

Abstract
Epstein–Barr virus (EBV) infection can engender severe B cell lymphoproliferative diseases1,2. The primary infection is often asymptomatic or causes infectious mononucleosis (IM), a self-limiting lymphoproliferative disorder3. Selective vulnerability to EBV has been reported in association with inherited mutations impairing T cell immunity to EBV4. Here we report biallelic loss-of-function variants in IL27RA that underlie an acute and severe primary EBV infection with a nevertheless favourable outcome requiring a minimal treatment. One mutant allele (rs201107107) was enriched in the Finnish population (minor allele frequency = 0.0068) and carried a high risk of severe infectious mononucleosis when homozygous. IL27RA encodes the IL-27 receptor alpha subunit5,6. In the absence of IL-27RA, phosphorylation of STAT1 and STAT3 by IL-27 is abolished in T cells. In in vitro studies, IL-27 exerts a synergistic effect on T-cell-receptor-dependent T cell proliferation7 that is deficient in cells from the patients, leading to impaired expansion of potent anti-EBV effector cytotoxic CD8+ T cells. IL-27 is produced by EBV-infected B lymphocytes and an IL-27RA–IL-27 autocrine loop is required for the maintenance of EBV-transformed B cells. This potentially explains the eventual favourable outcome of the EBV-induced viral disease in patients with IL-27RA deficiency. Furthermore, we identified neutralizing anti-IL-27 autoantibodies in most individuals who developed sporadic infectious mononucleosis and chronic EBV infection. These results demonstrate the critical role of IL-27RA–IL-27 in immunity to EBV, but also the hijacking of this defence by EBV to promote the expansion of infected transformed B cells.
Similar content being viewed by others


Main
EBV is a γ-herpes family virus with oncogenic potential8. EBV infection is one of the most widespread infections in humans with 90% of individuals being infected at the age of 20–252. EBV primarily infects epithelial cells and B lymphocytes in the oropharynx establishing thereafter a lifelong latent silent infection in B lymphocytes. However, EBV persistence may result in malignant and non-malignant proliferative diseases9, the most frequent of which being B cell lymphoid neoplasms. EBV infection is also sometimes associated with haemophagocytic lymphohistiocytosis (HLH), a severe inflammatory disorder.
Although it is usually asymptomatic in immunocompetent individuals, the primary infection can cause infectious mononucleosis (IM), a self-limiting lymphoproliferation of infected B cells and activated T cells, reflecting a strong immune response3. After infection, EBV latent growth-transforming genes are expressed in naive B cells driving their robust proliferation and potentially unlimited expansion8. The immune response to EBV is mainly dependent of specific cytotoxic T cells that strongly and rapidly expand to remove infected cells1,2. This expansion is important; up to 40% of blood circulating T cells can be specific to EBV10.
There is a group of inborn errors of immunity (IEIs) that is selectively associated with impaired immunity to EBV, predisposing to severe IM, HLH and to the development B cell proliferative disorders1,4. Genetic characterization of these rare diseases, mostly paediatric, has uncovered selective key pathways and factors that are required for an efficient immunity to EBV, notably those involved in the expansion of T cells such as the signalling molecules ITK and RASGRP1, the DNA metabolism enzyme CTPS1, and the co‐stimulatory TNF receptors CD27 and TNFSFR9 and their ligands CD70 and TNFSF9 that are expressed on infected cells. However, there are still a number of patients in whom the genetic basis of their susceptibility to developing severe EBV infection is unclear, representing unique in natura experiences to reveal components of T cell responses to EBV.
Loss-of-function mutations in IL27RA
We investigated three children (P1.1, P1.2 and P2; from two families) who had severe acute primary EBV infection. In family 1, P1.1 and P1.2 developed unusual early onset IM symptoms at the age of 20 and 8 months, respectively, while, in P2 from family 2, the primary infection occurred at the age 17 years (Fig. 1a). Severe EBV infection in the patients was characterized by persistent fever, hepatitis and splenomegaly associated with blood T cell lymphocytosis requiring hospitalization and treatment with anti-CD20 (P1.1) or corticosteroids (P2) with relapse. The severity of IM in P2 was evidenced by features of HLH. In P1.1, high blood loads of EBV (up to 1 × 105 EBV copies per ml) persisted without any clinical symptoms, for 12 months. No clinical manifestations were noted thereafter in P1.1, P1.2 and P2 (6, 17 and 3 years after IM, respectively). Immunological phenotyping after IM of P1.1, P1.2 and P2 PBMCs showed counts and proportions of the different leukocyte subpopulations within normal ranges, including naive and memory CD4+ and CD8+ T cells with the exception of effector memory CD8+ and CD4+ T cells, which were decreased in all patients (Extended Data Fig. 1a–c). In P1.1 and P2, memory CD27+ B cell counts were also decreased. Serum Ig levels were within age-matched reference ranges, except for IgA, which was low in P1.1. T cell proliferation in response to phytohaemagglutinin (PHA) mitogen and anti-CD3 antibodies was normal for P1.1, P1.2 and P2. On the basis of these features, it was suspected that these patients carried a genetic defect leading to an immunodeficiency associated with vulnerability to EBV-induced disease.

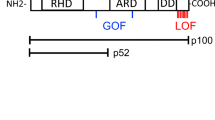
a, The pedigrees of the families in which mutations in IL27RA were identified. The black box and circle represent affected individuals, and the diagonal bars indicate deceased individuals. Each patient (P) is identified by a number. The arrow indicates the proband first identified. The genotypes shown in b are indicated with the mutated alleles in red. b, Sanger sequencing represented by DNA electropherograms of IL27RA regions containing the c.286C>T mutation in family 1 or the c.1336C>G and c.1142-2A>C mutations in family 2 and in healthy control donors (Ctrl). The arrows indicate the presence of heterozygous or homozygous mutations. c, The IL27RA intron–exon organization and protein domains. EC, extracellular; IC, intracellular; TM, transmembrane. The white boxes represent the coding exons. The mutations at the cDNA and protein levels are indicated by black arrows. d, Histograms from FACS analysis showing IL-27RA expression in T cells of a control individual, P1.1 and P1.2 stimulated by anti-CD3/CD28 or not (no stim.). e, The same as in d with T cells of P2, except that the histograms show surface plus intracellular (top) and surface alone (bottom) IL-27RA expression in T cells stimulated with anti-CD3/CD28 antibodies. Data from one representative experiment out of three. f,g Immunoblot analysis of IL-27RA and actin expression in T cell blasts from patients P1.1 and P1.2 (f) and P2 (g) and healthy control donors that were stimulated or not with anti-CD3/CD28 antibodies. Data from two independent experiments (exp. 1 and exp. 2). Molecular mass (kDa) values are shown on the left.
We performed whole-exome sequencing in P1.1 and P2. Genetic variants identified by whole-exome sequencing were filtered on the basis of their combined annotation-dependent depletion/mutation significance cut-off (CADD/MSC) scores and their allele frequencies in gnomAD11 and in the exome databases of our institute. Significant rare variants in the IL27RA gene were retrieved in both P1.1 and P2, consisting of a homozygous premature stop codon (g.19:14150387C>T (rs375317876), c.286C>T; p.Gln96X) in P1.1 and compound heterozygous variants in P2: a missense mutation (g.19:14160060C>G (rs201107107), c.1336C>G, p.Arg446Gly) and an essential acceptor splice mutation (g.19:14159791A>C (rs778365769), c.1142-2A>C). All variants were predicted to have a deleterious impact (Methods). None of these variants was detected as homozygous in public databases or in our own database, with the exception of the allele g.19:14160060C>G (rs201107107) encoding the p.Arg446Gly variant for which one homozygous allele was detected in gnomAD v.2.1.1 that is in the Finnish population. Accordingly, out of 165 alleles in gnomAD, 147 were found in the Finnish population (147 out of 25,114) (minor allele frequency = 0.0068). In the FinnGen database (https://www.finngen.fi/en) containing more than 400,000 individuals, 15 are homozygous for g.19:14160060C>G. Two of these individuals had hospital diagnoses of EBV IM (and none had disseminated BCG on vaccination). This represents a more than 50-fold significant (P < 0.001) enrichment over the age- and sex-matched population of the same database, indicating that this allele is associated with an increased risk of developing severe acute IM. The penetrance of this genotype appears to be partial, although this will require further investigation.
The segregation of the IL27RA variants was examined in family members using Sanger sequencing (Fig. 1b). Both parents in family 1 were heterozygous for the g.19:14150387C>T variant and the affected brother (P1.2) was, as expected, homozygous. In family 2, the mother and father were heterozygous carriers of the g.19:14160060C>G variant and the g.19:14159791A>C variant, respectively. These data were consistent with an autosomal recessive inheritance of the disease. g.19:14150387C>T causes a premature stop codon p.Gln96X that is predicted to remove most of the IL-27RA protein, whereas g.19:14160060C>G results in the missense mutation p.Arg446Gly in the extracellular domain of IL-27RA (Fig. 1c). The essential splicing site g.19:14159791A>C variant was found to cause abnormal splicing characterized by the use of an alternative acceptor site within exon 9 leading to an in-frame deletion of 45 nucleotides yielding a shorter protein lacking 15 amino acids (p.Gln381_Ala395del), while the other alleles had no effect on mRNA/cDNA expression (Extended Data Fig. 2a,b).
IL27RA encodes the α subunit of IL-27 receptor (IL-27RA) that, with gp130, forms a heterodimer belonging to the type 1 group 2 of cytokine receptor family6,12. The only known ligand for this receptor is the IL-27 cytokine. IL-27RA is mostly expressed in lymphoid tissues, specifically the thymus, spleen, lymph node and peripheral blood leukocytes12,13. To determine the impact of genetic variants in IL27RA identified in these patients, the expression of IL-27RA protein was examined in T cells (Fig. 1d–g). IL-27RA was upregulated in control T cells (from healthy donors) in response to stimulation with CD3 and CD28, whereas no or very weak IL-27RA expression was detected on the surface of activated T cells from P1.1, P1.2 and P2 (Fig. 1d,e). IL-27RA expression was also absent in T cell extracts from P1.1 and P1.2 as shown by western blotting, contrasting with the detectable amounts of IL-27RA in extracts from control T cells (Fig. 1f). In P2, IL-27RA protein was detected, although at a significantly lower molecular mass than wild-type IL-27RA detected in control T cells (Fig. 1g). This product with a low molecular mass was also weakly detected in control cells and probably corresponds to immature or degraded IL-27RA protein that accumulates in P2 cells as a result of both mutations p.Arg446Gly and p.Gln381_Ala395del mutations (Fig. 1e,g). Furthermore, although the IL-27RA mutant protein was not or weakly expressed at the surface of P2 cells, it was detectable intracellularly using flow cytometry and immunohistochemistry, showing IL-27RA accumulation in the cytoplasm (Fig. 1e and Extended Data Fig. 2c,d). T cells from both parents of P2 showed intracellular IL-27RA staining and decreased IL-27RA surface staining, consistent with their heterozygous status and indicating that both mutations have deleterious consequences (Extended Data Fig. 2d).
IL-27 when bound to the gp130–IL-27RA heterodimer activates the JAK–STAT pathway, predominantly STAT1 and STAT3. This activation is mediated by the intracytoplasmic domains of gp130-IL27RA that contains binding sites for JAK1/25,6,13. The ability of IL-27 to activate IL-27RA signalling was therefore investigated in T cell blasts of patients using flow cytometry and western blotting. STAT1 and STAT3 were rapidly phosphorylated from 2 min in lysates of control T cells in response to IL-27 and phosphorylation persisted at 30 min (Fig. 2a and Extended Data Fig. 3a). Phosphorylated STAT1 (p-STAT1) and p-STAT3 proteins were also detectable by intracellular staining after 15 min of stimulation with IL-27 (Fig. 2b and Extended Data Fig. 3b). By contrast, phosphorylation of STAT1 and STAT3 was weakly or not at all detectable in IL-27-stimulated T cell blasts from P1.1 and P2. Residual phosphorylation is probably explained by remaining IL-2 in the medium as it disappeared when cells were starved (Extended Data Fig. 3c). Notably, IL-27 did not trigger MAP kinase activation in both control and patient T cells (depicted by the phosphorylation of ERK1/2) in contrast to anti-CD3 stimulation (Fig. 2a). Thus, these data demonstrate that the genetic variants in IL27RA identified in the three patients result in a loss of function (LOF). However, the clinical incomplete penetrance observed in the Finnish population associated with the homozygous mutated IL-27RAArg446Gly allele strongly supported that this allele is hypomorphic, although we could not detect residual signalling in P2, probably because it is expressed in the presence of the second mutated allele (p.Gln381_Ala395del). To clarify this point, the two alleles of P2 were expressed separately in the mouse cell line NIH-3T3 (that did not respond to IL-27) and tested for their ability to activate the phosphorylation of STAT1 and STAT3 after IL-27 stimulation (Extended Data Fig. 4). Although the IL-27RAGln381_Ala395del allele was not expressed and did not trigger p-STAT1 nor p-STAT3 in response to IL-27, the IL-27RAArg446Gly-coding allele resulted in a weak but detectable IL-27RA expression associated with a significant increase in STAT1 and STAT3 phosphorylation in response to IL-27 stimulation. These data confirm that the IL-27RAArg446Gly allele is hypomorphic and therefore probably accounts for the incomplete clinical penetrance in the Finnish population.

a, Immunoblot analysis of p-STAT1, STAT1, p-STAT3, STAT3, p-ERK1 and p-ERK2 (p-ERK1/2), ERK1/2 and GAPDH expression in a healthy control individual (Ctrl) or patient P1.1 T cells stimulated with IL-27 for different periods of time as indicated (left) or IL-2, anti-CD3 or not stimulated for 5 min (right). b, FACS analysis of intracellular p-STAT1, p-STAT3 expression and isotype control in control and P1.1 T cells stimulated or not with IL-27 for 15 min. Analysis of total T cells after gating on CD3+. c, Overlaid FACS histograms showing T cell divisions by dilution of CellTrace Violet (CTV) dye in PBMCs of a control individual and P1.1 stimulated with incremental doses of coated anti-CD3 antibodies in the presence or not of IL-27 (50 ng ml−1) for 5 days. d, The same as in c except that PBMCs from a patient with STAT1 deficiency (STAT1 LOF) were tested. e–g, Control and P1.1 T cells were transduced with an empty lentivector (empty) or containing a cDNA encoding wild-type IL-27RA (pLVX-IL27RA) with mCherry as a reporter gene of transduced cells. e, Immunoblot analysis of p-STAT1, STAT1, p-STAT3, STAT3, actin and IL-27RA from lysates of transduced cells stimulated or not with IL-27 for 15 min. f, FACS analysis of p-STAT1, p-STAT3 and mCherry expression in cells stimulated with IL-27 for 15 min. g, Overlaid FACS histograms showing cell divisions by CTV dye dilution of transduced mCherry+ (filled) or not mCherry− (clear) T cells that were stimulated with incremental doses of coated anti-CD3 antibodies in the presence of IL-27. For a–c, the same results but for patient P2 are shown in Extended Data Fig. 3. For a and e, molecular mass values in kDa are shown at the middle (a) and left (e). One representative experiment of one (d) of two (a, b and e), three (f), four (g) and five (c) independent experiments.
