in vivo growth assays. The data are representative of three independent experiments each with two technical replicates. b, Full time-course of liposome leakage assays related to panel (a). c, Negative-stain EM micrographs of Bacteroidetes bGSDM and Vitiosangium bGSDM pores in E. coli liposomes (left) and plot comparing pore inner diameters of these bGSDMs in DOPC liposomes versus the same bGSDMs in E. coli liposomes (right). The number of pores measured (n) for each species in E. coli liposomes is Bacteroidetes (n = 171) and Vitiosangium (n = 100). The inner diameters values for pores in DOPC liposomes are the same as in Fig. 1c. The black bar represents the average inner diameter of measured pores. Scale bar = 50 nm. Micrographs are representative of >10 replicates with similar results.
Extended Data Fig. 3 Electron microscopy of Bacteroidetes bGSDM pores and Vitiosangium bGSDM pores and slinkies.
a, Representative cryo-EM micrograph and select 2D class averages of Bacteroidetes bGSDM pores from pore-liposome samples. Scale bars = 20 nm. Micrographs are representative of >4,000 replicates with similar results. b, Representative cryo-EM micrograph and select 2D class averages of DDM-extracted Bacteroidetes bGSDM pores. Numbers in the upper left-hand corner of 2D classes represent the number of bGSDM protomers observed in that class. Scale bars = 20 nm. Micrographs are representative of >2,500 replicates with similar results. c, HECAMEG detergent-extracted Vitiosangium bGSDM pores. Scale bar = 50 nm. Micrographs are representative of >10 replicates with similar results. d, Comparison of inner diameters measured from pore-liposome samples (Fig. 1 and Extended Data Fig. 2) and HECAMEG detergent-extracted Vitiosangium bGSDM pores. The number of pores measured (n) from each sample is as follows: pore-liposome (n = 56), extracted pore (n = 189). The black bar represents the average inner diameter of measured pores. e, DDMAB detergent-extracted Vitiosangium bGSDM slinkies. Scale bars = 50 nm. Micrographs are representative of >10 replicates with similar results.
Extended Data Fig. 4 Cryo-EM data processing of Vitiosangium bGSDM pores and slinkies.
a, Representative cryo-EM micrograph and 2D class averages of HECAMEG detergent-extracted bGSDM pores. Scale bars = 50 nm. Micrographs are representative of >8,000 replicates with similar results. b, Single-particle processing schematic of DDMAB detergent-extracted bGSDM slinkies. From top to bottom: representative cryo-EM micrograph and 2D class averages (as in Fig. 2b), particle classification and map refinement, and local resolution estimate of final map. c, Fourier shell correlation (FSC) curves versus resolution of bGSDM slinky map. Resolution was estimated at an FSC of 0.143. Scale bars = 50 nm. Micrographs are representative of >8,000 replicates with similar results. d, Representative cryo-EM micrograph and 2D class averages of HECAMEG detergent-extracted bGSDM pore-slinky mixture (as in Fig. 2a). Micrographs are representative of >4,000 replicates with similar results.
Extended Data Fig. 5 Cryo-EM model to map fitting of bGSDM slinky and pore.
a, 54-mer model of the Vitiosangium bGSDM in a slinky-like oligomerization. b, Examples regions of model to map fit quality for a single Vitiosangium bGSDM protomer. The map surface has been contoured to 16σ.
