

Abstract
The myriad microorganisms that live in close association with humans have diverse effects on physiology, yet the molecular bases for these impacts remain mostly unknown1,2,3. Classical pathogens often invade host tissues and modulate immune responses through interactions with human extracellular and secreted proteins (the ‘exoproteome’). Commensal microorganisms may also facilitate niche colonization and shape host biology by engaging host exoproteins; however, direct exoproteome–microbiota interactions remain largely unexplored. Here we developed and validated a novel technology, BASEHIT, that enables proteome-scale assessment of human exoproteome–microbiome interactions. Using BASEHIT, we interrogated more than 1.7 million potential interactions between 519 human-associated bacterial strains from diverse phylogenies and tissues of origin and 3,324 human exoproteins. The resulting interactome revealed an extensive network of transkingdom connectivity consisting of thousands of previously undescribed host–microorganism interactions involving 383 strains and 651 host proteins. Specific binding patterns within this network implied underlying biological logic; for example, conspecific strains exhibited shared exoprotein-binding patterns, and individual tissue isolates uniquely bound tissue-specific exoproteins. Furthermore, we observed dozens of unique and often strain-specific interactions with potential roles in niche colonization, tissue remodelling and immunomodulation, and found that strains with differing host interaction profiles had divergent interactions with host cells in vitro and effects on the host immune system in vivo. Overall, these studies expose a previously unexplored landscape of molecular-level host–microbiota interactions that may underlie causal effects of indigenous microorganisms on human health and disease.
Similar content being viewed by others


Main
The omics revolution has revealed innumerable associations between the microbiome and diverse human phenotypes. However, the causal mechanisms that underlie microbiota-mediated effects on human physiology remain largely undefined. A key barrier to uncovering these mechanisms is the lack of a comprehensive understanding of the transkingdom molecular circuitry linking indigenous microorganisms and their human hosts. Given that commensal microorganisms exist within the extracellular compartments of their hosts, engagement with the host exoproteome represents a potential major source of functional host–microbiota interactions. Indeed, classical pathogens encounter an array of human extracellular and secreted proteins during infection, and specific interactions with host ‘exoproteins’ are involved in nearly all aspects of microbial pathogenesis, including adhesion and invasion, extracellular matrix remodelling and immune evasion4,5,6,7,8,9. A handful of commensal bacteria have been similarly shown to engage with host exoproteins to enable niche colonization and immunomodulation, suggesting that interactions with host exoproteins may be used to infer microbiota functions10,11. Yet, aside from these anecdotes, direct interactions between the human microbiota and host exoproteins remain largely unmapped. Previous studies have used advanced proteomic approaches to reveal host–pathogen interactions12,13,14,15. However, inherent constraints of these technologies limit their scalability and applicability to the discovery of commensal interactions with host exoproteins16,17,18. New approaches that enable proteome-scale explorations of host–microorganism interactions across hundreds of microbial strains are thus required to determine the frequency, patterns and potential biological functions of microbiota-mediated host exoprotein engagement.
A tool to map transkingdom interactions
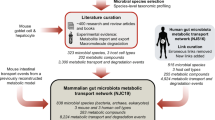
We developed a high-throughput technology to screen intact microbial cells for the ability to bind to human proteins, called BASEHIT (Bacterial Selection to Elucidate Host–Microbe Interactions in High Throughput) (Fig. 1a). In brief, we biotinylated individual bacterial strains before incubating each strain with a library of thousands of genetically barcoded yeast clones displaying individual human exoproteins. We then isolated bacteria–yeast complexes using streptavidin beads, enriching yeast clones interacting with the bacterial strain of interest. Finally, we identified and quantified the barcodes encoded by the enriched yeast clones via next-generation sequencing. Using these data, we derived a ‘BASEHIT score’ that represents the predicted degree of interaction between an individual microorganism and each host protein based on the enrichment of the corresponding yeast barcodes relative to the pre-selection yeast library (see Methods for a detailed description of the BASEHIT scoring model).

a, Overview of the workflow for assembling a host–microbiota interactome atlas. For the BASEHIT process, individual biotinylated microbial isolates are mixed with a library of barcoded yeast clones, each of which display a single human exoprotein (3,324 exoproteins total). Bacteria-bound yeast are isolated via magnetic selection, DNA barcodes are sequenced via next-generation sequencing (NGS) and a BASEHIT score is calculated for each protein based on the relative degree of barcode enrichment compared with the pre-selection library. The resulting host–microbiota interactome atlas comprises more than 500 individual host–microorganism interactomes and hundreds of previously undiscovered microbial interactions with host exoproteins. The graphics in a were created using BioRender (https://biorender.com). b, Microorganisms from all four major body tissues and six phyla (one strain from Deinococcus-Thermus is not depicted) exhibited a range of interaction profiles. Proteins with high background binding to streptavidin magnetic beads are excluded from these counts (see Methods for details). c, Validation examples of predicted interactions via flow cytometry of bacteria stained with recombinant host exoproteins. d, Network of predicted exoprotein–microorganism interactions collapsed by species. The circle colour indicates the genus and the circle size indicates the number of strains sampled for that species.
To enable proteome-scale interrogation of host–microorganism interactions, we expanded our exoproteome-scale yeast display library19 to encompass 3,324 well-displayed human extracellular and secreted proteins (Extended Data Fig. 1a and Supplementary Table 1), as well as 12 viral proteins that serve as internal ‘negative’ controls. All proteins were expressed as tagged C-terminal fusions to Aga2, leaving the native N termini exposed (Extended Data Fig. 1b). This library covers over 60% of the human exoproteome with a median of 20 barcodes (unique clonal replicates) per protein (Extended Data Fig. 1c,d) and spans a range of tissue expression patterns and protein folds (Extended Data Fig. 1e,f). The displayed proteins in this library are generally well folded and recapitulate known protein–protein interactions19.
We used adherent-invasive Escherichia coli (AIEC), which invade epithelial cells via interactions with the cell-surface proteins CD55 and CEACAMs20,21, to test the BASEHIT process. We observed robust enrichment of yeast expressing CD55 and CEACAM1 after selection with AIEC across a range of experimental conditions (Extended Data Fig. 2a,b), whereas no enrichment was observed with non-Dr family adhesin-expressing E. coli strains (Extended Data Fig. 2c). CD55 and CEACAM1 were also the most highly enriched proteins after selection with AIEC (Extended Data Fig. 2d), demonstrating the selectivity of BASEHIT. To further test the sensitivity of BASEHIT, we screened a panel of 33 previously described host–microorganism interactions (Supplementary Table 2). We focused on direct interactions between whole bacteria and surface proteins, as BASEHIT cannot detect other classes of host–microorganism interactions, such as host receptor engagement by shed or secreted bacterial products (for example, activation of Toll-like receptors by pathogen-associated molecular patterns). Overall, we successfully recapitulated 54% of these interactions (Extended Data Fig. 2e), suggesting that BASEHIT can accurately identify a substantial fraction of direct interactions between bacteria and human exoproteins.