Role of IL-27 in T cell responses
We next assessed the functional consequences of IL-27RA deficiency. Early studies showed that IL-27 could favour T helper type 1 (TH1) differentiation in mice14,15,16,17. Accordingly, the production of TH1-cell-associated cytokines (IFNγ and TNF) and IL-2 by naive CD4+ T cells from P1.1 and P1.2 was found to be significantly impaired (Extended Data Fig. 1d), whereas the numbers of TH1 and TH1* cells were within the normal ranges (Extended Data Fig. 1c). IL-27 is also known to promote proliferation of naive CD4+ and CD8+ T cells18,19,20. More recently, the expansion of memory-like CD8+ T cells in a mouse model of persistent viral infection was found to be IL-27 dependent7. We therefore postulated that the robust expansion of EBV-specific T cells that is a key step in the immune control of EBV infection2 could be IL-27 dependent. To assess this possibility, T cell proliferation was analysed after stimulation with anti-CD3 antibodies in the absence or presence of IL-27 (Fig. 2c and Extended Data Fig. 3d). Addition of IL-27 alone did not induce the proliferation of control T cells, but it provided a synergistic effect on proliferation induced by low doses of anti-CD3 antibodies (0.001–0.1 μg ml−1), an effect that is lost at the highest concentration of anti-CD3 antibodies (1 μg ml−1) and is inhibited by addition of blocking anti-IL-27 antibodies (Extended Data Fig. 3e). Notably, this synergistic effect of IL-27 was restricted to naive T cells (Extended Data Fig. 3f,g). By contrast, IL-27 had no effect on anti-CD3-induced proliferation of T cells from patients (P1.1 and P2) (Fig. 2c and Extended Data Fig. 3d). Furthermore, as expected from the critical role of STAT1 in IL-27RA signalling and function, T cells from a patient with STAT1 deficiency did not proliferate in response to IL-27 (Fig. 2d).
To formally prove that the variants in IL27RA directly impeded IL-27 function in T cells, T cells of P1.1 were transduced with a lentiviral vector (pLVX) containing a cDNA encoding wild-type IL27RA (pLVX-IL27RA) to restore expression of IL-27RA (Fig. 2e–g). The pLVX plasmid also contains a mCherry reporter gene that enables the sorting or tracking of transduced cells. Transduction of P1.1 T cells with pLVX-IL27RA, but not with empty pLVX vector, restored IL-27RA expression as assessed by western blotting (Fig. 2e (bottom blots)). When IL-27RA expression was restored in P1.1 T cells, increased amounts of phosphorylated STAT1 and STAT3 after IL-27 stimulation were found in cell lysates (Fig. 2e). p-STAT1 and p-STAT3 were also detected by intracellular staining in pLVX-IL27RA-transduced mCherry+ patient T cells, but not in mCherry− cells (non-transduced cells), whereas no phosphorylated STAT1 and STAT3 were found in patient T cells that were transduced with an empty pLVX vector (Fig. 2f). Enhancement of CD3-dependent proliferation by IL-27 was also restored in mCherry+ patient T cells to a comparable level to that observed in control T cells, but not in mCherry− patient T cells (Fig. 2g). No such an effect was observed in cells that were transduced with an empty pLVX vector. Notably, overexpression of IL-27RA in control T cells resulted in enhanced phosphorylation of STAT1 and STAT3 by IL-27, but this was not associated with increased proliferation. Taken together, these data indicate that IL-27 can effectively sustain TCR-mediated T cell proliferation, while this synergistic effect is abolished in patients with IL-27RA deficiency.
This function of IL-27 could therefore impact the expansion and differentiation of naive EBV-specific T cells. To address this, we first screened peripheral blood mononuclear cells (PBMCs) of P1.1 and P1.2, who both expressed class I MHC HLA-A02* molecules for the presence of EBV-CD8+ T cells using HLA-A2 pentamers containing EBV peptides. EBV-specific CD8+ T cells were detected among PBMCs of both P1.1 and P1.2, 9 months and 14 years after IM, respectively (Fig. 3a–c), therefore suggesting that, at first sight, IL-27 was not required for induction, expansion and/or survival of EBV-specific CD8+ T cells.

a–e, Expansion, phenotype and cytotoxic function of EBV-specific T cells from PBMCs of patients P1.1, P1.2 and a healthy control donor co-cultured with their corresponding autologous EBV-infected B cell lines (LCLs). EBV-specific T cells were detected with HLA-A2-EBV pentamers. a, The experimental plan for b–e with the following different steps: expansion (b,c), phenotyping (d) and the testing of cytotoxic function (e) of EBV-specific T cells. b, Representative dot plots (left) from FACS analysis showing the percentages of EBV-specific T cells from PBMCs of a control individual (Ctrl), P1.1 and P1.2 co-cultured or not (day 0, ex vivo) with LCLs at day 9. Samples of P1.1 at 9 months after IM with still persistent high EBV load and P1.2 at 14 years after IM. c, Graph bars from dot plots (as in b) showing the percentage of EBV-specific T cells at day 0 and day 9 of coculture with LCLs. d, FACS analysis of the percentage of EBV-specific CD8+ T cells expressing activation, differentiation and exhaustion T cell markers from PBMCs of a control individual, P1.1 or P1.2 cocultured with LCLs at day 0 (top) and day 9 (bottom) as in a. In b and c, the yellow squares correspond to P1.1 at the time that she had severe IM (P1.1-SIM). For b–d, data are from seven independent experiments for P1.1 and three experiment for P1.2. e, The cytotoxic response of EBV-specific T cells from a control individual and P1.1 at day 9 of expansion against autologous (circle) or HLA-A2 matched (square) LCLs expressing fluorescence resonance energy transfer (FRET)-activated caspase 3 (activated-CASP3+ LCLs). With control T cells, HLA-A2-matched LCLs correspond to IL-27RA-deficient LCLs (from P1.1) while, for P1.1 T cells, HLA-A2-matched LCLs correspond to control LCLs. The percentages of activated CASP3+ LCLs corresponding to LCLs under killing in which caspase 3 has been activated. Effector EBV-specific T cell to target LCL ratios are indicated on the x axis. One representative experiment of two independent experiments with three biological replicates. The same statistical significance was determined for the second experiment. f, Representative FACS analysis of the percentage of LCLs cells expressing FRET-activated CASP3 after being incubated with CD8+ T cell blasts from control individuals (n = 3), P1.1 or P1.2 with anti-CD3 antibodies in the presence or not of IL-27. The ratio of target (LCLs) and effector cells (CD8+ T cell blasts) is shown on the x axis. Data from one of two independent experiments. For c–f, data are median ± s.d., with each symbol corresponding to one independent biological sample. Statistical analysis was performed using two-tailed Mann–Whitney U-tests (c), two-tailed unpaired t-tests (d) and unpaired and paired two-tailed t-tests to compare the control versus patient and IL-27 versus no IL-27 groups, respectively (e).
However, in contrast to EBV-specific CD8+ T cells from HLA-A2-positive healthy individuals, EBV-specific T CD8+ cells from P1.1 and P1.2 did not expand in vitro when co-cultured with irradiated autologous EBV-transformed B cells (known as lymphoblastoid cell lines (LCLs)) (Fig. 3b,c). Furthermore, in contrast to control EBV-specific CD8+ T cells (from healthy donors), EBV-specific CD8+ T cells as well as non-specific CD8+ T cells of P1.1 and P1.2 expressed elevated levels of exhaustion and activation markers such as CD25, CD137, CD40L, PD1-L, KRGL1, CD57, LAG3 and 2B4/CD244 after 9 days of co-culture with LCLs (Fig. 3d and Extended Data Fig. 5a). Analysis of a blood sample from P1.1 (designated P1.1-SIM in Fig. 3c,d) at the time of the acute/severe IM (SIM) showed EBV-specific CD8+ T cells with a similar exhausted and activated phenotype consistent with the data from in vitro expansion experiments. When tested, exhausted and activated EBV-specific T CD8+ cells from P1.1 (that did not expand) exhibited a decreased ability to kill autologous LCLs or HLA-A2-matched LCLs compared with control EBV-specific T cells (Fig. 3e). Notably, LCLs of the patient were killed as efficiently as control LCLs by control EBV-specific T cells. These data suggest that IL-27RA deficiency in LCLs does not impair antigen-presenting ability. As P2 did not express class I MHC HLA-A02* molecules, we could not analyse EBV-specific T cells in this patient as only EBV-specific HLA-A2 pentamers are available to us. To circumvent this limitation, expanded CD8+ T cells from P2 were examined after in vitro co-culture of PBMCs (from P2) with autologous LCLs (Extended Data Fig. 5b). The resulting T cell population should contain most of the EBV-specific CD8+ T cells present in the PBMCs of the patient. Expanded CD8+ T cells from P2, in contrast to control expanded CD8+ T cells, also exhibited increased levels of activation and exhausted markers, similar to EBV-specific T cells from P1.1 and P1.2 (Fig. 3d).
Notably, LCLs of P1.1 and P1.2 expressed the same levels of HLA-A2 molecules as control LCLs (Extended Data Fig. 6a) and CD70 and CD137L expression was also similar between P1.1, P1.2, P2 and control LCLs (Extended Data Fig. 6b), while EBV-specific T cells from P1.1, P1.2 and P2 (at day 0) displayed CD137, CD27 and CD244 (also known as 2B4) expression levels comparable to control cells (Fig. 3d and Extended Data Fig. 5b). SAP, the product of the X-linked lymphoproliferative syndrome type 1 gene (SH2D1A) was normally expressed in natural killer (NK) and T cells of P1.1 (Extended Data Fig. 5c). These data suggest that there is no interference between IL-27 and the CD27–CD70 and CD137–CD137L pathways, which are known to be important for an efficient expansion of EBV-specific T cells, as well as the SAP–CD244 pathway, which is key to eliminating EBV-infected cells1,4. Collectively, these data show that, in the absence of IL-27RA, the proliferation and differentiation of EBV-specific T cells into potent effector T lymphocytes are impaired in association with an abnormal activation and exhausted-like phenotype of EBV-specific T cells. These data could also indicate a direct role of the IL-27–IL-27RA pathway in priming cytotoxic effector activity (independently of its role in proliferation) as suggested by a recent study21. To test this, we analysed cell cytotoxicity of PHA-derived CD8+ T cells from controls, P1.1 and P1.2 in response to TCR/CD3 stimulation after pre-incubation or not with IL-27 for 48 h. We observed that IL-27 enhanced cell cytotoxicity of control T cell blasts, whereas this positive effect was lost in IL-27RA-deficient PHA-derived T blasts (from P1.1 and P1.2) (Fig. 3f). In these conditions, PHA-derived T cell blasts from patients did not show an exhausted over-activated phenotype (Extended Data Fig. 7a). Strengthening these observations, a similar potentiating effect by IL-27 was observed on the TCR-dependent cell cytotoxicity of super-antigen TSST-1-derived T cell blasts (Extended Data Fig. 7b), while IL-27 in these conditions had no effect on the expansion and phenotype of these T cells (Extended Data Fig. 7c,d). Taken together, these observations indicate that IL-27 has the ability to directly enhance or prime the cytotoxic activity of CD8+ T cells. Thus, the absence of IL-27 signals may result in non-productive activation, accelerated exhaustion and decreased effector cytotoxic function of EBV-specific T cells subsequently impairing control of EBV-infected B cells.
IL-27 production by EBV-infected B cells
IL-27 is a heterodimeric cytokine composed of IL-27p28 (encoded by IL27) and the EBV-induced gene 3 (encoded by EBI3). IL-27 is mainly produced by antigen-presenting cells, namely macrophages, dendritic cells and B cells22,23,24. Most of these observations were made in mice in the context of persistent viral infection. Notably, B-cell-derived IL-27 appears to be important for the control of persistent LCMV infection23. As EBI3 was initially identified in EBV-transformed B cell lines or LCLs25 and found to be expressed in EBV-positive lymphomas25,26,27, we hypothesized that B cells could be a major source of IL-27 after infection by EBV. This was also supported by the IL-27RA-dependent proliferation of EBV-specific T cells when co-cultured with LCLs (Fig. 3b,c). To investigate this further, B cells were infected with GFP-tagged EBV and analysed for IL-27 production. In GFP-positive EBV-infected B cells, IL-27 expression was upregulated, but this was not observed in GFP− B cells (Fig. 4a). Furthermore, IL-27 accumulated in the supernatant of PBMCs after infection by EBV, as well as in the supernatants of LCLs that arose from long-term cultures of PBMCs infected by EBV (Fig. 4b). Intracellular expression of IL-27p28 and EBI3 was also detectable in LCLs (Extended Data Fig. 6c). Taken together, these results show that, after infection and transformation by EBV, B cells are capable of producing substantial amounts of IL-27. Thus, this direct production of IL-27 by EBV-infected B cells may represent an important source of IL-27 required for the initiation and support of proliferating and differentiating EBV-specific T cells into potent effector T cells. Consistent with this hypothesis, neutralization of IL-27 in co-culture experiments with LCLs resulted in a significant impairment of EBV-specific T cell expansion (Extended Data Fig. 5d–g).

a, FACS dot plots of CD19 and GFP expression of PBMCs at day 3 after infection or not with EBV-GFP (top). Bottom, histograms of IL-27 expression in infected CD19+GFP+ B cells (right) or in non-infected CD19+GFP− B cells (left). b, IL-27 quantification in the supernatants of control PBMCs (donors, n = 12) at different days after EBV infection or different EBV-transformed B LCLs (n = 12) (right bar). Each circle corresponds to an independent biological sample. c–f, Growth of LCLs from different control donors (n = 5), patients with IL-27RA deficiency (P1.1, P1.2 and P2) (n = 3) and patients with STAT1 LOF (n = 4). c, The percentages of living LCLs at different days of culture were calculated from direct cell counts. Values were normalized to cell numbers at day 0. Values are from three independent experiments, except for day 12 and day 14, for which there was one experiment. At day 12 and 14, the percentages are 16% and 10%, respectively. d, FACS histograms of IL-27RA expression in LCLs. The dashed line shows the isotype control. e, Immunoblot analysis of p-STAT1, STAT1, p-STAT3, STAT3 and actin expression in LCLs stimulated with IL-27 or not during 15 min. Molecular mass values in kDa are shown on the right. f, Expansion indexes calculated from cell divisions analysed by CellTrace Violet dilutions at day 0, 3 and 5 of culture of LCLs. g, The same as in f, except that LCLs of P1.1, P1.2 and P2 or control LCLs were transduced with a lentiviral vector for IL-27RA (pLVX-IL27RA) or an empty pLVX vector. h, The percentages of living LCLs as in c, cultured in the presence or not of neutralizing anti-IL-27 or anti-IL-2 antibodies. Values are from three independent experiments. For c and h, data are median ± s.d. of the percentages of living cells in culture. Statistical analysis was performed using two-tailed Mann–Whitney U-tests (c and h) and two-tailed unpaired t-tests (b, f and g). One representative experiment of two (e), three (a) or five (d) independent experiments. Data from two (g) or three (b, c, f, h) independent experiments.