Extended Data Fig. 6 Functional conservation of the GSDM active state.
a, Predicted bGSDM structures are organized from left to right based on percent sequence identity to the Vitiosangium bGSDM: Runella bGSDM (18%), Bradyrhizobium bGSDM (19%), and Lysobacter bGSDM (28%). Phyre homology model threading utilized the active hGSDMD structure (PDB ID 6VFE, left model) or the active Vitiosangium bGSDM structure (this study, right model). Each bGSDM structure was also predicted using AlphaFold after deleting ~20 amino acids from the C-termini and yielded inactive-like structures (center). The modeled sequences are as follows: Runella bGSDM (1–247), Bradyrhizobium bGSDM (1–237), and Lysobacter bGSDM (1–240). b, Structure-based alignment of bacterial and mammalian gasdermins. The Vitiosangium bGSDM active structure was aligned to the active mGSDMA3 (PDB ID 6CB8) and hGSDMD (PDB ID 6VFE). Secondary structures are indicated below the sequences for each structure, in addition to secondary structures from the Vitiosangium bGSDM inactive state crystal structure. Residues of the Vitiosangium bGSDM that surround the palmitoyl in the inactive state are boxed in black, asterisks indicate residues that have been mutated to test their effect on bGSDM-mediated bacterial cell death, and green boxes indicate resides that align with the conserved glycines in MACPF/CDC proteins. c, Topology diagrams showing the transitions from inactive to active structures of the Vitiosangium bGSDM (left) and mGSDMA3 (right). Conserved α-helices and β-strands are outlined in black, the positions of residues universally conserved in charge, identity, or aromaticity are indicated in active state topology diagrams, and green arrows indicate the sites of conserved glycines in MACPF/CDC protein structures.
Extended Data Fig. 7 Evidence for a possible divergent evolution of gasdermins and cytolysins.
a, Structure-based alignment of the Vitiosangium bGSDM (aa 6–229) and the pneumolysin (PLY) pore-forming domain (aa 4–355). The Vitiosangium bGSDM active state structure (this study, PDB 8SL0) was aligned to the active state PLY structure (PDB ID 5LY6). Secondary structures are indicated below the sequences for each structure and numbered according to prior conventions19. Conserved glycine residues of the PLY structure that are present in other MACFP/CDC proteins are boxed in green. b, A query of the AlphaFold database with experimentally determined bGSMD structures yielded putative bGSDM-like proteins with cytolysin-like features. A representative structure from a Pseudomonas species is shown on the right, indicating the N-terminal bGSDM-like domain in salmon color and the C-terminal immunoglobulin-like β-sandwich domain in green color with similarity to the membrane binding domain of PLY and other cytolysins. The sequence alignment shows the highly conserved undecapeptide present in the β-sandwich domains of multiple cytolysins c, Structure-based alignment of the Vitiosangium bGSDM (aa 2–229) and the Pseudomonas bGSDM-like protein pore-forming domain (aa 1–245). The Vitiosangium bGSDM inactive state structure (PDB 7N51) was aligned to the putative inactive state structure of the bGSDM-like protein (AF-A0A2N3PBA1). Secondary structures are indicated below the sequences for each structure, using α-helix and β-sheet numbering for the bGSDM-like protein that reflect homology to the bGSDM.
Extended Data Fig. 8 Extraction and cryo-EM data processing of Vitiosangium bGSDM pores with sideviews.
a, Fractions from the detergent extraction of Vitiosangium bGSDM pores from E. coli liposomes analyzed by SDS-PAGE and Coomassie staining. The bGSDM sample extracted at 50 nm HECAMEG was subsequently used for cryo-EM analysis b, Representative cryo-EM micrograph of HECAMEG detergent-extracted bGSDM pores from (a). Scale bar = 50 nm. Micrograph is representative of >30,000 replicates with similar results. c, 2D class averages from processing micrograph indicated in (b) and single-particle processing schematic of HECAMEG detergent-extracted bGSDM closed-ring pores and slinky-like oligomers. Scale bars = 50 nm. Data was processed using homogeneous (homo.) or non-uniform (NU) refinements. d, Fourier shell correlation (FSC) curves versus resolution of bGSDM slinky map. Resolution was estimated at an FSC of 0.143. e, Docking of the 52-mer elliptical pore-model into the 6.5 Å resolution map from (c). The left model represents a geometric model based on the structure of the slinky-like oligomer, with an eccentricity of 0.86. The right model represents the maximum eccentricity pore undulation observed in during the MD simulations of the 52-mer pore, with an eccentricity of 0.67.