We next assessed the effects of biotinylation and bacterial cell density on the detection of an array of exoprotein–commensal interactions identified in a pilot BASEHIT screen. Most interactions showed an ‘inverse U’ relationship between enrichment and biotinylation, as over-biotinylation may impair binding by obscuring a binding interface, and under-biotinylation may impair selection by streptavidin beads (Extended Data Fig. 3a). However, in all cases, 5 µM biotin showed enrichment within twofold of the optimal concentration. Furthermore, we found that 50 µl of bacteria at 5 OD ml−1 was within threefold of the optimal identified bacterial cell quantity across all tested strains (Extended Data Fig. 3b). Of note, we did not observe any substantial enrichment of negative control viral proteins under any tested condition. On the basis of these data, we selected optimal experimental conditions for all subsequent BASEHIT screens.
A host exoprotein–microbiota interactome
To assess the frequency and patterns of interactions between indigenous human microorganisms and human exoproteins across diverse niches, we performed BASEHIT on a collection of 519 human-associated bacterial strains isolated from four major human barrier tissues (the skin, gut, oral cavity and female reproductive tract) (Extended Data Fig. 1g–i and Supplementary Table 3). The microorganisms in this collection span 6 phyla, 45 families, 59 genera and 261 species and include multiple strains assigned to the same species (1–105 strains per species). In total, we interrogated more than 1.7 million potential binary interactions between individual host proteins and unique bacterial strains via BASEHIT.
We developed a negative binomial model to predict exoprotein–microorganism interactions from BASEHIT data (Fig. 1a, BASEHIT score; additional details are in the Methods and Extended Data Fig. 4). Using this model, we identified 3,650 predicted binary strain–protein interactions involving 631 exoproteins, 184 species and 383 strains (Supplementary Table 4). These interactions were broadly distributed across bacterial phylogenies and tissues of origin, and host exoprotein expression patterns and protein folds (Fig. 1b and Extended Data Fig. 5a–d). Most interacting exoproteins exhibited highly restricted binding patterns: 270 exoproteins interacted with a single bacterial strain, and another 260 proteins only interacted with strains from the same genus, implying shared mechanisms of interaction. By contrast, 35 human proteins interacted with more than 10 bacterial species, suggesting potential roles in host-mediated microbial recognition or convergent targeting of host proteins by diverse bacteria; alternatively, such exoproteins may exhibit inherent promiscuity or poor behaviour in BASEHIT. After excluding proteins with high background binding to streptavidin magnetic beads, we found that many bacterial strains (69 out of 519) had only one interaction partner and more than one-quarter of all strains (169 out of 519) had no predicted interactions. By contrast, 62 bacteria exhibited broad interaction profiles (10 or more interactions). Among these ‘superbinders’, a Proteus mirabilis strain that elicits chronic intestinal inflammation in mice22 exhibited the largest number of interactions (76) (Extended Data Fig. 5b). We validated select interactions via bacterial flow cytometry using recombinant proteins produced in mammalian cells (Fig. 1c). Finally, we collapsed all sampled strains into their parent species to visualize the broad host exoprotein–microbiota interactome network (Fig. 1d).
Interactome organizational principles
We applied feature-set enrichment methods to identify characteristics of tested bacterial species associated with the number of bound human proteins (‘binding degree’). We considered three different types of attributes — (1) higher-level taxonomic labels, for example, genera; (2) physiological traits, for example, ability to grow on a specific nutrient; and (3) host environment, for example, the human gut — which revealed 28 statistically significant trends (Wilcoxon rank-sum tests; minimum n = 5 species and two-tailed nominal P values with false discovery rate (FDR) q = 0.1; Supplementary Table 5). Because attributes often overlapped heavily in their associated species, we collapsed attributes with Jaccard overlap scores of more than 0.5 into individual groups and selected the most significantly binding-associated member from each group as its representative. This process identified 11 non-redundant species attributes associated with binding degree (Fig. 2a). Among these, species annotated in ProTraits23 as growing on trehalose, including E. coli and Klebsiella pneumoniae, exhibited the strongest positive enrichment for interaction degree (n = 6 species; median = 17 binding partners; FDR q = 0.0164). These species also contributed to significant enrichments for the Enterobacteriaceae family (n = 11; median = 10.8; FDR q = 0.018) and the ProTraits category ‘pathogenic in mammals’ (n = 35; median = 9.5; FDR q = 0.016). Microorganisms native to the human oral cavity (n = 53; median = 7.0; FDR q = 0.064), including many Streptococcus species (n = 23; median = 14.0; FDR q = 0.0043), were also positively enriched for host exoprotein binding. Together with the Enterobacteriaceae members, these species contributed to a highly significant trend towards elevated binding among facultative anaerobes (n = 34; median = 12.5; FDR q = 0.0003). Conversely, species associated with strict anaerobicity (n = 52; median = 2.6; FDR q = 0.0056), including the Bacteroidaceae family (n = 18; median = 1.6; FDR q = 0.030) and residents of the human gut (n = 98; median = 4.0; FDR q = 0.0022), were significantly depleted for interactions with human proteins. This difference may reflect a tendency for species residing in the lumen of the human gut to interact with the host through secreted metabolites rather than directly via cell-based interactions. These observed tendencies, although statistically significant, reflect average differences between classes of microorganisms, and strains from each category still exhibit a range of binding degrees, and specific binding capabilities are not necessarily determined solely by one functional category.

a, Species attributes (taxa and phenotypes) with atypically high or low numbers of human protein-binding partners (Wilcoxon rank-sum test; FDR q < 0.1). These examples were selected to have minimal overlap in their member species (Jaccard similarity < 0.5). b, Biological processes enriched among the human protein-binding partners of microbial genera (the top 20 FDR-significant examples are ranked on fold enrichment; degree-preserving network permutation test). GO, Gene Ontology. c, More closely related strains tend to have more similar sets of human protein-binding partners (degree-preserving network permutation test). d, Taxonomy of selected taxa from c; up to three species per genus are included, ranked by strength of within-species conservation, within-genus conservation or conspicuous lack of conservation (for example, non-conserved genera and species with two or more daughters). In a,c, boxplot ‘boxes’ indicate the first, second and third quartiles of the data (Q1, Q2 and Q3, respectively), ‘whiskers’ indicate the inner fences of the data (1.5 times the interquartile range above Q3 or below Q1) and values outside the inner fences are plotted separately as points. Grey bars in a indicate the IQR of the background set. Statistics were derived from n = 204 species with non-zero degree in the interaction network; FDR adjustments were made.