IL-27 and EBV-transformed B cell growth
In the course of these experiments, we noticed that the generation of EBV-transformed B cell lines (LCLs) from PBMCs of all three IL-27RA-deficient patients was delayed and, when established, these cells did not expand as efficiently as control LCLs (from healthy donors) (Fig. 4c and Extended Data Fig. 6d). Notably, IL-27RA-deficient LCLs, like control LCLs, exhibit a classical pattern of EBV type III latency gene expression associated with LCLs28 (Extended Data Fig. 8a). These findings exclude the possibility that an abnormally high viral replication (associated with expression of EBV lytic genes) in these cells could have had a role in their decreased ability to expand. These observations suggest an additional role for IL-27 in promoting the growth of LCLs through an autocrine loop. Indeed, control LCLs were found to express IL-27RA, while no expression of IL-27RA was detectable on the surface of LCLs from P1.1, P1.2 and P.2 (Fig. 4d). Notably, STAT1 and STAT3 phosphorylation were detectable in control LCLs after incubation with IL-27, indicating that IL-27RA is functional in LCLs. By contrast, phosphorylation of STAT1 and STAT3 was absent in IL-27-stimulated IL-27RA-deficient LCLs from P1.1, P1.2 and P2 (that are IL-27RA-deficient). Furthermore, IL-27-induced STAT1, but not STAT3, was abolished in LCLs derived from patients with autosomal recessive complete STAT1 deficiency (Fig. 4e). As a positive control, IL-6-induced STAT1 and STAT3 phosphorylation in IL-27RA-deficient LCLs (like in control LCLs) was preserved, indicating that intrinsic activation of STAT1 and STAT3 is functional in IL-27RA-deficient LCLs (Extended Data Fig. 9a). In support of the existence of an autocrine loop, high-sensitivity western blots revealed basal STAT1 and STAT3 phosphorylation (without adding exogenous IL-27) in control LCLs that was diminished in IL-27RA-deficient LCLs (Extended Data Fig. 6f).
STAT1-deficient LCLs also failed to expand in vitro (Fig. 4c). The decreased expansion of IL-27RA-deficient (and STAT1 LOF) LCLs was associated with a significant diminution of indexes of expansion compared with those of control LCLs (Fig. 4f and Extended Data Fig. 9b). The low proliferation rate associated with defective STAT1 phosphorylation of IL-27RA-deficient LCLs was amended to levels comparable to those of controls LCLs when IL-27RA expression was restored by transducing the expression vector pLVX-IL27RA. No correction was observed in cells that were transduced with an empty pLVX vector (Fig. 4g and Extended Data Fig. 9c–e). Notably, IL-27RA and STAT1-deficient LCLs produced amounts of IL-27 that were similar to those produced by control LCLs, thereby excluding a defect in IL-27 production as a factor accounting for the poor cell growth (Extended Data Fig. 6e). These data demonstrate that impaired proliferation and STAT1 phosphorylation of IL-27RA-deficient LCLs directly result from IL-27RA deficiency. Furthermore, the addition of a blocking antibody to IL-27, but not to IL-2, significantly decreased the growth of control LCLs (Fig. 4h and Extended Data Fig. 10a) and abolished IL-27-induced STAT1 and STAT3 phosphorylation (Extended Data Fig. 10b). Accordingly, CRISPR–Cas9 genome editing that selectively inactivated IL27 in control LCLs by targeting exon 2 of IL27 resulted in a rapid accumulation of DAPI+ dead cells in the cultures, whereas genome editing using an empty CRISPR–Cas9 vector moderately reduced cell viability (Extended Data Fig. 10c). As expected, addition of exogenous IL-27 prevented the death of IL27-inactivated LCLs. However, cells did not grow in these conditions. This may be explained by decreased IL-27RA expression in the absence of IL-27; IL-27 when bound to IL-27RA could be required to stabilize IL-27RA cell surface expression and/or to activate IL27RA gene expression. This assumption was confirmed by the study of control LCLs that were engineered to overexpress IL-27RA (after transduction with pLVX-IL27RA), in which IL27 was inactivated by CRISPR–Cas9 resulting in decreased IL-27 production (Extended Data Fig. 10d). In contrast to control LCLs in which IL27 was inactivated, LCLs overexpressing IL-27RA (and in which IL27 was inactivated) survived when exogenous IL-27 was added (Extended Data Fig. 10e). Importantly, deprivation of exogenous IL-27 rapidly triggered cell death. No such effect was observed in cells that were transduced with an empty CRISPR–Cas9 vector. Thus, these results show that autocrine IL-27 production is required for cell growth of EBV-transformed B cells. The effect of the IL-27–IL-27RA pathway on gene expression in LCLs was further examined in response to IL-27 stimulation (Extended Data Fig. 8b). Analysis of transcript expression signatures showed that data from IL-27-stimulated IL-27RA-deficient LCLs clustered with those from non-stimulated control LCLs, suggesting that most of the transcriptional changes triggered by IL-27 in control LCLs are lost in IL-27RA-deficient cells, although IL-27 can still induce a subset of genes in IL-27RA-deficient cells. Differentially expressed genes by the IL-27–IL-27RA pathway involved genes that are known to be important for that survival of B lymphocytes such as BCL6.
Notably, IL-6, which belongs to the same cytokine family as IL-27, is known to be a growth factor for LCLs29,30, and EBI3 has been reported to trigger IL-6 trans-signalling31. Thus, IL-6–IL-6 receptor signalling through IL-6 or EBI3, which is also STAT1 dependent could account for some residual growth (Fig. 4c) and gene expression (after IL-27 stimulation) of IL-27RA-deficient LCLs as compared to STAT1-deficient LCLs (Extended Data Fig. 8b).
Neutralizing anti-IL-27 autoantibodies
Neutralizing anti-cytokine autoantibodies have been shown to underlie various infectious diseases32. Some of these anti-cytokine autoantibodies phenocopy genetically determined immunodeficiencies such as defects in type I and type II IFN32, IL-17A/F33,34 or IL-635,36. Autoantibodies neutralizing type I IFN are particularly relevant to our study, as they underlie at least three viral diseases—critical COVID-19 pneumonia37, adverse reaction to the live-attenuated yellow fever viral vaccine38 and critical influenza pneumonia39. We therefore hypothesized that anti-IL-27 autoantibodies might exist and be involved in sporadic acute severe EBV-driven IM as observed in few adolescents and adults3. To test this possibility, we developed an enzyme-linked immunosorbent assay (ELISA) to detect anti-IL-27 autoantibodies to screen serum samples of patients with severe IM requiring hospitalization and/or immunosuppressive treatments for most of them (Extended Data Table 1), T/NK EBV+ lymphoproliferative diseases such as T/NK-cell type chronic EBV active infection (CAEBV) (Extended Data Table 2), a severe viral disease characterized by persistent infection of T and/or NK cells40,41, patients with IEIs previously associated with and high titres of various anti-cytokine autoantibodies (such as STAT1 gain-of-function (GOF) mutations and AIRE deficiency known to cause autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) syndrome) (Fig. 5a and Extended Data Fig. 11a). High titres of autoantibodies that positively reacted with recombinant IL-27 in ELISA-like experiments were detected in serum samples from most patients with IM, CAEBV and STAT1 GOF. By contrast, very low titres or no autoantibodies were detectable in the sera of patients with APECED or in most of healthy control individuals. Notably, the presence of anti-IL-27 autoantibodies was significantly correlated with high levels of IL-27 in the sera of patients, and autoantibodies complexed with IL-27 were consistently detected (Fig. 5a and Extended Data Fig. 11a). Notably, in the four tested patients with IM, anti-IL-27 autoantibodies decreased over time after IM, while in the three CAEBV patients tested, anti-IL-27 autoantibodies remained stable (Extended Data Fig. 11b).

a, Detection and quantification of anti-IL-27 IgG and IgA antibodies (left) and IL-27 (right) in the serum of healthy control individuals (n = 35, including 4 controls negative for EBV), patients with EBV-associated pathologies including IM (n = 25) and chronic active EBV infection (CAEBV) (n = 15), patients carrying STAT1 GOF mutations (n = 24) or patients with APECED syndrome (n = 6). The arbitrary optical density at 450 nm (OD450) was used for anti-IL-27 antibody quantification. Each black circle corresponds to one healthy donor or one patient. Data are from six independent experiments for anti-IL-27 autoantibodies and IL-27. b, Representative histograms showing cell divisions by CellTrace Violet dye dilution of naive CD4+ T cells stimulated with coated anti-CD3 antibodies in the presence or not of IL-27 incubated with serum samples of a control individual (Ctrl), a patient with IM or a patient carrying STAT1 GOF, or blocking anti-IL-27 antibody (right). Left, the index of expansion of naive CD4+ T cells calculated from FACS histograms as shown on the right (with the same colour code). Each white circle corresponds to one experimental condition without serum (n = 7), with one serum sample or the anti-IL-27 monoclonal antibody (n = 5). Different healthy control individuals (n = 9), patients with IM (n = 10; IM1, IM2, IM3, IM4 (×2), IM6, IM7, IM11, IM13, IM22), patients with chronic active EBV infection (CAEBV) (n = 4; CAEBV2, CAEBV4, CAEBV5, CAEBV8) or patients with STAT1 GOF (n = 5; STAT1 GOF 5, STAT1 GOF 7, STAT1 GOF 9, STAT1 GOF 14) were tested. Data are from five independent experiments. For a and b, data are median ± s.d., with each symbol corresponding to one independent biological sample. Statistical analysis was performed using two-tailed Mann–Whitney U-tests (a) and two-tailed paired t-tests (b).
Consistent with the ELISA data, sera from patients with IM or STAT1 GOF mutations, but not from serum from a healthy donor and an APECED patient, were able to detect the IL-27p28 subunit of recombinant IL-27 by western blotting (Extended Data Fig. 11c,d). These results are therefore consistent with the presence of autoantibodies specific to IL-27 in patients with IM, CA-EBV and STAT1 GOF mutations. Importantly, all sera tested from patients (IM (n = 8), CAEBV (n = 3) and STAT1 GOF (n = 4)) that contained anti-IL-27 autoantibodies were found to be neutralizing as they significantly inhibited the synergistic CD3-dependent cell proliferation triggered by IL-27 in T cells (Fig. 5b). Autoantibodies from IM and CAEBV also neutralized IL-27-dependent p-STAT1 induction in T cells and LCLs (Extended Data Fig. 12a,b). This inhibition was similar to that obtained using a blocking anti-IL-27 monoclonal antibody, whereas sera from healthy donors (n = 9) caused no inhibition (Fig. 5b). Finally, IgG-depleted sera of patients did not retain any neutralizing activity, while the purified enriched IgG fraction contained the neutralizing activity (Extended Data Fig. 12c), confirming that the neutralization is caused by autoantibodies that are mostly contained in the IgG fraction. Dosage of the neutralization activity showed variability, with some sera being strongly neutralizing (Extended Data Fig. 12d,e). Notably, patient IM13, who did not receive immunosuppressive treatment indicating a lesser severity disease (Extended Data Table 2), had the lowest neutralization titre. Along these lines, in LCLs, the neutralization activity on p-STAT1 of three serum samples (out of four) of patients with CAEBV was stronger than that of samples of patients with IM (Extended Data Fig. 12b). These elements suggest a correlation between neutralization titre/activity with the persistence and the severity of EBV infection. Collectively, these data demonstrate the presence of neutralizing anti-IL-27 autoantibodies in the serum of patients with IM, CAEBV and STAT1 GOF.
Discussion
Here we report an IEI characterized by restrictive susceptibility to EBV, revealing the importance of IL-27 in anti-EBV immunity. We also showed that EBV triggers IL-27 production by B cells, further emphasizing the link between EBV and IL-27. Previous studies suggested that this production is directly activated by EBV. Indeed, production of the IL-27-subunit EIB3 by EBV-infected B cells is dependent of the EBV coding protein LMP-125. IL-27RA deficiency is undoubtedly associated with a selective susceptibility to EBV. However, the clinical penetrance appears to be incomplete in the Finnish cohort. This is probably explained by the residual expression and activity of the IL-27RA(Arg446Gly) variant (Extended Data Fig. 4). Incomplete penetrance of EBV susceptibility has been reported in patients with TNFRSF9 deficiency, with more than 30% being asymptomatic42,43,44. We also cannot exclude the role of additional genetic factors as was shown for TNFRSF9 deficiency43.
No other notable infections were reported in the three patients (17 years after IM for P1.2), with the exception of P1.1 who had symptomatic VZV infection (before IM; Methods), but as it is a frequent symptomatic infection in infancy, it could not be a consequence of the IL-27RA deficiency. When further patients are identified, we cannot exclude that additional clinical phenotypes will be found to be associated with IL27RA deficiency, including infections to other pathogens. This is often the case for newly discovered IEIs.
A role for IL-27 in antiviral immunity in humans has been previously suspected7. Here we show that IL-27 is required for the expansion of naive EBV-specific T cells and their differentiation and/or priming into potent effector cytotoxic T cells. IL-27 can be viewed as providing an additional costimulatory signal that contributes to the required major amplification of the T cell immune response against EBV-infected B cells, as is the case for other co-stimulatory pathways involving SLAM/2B4, CD27 and CD137 receptors1. Indeed, vigorous expansion and differentiation of EBV-specific cytotoxic T cells is needed to efficiently control and eliminate expanding EBV-infected cells during primary EBV infection10,45. The effect of IL-27 was observed with weak CD3 stimulation in vitro suggesting that IL-27 is important at the early steps of the immune response when available viral antigens or infected B cells are low.
IEIs that cause cytotoxicity defects of T and NK cells have been associated with severe or fulminant IM1, resulting from the accumulation of T cells that are unable to kill EBV-infected cells by being overactivated by the persistence of infected cells. Patients with impaired expansion of EBV-specific T cells, as it is observed in inherited deficiencies of CTPS1 or CD274,46, can also develop IM that is thought to be dependent on bystander activation of T cells through the release of inflammatory cytokines. In the case of IL-27 deficiency, an impaired cytotoxic T cell response together with bystander T cell activation probably contributes to the severe IM phenotype. Furthermore, considering the regulatory anti-inflammatory functions also assigned to IL-276,16, IL-27 might limit the expansion of EBV-specific T cells once the expansion reaches a certain threshold to control excessive/deleterious inflammation or/and exert inhibitory functions on other inflammatory cells.
The non-redundant protective role of IL-27 in the primary EBV infection is also supported by the detection of neutralizing anti-IL-27 autoantibodies in individuals presenting acute sporadic severe IM and CAEBV that could inhibit IL-27-dependent expansion of EBV-specific T cells in vivo. Patients with CAEBV infection also exhibit high titres of anti-IL-27 autoantibodies with a higher neutralizing activity in some of them (than in patients with IM) and that seems to persist over time (in contrast with patients with IM). This suggests a correlation between neutralization activity or/and persistence of autoantibodies and severity/persistence of the EBV disease. Additional studies are needed to establish the level of these correlations. Patients with immune deficiency caused by STAT1 GOF also exhibited significant levels of autoantibodies raised to IL-27 but also to other cytokines47. Few patients with STAT1 GOF experienced severe viral diseases, including EBV48, although these patients are not particularly susceptible to EBV infection. Effector T cell functions in these patients are exacerbated, which might compensate for a defective IL-27 response. However, these anti-IL-27 autoantibodies might actually represent a natural regulation of IL-27 activity that is exacerbated in patients with IM and might therefore be a secondary non-causal event. Like other anti-cytokine autoantibodies, anti-IL-27 autoantibodies may exist before infection in individuals who develop IM or CAEBV once infected by EBV32. Notably, anti-IL-27 autoantibodies were detected concomitantly to a high level of IL-27 in patients, suggesting that they are induced by IL-27 secretion, even if they may pre-exist at low levels before EBV infection. Further study is warranted to better understand the role of anti-IL-27 autoantibodies in EBV infection and whether they could be associated with other viral infections.