Extended Data Fig. 9 Cell death and liposome rupture by the Vitiosangium bGSDM is robust to single mutations.
a, Residue-residue contacts between neighboring subunits occurring with a frequency of >0.01 over the last 900 ns of an MD simulation of the 52-mer pore with C4-palmitoylation. b, Structural representations of the Vitiosangium bGSDM oligomer. Center, an electrostatic charge model of a 4-mer of the slinky-like oligomer. Left inset, sites targeted for mutation on the ‘positive patch’ and α1 thumb on the pore exterior. Right, dimer of the Vitiosangium bGSDM along the interface shaded to indicate the frequency of contact occurring over the course of the MD simulation described in panel (a). Right inset, residues targeted for mutagenesis at the subunit interface. c, Colony forming units (CFU) per mL of E. coli derived from spot assays shown in panel (d). Growth assays test single charge-swap mutations to residues at select interfaces in the active model. The experiment was performed with n = 2 biologically independent samples and the data are representative of two or three independent experiments. d, E. coli harboring plasmids encoding full-length GSDMs (full) or the N-terminal pore-forming domain alone (∆CTD) were grown on LB-agar plates in duplicate. LB-agar contained either 1% glucose or 0.2% arabinose to repress or induce expression, respectively. Cells were serially diluted and plated out from left (100) to right (10−7) with 5 µL per spot.
Extended Data Fig. 10 Liposome rupture and cell death by the Vitiosangium bGSDM is sensitive to mutation of the palmitoylated cysteine but not other single amino acid residues.
a, Relative fluorescent units (RFUs) at six hours from liposome rupture experiment testing single amino acid mutants of the Vitiosangium bGSDM with DOPC liposomes. b, Full time-course RFU data for the plot shown in panel (a). c, RFU at six hours for liposome rupture experiment testing single amino acid mutants of the Vitiosangium bGSDM with DOPC liposomes. The data are representative of three independent experiments each with n = 2 technical replicates. d, Full time-course RFU data for the plot shown in panel (c). e, Bacterial spot assays testing mutation of residues proximal to the N-terminal cysteine of the Vitiosangium bGSDM. TOP10 E. coli harboring plasmids encoding full-length GSDMs (full) or the N-terminal pore-forming domain alone (∆CTD) were grown on LB-agar plates in triplicate. LB-agar contained either 1% glucose or 0.2% arabinose to repress or induce expression, respectively. Cells were serially diluted and plated out from left (100) to right (10−7) with 5 µL per spot.
Extended Data Fig. 11 MD simulations of small membrane pores formed by active 1, 2, and 3-mer bGSDM assemblies.
Snapshots of palmitoylated (left) and non-palmitoylated (right) mono- and oligomers after 3.3 μs of simulation in a bacterial membrane in top view and side view. Protein shown in purple cartoon representation, membrane phosphates shown as tan spheres, palmitoylated C4 shown in cyan opaque licorice and transparent surface representation. In the side views, water inside the small pores is shown in blue transparent surface representation. Otherwise, solvent molecules and lipid fatty acid tails are omitted for clarity.
Supplementary information
Supplementary Table 1
Restraints used during energy minimization (EM) and stepwise equilibration (EQ) in kJ mol−1 nm−2.
Supplementary Table 2
Overview of bGSDM production simulations.
Supplementary Video 1
Atomistic molecular dynamics simulation of the fully assembled, palmitoylated 52-meric Vitiosangium bGSDM pore in a bacterial lipid membrane.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Johnson, A.G., Mayer, M.L., Schaefer, S.L. et al. Structure and assembly of a bacterial gasdermin pore. Nature 628, 657–663 (2024). https://doi.org/10.1038/s41586-024-07216-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07216-3
- Springer Nature Limited
This article is cited by
-
Renal macrophages and NLRP3 inflammasomes in kidney diseases and therapeutics
Cell Death Discovery (2024)