We similarly quantified biophysical enrichments among microbially bound human proteins. The vast majority (81%) of screened human proteins had no predicted microbial interactions. We therefore focused on potential differences between proteins with one or more interactions (‘targets’) versus non-interacting tested proteins (‘non-targets’). Among physical properties, the most significant enrichment was decreased mass among targets (median = 34.5 kDa) relative to non-targets (median = 45.4 kDa; Wilcoxon rank-sum test, two-tailed nominal P value with FDR q < 10−12; Extended Data Fig. 6 and Supplementary Table 6). In addition, targets were less hydrophobic than non-targets (median hydrophobicity = −0.26 versus −0.21; FDR q = 0.0046), although this may be a consequence of the preceding size effect (smaller proteins contain proportionally more solvent-accessible surface residues, which tend to be hydrophilic).
We next quantified potential enrichments in the biological functions of targets versus non-targets using a collection of non-overlapping Gene Ontology biological process terms. Significant global functional enrichments among bound proteins were rare, with only three processes achieving FDR significance (Fisher’s exact test; two-tailed nominal P values with FDR q < 0.1; Supplementary Table 7). The most common process was negative regulation of WNT signalling (GO:0090090), which was annotated to 17 binders (2.4-fold enrichment; FDR q = 0.085). WNT signalling is widely involved in homeostasis, growth and repair across many tissues, and manipulation of this pathway may enable modulation of the tissue microenvironment in multiple host niches.
The dearth of global functional enrichments among targets suggested a lack of ‘universal’ exoprotein interaction strategies. Instead, interaction strategies may be shared within taxonomically and environmentally related strains. We therefore applied degree-preserving network permutation tests to determine whether related groups of microorganisms bound human proteins involved in specific biological processes. This process identified 254 FDR-significant (q < 0.1) instances of congeneric strains binding functionally related human proteins (based on one-tailed nominal P values; Supplementary Table 8). In 124 of these overrepresented ‘binding strategies’, the associated genus bound at least five distinct proteins in the associated biological process, corresponding to a twofold enrichment over the random expectation in all but one case (see Fig. 2b for the top 20 examples). For example, Bacteroides strains bound ten proteins in the ephrin receptor signalling pathway (GO:0048013; a 6.8-fold enrichment) including EPHB2, which is highly expressed in intestinal stem cells24. Binding of EPHB2 may enable localization to, or manipulation of, particular stem cell populations by crypt-dwelling Bacteroides strains25. Proportionally fewer functional classes were preferentially targeted by bacteria from the same host niche (for example, gut versus skin; Supplementary Table 9); the few significant niche-based functional associations largely reflected the strategies adopted by the major genera from that environment (for example, Bacteroides in the gut). Thus, microbial exoprotein-binding strategies appear to be conserved within taxonomic groups but are less universal to specific human body sites or the human microbiome as a whole.
The observation that many functional categories were enriched among human proteins bound by specific microbial genera led us to investigate the general evolutionary conservation of the interaction profile of strains. Using degree-preserving network permutation tests, we investigated whether pairs of strains from the same parent species tended to share more binding partners than expected at random (as quantified by Jaccard similarity). This hypothesis proved true to a striking extent: of 38 species with two or more tested strains with interactions, 25 (66%) shared more interactions than expected by chance (one-tailed nominal P values with FDR q < 0.1; Fig. 2c and Supplementary Table 10). This resulted in an average Jaccard similarity of 0.16 among conspecific strains: fourfold higher than expected. A similar pattern was observed among congeneric species: of 27 applicable genera, 15 (56%) exhibited significantly conserved interaction profiles among member species, with an average Jaccard similarity of 0.13. Of note, this is not due to intraspecies conservation, as our analysis at the genus level only considers comparisons between strains from different species. Klebsiella and Staphylococcus exhibited high conservation within and among multiple member species of these genera, whereas Lactobacillus and Prevotella exhibited genus-level conservation but inconsistent within-species conservation (Fig. 2d). Bacteroides profiles were notably not significantly conserved, despite preferentially binding proteins in specific functional categories (Fig. 2b), although this may reflect a lack of sensitivity in our statistical analysis due to variable taxonomic classification of the strains in our screen. Conservation among genera in the same family was considerably lower (mean Jaccard similarity = 0.046) and not statistically significant (P = 0.093). This pattern continued for most higher-level taxonomic groups. However, high conservation among the proteobacteria (Jaccard score = 0.29; FDR q < 0.001) slightly inflated the average conservation at higher taxonomic ranks. In general, these analyses are intended to detect significant evidence of conservation and may be limited when the taxonomic group is sparsely sampled or exhibits high phylogenetic diversity. To assess the sensitivity of our analyses using shared taxonomy as a proxy for the evolutionary relatedness of strains, we conducted a parallel analysis for a subset of fully sequenced strains incorporating an explicit measure of phylogenetic distance. Consistent with our findings based on taxonomy, the phylogenetic distance of strains was inversely correlated with similarity of interaction partners (Spearman r = −0.416; Mantel test, two-tailed P < 10−4; Extended Data Fig. 7a).
Strain variation in exoprotein binding
Bacterial strains assigned to the same species can exhibit divergent effects on host physiology26 and strain-specific interactions with the exoproteome could contribute to such functional variation. Although global network analysis revealed a significant degree of phylogenetic conservation in interaction profiles, we still observed substantial variation in binding patterns among both congeneric and conspecific bacterial strains (average within-species Jaccard similarity of human protein binding partners of 0.16; Fig. 2c).
To explore this variation, we focused on two deeply sampled skin commensal taxa: Staphylococcus (199 strains from 17 species) and Cutibacterium acnes (also known as Propionibacterium acnes; 40 strains). These genera both showed relatively high levels of within-species conservation of interaction profiles (Fig. 2c) but exhibited suitable variability for further analysis (within-species Jaccard similarities of less than 0.6 for all species within these genera; Fig. 2d).