Finally, we showed that IL-27 is also a critical autocrine growth factor of established EBV-transformed B cells. Thus, we postulate that, in vivo, IL-27 is important to maintain the fitness of EBV-infected B cells and their transformation (which depends on the EBV latency III genes). This could also explain why patients with IL-27RA deficiency did not further develop until today (after their severe IM; 6, 17 and 3 years after IM for P1.1, P1.2 and P2, respectively) protracted EBV+ B cell lymphoproliferation or lymphoma in contrast to other immune deficiencies associated with EBV infection4,42,43,44,46,49. Nevertheless, in vivo, the initial expansion of infected B cells appears not to be fully dependent on IL-27 as the patients developed severe IM, suggesting that the initial EBV latency program is sufficient to sustain B cell proliferation independently of IL-27. Moreover, patients exhibited a high EBV load, indicating that viral replication does not require the IL-27RA–IL-27 pathway in B cells and epithelial cells in which viral replication/load originally occurs during primary infection. In B cells, the role of the IL-27–IL-27RA pathway seems negligible as Ig levels were normal in the patients and the patients were not prone to infections associated with B cell defects50. Additional investigations are required to assess the mechanism(s) of IL-27 in the proliferation and maintenance/survival of EBV-infected B cells. Notably, IL-27 could be viewed as a therapeutic target to inhibit the growth of EBV+ B cells in immunocompromised individuals that occurs when EBV is reactivated or in patients with EBV+ B cell lymphomas in which the immune response can be considered to be inefficient.
Methods
Human participants and samples and study approval
The patients and the relatives studied here were living in and followed up in France. Informed and written consent was obtained from donors, patients and families of patients. The study and protocols conform to the 1975 declaration of Helsinki as well as to local legislation and ethical guidelines from the Comité de Protection des Personnes de l’Ile de France II and the French advisory committee on data processing in medical research. Experiments using samples from human participants were conducted in France and Australia in accordance with local regulations and with the approval of the IRBs of corresponding institutions. The healthy donors and patients characteristics (age, treatments, genetic information) are listed in Extended Data Tables 1–4. All healthy control individuals had no symptomatic EBV infection. Plasma and serum samples from the patients and controls were frozen at −80 °C immediately after collection.
Patient case reports
P1.1 was born from Caucasian/European consanguineous parents. Her past medical history was unremarkable, except for two benign episodes of bronchiolitis and a short hospitalization for super-infection of a chickenpox skin lesion at 13 months old. At the age of 20 months, she was hospitalized for a 7-day persistent fever with a sore throat. Physical examination showed hepatosplenomegaly (HSM) (at 3 and 5 cm from the costal margin). Blood count showed marked hyper-leucocytosis (maximum, 62 g l−1) with lymphocytosis (maximum, 45 g l−1) and excess of basophil cells, mild anaemia and a normal platelet count. LDH was increased, while bilirubin was normal. Triglycerides were increased but ferritin and fibrinogen were in the normal ranges. EBV serology was compatible with EBV primo-infection (anti-VCA IgM and IgG positive, anti-EBNA IgG negative), EBV PCR was positive between 3.56 and 4.33 log copies per ml. Peripheral immunophenotype showed sharp CD8+ lymphocytosis with an activated profile. Blood smear displayed rare cytophagocytosis and excluded malignant disease. After 15 days, due to persistent symptoms (spiking fever, increased HSM) and substantial biological abnormalities recapitulating incomplete haemophagocytosis (3 out of 8 positive criteria), the patient received steroids (methylprednisolone 2 mg per kg). Partial response and development of hyper B cell lymphocytosis prompted to start rituximab, an anti-CD20 monoclonal antibody (4 injections, 375 mg m2 per week), allowing complete clinical and biologic normalization. Owing to transient B cell depletion, Immunoglobulin substitution was started for 12 months. Three years later, at the age of 5.5 years, partial B cell deficiency was diagnosed because of absent anti-tetanus antibodies despite completed vaccination and persistent memory B cell lymphopenia. Owing to recurrent upper respiratory tract infections, weekly subcutaneous immunoglobulin substitution was restarted. At the last follow-up, at the age of 8 years, the patient is asymptomatic and is doing well. Her immunophenotype is in the normal range including memory B cells. Blood EBV viral load was low.
P1.2, the oldest brother was hospitalized at the age of 8 months for severe mononucleosis compatible with primo-infection (positive anti-VCA IgM and IgG antibodies but negative anti-EBNA IgG) fever, HSM, lymphadenopathies, hyperleukocytosis (40 g l−1), mild increase in liver enzymes (AST, 332 UI l−1; ALT, 357 UI l−1; GGT, 1,651 UI l−1; and LDH, 1,808 UI l−1). He spontaneously improved within 2 weeks, without treatment. He presented benign bronchiolitis and pneumonitis in infancy and few episodes of upper respiratory infection during childhood. At the last follow up, at the age of 18, he is healthy and well. His immunophenotype shows a mild CD4 and CD8 lymphopenia. Humoral immunity is normal (normal immunoglobulin dosage, positive post-vaccine response, normal memory B cells). Blood EBV viral load is negative. Two children of the family died in infancy (neonatal anoxia in a preterm baby and dysmorphic syndrome and hypertrophic cardiomyopathy in a boy at 4 months of age).
P2 was hospitalized at the age of 17 years for severe mononucleosis with hepatitis and HLH symptoms compatible with a primary infection (blood EBV load at 5.56 log copies per ml, positive anti-VCA IgM and IgG antibodies but negative anti-EBNA IgG). She had no familial and medical previous histories. She presented with fever, sore throat, hepatic insufficiency, HSM, lymphadenopathies, bi-cytopenia with anaemia (7.7 g dl−1), hyperleukocytosis (40 g l−1), increase in liver enzymes (AST, 140 UI l−1; ALT, 172 UI l−1; GGT, 462 UI l−1; PAL, 966 UI l−1; and LDH, 1,000 UI l−1). Triglycerides (7.25 g l−1) and ferritin (1,086 ng ml−1) were increased and fibrinogen (1.32 g l−1) decreased. The myelogram showed an accumulation of macrophages with rare haemophagocytosis images. Lymphocyte immunophenotyping showed hyperlymphocytosis (11,808 per mm3) with a high proportion of activated HLA-DR+CD8+ T cells (70%). She was treated by corticosteroid therapy with methylprednisolone leading to a rapid improvement in a few days. After corticosteroid therapy was stopped, she had a relapse 1 month later characterized by hepatic cytolysis with a massive infiltration of T lymphocytes (assessed after liver biopsy and histological analysis). One year and a half later blood EBV load was strongly reduced to 2.95 log copies per ml. However, the serology remained abnormal with no detectable IgG anti-EBNA antibodies. Two and half years later, at the last follow-up, she is well.
Features of patients and controls tested for anti-IL-27 autoantibodies
The median age of the 25 patients diagnosed with EBV+ IM was 12 years (range, 1–32 years) with a ratio of female/male R = 0.9. Nineteen required hospitalization and were diagnosed during hospitalization (Extended Data Table 1). All of the patients had EBV seroconversion indicating primary EBV infection. The median age of the 15 patients diagnosed with systemic T/NK-cell type chronic active EBV (CAEBV group) was 23 years (range, 5–50 years) with a ratio female/male R = 0.36. All patients had high persistent EBV viral load (>105 EBV copies) in the blood despite anti-CD20 (rituximab) treatment (Extended Data Table 2). The presence of circulating EBV-infected T or/and NK cells was confirmed by flow-FISH analysis using specific EBV probe51 in all of the patients. The median age of the 19 patients with STAT1 GOF mutations (STAT1 GOF group) was 27 years (range, 5–50 years) with a ratio female/male R = 0.9, and the median age of the 6 patients with AIRE mutations (APECED group) was 11 years (range, 2–18 years) with a ratio female/male R = 5. The clinical features of patients from these groups have been previously described and referred to the Extended Data Table 3. The median age of the 35 healthy donors (controls) was 41 years (range, 21–71 years) with a ratio female/male R = 1.2. The control samples were collected from blood of healthy donors recruited by the Etablissement francais du sang (EFS) and healthy volunteer donors from our institute. All controls showed EBV seroconversion, except for four individuals who were negative for EBV (Extended Data Table 4). Anti-IL-27 autoantibodies were not detected or were very low in these four individuals like in the other controls positive for EBV.
No significant variations in the IL27RA gene were detected in the genomic DNA of patients IM1, 2, 4, 8, 9, 10 and 11 and CAEBV4 by direct Sanger sequencing, and by exome sequencing in patients IM5, 6 and 7 and CAEBV1, 2, 3, 5, 6, 7 and 8 tested in this study.
Notably, only serum samples of IL-27RA-deficient patients P1.1 and P2, 6 years and 14 years after they developed IM, respectively, were available and have been tested for anti-IL-27 autoantibodies and IL-27. No anti-IL-27 autoantibodies and IL-27 were detectable in these samples.
Exome sequencing and analysis
Exome capture and analysis were performed as previously described46,49. All variants in IL27RA were predicted to be deleterious on the basis of CADD/MSC scoring (MSC of 3.3 for IL27RA) and allele frequencies (AF) in gnomAD v.2.1.1, gnomAD v.3.1.2 (https://gnomad.broadinstitute.org) and in our own data base of 2,1472 exomes and 3,701 genomes (p.Gln96X, CADD: 33, gnomAD AF: 7.96 × 10−6 (2 out of 251,336), own database AF: 0 (0 out of 50,346); p.Arg446Gly, CADD: 8.7, gnomAD AF: 5.84 × 10−4 (165 out of 282,746), own database AF: 0 (0 out of 50,346); c.1142-2A>C, CADD: 19.1, gnomAD AF: 7.96 × 10−6 (2 out of 251,336); own database AF: 1.98 × 10−6 (1 out of 50,346)).
Sanger sequencing
Genomic DNA from peripheral blood cells of the patients and family members was isolated according to standard methods. Oligonucleotide primers flanking the exon 3 (to assess c.286C>T variation in family 1: (forward 5′-GAAGCAGCAGAGGTGGGATT-3′ and reverse 5′-TCACTGGGGAAGGGGAAACT-3′) and exons 9–10 (to assess c.1336C>G and c.1142-2A>C variations in family 2: (forward 5′-ACCTGTTCCTTCCCCATTTC-3′ and reverse 5′-CTGACTGATGGGTGGGTCTT-3′) of IL27RA were used to identify variants by genomic DNA amplification. Oligo primers flanking exon 1 (forward 5′-GGACCCTGAGCGAAAGGTG-3′ and reverse 5′-CCATTCCCCAGTGTCGCC-3′), exon 2 (forward 5′-TTCGACCCCCTTTCCTGAGA-3′ and reverse 5′-CAGGCTTGACTCCTGGAACC-3′), exons 3–4 (forward 5′-AGCCGAGTGTGCATCTCATC-3′ and reverse 5′-GGCACATGCAGGCATTTTGA-3′), exons 5–6 (forward 5′-AATACCCTTGACTCAGGCCC-3′ and reverse 5′-GGGTTTCCTAACACGTGCAGA-3′), exons 7–8 (forward 5′-AGGTGACCTCAGGGAGACAC-3′ and reverse 5′-TCTAACAGCGGGGATGGAGA-3′), exons 9–10 (forward 5′-ATGAGCATGGGAAGGAGAGC-3′ and reverse 5′-ACACCATGTATGGTCCCTGG-3′), exon 11 (forward 5′-GGAAGAGCACATGCAAAGGC-3′ and reverse 5′-GACTCCAGATGCTACCTCTCC-3′), exon 12 (forward 5′-TCCACAGTGGGGAGAGGTAG-3′ and reverse 5′-GTAGTCAGGGCAGCAGATCC-3′), exon 13 (forward 5′-TTGTGGGGCTTGTTCCTGTT-3′ and reverse 5′-GAAGAGATGAGCCTGTAATCCTG-3′) and exon 14 (forward 5′-GATCCTGCCAACAGCAGTTC -3′ and reverse 5′-AAATTCAGTTCTGTTACAGCGGA-3′) of IL27RA were used to amplify and sequence the IL27RA gene from DNA of patients IM1–9. PCR products were amplified using high-fidelity Platinum TaqDNA Polymerase (Invitrogen) according to the manufacturer’s recommendations, purified using the QIAquick gel extraction kit (Qiagen) and sequenced using the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit (PerkinElmer) according to the manufacturer’s recommendations. All sequences were collected on the 3500XL genetic analyzer (Applied biosystem) using 3500 Series Data collection software v.2 (Thermo Fisher Scientific). Collected sequences were analysed using 4peaks software (v.1.8; A. Griekspoor and T. Groothuis, http://nucleobytes.com/index.php/4peaks).
Cell culture
PBMCs collected from patients and healthy donors were isolated by Ficoll–Paque density gradient (Lymphoprep, Proteogenix) from blood samples using standard procedures. Expansion of T cell blasts was obtained by incubating PBMCs for 72 h with phytohaemagglutinin (PHA) (2.5 μg ml−1, Sigma-Aldrich) or the toxic shock syndrome toxin-1 super antigen (TSST-1) (1 µg ml−1, Toxin Technology) in Panserin 401 (Pan Biotech) supplemented with 5% human AB serum (Bio West), penicillin (100 U ml−1) and streptomycin (100 μg ml−1). After 3 days, dead cells were removed by Ficoll–Paque density gradient and blasts were maintained in culture with IL-2 (100 UI ml−1) for 3 to 5 days before phenotyping analysis for CD3, CD4, CD8, CD25, CD27, CD45RA and CD57 expression, and to be tested in the various assays. For the TSST1 condition, half of the cells were supplemented and maintained with IL-27 (50 ng ml−1) and IL-2 (100 UI ml−1) every 2 days. Phenotypes of T cell blasts from healthy donors and the patient were comparable for the expression of these different markers. NIH 3T3 cells (ATCC, CRL-1658) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO-BRL (Gibco, Thermo Fisher Scientific)) supplemented with 10% (v/v) fetal calf serum (Gibco, Thermo Fisher Scientific), penicillin (100 U ml−1) and streptomycin (100 μg ml−1) in a humidified 37 °C, 5% CO2 incubator. Cells were plated in 10 cm dishes at 1 × 106 cells per plate after transfection.
EBV-transformed LCLs and EBV-specific T cell lines
EBV-transformed LCLs were obtained according to procedures previously described42,43. After obtention, LCLs were verified for the absence of mycoplasma contamination. LCLs of patients and controls were cultured in RPMI 1640 (Life Technologies) supplemented with 10% heat-inactivated fetal calf serum (Gibco), penicillin (100 U ml−1) and streptomycin (100 μg ml−1). EBV-specific T cell lines were obtained from the patient and control healthy donors using PBMCs that were co-cultured with 45 Gy irradiated autologous LCLs at a PBMC/LCL ratio of 40:1. After 8–10 days, viable cells were stimulated with 45 Gy irradiated autologous LCLs at a PBMC/LCLs ratio of 4:1. The cells were weekly restimulated with 45 Gy irradiated autologous LCLs in the presence of IL-2 (40 U ml−1). To inhibit the IL-27–IL-27RA pathway, neutralizing anti-IL-27 (2 µg ml−1) (BioLegend, MM27-7B1) and recombinant human IL-27RA Fc chimera protein (2 µg ml−1) from R&D systems were added during the co-cultures every 3 days. All LCLs from patients were verified by western blotting.