Within the Staphylococcus genus, S. aureus, S. haemolyticus and S. pasteuri exhibited the highest species-level conservation in interaction profiles (Fig. 2d). Staphylococcus isolates also showed high levels of similarity at the genus level (Fig. 2d and Supplementary Table 10), highlighting the existence of shared interactions across species. Overall, 112 host proteins were bound by more than one Staphylococcus strain. Network visualization of these interactions revealed at least three major binding patterns (Fig. 3a) — ‘subspecies’ groups defined by preferential binding of one or several host proteins (cluster 1, exemplified by S. epidermidis), proteins bound by a subset of strains from multiple species (cluster 2, including highly overlapping S. aureus and S. haemolyticus) and a ‘superbinder’ group, consisting of two distinct subgroups (cluster 3, one of S. pasteuri and the other spanning four species) (Extended Data Fig. 8a,b). These superbinder strains were all highly promiscuous, with 6–47 total host interaction partners per strain, including 36 proteins bound by at least 5 strains. Several proteins in the latter group are highly expressed in the skin and were either uniquely (CDSN and FAT2) or predominantly (XG) bound by skin-resident Staphylococcus strains, suggesting a potential role in tissue-specific colonization or adaptation (Extended Data Fig. 8c,d).

a, Network of Staphylococcus–exoprotein interactions including all proteins that interacted with at least two Staphylococcus strains. The coloured shapes denote three clusters demonstrating divergent patterns of strain-variable and species-variable interactions. Specific interactions underlying each cluster are shown in the heatmaps on the right. b, Heatmap depicting interactions between 37 C. acnes strains and eight proteins bound by at least 10 C. acnes strains; three strains without interactions are not displayed. Two subspecies clusters of C. acnes are defined primarily by binding to SLC22A31 or OPRK1 and SPINT3. c, Density correlation plot of gene content versus exoprotein-binding patterns across all C. acnes strains. One-tailed Mantel test was performed, n = 40 independent C. acnes strains.
We next examined C. acnes, which was deeply sampled (40 strains) and showed a broad interaction profile (median of 6 interactions per strain). C. acnes strains formed two clear subspecies groups based on their exoprotein interactions (Fig. 3b). Although nearly all strains bound FAM174A, TMEM119, PDCD1 and CNGB3, two groups of strains could be defined based on SLC22A31 (group 1) or OPRK1 and SPINT3 (group 2) binding. CST6 was bound by some strains within both groups. The first class of conserved interactions (that is, SLC22A31 or OPRK1 and SPINT3) may reflect critical processes for niche occupancy and survival that are maintained across all strains within the distinct groups, whereas strain-variable interactions (for example, CST6) may drive functional variability between isolates. As specific exoprotein interactions may be encoded by conserved genes or large co-varying gene sets, we next asked whether overall gene content similarity (UniRef90 gene family presence within genomes) between pairs of strains of C. acnes was related to similarity in host interaction profiles. Indeed, we observed a striking correlation between overall genomic similarity and shared host interactions (Fig. 3c; Spearman correlation between Jaccard-based distances = 0.693; Mantel test, one-tailed P < 0.0001), suggesting that host interaction patterns can delineate genomically distinct subspecies groups of C. acnes. Expanding on this observation, we investigated the relationship between genomic and exoprotein binding profiles at the intraspecies level across a diverse set of bacteria. Consistent with our findings for C. acnes and our global phylogenetic analysis (Fig. 3c and Extended Data Fig. 7a), we observed a strong negative and statistically significant correlation between phylogenetic distance and interaction profile, demonstrating that this trend extends to the intraspecies level (Spearman r = −0.513; Mantel test, two-tailed P < 10−4; Extended Data Fig. 7b). Finally, using comparative genomic approaches, we identified candidate genes that may be involved in novel Staphylococcus–host and Cutibacterium acnes–host exoprotein interactions discovered by BASEHIT (Supplementary Tables 11–20 and Methods).
Commensal modulation of host biology
Individual interactions discovered via BASEHIT may explain specific microbial effects on host physiology. To explore this possibility, we examined five genera associated with human health and disease whose isolates were primarily associated with the gastrointestinal tract: Bacteroides, Fusobacterium, Prevotella, Ruminococcus and Streptococcus (Fig. 4a). Numerous strains from these genera exhibited specific interactions with host proteins that spanned a range of epithelial cell-surface proteins, proteases, extracellular matrix-associated proteins, and immune cell receptors or cytokines. These proteins can be segregated into three broad functional categories potentially associated with key steps of microbial colonization: niche colonization and adhesion, remodelling of the local tissue microenvironment and immune evasion. Of note, many of these interactions were highly specific to a small number of related strains or were unique within the broader host–microorganism interactome (Extended Data Fig. 9 and Supplementary Table 21). From this set of strains, we focused on two genera, Fusobacterium and Ruminococcus, for which there is clear evidence of strain-specific involvement in human disease27,28,29,30,31,32,33,34.

a, Putative protein functions and associated interactions with select members of the human gastrointestinal microbiome. The schematic was created using BioRender (https://biorender.com). b, R. gnavus exhibits strain-specific interactions. The heatmap shows interactions between R. gnavus and host exoproteins. c, Validation of strain-specific R. gnavus–CD7 interactions. Indicated strains were stained with recombinant CD7–Fc fusion protein (teal, violet or magenta histograms) or Fc alone (grey histograms), and binding was measured by bacterial flow cytometry. d, Fusobacterium strains interact widely with immune and epithelial proteins. The heatmap shows interactions between gastrointestinal tract or tumour-resident Fusobacterium strains and exoproteins with known roles in immune signalling or regulation of the WNT pathway (annotated with teal or magenta circles, respectively). The star denotes HM758 and the diamond denotes HM997. e, Validation of strain-specific F. nucleatum–DKK1 interactions. The indicated strains were stained with recombinant biotinylated DKK1 (B-DKK1) tetramers (prepared with streptavidin–phycoerythrin (blue or purple histograms)) or streptavidin–phycoerythrin alone (grey histograms), and binding was measured by bacterial flow cytometry.
Ruminococcus gnavus is enriched in patients with Crohn’s disease, and strain-level variation and ‘clade switching’ in R. gnavus are closely associated with disease flares27,28. We identified strain-variable interactions between R. gnavus and the mucus-associated trefoil factors TFF1, TFF2 and TFF3, as well as the T cell co-stimulatory molecule CD7 (Fig. 4b). Although TFF1–TFF3 were also bound by a range of other bacteria, CD7 was uniquely bound by a subset of R. gnavus strains (Extended Data Fig. 9). We confirmed strain variation in CD7 binding via staining with recombinant CD7 (Fig. 4c). This combination of mucosal and immune protein binding may contribute to the unique inflammatory potential of specific R. gnavus strains.
Fusobacterium species are enriched in human colorectal tumours29,30,31 and specific Fusobacterium nucleatum strains can interact with defined host protein targets (TIGIT and CEACAM1)11,32. In addition to these previously described Fusobacterium–exoprotein interactions (Extended Data Fig. 2e), we observed multiple previously unreported strain-specific interactions between Fusobacterium isolates and immune-associated or tumour-associated proteins (Fig. 4d); for example, a single Fusobacterium strain bound to the γδ T cell regulator BTN3A3 and a subset of oral strains bound to the neutrophil-derived antibacterial protein LCN2. Fusobacterium strains derived from oral mucosa or tumours also bound two key modulators of the WNT pathway, DKK1 and SOST, and a range of immune receptors, including immunoregulatory receptors such as LAIR1 (bound by one gut and one oral strain) and SIRPα (bound by a single oral strain) (Fig. 4d). We confirmed DKK1, SIRPα and LAIR1 binding to select Fusobacterium strains via staining with recombinant proteins (Figs. 1c and 4e). Oral, gut and tumour-associated Fusobacterium isolates displayed both shared and divergent binding partners across tissue origins. As oral Fusobacterium strains can seed tumour-resident populations and exacerbate cancer33,34, the identification of shared host protein-binding capabilities between isolates may reveal specific strains from different tissues that have the potential to translocate to and subsequently modulate the tumour–immune microenvironment and promote cancer progression.
To test the potential effects of these newly discovered interactions on host biology, we identified phylogenetically related strains with divergent interaction profiles and examined their effects on host cell binding or immune responses in vitro or in vivo. First, we tested whether CD7 expression in mammalian cells was sufficient to enable host cell binding by R. gnavus; we found that a CD7-binding R. gnavus, but not a non-CD7-binding strain, bound specifically to CD7-expressing Expi293 cells (Fig. 5a,b and Extended Data Fig. 10a). Second, we examined the effects of SIRPα-binding and non-binding Fusobacterium strains on macrophage phagocytosis (Fig. 5c). We observed that the SIRPα-binding Fusobacterium strain was phagocytosed at lower levels than six distinct non-binding Fusobacterium strains (Fig. 5d and Extended Data Fig. 10b). Furthermore, co-incubation with the SIRPα-binding strain led to lower levels of phagocytosis of labelled bioparticles relative to co-incubation with a non-binding strain (Fig. 5e and Extended Data Fig. 10b). Finally, we monocolonized germ-free mice with six Bacteroides strains with divergent binding patterns: three strains that bound a common set of five host proteins, including multiple ephrin receptors, the phosphatase PTPRJ and the adhesion molecule MCAM (binders), and three that lacked binding to all of these proteins (non-binders) (Fig. 5f). RNA sequencing of bulk colonic tissue revealed that mice colonized with the binders showed lower levels of multiple immunoglobulin gene transcripts, including the genes encoding the heavy chain (Igha) and joining chain (Jchain) of dimeric secretory IgA (Fig. 5g,h). Furthermore, germ-free mice colonized with a representative binder strain exhibited lower levels of faecal IgA than mice colonized with a representative non-binder strain, demonstrating that binder and non-binder strains have divergent effects on the mucosal immune response (Fig. 5i).

a, Schematic of carboxyfluorescein succinimidyl ester (CFSE)-labelled R. gnavus strains binding to CD7-expressing cells. b, Quantification of the per cent CFSE-positive population. Two-way analysis of variance (ANOVA) with Dunnett’s test, P < 2.2 × 10−16, n = 4 independent experiments. c, Schematic of macrophage phagocytosis of CFSE-labelled Fusobacterium spp. strains and fluorescent bioparticles. FITC, fluorescein isothiocyanate. The graphics in a,c were created using BioRender (https://biorender.com). d, Mean fluorescence intensity (MFI) of THP-1 phagocytosis of CFSE-labelled SIRPα-binding and non-binding Fusobacterium spp. (bacteria to THP-1 ratio of 5:1). One-way ANOVA with Dunnett’s test; for comparisons with HM556 with P < 0.0001: P = 1.32 × 10−14 (HM997), P = 7.86 × 10−5 (HM758), P < 2.2 × 10−16 (HM260) and P = 1.88 × 10−6 (HM1289). Representative data from n = 3 biologically independent samples. e, Phagocytosis of E. coli K12 bioparticles by THP-1 cells incubated with unlabelled SIRPα-binding or non-binding Fusobacterium spp. (bioparticles to THP-1 ratio of 1:1; Fusobacterium spp. to THP-1 ratio of 2:1). One-way ANOVA with Dunnett’s test, P = 3.23 × 10−14. Representative data from n = 4 biologically independent samples. f, Three binder and three non-binder Bacteroides strains were used to monocolonize germ-free C57BL/6 mice. n = 2 mice per strain. g, Differentially expressed genes in bulk colonic tissue from monocolonized mice as identified by RNA sequencing. Genes with log2-transformed fold change of more than 1 or less than 1 (vertical dashed lines) and −log10(P value) > 2 (horizontal dashed line) were identified as significantly upregulated or downregulated by gene-specific analysis with FDR step-up adjustments. h, Normalized transcript counts for Jchain (left) and Igha (right). Two-tailed unpaired Student’s t-tests. In g,h, n = 6 mice per binding group. i, Germ-free C57BL/6 mice were monocolonized with one binder and one non-binder Bacteroides strain, and faecal IgA was measured by ELISA and normalized by total protein content. One-way ANOVA with a Šidák’s test, n = 5 mice per bacterial strain. Data in b,d,e,h,i representmean ± s.d.
Discussion
Here we developed a novel technology that enables comprehensive mapping of host–microorganism interactions across the majority of all human exoproteins and used this technology to assemble more than 500 strain-specific host–microorganism interactomes across 59 genera and 4 barrier tissues. We uncovered an extensive network of transkingdom connectivity between human exoproteins and indigenous microorganisms, which revealed shared host–microorganism interaction patterns across phylogenetically and functionally related strains and myriad examples of strain-specific host–microorganism interactions that may mediate microbial effects on diverse human phenotypes.