EBV-specific T cell detection
HLA genotyping of patient P1 showed that he was a carrier of HLA-A*2, for which HLA-A*2 reagents were available to assess EBV-specific T cells, while patient P2 was HLA-A*03:02/A*68:01 and HLA-B*35:03/B*38:01, for which there is no validated and commercially available reagents to assess EBV-specific T cells. EBV-specific CD8+ T cells from PBMCs of P1 and a healthy control carrier of HLA-A*2 were detected using a mix of unlabelled EBV HLA-A2:01 Pro5 Pentamers (Proimmune) mixed with R-PE Pro5 Fluorotag in addition with BV785-anti-CD3, APC-anti-CD19, BV510-anti-CD4 and BV650-anti-CD8 antibodies according to the manufacturer’s instructions. The EBV HLA-A2:01 Pro5 Pentamers mix contains four different pentamers presenting GLCTLVAML (residues 259–267 from BMLF1), FLYALALLL (residues 356–364, from LMP2), CLGGLLTMV (residues 426–434 from LMP2) or YLLEMLWRL (residues 125–133 from LMP1) peptides derived from BMLF1, LMP1 and LMP2 proteins of EBV. Non-specific HLA-A*02:01 pentamers assembled with an irrelevant peptide known to have no T cell response (Pro5 MHC Pentamer A*02:01 Negative Control Pentamer, Proimmune) were used as a negative staining control.
Flow cytometry
Cell staining and the flow-cytometry-based phenotypic analyses of PBMCs and cells were performed according to standard flow cytometry methods. The following monoclonal antibodies conjugated to fluorescein isothiocyanate (FITC), R-phycoerythrin (PE), phycoerythrin-cyanin5 (PE-Cy5), phycoerythrin-cyanin5.5 (PE-Cy5.5), phycoerythrin-cyanin7 (PE-Cy7), peridinin-chlorophyll (PerCP), peridinin-chlorophyll-cyanin5.5 (PerCP-Cy5.5), allophycocyanin (APC), allophycocyanin-Cyanin7 (APC-Cy7), allophycocyanin-Vio7 (APC-Vio7), Alexa-700, Brilliant Violet 421 (BV421), Brilliant Violet 510 (BV510), Brilliant Violet 605 (BV605), Brilliant Violet 711 (BV711), Brilliant Violet 650 (BV650) or Brilliant Violet 785 (BV785) were used: anti-CD3 (UCHT1), anti-CD4 (OKT4), anti-CD8 (RPA-T8), anti-CD11c (3.9), anti-CD14 (M5E2), anti-CD16 (3G8), anti-CD19 (HIB19), anti-CD25 (BC96), anti-CD27 (LG.3A10), anti-CD28 (CD28.2), anti-CD31 (WM59), anti-CD45RA (HI100), anti-CD45RO (UCHL1), anti-CD56 (HCD56), anti-CD57 (HNK-1), anti-CD70 (113-16), anti-CD127 (A7R34) anti-CD137 (4B4-1), anti-CD161 (HP-3G10), anti-CD183 (G025H7), anti-CD184 (12G5), anti-CD185 (J252D4), anti-CD196 (G034E3), anti-CD197 (G043H7), anti-CD279 (EH12.EH7), anti-CD303 (BDCA-2), anti-TCRαβ (IP26), anti-TCRγδ (B1), anti-IgM (MHM-88), anti-IgD (IA6-2) and anti-HLA-DR (LN3), all purchased from BioLegend and anti-IL-27RA (FAB14791P) from R&D.
Invariant natural killer T cells were detected by staining with anti-Vα24-Jα18 (6B11-BioLegend) and anti-Vβ11 (C21- Beckman Coulter) antibodies. MAIT cells were detected by staining with anti-Vα7.2 (3C10- BioLegend) and anti-CD161 (HP-3G10 -BioLegend) or using 5-OP-RU-loaded MR1 tetramer (NIH Tetramer Core Facility).
For intracellular staining of p-STAT1 and STAT3 proteins, cells were stimulated for 15 to 20 min with human recombinant IL-27 protein (Preprotech) (50 ng ml−1) and then fixed and permeabilized using the BD Phosflow kit (BD biosciences) according to the manufacturer’s instructions. The following antibodies were used: anti-p-STAT1 (pTyr701; 14 and 4a) and anti-p-STAT3 (pTyr705; 4 and p-STAT-3), all from BD Biosciences.
For intracellular staining of SAP protein, control and patient PBMCs were labelled for cell surface antigens and then fixed and permeabilized using BD FACS lysing solution and FACS permeabilizing solution 2 (BD biosciences) according to the manufacturer’s instructions and stained with the R-phycoerythrin (PE)-conjugated anti-SAP antibodies (1A9) from BD Biosciences. A list of all features/dilutions of used antibodies is provided in Extended Data Table 5. Data were collected on LSR-Fortessa cytometer (from BD Biosciences) using BD FACSDiva software v.9.0 (BD Biosciences) and analysed using FlowJo v.10.8.0 software (Tree Star).
Cytokine assays
PBMCs were incubated with monoclonal antibodies against CD8, CD4, CD45RA, CCR7, CD127 and CD25. Naive and memory CD4+ and CD8+ T cells were isolated by first excluding regulatory T cells (CD4+CD45RA−CD25highCD127low) and then sorting CD4+ or CD8+CD45RA+CCR7+ or CD45RA−CXCR5−CCR7+/− cells, respectively. Isolated naive and memory CD4+ or CD8+ T cells were then cultured in 96-well round-bottom plates (30–40 × 103 cells per well) with T cell activation and expansion beads coated with monoclonal antibodies against CD2/CD3/CD28 (Miltenyi Biotech) alone (for TH0 conditions) or supplemented with IL-12 (for TH1 differentiation) or IL-27. After 5 days, the supernatants were collected and the production of IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, IL-13, IL-17A, IL-17F, TNF and IFNγ was determined by cytometric bead arrays (Becton Dickinson). IL-22 secretion was measured by ELISA (eBioscience). For intracellular cytokine detection, activated CD4+ and CD8+ T cells were restimulated with incremental doses of anti-CD3 antibody (OKT3), anti-CD3/28-coated beads or PMA (100 ng ml−1)/ionomycin (750 ng ml−1) for 6 to 12 h in the presence of monensin (Golgistop from BD-Biosciences) and brefeldin A (Golgiplug from BD-Biosciences) added after 1 h of stimulation. Cells were then collected, stained with extracellular markers, fixed and permeabilized using the Cytofix/Cytoperm kit according to the manufacturer’s instructions (BD-Biosciences). Cells were then stained with specific antibodies to quantify intracellular levels of cytokines using anti-IL-4 (BD biosciences, 8D4-8), anti-IL-9 (BD biosciences, MH9A3), anti-IL-13 (R&D Systems, MAB213-100), anti-IL-10 (Miltenyi Biotec, REA842), anti-IL-17A (BD Biosciences, N49-653), anti-IL-17F (BD Biosciences, O33-782), anti-IL-22 (BD biosciences, MH22B2), anti-IL-21 (BD biosciences, 3A3-N2.1), anti-IFNγ (BioLegend, B27), anti-IL-2 (BioLegend, MQ1-17H12) and anti-TNF in CD4+ T cells and anti-IFNγ (BioLegend, B27), anti-IL-2 (BioLegend, MQ1-17H12), anti-TNF (BioLegend, W19063E), anti-granzyme B (BioLegend, QA16A02) and anti-perforin (BioLegend, DG9) in CD8+ T cells.
Secretion of IL-27 was assessed by ELISA (R&D Systems, DY2526), according to the manufacturer’s instructions. For intracellular staining of EBI3 and IL-27p28 subunits of IL-27 in PBMCs, LCLs and THP-1 cells, cells were first incubated with BD GolgiStop and BD GolgiPlug (both from BD Biosciences, 554724, 555029) for 3 h. Fc receptors were blocked using Human TruStain FcX (BioLegend, 422302) and cells were then fixed and permeabilized using BD Cytofix/Cytoperm (BD Biosciences, 554715) according to the manufacturer’s instructions. Afterwards, cells were incubated with anti-EBI3 (BioLegend, A15058A, 684602) or anti-IL-27p28 (BioLegend, B0315A10, 676902) antibodies or the corresponding isotype control (mouse IgG2b kappa isotype control, MPC-11, 400312) at 4°C for 30 min. Finally, cells were stained with anti-mouse IgG (BioLegend, 405307) at 4°C for 30 min. Data were collected on the LSR-Fortessa cytometer (from BD Biosciences) and analysed using FlowJo v.10.8.0 (Tree Star).
T cell proliferation assays
Direct cell proliferation of T cell blasts, PBMCs and LCLs was monitored by labelling the cells with the violet dye (Violet Proliferation Dye 450, BD Biosciences), also designated as Cell Trace Violet dye (CTV). T cell blasts or PBMCs were labelled with the violet dye and then cultured respectively for 3 and 6 days in complete medium alone or with incremental doses of coated anti-CD3 antibody (OKT3 functional grade, eBiosciences) or anti-CD3 + CD28 coated beads (Invitrogen) with or without adjunction of human recombinant IL-27 (PeproTech) at the final concentration of 50 ng ml−1. LCLs were washed three times in PBS and starved overnight in RPMI medium supplemented with 0.1% of foetal bovine serum. Then, LCLs were labelled with violet dye and cultured for 3 and 5 days in complete medium. After 3, 5 or 6 days of culture, cells were collected and Violet dye dilution was assessed using flow cytometry.
Proliferation, replication and expansion indexes were calculated using Flowjo as the total number of divisions/cells that went into division (for proliferation), the total number of divided cells/cells that went into division (for replication) and total number of cells/cells at start of culture (for expansion).
LCL growth/expansion monitoring assay
A method based on flow cytometry, which enables cell numeration, has been developed for the evaluation of cellular expansion. LCLs were seeded at 125,000 cells per well in 96-well plates or at 1 million per ml in T25. Cells in the wells were counted every 2 days using flow cytometry using fluorescent beads (Precision Count Beads, BioLegend 424902) according to the provider’s instructions and stained with DAPI or annexin V and 7-AAD (BioLegend). DAPI+ or annexin V+7-AAD+ cells were excluded. The percentage of cells every 2 days was calculated and normalized to the initial numbers of cells at day 0. Notably, as STAT1-deficient LCLs grew very slowly, they were maintained at a high cell concentration (5 million per ml) in culture before the experiment. When specified, cells were cultured with antagonist blocking antibodies to IL-27 (Ultra-Leaf purified anti-human IL-27p28 BioLegend, MM27-7B) or anti-IL-2 (Ultra-Leaf purified anti-human IL-2 BioLegend, MQ1-17H12) at 2 and 3 μg ml−1 respectively every 2 days (three times a week).
Cytotoxicity assay
EBV-transformed LCLs of patient P1.1 and one healthy donor expressing HLA-A2 were infected with a lentivirus containing pMSCV-CFP-DEVD-YFP construct (gift from P. Bousso) which enables the expression of CFP and YFP protein linked by a caspase 3 cleavage sequence (DEVD)52. Then, 3 days after infection, infected cells were sorted using GFP reporter protein expression. These LCL lines expressing CFP-DEVD-YFP were then used to measure T cell cytotoxicity when co-cultured with autologous or HLA-matched EBV-specific T cells, PHA- or TSST-1-derived T cell blasts at a different ratio of effector/target in a 96-well round-bottom plate. The number of effector T cells (from the different T cell blast cultures) to be added in the cytotoxic assays was evaluated, calculated and normalized (before their addition in the assays) using the percentage of EBV-pentamer-positive T cells for EBV-specific T cells (after 9 days of co-culture with autologous LCLs), the percentage of TCR Vβ2 CD8+ T cells for TSST-1-derived T cell blasts (after 8 days of culture) or the percentage of CD8+ T cells for PHA-derived T cell blasts (after 8 days of culture), assessed using FACS analysis (as described below). PHA-derived T cell blasts were incubated for 48 h before and during the assay with or without IL-27 (50 ng ml−1). For TSST-1-derived T cell blasts in the presence of IL-27, IL-27 (50 ng ml−1) was added during the assay. PHA and TSST-1 T-cell-blast-mediated cytotoxicity was analysed by co-incubation with LCLs plus anti-CD3 antibody (1 µg ml−1) or forehand LCLs pulsed (or loaded) with TSST-1, respectively. For loading with TSST-1, LCLs were pulsed with TSST-1 (1 µg ml−1) for 1 h at 37 °C, then washed twice before being co-incubated with T cell blasts. Mixtures of effector and LCL cells were pelleted by centrifugation and incubated for 6 h at 37 °C. Cell death of LCLs was quantified by FRET using flow cytometry analysis.
Immunoblotting
T cell blasts were stimulated at different times with human recombinant IL-27 protein (R&D) or anti-CD3/28 beads as previously described46. For experiments with LCLs, cells were washed twice and maintained at 4 °C before stimulation, and stimulated for 10 min with complete RPMI medium with IL-27 (40 ng ml−1) from Peprotech or IL-2 (10 U ml−1) or recombinant IL-6–IL-6Ra chimera protein (20 ng ml−1) from R&D Systems.
The following antibodies were used for immunoblotting: anti-p-STAT1 (anti-phosphorylated Tyr701, D4A7), anti-phosphorylated STAT3 (anti-phosphorylated Tyr705, D3A7), anti-STAT1 (D4Y6Z) and anti-STAT3 (D3Z2G) and anti-actin (D18C11) purchased from Cell Signaling Technology, anti-IL-27RA (MAB1479 191106 raised against recombinant IL-27RA Gly34-Lys516 as an immunogen) from R&D Systems. All features of used antibodies are listed in Extended Data Table 5. For IL-27 western blotting, 500 ng of IL-27 from Peprotech or BioLegend was analysed by SDS–PAGE and revealed using the neutralizing anti-lL-27 antibody (MM27-7B1), anti-IL-27 p28 (B0315A10, BioLegend) or anti-EBI3 (A15058A, BioLegend). Membranes were then washed and incubated with anti-mouse/anti-rabbit HRP-conjugated antibodies from Cell Signaling Technology. Pierce ECL western blotting substrate was used for detection.
Plasmid constructs, CRISPR–Cas9 genome editing and infections
A full-length IL27RA cDNA was obtained by PCR with reverse transcription from blasts using the forward 5′-GGACCCGGCAAGGCTG-3′ and reverse 5′-CATGAGCATCCCTCTAGCCTG-3′ specific primers. The cDNAs were verified by sequencing and inserted into a bicistronic lentiviral expression vector encoding mCherry as a reporter (pLVX-EF1α-IRES-mCherry Vector, Clontech). Viral particles for infection were obtained by co-expression of the lentiviral vector containing IL-27RA with third-generation lentiviral plasmids containing Gag-Pol, Rev and the G protein of the vesicular stomatitis virus (VSVG) into HEK293T using calcium phosphate. The viral supernatants were collected every 12 h on 2 consecutive days, starting 48 h after transfection, and the viral particles were concentrated by ultracentrifugation at 49,000g for 1.5 h at 12 °C. The control and patient T cell blasts were infected with viral particles at a minimal titre of 108 TU ml−1 (multiplicity of infection, 10) and, 48 h after infection, cells were maintained in IL-2 in culture before STAT phosphorylation analysis or proliferation assays.
cDNAs encoding IL-27RA(Arg446Gly) and IL-27RA(Gln381_Ala395del) were obtained by mutagenesis from the full-length IL27RA cDNA cloned into the pLVX lentiviral expression vector using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) according to manufacturer’s instructions. The following pairs of oligonucleotides designed using an online tool (https://nebasechanger.neb.com) were used for the mutagenesis (IL-27RA(Arg446Gly), forward 5′-GCACCAGCTTGGAGGCCACCT/reverse 5′-CTTGGGACCTCTCCCCAC; and IL-27RA(Gln381_Ala395del), forward 5′-TCTCTGCTTCAGGC/reverse 5′-CTGGTAACAGAGCAC). The presence of mutations was confirmed by Sanger sequencing using forward 5′-CATGTAGTGGACTGGGCTCG-3′/reverse 5′-TGTCCAGCGATGGTAGATGC-3′ oligonucleotides. pLVX vectors encoding wild-type IL-27RA, IL-27RA(R446G) and IL-27RA(G381_A395del) were introduced into NIH-3T3 cells using lipofectamin transfection reagent (Invitrogen). Then, 36 h after, cells were tested for IL-27RA expression and for phosphorylation of STAT1 and STAT3 after stimulation with IL-27 (50 ng ml−1) for 15 min.