Our data imply new roles for host exoprotein–microorganism interactions in microbial colonization, niche remodelling and immunomodulation. For example, we observed unique interactions between defined tissue isolates and corresponding tissue-enriched proteins: multiple skin-derived Staphylococcus isolates bound to highly skin-expressed proteins, including CDSN, FAT2 and XG, whereas isolates from all other tissues failed to bind to CDSN and FAT2, and sparsely bound XG. FAT2 and CDSN regulate keratinocyte junctions and corneodesmosomes, respectively35,36, and the blood group antigen XG37 is highly expressed in keratinocytes and melanocytes38. We also observed strain-specific interactions that may underlie the unique effects of select microorganisms on human biology. For example, multiple strains from the inflammatory bowel disease-associated species R. gnavus28 bound the T cell co-stimulatory receptor CD7, which is highly expressed on intraepithelial lymphocytes, and CD7 expression was sufficient to enable host cell binding by CD7-binding R. gnavus strains38. These data suggest that select R. gnavus strains may colonize mucosal tissues or modulate intestinal immunity via binding to CD7. Phylogenetically related bacterial strains also sometimes bound multiple host proteins with related biological functions. Select strains from the colorectal cancer-associated genus Fusobacterium29,30,31 bound DKK1 and SOST, both of which antagonize WNT signalling39. Given the pleiotropic activity of WNT proteins and their diverse roles in tumour development, progression and metastasis40,41, such interactions may explain the varied impacts of Fusobacterium in cancer. Select Fusobacterium strains also bound either LAIR1 or SIRPα, both of which inhibit myeloid cell activity and may thus facilitate evasion of phagocyte-mediated bacterial clearance42,43. Accordingly, we observed that a SIRPα-binding Fusobacterium strain exhibited reduced macrophage engulfment compared with non-binding strains. Together with previous studies showing that Fusobacterium can directly engage the T cell co-inhibitory receptor TIGIT11, these data suggest that Fusobacterium species may evade both innate and adaptive immunity through specific exoprotein interactions; such interactions may explain the persistence of Fusobacterium strains within tumours. Finally, mice colonized with Bacteroides strains that exhibited shared interactions with a suite of immune-expressed and epithelial-expressed proteins displayed reduced intestinal IgA responses compared with mice colonized with non-binder strains. Together, these data suggest that commensal interactions with host exoproteins facilitate microbial colonization and modulate host immunity.
Despite its utility, BASEHIT also has notable limitations that may result in both false-negative and false-positive results. By design, BASEHIT identifies bacterial interactions with individual human proteins; however, microorganisms may also bind to specific host lipid or glycan structures44,45, which BASEHIT will fail to capture or may lead to aberrant bacterial binding to proteins that exhibit altered glycoforms in yeast display. Although our display library is selected based on epitope-tag expression (which implies successful protein folding and display), it is still possible that some ‘successfully’ displayed proteins will not assume correctly folded conformations. Such proteins could fail to bind to cognate ligands or bind to bacteria in a nonspecific manner, which may introduce false negatives or false positives, respectively, within the presented interactome. Furthermore, the biochemical nature of each new interaction identified via BASEHIT cannot be imputed using BASEHIT data alone. Previously characterized host–pathogen interactions range from highly co-evolved and host species-specific binding of individual host proteins by microbial adhesins46,47 to highly promiscuous binding of linear peptide motifs48 or common carbohydrate moieties5. Similarly, in addition to the pairwise microbial-binding events described above, a subset of microorganisms in our collections, including several putative pathogenic strains, simultaneously bound to more than ten structurally and functionally diverse host proteins. These binding patterns were often phylogenetically conserved and may reflect microbial targeting of shared host protein features (for example, glycans), or binding of select bacterial surface features by multiple host proteins. Future biochemical and structural characterizations will be needed to establish the molecular details of these newly discovered interactions.
By assigning host-centric functions to hundreds of microbiome strains, these studies establish a foundational resource that will seed a wide range of future studies of this previously unmapped landscape, including detailed studies of the genetic, biochemical and structural features underlying specific interactions of interest as well as explorations of the broader principles that dictate the evolution and physiological functions of host–microbiota interactions. For example, integrating the functional annotations provided by BASEHIT with comparative genomic approaches may eventually allow for the annotation of critical components of the ‘metagenomic dark matter’ that enable microbial interactions with the host. These new annotations may expose key associations between microbial functions and human phenotypic outcomes that are currently hidden in diverse metagenomic datasets. Overall, the host–microbiome interactome reported here provides new insights into the molecular details that may underlie diverse microbiota-mediated effects on human health and reveals myriad targets for potential therapeutic manipulation of both pathogenic and beneficial host–microbiota interactions.
Methods
Human exoproteome library construction
The Aga2 yeast display library was constructed as previously described19. In brief, proteins annotated as secreted, membrane associated or otherwise associated with an extracellular region based on UniProt identifiers49 (4,855 total human proteins) were identified, and the extracellular portions were manually selected based on existing UniProt topology annotations. Because the majority of these exoproteins included are type I membrane or classically secreted proteins, we chose a C-terminal tag to leave the native N terminus following signal peptide cleavage to be closer to the ‘endogenous’ form of the protein. Sequences encoding these extracellular regions were amplified from commercially available cDNAs or synthetic gene fragments (Twist Biosciences) (3,461 proteins with extracellular regions suitable for yeast display were identified and cloned, along with 12 coronavirus proteins as internal negative controls) and electroporated together with the linearized yeast display vector pDD003 (Supplementary Table 1 and Supplementary Figs. 1 and 2) into JAR300 yeast in a 96-well format. After growth, all wells were pooled and yeast with surface-displayed protein constructs were isolated by staining for the C-terminal FLAG epitope tag using a Sony SH800Z sorter. The sorted library was expanded and frozen. Barcode–gene pairings were identified with a Tn5-based protocol as previously described19, with 3,324 human exoproteins and 12 negative control viral proteins included in the final library. Plasmid design and sequence analyses were performed using SnapGene 4.1.3.
BASEHIT screening
Bacterial growth and labelling
Bacterial strains were grown in media and atmospheric conditions as indicated in Supplementary Table 3. Approximately 5 optical density (OD) units of saturated bacterial culture were pelleted and washed three times with 1 ml PBS. Washed bacteria were resuspended in 1 ml PBS with 5 µM Sulfo-NHS-biotin (Thermo Scientific) (or other concentrations where specifically noted for optimization) and incubated at 37 °C for 30 min. Excess unreacted biotinylation reagent was quenched by addition of 10 µl of 1 M Tris (pH 8.0), and cells were washed with 1 ml PBS. Labelled cells were resuspended in 1 ml PBS with 10% (v/v) glycerol and frozen at −80 °C for selections. Volumetric bacterial particle counts were performed by diluting the bacterial samples 10× in sterile filtered PBS + 0.5% BSA + 0.5 mM EDTA (PBE), quantifying the total number of particles within 30 µl of sample on a Cytoflex (Beckman Coulter) to calculate particles per millilitre.