For IL-27 gene knockdown by CRISPR–Cas9, the SpCas9(BB)-2A-eGFP (PX458) plasmid (48138, Addgene) was used for genome editing. Single-guide RNAs (sgRNAs) were designed according to a previously reported procedure49. sgRNA sequences targeting the IL27 gene is CACCGGTGCCGCTGCAGATCGCGG for exon 2. Pairs of synthetized oligonucleotides were designed online (http://crispor.tefor.net/; forward 5′-CACCGAAGCTGCTCTCCGAGGTTCG/reverse 5′-AAACCGAACCTCGGAGAGCAGCTTC), annealed, phosphorylated, ligated to linearized PX458 plasmid and transferred into Stabl3 bacteria (Thermo Fisher Scientific). sgRNA insertion in the plasmid was confirmed by Sanger sequencing using forward 5′-GGAAACATCAGGGAGCTGCT-3′/reverse 5′-CAAGCTGGTGTCTGGGGATT-3′ primers. The efficiency of sgRNAs was tested in EBV-transformed LCLs, Jurkat and HEK293T cell lines. Genomic DNA of cells transfected by electroporation with the PX458 plasmid was obtained using the Agarose Gel DNA extraction kit (Roche) and used as a template to amplify genomic fragments containing target sites. Specific primers, described below for sanger sequencing, allowed us to PCR-amplify a 291 bp product containing exon 2 target sites. PCR products were purified and analysed by Sanger sequencing. Then, the Cas9 cutting efficiency and indel occurrence were evaluated by analysing the PCR product sequences using the http://crispid.gbiomed.kuleuven.be/ and https://tide.nk.nl/ websites. The percentage of insertions and deletions was 40.8% in Jurkat cells; insertions and deletions were detected in HEK293T and LCL cells, but were not precisely evaluated.
EBV-transformed LCLs of healthy donors transduced or not with a lentiviral vector containing IL-27RA were transfected twice by electroporation using the Nepa21 electroporator (Nepagene) with PX458 plasmids, sorted on eGFP expression and maintained in culture with human recombinant IL-27 (100 ng ml−1). IL-27 expression and production were analysed using intracellular flow cytometry and ELISA in culture supernatants of LCLs, respectively. Clones lacking IL-27 expression were sequenced for IL27 by Sanger sequencing using specific primers to determine the mutations that have been introduced by CRISPR Cas9 genome. To monitor the survival of CRISPR IL27 or empty LCLs, IL-27 supplementation was stopped and cell death was assessed by flow cytometry using Annexin V and DAPI or 7-AAD (BioLegend) labelling according to the manufacturer’s instructions. All constructs were validated by Sanger sequencing using the BigDye Terminator v.3.1 Cycle Sequencing Kit (Life Technologies) and the 3500xL Genetic Analyzer (Applied Biosystems) according to the manufacturer’s instructions. Sequence analysis was performed using DNADynamo version v.1.63 (BlueTractorSoftware).
Splicing product analysis
Total RNA was extracted from activated T cell blasts using the RNeasy Mini kit (QIAGEN), and cDNA was prepared by Superscript II First Strand Synthesis System (Invitrogen). PCR amplification of exon 7 to exon 11 of IL27RA was performed using the following primers: forward 5′-CCTCTCTTTGGTCTGCTTGG-3′ and reverse 5′-CCTGTCCAGCGATGGTAGAT-3′. PCR products were verified by Sanger sequencing.
Anti-IL-27 autoantibody detection
ELISA was performed as previously described34,36. In brief, 96-well ELISA plates (Maxisorp; Thermo Fisher Scientific) were coated by incubation overnight at 4 °C with 1 μg ml−1 rIL-27 (PeproTech). The plates were then washed (PBS-Tween 0.05%), blocked by incubation with the same buffer supplemented with 5% non-fat milk powder, washed and incubated with serum or plasma samples (diluted 1/50) from patients and controls for 2 h at room temperature. The plates were thoroughly washed. Fc-specific HRP-conjugated IgG fractions of polyclonal goat antiserum against human IgG, IgA and IgM (Nordic Immunology Laboratories) were added to a final concentration of 2 μg ml−1. The plates were incubated for 1 h at room temperature and washed. The substrate was added, and the optical density was measured (Victor X4; Perkin Elmer). To detect complexed anti-IL-27 autoantibodies with IL-27 in the sera, the same plates were coated by incubation overnight at room temperature with 800 ng ml−1 capture antibody from the ELISA kit (Human IL-27 DuoSet ELISA, DY2526, R&D). The plates were then washed (PBS-Tween 0.05%), blocked by incubation with PBS-BSA 1% filtered, washed and incubated with serum or plasma samples (diluted 1/50) from patients for 2 h at room temperature and revealed as described above with HRP-polyclonal goat antiserum against human IgG, IgA and IgM.
The presence of anti-IL-27 autoantibodies was confirmed by western blotting according to the following method. A total of 500 ng of human recombinant IL-27 (PeproTech) was separated by SDS–PAGE (10% acrylamide) under reducing conditions and transferred to PVDF membranes (Millipore). The membranes were blocked by incubation with PBS supplemented with 5% BSA and 0.05% Tween-20 and were washed and incubated overnight at 4 °C with plasma samples of patients or controls diluted 1:100 in PBS with 5% BSA and 0.01% Tween-20. The membranes were then washed three times and incubated for 1 h at room temperature with HRP-coupled anti-human IgG, IgA and IgM secondary antibodies (GAHu/Ig(Fc)/PO, Nordic MUbio), used at a final concentration of 1:10,000. The membranes were washed three times and Pierce ECL western blotting substrate was used for detection.
Neutralizing assay
The blocking activity of anti-IL-27 autoantibodies contained in the serum of patients was determined by assessing intracellular STAT1 phosphorylation and the proliferation of healthy control T cells after stimulation with IL-27 (50 ng ml−1) or IL-27 + anti-CD3 (OKT3; 0.1 μg ml−1) or by assessing intracellular STAT1 phosphorylation in LCLs stimulated with IL-27 (5 ng ml−1) in the presence of 2.5% of serum (final dilution 1:40). For STAT1 phosphorylation, cells were stimulated for 15 min. Additional dilutions of serum (final 1:80 and 1:160) were also tested in the proliferation assay. Anti-IL-27 commercial antibody (Ultra-Leaf purified anti-human IL-27p28 BioLegend, MM27-7B) was used at a concentration of 2 µg ml−1 as a positive neutralizing control. IgG from sera were purified using Nab Protein G spin columns (Thermo Fisher Scientific) according to the manufacturer’s instructions. In brief, 200 µl of serum was immobilized on protein G columns using binding buffer (Pierce, protein G IgG binding buffer, 21011). After 10 min of incubation, the non-bound fraction (IgG-depleted fraction) was recovered after centrifugation and the column was washed three times with binding buffer. The IgG-positive bound fraction was then eluted by centrifugation using elution buffer (0.1 M glycine solution, pH 2–3) and neutralized with 10% of alkaline solution (1 M Tris solution, pH 8–9). Both fractions were concentrated by centrifugation using the Amicon Ultra 0.5 ml centrifugal filter (Merck Millipore). The protein concentration of both fractions was determined using a spectrophotometer (Nanodrop, Thermo Fisher Scientific) and fixed at 80 µg ml−1 by dilution in phosphate buffer, pH 7.4). The fractions were then tested for neutralization at the final dilution of 1:40 (2 µg ml−1).
IL-27RA intracellular and surface staining
T cell blasts were stimulated with anti-CD3/CD28 activating Dynabeads (LifeTechnologies) for 3 days. Surface IL-27RA was stained with purified anti-IL-27RA antibodies (R&D Systems, 191106) followed by Alexa Fluor 488-conjugated secondary antibodies (Invitrogen). Cells were then fixed and permeabilized using the FOXP3 staining kit (eBioscience), and incubated with PE-conjugated anti-IL-27RA antibody (R&D Systems, 191106). Stained cells were analysed on the BD LSRII-Fortessa (BD Biosciences) system, and data were processed using FlowJo (Tree Star).
Immunofluorescence
T cell blasts were stimulated with anti-CD3/CD28 activating Dynabeads (LifeTechnologies) for 3 days. Surface IL-27RA was stained with anti-IL-27RA antibodies (R&D Systems, 191106) followed by Alexa Fluor 488-conjugated secondary antibody (Invitrogen). Cells were then fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and incubated with anti-IL-27RA antibodies (R&D Systems, 191106) followed by Alexa Fluor 546-conjugated secondary antibodies (Invitrogen). Cell nuclei were stained with DAPI. Images were acquired using the Leica SP8 STED confocal microscope (Leica Microsystems).
RNA-seq library of LCLs
Total RNA was isolated from LCLs stimulated or not with IL-27 for 6 h using the RNeasy Kit (QIAGEN) including DNase treatment. RNA quality was assessed by capillary electrophoresis using the Fragment Analyzer with High Sensitivity RNA reagents (Agilent Technologies) and the RNA concentration was measured by spectrophotometry with Xpose (Trinean) and the Fragment Analyzer. RNA-seq libraries were generated from 600 ng of RNA using Universal Plus mRNA-seq with NuQuant (NuGEN) and sequenced (paired-end reads 100 bases + 100 bases) on the Illumina NovaSeq 6000 system. A total of around 90 million of filter-passing paired-end reads was produced per library. Reads were mapped and aligned to the human reference genome (GRCh37) and to EBV1 and EBV2 reference genomes using STAR53, and the number of uniquely mapped reads for each gene was counted using HTSeq54. Only genes with an average ≥1 read per sample were used in our analysis. DESeq255 was used for normalization and the negative binomial model was used for differential expression analysis. The RNA-seq data discussed in this publication have been deposited in NCBI Gene Expression Omnibus56 and are accessible through GEO series accession number GSE253756.
Statistical analysis
P values were calculated using Student’s unpaired or paired t-tests or Mann–Whitney U-tests with a two-tailed distribution. P < 0.05 was considered to be significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the Article and its Supplementary Information. Source data are available at Figshare (https://doi.org/10.6084/m9.figshare.25062611). Original data are available from the corresponding author and E.M. (emmanuel.martin@inserm.fr) on reasonable request. Exome-sequencing data of patients are not publicly available due to ethical restrictions, including the possibility of revealing identifying information, to which the patients have not consented. Source data are provided with this paper.
References
Latour, S. & Fischer, A. Signaling pathways involved in the T-cell-mediated immunity against Epstein-Barr virus: lessons from genetic diseases. Immunol. Rev. 291, 174–189 (2019).
Taylor, G. S., Long, H. M., Brooks, J. M., Rickinson, A. B. & Hislop, A. D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 33, 787–821 (2015).
Dunmire, S. K., Hogquist, K. A. & Balfour, H. H. Infectious mononucleosis. Curr. Top. Microbiol. Immunol. 390, 211–240 (2015).
Tangye, S. G. & Latour, S. Primary immunodeficiencies reveal the molecular requirements for effective host defense against EBV infection. Blood 135, 644–655 (2020).
Hirahara, K. et al. Asymmetric action of STAT transcription factors drives transcriptional outputs and cytokine specificity. Immunity 42, 877–889 (2015).
Kastelein, R. A., Hunter, C. A. & Cua, D. J. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu. Rev. Immunol. 25, 221–242 (2007).
Huang, Z. et al. IL-27 promotes the expansion of self-renewing CD8+ T cells in persistent viral infection. J. Exp. Med. 216, 1791–1808 (2019).
Munz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 17, 691–700 (2019).
Shannon-Lowe, C. & Rickinson, A. The global landscape of EBV-associated tumors. Front. Oncol. 9, 713 (2019).
Callan, M. F. et al. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus In vivo. J. Exp. Med. 187, 1395–1402 (1998).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Sprecher, C. A. et al. Cloning and characterization of a novel class I cytokine receptor. Biochem. Biophys. Res. Commun. 246, 82–90 (1998).
Pflanz, S. et al. WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J. Immunol. 172, 2225–2231 (2004).
Chen, Q. et al. Development of Th1-type immune responses requires the type I cytokine receptor TCCR. Nature 407, 916–920 (2000).
Owaki, T. et al. A role for IL-27 in early regulation of Th1 differentiation. J. Immunol. 175, 2191–2200 (2005).
Yoshida, H. & Hunter, C. A. The immunobiology of interleukin-27. Annu. Rev. Immunol. 33, 417–443 (2015).
Artis, D. et al. The IL-27 receptor (WSX-1) is an inhibitor of innate and adaptive elements of type 2 immunity. J. Immunol. 173, 5626–5634 (2004).
Schneider, R., Yaneva, T., Beauseigle, D., El-Khoury, L. & Arbour, N. IL-27 increases the proliferation and effector functions of human naive CD8+ T lymphocytes and promotes their development into Tc1 cells. Eur. J. Immunol. 41, 47–59 (2011).
Charlot-Rabiega, P., Bardel, E., Dietrich, C., Kastelein, R. & Devergne, O. Signaling events involved in interleukin 27 (IL-27)-induced proliferation of human naive CD4+ T cells and B cells. J. Biol. Chem. 286, 27350–27362 (2011).
Pflanz, S. et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity 16, 779–790 (2002).
Pagano, G. et al. Interleukin-27 potentiates CD8+ T-cell-mediated anti-tumor immunity in chronic lymphocytic leukemia. Haematologica https://doi.org/10.3324/haematol.2022.282474 (2023).
Harker, J. A. et al. Interleukin-27R signaling mediates early viral containment and impacts innate and adaptive immunity after chronic lymphocytic choriomeningitis virus infection. J. Virol. https://doi.org/10.1128/JVI.02196-17 (2018).
Pratumchai, I. et al. B cell-derived IL-27 promotes control of persistent LCMV infection. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.2116741119 (2022).
Devergne, O., Birkenbach, M. & Kieff, E. Epstein-Barr virus-induced gene 3 and the p35 subunit of interleukin 12 form a novel heterodimeric hematopoietin. Proc. Natl Acad. Sci. USA 94, 12041–12046 (1997).
Devergne, O. et al. A novel interleukin-12 p40-related protein induced by latent Epstein-Barr virus infection in B lymphocytes. J. Virol. 70, 1143–1153 (1996).
Niedobitek, G., Pazolt, D., Teichmann, M. & Devergne, O. Frequent expression of the Epstein-Barr virus (EBV)-induced gene, EBI3, an IL-12 p40-related cytokine, in Hodgkin and Reed-Sternberg cells. J. Pathol. 198, 310–316 (2002).
Larousserie, F. et al. Analysis of interleukin-27 (EBI3/p28) expression in Epstein-Barr virus- and human T-cell leukemia virus type 1-associated lymphomas: heterogeneous expression of EBI3 subunit by tumoral cells. Am. J. Pathol. 166, 1217–1228 (2005).
Kang, M. S. & Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 47, e131 (2015).
Tosato, G. et al. Monocyte-derived human B-cell growth factor identified as interferon-β2 (BSF-2, IL-6). Science 239, 502–504 (1988).
Tosato, G., Tanner, J., Jones, K. D., Revel, M. & Pike, S. E. Identification of interleukin-6 as an autocrine growth factor for Epstein-Barr virus-immortalized B cells. J. Virol. 64, 3033–3041 (1990).
Chehboun, S. et al. Epstein-Barr virus-induced gene 3 (EBI3) can mediate IL-6 trans-signaling. J. Biol. Chem. 292, 6644–6656 (2017).
Puel, A., Bastard, P., Bustamante, J. & Casanova, J. L. Human autoantibodies underlying infectious diseases. J. Exp. Med. https://doi.org/10.1084/jem.20211387 (2022).
Kisand, K. et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 207, 299–308 (2010).
Puel, A. et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 207, 291–297 (2010).
Nanki, T. et al. Suppression of elevations in serum C reactive protein levels by anti-IL-6 autoantibodies in two patients with severe bacterial infections. Ann. Rheum. Dis. 72, 1100–1102 (2013).
Puel, A. et al. Recurrent staphylococcal cellulitis and subcutaneous abscesses in a child with autoantibodies against IL-6. J. Immunol. 180, 647–654 (2008).
Bastard, P. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science https://doi.org/10.1126/science.abd4585 (2020).
Bastard, P. et al. Auto-antibodies to type I IFNs can underlie adverse reactions to yellow fever live attenuated vaccine. J. Exp. Med. https://doi.org/10.1084/jem.20202486 (2021).
Zhang, Q. et al. Autoantibodies against type I IFNs in patients with critical influenza pneumonia. J. Exp. Med. https://doi.org/10.1084/jem.20220514 (2022).
Fournier, B. & Latour, S. Immunity to EBV as revealed by immunedeficiencies. Curr. Opin. Immunol. 72, 107–115 (2021).
Kawamoto, K. et al. A distinct subtype of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disorder: adult patients with chronic active Epstein-Barr virus infection-like features. Haematologica 103, 1018–1028 (2018).
Alosaimi, M. F. et al. Immunodeficiency and EBV-induced lymphoproliferation caused by 4-1BB deficiency. J. Allergy Clin. Immunol. 144, 574–583 (2019).
Rodriguez, R. et al. Concomitant PIK3CD and TNFRSF9 deficiencies cause chronic active Epstein-Barr virus infection of T cells. J. Exp. Med. 216, 2800–2818 (2019).
Somekh, I. et al. CD137 deficiency causes immune dysregulation with predisposition to lymphomagenesis. Blood 134, 1510–1516 (2019).
Hoshino, Y., Nishikawa, K., Ito, Y., Kuzushima, K. & Kimura, H. Kinetics of Epstein-Barr virus load and virus-specific CD8+ T cells in acute infectious mononucleosis. J. Clin. Virol. 50, 244–246 (2011).
Martin, E. et al. CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature 510, 288–292 (2014).
Yamazaki, Y. et al. Two novel gain-of-function mutations of STAT1 responsible for chronic mucocutaneous candidiasis disease: impaired production of IL-17A and IL-22, and the presence of anti-IL-17F autoantibody. J. Immunol. 193, 4880–4887 (2014).
Toubiana, J. et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 127, 3154–3164 (2016).
Izawa, K. et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J. Exp. Med. 214, 73–89 (2017).
Durandy, A., Kracker, S. & Fischer, A. Primary antibody deficiencies. Nat. Rev. Immunol. 13, 519–533 (2013).
Fournier, B. et al. Rapid identification and characterization of infected cells in blood during chronic active Epstein-Barr virus infection. J. Exp. Med. https://doi.org/10.1084/jem.20192262 (2020).
McStay, G. P., Salvesen, G. S. & Green, D. R. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 15, 322–331 (2008).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Acknowledgements
We thank the patients and the families for their contribution and the healthy donors for blood gifts; M. Zarhrate, C. Bole and N. Cagnard for technical assistance; and the staff at the Service de Biochimie Générale, Hospital Necker-Enfants Malades for testing EBV antibodies in sera. S.L. is a senior scientist at the Centre National de la Recherche Scientifique-CNRS (France). C.G. is supported by the Agence Nationale de Recherche (ANR, France). This work was supported by grants from the Ligue Contre le Cancer-Equipe Labélisée (to S.L.), the Institut National de la Santé et de la Recherche Médicale, the Agence Nationale de Recherche (ANR-18-CE15-0025-01 to S.L., ANR-10-IAHU-01 to Institut Imagine and ANR-21-CE15-0018 to E.M.), the Institut National du Cancer (PLBIO-2020_LATOUR, PEDIAC consortium INCa_15670, PEDIAHR22_017), the Société Française de Lutte contre les Cancers et Leucémies de l’Enfant et de l’Adolescent, AREMIG (to S.L.) and the Fédération Enfants et Santé (to S.L.). Exome sequencing was funded by the Rare Diseases Foundation (to S.L.). C.S.M. and S.G.T. are supported by Investigator grants awarded by the National Health and Medical Research Council of Australia.
Author information
Authors and Affiliations
Contributions
E.M. and S.W. designed, performed experiments and analysed data. E.M. participated in the writing of the manuscript. A. Fernandes, C.G., C.L., A.-L.R., Y.B.A., A.H., C.S.M., G.R., K.T. and S.G.T. performed experiments and analysed the data. M.D. and M.S. provided and analysed data from FinnGen. M. Simonin, T.L.V., P.B., M.-L.V., M.B., B.F., C.M., M.M., D.B., J.B., C.S., A.P. and J.-L.C. provided samples and/or reagents and/or analysed data. B.F., N.C. and B.N. identified the patients provided and analysed clinical data. A.F. analysed data and participated in the writing of the manuscript. S.L. wrote the manuscript, designed and supervised the research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Immunophenotyping of IL-27RA-deficient patients.
a, Frequencies of the different lymphocyte populations from PBMCs of patients (P1.1, P1.2 and P2) and controls represented by graph bars from data obtained From FACS analysis after cell-specific staining. T, B and NK subpopulations are shown in upper panels. αβ, γδ, CD4, CD8 T cells and innate-like T cells (MAIT and iNKT) gated in CD3+ or Treg gated in CD4 + T cells are shown in middle panels. Naïve (CD31+CD45RA+CCR7+), central memory (CD45RA−CCR7+CD27+), effector memory (CD45RA−CCR7−CD27−) and exhausted effector memory/TEMRA (CD45RA+CCR7−CD27−) compartments in CD4+ and CD8+ T cells are shown in lower panels. b, Representative dot plots from FACS analysis of follicular helper T cell (Tfh) population (left panel). Graph bars (right panel) representing the frequencies in percent of Tfh cells (CD4+CD45RA− CD127 + CD25- CXCR5 +) gated on CD3+ cells. Data obtained from FACS analysis as shown in the left dot plots. c, Representative dot plot FACS analysis of non-follicular (CXCR5−) T helper cell subpopulations, Th1 (CCR4−CXCR3+CCR6−), Th1* (CCR4−CXCR3+CCR6+) Th2 (CCR4+CXCR3−CCR6−) and Th17 (CCR4+CXCR3+−CCR6+) populations (left panel). Graph bars (right panel) representing the frequencies in percent of Th1/1*/2/17 cells gated on memory CD4+ T cells (no Tfh). Data obtained from FACS analysis of dot plots as shown in the left panel. (a,b,c) Data are medians ± s.d. with each symbol/circle represents an independent biological/experimental sample. Ten to 15 age-matched controls and 3 patients were analysed. The horizontal bars represent the median±sd. Data obtained from six independent experiments. Two-tailed Mann-Whitney tests were used. d, Graph bars corresponding to medians ± s.d. of IL-2, IL-22, IFN-γ and TNF-α cytokines levels from coated bead array (CBA) quantifications in culture of naïve and memory sorted CD4 T cells of grouped P1.1 and P1.2 IL27RA-deficient patients (red, n = 5) compared to controls (grey, n = 5) stimulated with CD3/28/2 beads (Th0), CD3/28/2 beads and IL-12 (Th1) or CD3/28/2 beads and IL-27 (IL-27) during 5 days. Data were obtained from FACS analysis. Each symbol/circle represents an independent biological/experimental sample. Two-tailed Wilcoxon-Mann-Whitney tests were used.
Extended Data Fig. 2 Effect of the c.1142-2A>C and c.1336C>G mutations in IL27RA gene of family 2.
a, 45 nucleotide in-frame deletion resulting from the heterozygous c.1142-2A>C mutation in P2. On the left, PCR products from the amplification of cDNAs obtained from activated P2 and two control T cell blasts with forward and reverse oligonucleotides are depicted on a diagram corresponding to IL27RA cDNA from exons 7 to 11 (right panel). The size and structures of the potential PCR products are listed. The wild-type and the 45 nucleotide deleted products are indicated by black arrows on the right side of the gel. The lower panel shows the electropherogram of SANGER sequencing of the 515 bp product focused on the region containing the 45 nucleotide deletion. The sequence is depicted on the top with the deleted sequence (45 bp) in red and the new acceptor splice site in red bold. Data from one experiment. b, Same as in (a) excepted that PCR products from amplification of cDNAs were obtained from two control LCLs (Ctrl.1, Ctrl.2) or LCLs of P1.1, P1.2 and P2. Data from one experiment. c-d, Analyses of IL27RA expression in control T-cell blasts and T-cell blasts from P2 and her parents. c, pictures showing representative immunofluorescent stainings of surface IL27RA (green) and intracellular+ surface IL-27RA (red) expression. Staining of the nucleus was performed using DAPI labelling (blue). Scales 4 μM. d, Histograms from flow cytometry analysis. Data from one representative experiment of two.
Extended Data Fig. 3 Defective IL-27-induced STAT1 and STAT3 phosphorylation and proliferation of T-cells in Patient P2.
a, Immunoblots for phospho-STAT1 (P-STAT1), STAT1, phospho-STAT3 (P-STAT3), STAT3 and ACTIN expression in T-cell blasts of control 1 (Ctrl.1), control 2 (Ctrl.2) or IL27RA-deficient patient P2 (P2) stimulated with 50 ng.ml−1 of human recombinant IL-27 for different periods of time. Data from one representative experiment of two. b, Histograms from FACS analysis showing intracellular phospho-STAT1, phospho-STAT3 expression and corresponding isotypes (clear) in controls (grey) or P2 (red) T-cell blasts stimulated (dark) or not (light) with IL-27 cytokine during 15 min. Data from one representative experiment of two. c, Residual STAT1 and STAT3 phosphorylations of control and IL27RA deficient T-cell blasts when cultured in the presence of IL-2. Immunoblots for phospho-STAT1, STAT1, phospho-STAT3, STAT3, and GAPDH expressions in non-stimulated T-cell blasts of control healthy donors (Ctrl.1 and Ctrl.2) and IL27RA-deficient patients (P1.1 and P1.2) maintained in culture with IL-2 or washed 3 times and then starved during 2 days (IL-2 starvation). Data from one representative experiment of two. d, Overlaid FACS histograms showing cell divisions by dilution of Cell Trace Violet (CTV) dye of T cells from a healthy control (Ctrl.) (grey), parents of P2 (grey) and IL27RA-deficient patient P2 (red) stimulated with allow concentration of coated anti-CD3 antibody (0.1 µg.ml−1) in presence (dark) or not (light) of 50 ng.ml−1 of IL-27 during five days. Data from one representative experiment of two. e-g, IL-27 promotes the proliferation of naive T cells. T cells from control PBMCs stimulated with anti-CD3 antibody (0.1μg.ml−1) in presence or not of IL-27 (50 ng.ml−1) during five days. e, Overlaid FACS histograms showing cell divisions by dilution of cell trace violet (CTV) dye of T cells that have been incubated or not with anti-IL-27 blocking antibody. One representative experiment of four. f, Overlaid FACS histograms on left panels showing cell divisions by dilution of cell trace violet (CTV) dye of naïve CD45RA+CD27+ (upper panels) and memory CD45RA- CD27 + T cells (lower panels). Data from one representative experiment of three. g, Graphs corresponding to replication index calculated from FACS histograms as in (f) of three independent experiments in which PBMCs of healthy donors (n = 4) have been tested. Stimulations with anti-CD3 alone or with anti-CD3 plus IL-27 correspond to white or black circles respectively. Black line represents the medians and each symbol/circle represents an independent biological/experimental sample. Two-tailed paired t-tests were used.
Extended Data Fig. 4 The IL27RA R466G mutant is weakly expressed and is able to activate STAT1 and STAT3.
NIH-3T3 cells have been transfected with lentiviral expression vectors containing a cDNA coding wild type IL27RA (black), IL27RA Arg446Gly (red), IL27RA G381_A395del mutant (orange) or an empty vector (white) with the mCherry as a reporter gene of transfected cells. a, Overlaid histograms from FACS analysis showing IL-27RA expression in mCherry+ cells. Dashed line histograms for isotype control. One representative experiment of 3 independent experiments. b, Histograms from FACS analyses in the left depicting intracellular phospho-STAT1 (P-STAT1) expression in cells stimulated with human IL-27 or not (No. stim.). The bar graphs on the right correspond to P-STAT1 mean fluorescence intensities (MFI) normalized on the corresponding isotype MFI. MFI were calculated from histograms as shown on the left. c, same as b, except that phospho-STAT3 (P-STAT3) expression was analysed. (b-c). Errors bars represent the median ± s.d. The symbols correspond to three experimental replicates from two independent experiments (triangles and circles). Two-tailed paired t tests were used.
Extended Data Fig. 5 Impaired differentiation of IL27RA-deficient CD8 + T cells toward EBV-infected B cells (LCLs) and normal expression of SAP in PBMCs of P1.1.
a, Graph bars corresponding to medians ± s.d. of percentages of healthy donors (black, n = 7), P1.1 (dark red, n = 5) or P1.2 (light red, n = 1) EBV-pentamers negative CD8+ T expressing activation, differentiation and exhaustion T-cell markers from PBMCs co-cultured with LCLs at day 0 (upper graph) and day 9 (lower graph). Yellow squares correspond to P1.1 at the time she had severe infectious mononucleosis (P1.1-SIM). Data were obtained from FACS analysis of seven independent experiments. Each symbol represents an independent biological sample. Two-tailed paired t-tests were used. b, Graph bars corresponding to the median ± s.d. of percentages of EBV-specific T cells from P2 (red, n = 1) and controls (Ctrl.) (black, n = 4) expressing activation, differentiation and exhaustion T-cell markers and gated on CD8+ T cells at day 0 or day 9 of co-culture with autologous EBV-infected B cells (LCLs). Each symbol/circle represents one independent biological/experimental sample. c, Intracellular staining for SAP in PBMCs of one control (Ctrl.) and patient P1.1. Gating on B cells, NK cells and T cells. Empty and filled histograms correspond to staining with control-matched isotype or anti-SAP antibody respectively. One representative experiment of two. d-g, Blockage of the IL-27-IL27RA pathway is associated with an impaired expansion of EBV-specific T cells towards LCLs. FACS analyses of expansion, apoptosis, proliferation and phenotypes of EBV-specific CD8+ T cells from PBMCs of a healthy control co-cultured during 15 days with autologous LCLs in the presence (red) or not (black) of neutralizing anti-IL-27 antibody and recombinant IL-27RA protein. One representative experiment of two. d, Dot plots showing percentages of EBV-specific T cells. e, Overlaid histograms showing activation, differentiation and exhaustion markers in EBV-specific T cells. f, Dot plots showing early (annexin V+, 7-AAD−) and late (annexin V+, 7-AAD−) apoptotic cells. g, FACS histograms showed cell divisions by CTV dye dilution of EBV-specific T cells after 5 days of re-stimulation with autologous LCLs.