Yeast library selections
The yeast library was expanded in SDO-Ura at 30 °C. One day later, yeast were induced by resuspension at an OD of 1 in SGO-Ura supplemented with 10% SDO-Ura and culturing at 30 °C for 24 h. Before selection, plasmid DNA was extracted from 400 µl of the pre-selection library to allow for comparison to post-selection libraries using a Zymoprep Yeast Plasmid Miniprep II kit (Zymo Research) according to standard manufacturer protocols. Of induced yeast cells, 3 × 107 were pelleted in sterile 96-well v-bottom microtitre plates. Yeast cells were resuspended in 100 µl PBE, 50 µl biotinylated bacteria (at 5 OD ml−1 unless otherwise specified for optimization) were added, and the yeast were incubated with shaking for 2 h at 4 °C. Yeast cells were washed once with 200 µl PBE, resuspended in 100 µl PBE with a 1:100 dilution of streptavidin microparticles (0.29 µm; Spherotech) and incubated with shaking for 1 h at 4 °C. Yeast cells were washed once with 200 µl PBE. Yeast cells were then pelleted and kept on ice. All of the following steps were carried out at room temperature. Yeast cells were resuspended in 150 µl PBE. A custom 96-well magnet was used to remove bead-bound yeast, which were washed twice with 150 µl PBE by removal and subsequent re-application of the magnet. Washed yeast cells were eluted into 150 µl PBE by removal of the magnet. Selected yeast were pelleted and expanded by growth in 1 ml SDO-Ura supplemented with chloramphenicol at 30 °C for 44 h.
Next-generation sequencing library preparation and sequencing
DNA was extracted from yeast libraries using Zymoprep-96 Yeast Plasmid Miniprep kits or Zymoprep Yeast Plasmid Miniprep II kits (Zymo Research) according to standard manufacturer protocols. A first round of PCR was used to amplify a DNA sequence containing the protein display barcode on the yeast plasmid. PCRs were conducted using 1 µl plasmid DNA, 159_DIF2 and 159_DIR2 primers, and the following PCR settings: 98 °C denaturation, 58 °C annealing, 72 °C extension and 20 rounds of amplification. The PCR product was directly used as template for the second round of PCR. A second round of PCR was conducted using 1 µl PCR product, Nextera i5 and i7 dual-index library primers (Illumina) and the following PCR settings: 98 °C denaturation, 58 °C annealing, 72 °C extension and 20 rounds of amplification. PCR products were pooled and run on a 2% agarose gel. The band corresponding to 257 base pairs was cut out, and DNA (next-generation sequencing library) was extracted using a QIAquick Gel Extraction Kit (Qiagen) according to standard manufacturer protocols. The next-generation sequencing library was sequenced using a full Illumina NovaSeq S4 2 × 150 lane, according to standard manufacturer protocols, to obtain 2 billion total reads by the Yale Center for Genome Analysis (YCGA).
Data analysis
To identify the exoprotein composition, samples were demultiplexed and sequenced barcodes were mapped, only accepting exact barcode matches to ensure that correct calls were used in the analysis. The input to the statistical model consists of counts of 69,894 barcodes distributed across 3,324 human and 12 viral proteins in an ‘input’ sample, 3 ‘beads-only’ samples and 3 output samples for each strain. The input sample quantifies the input concentration of each barcode in the living library of barcoded proteins. The beads-only samples are used to identify proteins that inherently stick to the streptavidin beads in the absence of microorganisms. The output samples are used to quantify the level of interaction between each strain and proteins that do not adhere to the streptavidin beads alone. To estimate the interaction scores from the sequenced barcode counts of the input, output and beads-only samples, we applied a zero-inflated negative binomial (ZINB) model to each protein separately. The model was evaluated with Stan (v2.29.2)50. The ZINB model is as follows:
Counti ~ ZINB(θi, μi, φ)
μi = exp(offset + protein + interactioni)
θi ~ Beta(1,1)
The offset term, which acts as a covariate in the linear predictor with a known slope of one, is a pre-computed number that adjusts for sequencing depth and the input concentration of each barcode (as assessed with the ‘Pre’ sample sequencing). It essentially normalizes the output counts but also propagates the additional precision that comes with more data (in the case of proteins with high input). The protein term is an estimate of the baseline output for each protein to account for proteins that show universally high or low output across all strains. Finally, the interaction term accounts for residual variation in the output counts caused by the interaction between the protein and individual strains. Each sample in the assay has its own zero-inflation fraction to account for the expected variation in output among strains.
We used the posterior mean of this interaction term as our interaction score. We included weak priors for the protein baseline term, the strain-wise zero fractions and the dispersion parameter φ. We included a strong regularizing prior (Normal(μ = 0, σ = 0.15)) on the interaction terms as a way of shrinking estimates towards zero, reducing false positives, and encoding our prior belief that only a very small fraction of interactions are truly non-zero. To validate our choice of σ = 0.15, we performed a simple prior sensitivity analysis that varied the standard deviation parameter of the interaction term prior from 0.075 to 0.3. We found that although this affected the number of hits identified as well as the absolute value of the interaction scores (as expected), the rank order and relative sizes of interaction scores were mostly unaffected (Extended Data Fig. 4f). Because the scores are ultimately derived from regression coefficients using a log-link function, they can be interpreted in typical log fold change units, for example, the posterior mean threshold of 0.5 corresponds to interactions that induce at least an exp(0.5) = 1.65-fold increase in output when other factors are held equal. After fitting the model, we also computed the ‘concordance’ of each interaction across replicates as the negative entropy of the input-adjusted output counts. The concordance threshold ensures that the observed output is relatively consistent across the triplicates. The thresholds were set by examining the global distribution of concordance scores (Extended Data Fig. 4d). To binarize our continuous interaction scores into a set of hits and non-hits, we used a hit-calling procedure that combined posterior intervals, score magnitude and cross-replicate concordance (Fig. 1a). To be called as a hit, an interaction needed to have: (1) a 95% posterior interval excluding zero, (2) have a posterior mean score exceeding 0.5, and (3) a cross-replicate concordance exceeding 0.75. We included notation on all interactions involving proteins where a normalized enrichment greater than 1 was observed in the beads-only control samples, to provide for additional caution due to high background (that is, nonspecific to bacterial cells) enrichment. A Venn diagram plotting function included in the R package allows the user to better understand the interacting effects of each of the hit-calling criteria by being able to inspect the number of interactions removed as potential hits with a given set of model estimates and thresholds.
To evaluate the posterior for each protein, we used Stan (v2.29.2)50 with 4 Markov chains, 1,000 warm-up samples and 50,000 post-warm-up samples per chain.
We have made our model available as an R package named basehitmodel at Zenodo https://doi.org/10.5281/zenodo.10606150 (ref. 51).
Orthogonal validation experiments
Protein purification
Protein ectodomains were cloned into pCER243, a derivative of pD2610-v2 (ATUM Bio). Protein ectodomains were cloned in-frame with an upstream H7 leader sequence and downstream 3×(GGGGS) linker and hIgG1 Fc fragment with an N297A mutation to abolish all effector functions. Expi293 cells (Thermo Fisher; no additional authentication or mycoplasma testing was performed following purchase) were transfected with the expression plasmids using the ExpiFectamine 293 Transfection Kit according to manufacturer protocols (Thermo Fisher). Proteins were purified from clarified media 4 days after transfection, using magnetic Protein A beads (Lytic Solutions). Proteins were eluted with 100 mM glycine (pH 3.0), immediately neutralized with 1 M Tris pH (8.0) and buffer exchanged into PBS. Protein purity was verified by SDS–PAGE. Protein concentration was measured by absorbance at 280 nm. Proteins were snap-frozen in liquid nitrogen and stored at −80 °C until use.