Extended Data Fig. 6 Characterization of IL27RA-deficient and STAT1-deficient LCLs.
a, Overlaid FACS histograms of HLA-A2 expression in LCLs from two controls (dark grey) and IL27RA-deficient patients (P1.1, P1.2 and P2 in red). Isotype staining corresponds to dashed line histograms. Data from one representative experiment of two. b, Overlaid FACS histograms of CD70 and CD137L (4-1BB/TNFSF9) expression in LCLs from three controls (Ctrl. in black), IL27RA-deficient patients (P1.1, P1.2 and P2 in red) and one STAT1-deficient patient (STAT1 LOF in green). Isotype staining corresponds to dashed line histograms. Data from one representative experiment of two. c, Overlaid FACS histograms showing of EBI3 expression (anti-EBI3 antibody, clone A15058A, Biolegend) and IL-27p28 (anti-IL-27p28 antibody, clone B0315A10, Biolegend) subunits of IL-27 in LCLs from six controls (dark grey), the three IL27RA-deficient patients (P1.1, P1.2 and P2 in red) and two STAT1-deficient patient (STAT1 LOF in green) and the monocyte cell line THP-1 (in blue). Isotype staining corresponds to empty histograms. The specificity of anti-EBI3 and p28 have been verified by WB in Extended Data Fig. 11d. Data obtained from three independent experiments. d, Graphs depicting the time necessary to obtain immortalization of B cells corresponding to LCLs from PBMCs of healthy controls (Ctrl., n = 10) and patients (P1.1 (n = 2), P1.2 (n = 1) and P2 (n = 1)) after incubation with EBV. P1.1 was tested two times independently. Data obtained from three independent experiments. e, Graphs depicting the quantification of IL-27 by ELISA in culture supernatants of LCLs in culture. Each symbol corresponds to the tested LCL of different control donors (Ctrl., n = 8), the three patients with IL27RA deficiency (P1.1, P1.2 and P2, n = 3) and patients with STAT1 deficiency (STAT1 LOF, n = 3). Data from three independent experiments. There is no correspondance between controls LCLs (Ctrl.) in d and e. f, Immunoblots for phospho-STAT1 (P-STAT1), STAT1 and ACTIN expressions in LCLs of controls (Ctrl.) and patients (P1.2, P1.2 and P2) directly prepared from the culture without washing that showed decreased basal P-STAT1 in LCLs of patients compared to control LCLs. One representative experiment of three.
Extended Data Fig. 7 IL-27 potentiates cell-cytotoxicity of TSST-1 induced T-cell blasts.
a, Representative graph bars from FACS analysis showing the median ± s.d. of CD8+ T cell percentages from healthy control blast donors (Ctrl., n = 3) and patients (P1.1, n = 1 and P1.2, n = 1) (red) expressing activation and exhaustion markers after incubation (chequered) or not (fully filled) with IL-27 for 48 h. Each symbol represents one independent biological sample. These cells were tested for their cytotoxicity activity shown in Fig. 3d. b-c, Analyses of expansion (a), cytotoxicity (b) and phenotyping (c) of TSST-1 induced T-cell blasts from three healthy control donors. b, Cytotoxic response of TSST-1 induced T cell blasts from healthy control donors (n = 3) at day 8 of culture against TSST-1 pulsed LCLs (black) or not (grey) in the presence (chequered) or not (fully filled) of IL-27. Representative graph bars shows the percentages of FRET activated CASP3+ LCLs corresponding to LCLs killing in which caspase 3 has been activated. The target cells (LCLs)/effector cells (T-cell blasts) ratios are indicated on the x-axis. One representative experiment of two with the same statistical significance. c, Representative graph bars from FACS analysis showing the medians ± s.d. of TCR Vβ2 + CD3 + T cell percentages from unstimulated (Unstim., n = 3) and stimulated healthy control donor PBMCs with the superantigen TSST-1 after 8 days in the presence (chequered grey, n = 3) or not (black, n = 3) of IL-27. d, Representative graph bars from FACS analysis showing the percentages of TCR Vb2+ CD8+ T cells expressing activation and exhaustion markers after 8 days of culture in the presence (chequered) or not (fully filled) of IL-27. (a-d). The errors bars represent the median ±s.d. and each symbol/circle corresponds to one biological/experimental sample. (b) two-tailed paired t tests were used.
Extended Data Fig. 8 Gene expression by transcriptome analysis including EBV genes of two control LCLs and in two IL27RA-deficient LCLs from two patients P1.1 and P.2.
a, Control LCLs (n = 2; CTL1_basal, CTL2_basal) and IL27RA-deficients LCLs (n = 2; P1.1_basal and P2_basal) display comparable EBV gene expression profiles in basal conditions. The left panel shows boxplots displaying the distribution of gene expression (Log2 normalized) of 10 EBV genes according to IL27RA vs CTL. The boxes in terms of minima, maximun, centre and bounds represent the interquartile range (IQR), demarcating the range between the first and third quartiles. Within this box, a horizontal line denotes the median value of the gene expression. The whiskers extending from the box represent the minimum and maximum values. b, Control (CTL1, CTL2) and IL27RA-deficient (P1.1 and P2) LCLs display different gene expression profiles in response to IL-27 (_stim.) compared to non-stimulated cells (_basal). Left panel, z-score hierarchical heat map visualization of the top 98 genes differentially expressed in each LCL and condition. Gene’s name is on the right. Upper right panel, Gene Ontology analysis (GO; http://geneontology.org/) of the top 200 genes of differentially expressed showing the most significant associated biological pathways. The 15 genes of the « cytokine-mediated signalling pathway » are highlighted. Lower left panel, Volcano plots comparing transcriptome data from control LCLs (left) stimulated with IL-27 versus basal (Stim vs Basal) (left panel) and IL27RA-deficient LCLs (right panel). The 15 genes enriched in the « cytokine-mediated signalling pathway » from the Gene Ontology analysis are indicated.
Extended Data Fig. 9 Defective proliferation and STAT1/3 phosphorylations restored by IL27RA expression in LCLs of patients.
a, Immunoblots for phospho-STAT1 (P-STAT1), STAT1, phospho-STAT3 (P-STAT3), STAT3 and GAPDH expressions in LCLs that were stimulated or not (no stim.) with IL-27 (40 ng.mL−1), IL-2 (10 U.mL−1) or IL-6 (20 ng.mL−1) for 10 min. LCLs from control healthy donors (Ctrl.#1 and Ctrl.#2), IL27RA-deficient patients (P1.2 and P2) and patients with STAT1 loss of function mutations (STAT1 LOF #1 and #2). One representative experiment of two. b, Representative overlaid FACS histograms showing cell divisions by cell trace violet (CTV) dye dilution of healthy control LCLs (Ctrl. 1 and Ctrl.2 in black), LCLs of patients P1.2 and P2 (in red) and LCLs of two patients with STAT1 loss of function (STAT1 LOF#1 and STAT1 LOF#2 in green) at day 0, 3 and 5 of culture. Data were obtained from one of three independent experiments in which additional controls and patients were tested. Calculated expansion indexes are shown in Fig. 4f. c, Histograms from FACS analysis showing IL-27RA expression (upper panel) and corresponding isotype (lower panel) in LCLs of healthy controls (Ctrl.1 and Ctrl.2 in black) and patients (P1.1, P1.2 and P2 in red) transduced with an empty lentivector (Empty pLVX) or a lentivector containing a cDNA coding wild-type IL27RA (pLVX-IL27RA) in filled histograms. Data from one representative experiment of two. d, same as b, except that LCLs from a control and patient P1.2 in which IL27RA expression was restored (pLVX-IL27RA) or not (Empty pLVX) shown in (c) were tested. Data from two independent experiments. Calculated indexes of expansion are shown in Fig. 4g. e, Immunoblots for phospho-STAT1, STAT1 and ACTIN in control LCLs (Ctrl.1, Ctrl.2) and patients LCLs (P1.1, P1.2 and P2) in which IL27RA expression was restored (pLVX-IL27RA) or not (Empty pLVX) shown in (c) that were stimulated or not with IL-27 for 5 min. Data from one of two independent experiments.
Extended Data Fig. 10 IL-27 neutralization or IL27 inactivation in LCLs inhibit proliferation and IL27RA signalling by triggering rapid cell death.
a, Curves of percentages of alive LCLs from three different controls cultured during 14 days in the presence of blocking anti-IL-27 (red, n = 2), anti-IL-2 (grey, n = 1) antibodies or no antibody (black, n = 2). For Ctrl.#1 and #2 curves of two independent experiments for which symbol represent the median ± s.d. of cell percentages. b, Immunoblots for phospho-STAT1, STAT1, phospho-STAT3, STAT3 and ACTIN expressions in LCLs of two controls (Ctrl.1 and Ctrl. 2) stimulated with IL-27 (+) or not (−) in the presence or not of anti-IL-27 or anti-IL-2 blocking antibody at 3 μg.mL−1 for anti-IL-2 (+), 2 μg.mL−1 (+) or 20 μg.mL−1 (++) for anti-IL27. Data from one representative experiment of two. c, Representative FACS density plots (left panels) of DAPI expression of control LCLs in which IL-27 was inactivated or not by transduction with an empty CRISPR-Cas9 vector (left) or CRISPR-Cas9 vector containing gRNAs targeting exon 2 of IL27 (right) and cultured or not (no IL-27) with IL-27 (+ IL-27) for 6 h, left panels. Bar graphs in the right showing percentages of DAPI+ dead control LCLs (grey, n = 3) or LCLs inactivated for IL27 (IL-27 CRISPR in red, n = 3) after 6 h of IL-27 deprivation (dark) or not (light). Each symbol corresponds to one control LCLs. Two independent experiments. Error bars represent median ± s.d., Mann-Whitney two-tailed tests. d, Density plots of intracellular IL-27 expression of control LCLs overexpressing ectopic IL27RA in which IL27 was inactivated by CRISPR-Cas9 targeting the exon 2 of IL27 (IL27 CRISPR), CRISPR-Cas9 empty (Empty CRISPR) vector as negative control. Cells were cultured in the presence of IL-27. Numbers correspond to % of cells in the gates. e, Representative FACS density plots of annexin V and 7-AAD expression of control LCLs transduced with pLVX-IL27RA, in which IL-27 was inactivated (IL27 CRISPR exon2) or not (Empty CRISPR) by CRISPR-Cas9 containing vectors and cultured with IL-27 (+ IL-27) or not (no IL-27) for 8, 24 and 48 h. 7-ADD-Annexin V+ and 7-ADD+Annexin V+ correspond to early and late apoptotic cells respectively. Percentages of cells in each gate. Data from one representative experiment of two.
Extended Data Fig. 11 Characterization of anti-IL-27 autoAbs detected in sera of patients with IM.
a, The upper left panel depicts principles of the ELISA to detect anti-IL-27 autoAbs (up) and complexed anti-IL-27 autoAbs with IL-27 (down). Dot plots of levels of anti-IL-27 autoAbs versus IL-27 (upper right panel), IL-27 versus complexed anti-IL-27 autoAbs (lower left panel) and anti-IL-27 autoAbs versus complexed anti-IL-27 autoAbs (lower right panel) in sera of patients with IM (red points) and controls (black points) with regression lines showing significant correlations. b, Detection and quantification of anti-IL-27 IgG and IgA antibodies in serum of four patients (IM#1, IM#2, IM#3, IM#8) with infectious mononucleosis (IM) (upper panel) and three patients (CAEBV#1, CAEBV#2, CAEBV#5) with chronic active EBV infection (CAEBV) over time after their IM (x-axis). In a and b, arbitrary optical density (O.D.) for anti-IL-27 autoAbs, complexed anti-IL-27 autoAbs and IL-27 quantifications. c, Immunoblots of recombinant IL-27 from Peprotech revealed using plasma from one healthy donor (Ctrl.), from two patients with IM (IM #1, IM #2) or from one patient carrying a STAT1 GOF mutation or anti-IL-27 p28 monoclonal antibody (MM27-7B, Biolegend) (anti-IL-27) in the right. Representative data of five independent experiments are shown. The arrows indicate the p28 subunit of recombinant IL-27 that resolves as a doublet. Size markers in kDa on the left. d, Characterization of recombinant IL-27 proteins and anti-p28 and anti-EBI3 antibodies used in this study. Immunoblots of IL-27 recombinant proteins from Peprotech and Biolegend. IL-27 was revealed using the anti-IL-27 p28 neutralizing monoclonal antibody (clone MM27-7B, Biolegend), anti-IL-27 p28 monoclonal antibody (B0315A10, Biolegend) or anti-IL-27 EBI3 (clone A15058A, Biolegend). The arrows on the left indicate the p28 (resolving as a doublet) and EBI3 subunits of IL-27 protein (Peprotech) or uncleavable recombinant IL-27 protein (EBI3+p28) (Biolegend). Size markers in kDa on the right. Data from one representative experiment of two.
Extended Data Fig. 12 Characterization of the neutralization effect of anti-IL-27 autoAbs detected in sera of patients with IM.
a, Representative FACS histograms depicting intracellular pSTAT1 expression in healthy control T cells stimulated for 15 min with a low concentration of anti-CD3 (0.1 µg.mL−1) or not (no stim.) with IL-27 (50 ng/mL−1) or not in the presence or not of serum samples of healthy control (Ctrl.), a patient with IM, a patient carrying STAT1 GOF mutation or a blocking anti-IL-27 monoclonal antibody. One representative experiment of three independent experiments. b, Left panel, mean fluorescence intensity (MFI) from FACS analysis of intracellular pSTAT1 expression in control LCLs stimulated or not with IL-27 (5 ng/mL−1) for 15 min in the presence or not of serum samples of healthy controls (Ctrl.) (n = 5), patients with IM (n = 4) or CAEBV (n = 4). One representative experiment of three independent experiments. The horizontal bars represent the median ± s.d. of pSTAT1 MFI. Two tailed paired t tests were used. Right panel, representative FACS histograms depicting intracellular pSTAT1 expression in LCL from which MFI values were calculated and shown in the left panel. Grey histograms correspond to no stimulation, while colour lines histograms correspond to stimulation with IL-27 in the presence of control (Ctrl.), IM or a CAEBV serum sample. (c-e), Characterization of the neutralizing activity of anti-IL-27 autoAbs with representative overlaid FACS histograms showing cell divisions by CTV dye dilution of naive CD4 T cells stimulated with coated anti-CD3 (0.1 µg.ml−1) in the presence of IL-27 (50 ng.ml−1) incubated with the indicated dilution of serum (d, e), purified IgG or depleted IgG fractions from serum (c). In d, control sera from healthy donors (Ctrl.#1, Ctrl.#2) and commercial human serum (AB). In e, sera of four IM patients (IM#1, IM#2, IM#4 and IM#22). Data from one experiment. In c, depleted (line) or purified (shaded) IgG fractions of sera from a healthy control (Ctrl.) (in grey), two patients with IM#1, IM#2 (in red), on patient STAT1 GOF (in green) or blocking anti-IL27 antibody (in black). Data from one experiment.
Supplementary information
Supplementary Figures
Supplementary Figs. 1–3.
Source data
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Martin, E., Winter, S., Garcin, C. et al. Role of IL-27 in Epstein–Barr virus infection revealed by IL-27RA deficiency. Nature 628, 620–629 (2024). https://doi.org/10.1038/s41586-024-07213-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07213-6
- Springer Nature Limited
This article is cited by
-
The ouroboros of autoimmunity
Nature Immunology (2024)