Bacterial flow cytometry
Bacteria were grown as described in Supplementary Table 3. Bacteria from saturated cultures (0.5–10 µl of culture volume) were pelleted and resuspended in 50 µl PBE with recombinant protein (0.05–5 µM final concentration of staining protein) and incubated with shaking for 1 h at 4 °C. Bacteria were washed twice with 200 µl PBE, resuspended in 50 µl PBE with 1:100 anti-hIgG–phycoerythrin secondary antibody (BioLegend) and incubated with shaking for 30 min at 4 °C. Bacteria were washed once with 200 µl PBE, resuspended in 200 µl PBE and analysed on a Sony SA3800 spectral analyser (Sony Biotechnology). Binding of Fc fusion proteins was compared with binding of Fc alone. Biotinylated DKK1 (DK1-H82F5, Acro Biosystems) was pre-incubated with streptavidin–phycoerythrin (405245, BioLegend) in a 4:1 ratio to form tetramers for 20 min at room temperature. A total of 0.1 OD Fusobacterium spp. culture and a total of 1.5 µg of DKK1 monomer per staining reaction was used. Bacterial pellets were resuspended in 50 µl precomplexed tetramer and incubated with shaking for 1.5 h at 4 °C. Bacteria were washed once with 200 µl PBE, resuspended in 200 µl PBE and analysed on a BD LSRII. Data were analysed using FlowJo (v10).
Statistical analysis of global network properties
We assigned tested strains (and their parent species) to taxonomic and phenotypic groups to enable statistical enrichment testing relative to network properties. For strains with sequenced isolate genomes, we determined a corresponding species taxonomy from the genome’s ‘Species Genome Bin (SGB)’ as defined previously52 (http://segatalab.cibio.unitn.it/data/Pasolli_et_al.html; January 2019 release). For all other strains, we estimated the species taxonomy from the given genus and species name in the corresponding strain collection. We excluded strains with ambiguous taxonomy from enrichment testing. We assigned species to phenotypic categories (for example, morphologies and growth conditions) based on annotations from ProTraits23 (http://protraits.irb.hr/; downloaded 17 December 2020), specifically those with at least 90% confidence.
Human proteins assayed as potential microbial binding partners were similarly assigned to functional categories and physical properties for enrichment testing. We extracted raw human proteome annotations from UniProt49 (https://www.uniprot.org/; downloaded 17 December 2020). We focused on assignments of human proteins to Gene Ontology Biological Process terms53 and on all implied assignments to higher-level biological process terms inferred from the Gene Ontology hierarchy (https://geneontology.org/). To isolate a set of largely non-overlapping Gene Ontology terms of a reasonable size for enrichment testing, we identified biological process terms with at least k = 100 members that did not contain any descendant terms with k or more members (‘informative’ Gene Ontology terms54); 896 biological process terms satisfied these criteria. We additionally characterized human proteins according to their physical properties (for example, size and hydrophobicity) as assembled in ref. 55.
Human proteins were categorized as ‘targets’ if they had one or more microbial interactions in the network; all other assayed proteins were classified as ‘non-targets’. We tested for functional enrichments among binders by comparing them with the sets of proteins annotated to each informative Gene Ontology term using Fisher’s exact test (treating the union of binders and non-binders as the background). Importantly, the test background only included proteins from the library, thus avoiding biases arising from the composition of the library (for example, its focus on exoproteins). We tested for associations between binding and physical properties by Wilcoxon rank-sum tests: specifically, evaluating whether each property differed significantly between binders and non-binders. We applied a similar non-parametric procedure to test for interaction between species groups (taxa and phenotypes with five or more members) and for the extent of interaction with human proteins. Each species was assigned the average degree of its member strains from the high-coverage network, and then tested members of a group were compared with tested non-members for statistically significant differences. In each case, we computed nominal two-tailed P values, which we then subjected to multiple hypothesis testing correction using the Benjamini–Hochberg FDR method56. Associations with adjusted P values (FDR q values) less than 0.1 are reported as FDR-significant in this work.
We performed additional statistical enrichment testing within the global network using degree-preserving network permutation. Here we first computed a property of interest (test statistic) based on the structure of the true network. The network was then repeatedly permuted (randomized) in a degree-preserving manner to evaluate the statistical significance of the test statistic. Specifically, starting from a given microbial strain–human protein interaction (edge), ma:ha, in the network, a second edge, mb:hb, is selected at random, and the human proteins of the two edges are swapped to produce two new candidate edges: ma:hb and mb:ha. If neither candidate edge duplicates an existing edge in the network, then the swap is accepted (thus conserving the interaction degree of the involved microorganisms and human proteins). Otherwise, the swapping procedure is repeated for another randomly selected second edge. This procedure is repeated until each original edge has participated in at least one swap. The test statistic is then recomputed from the permuted network and compared with the original (non-permuted) value. A one-tailed P value is computed for the test statistic as the fraction of n permuted networks in which the test statistic met or exceeded its value from the original network (with n = 104 used in this work). We corrected the resulting nominal P values for multiple-hypothesis testing using the FDR procedure described above.
We used the above-described network permutation procedure in two ways. First, we assessed whether particular groups of species were enriched for interactions with human proteins in specific functional categories. This involved counting pairs of strain–protein labels (for example, ‘genus’ as a strain label and a specific Gene Ontology term as a protein label) observed in edges from the original network and comparing those counts with analogues from the permuted networks. Among FDR-significant over-enrichments (q < 0.1), we focused on cases in which a given species group interacted with at least five proteins in a given functional category, and for which the observed count was at least two times larger than the mean of the permuted counts. This effect size is not influenced by the size of the Gene Ontology term pathway.
We additionally used the network permutation approach to measure conservation of human protein-binding partners among strains of variable taxonomic relatedness. For a pair of strains with at least one human binding partner each, we measured interaction conservation as the Jaccard similarity between the binding partners of the two strains. For species with two or more strains assayed, conservation was defined as the average Jaccard similarity over all pairs of daughter strains. The similarity between a pair of species was defined as the average Jaccard similarity between each daughter of the first species and each daughter of the second species. This logic was then used to compute within-taxon conservation recursively for progressively higher-level taxa (genera, families, and so forth). The entire procedure was repeated in each of 104 permutations of the original network to establish the statistical significance of conservation within specific taxa and the average conservation of each taxonomic rank.
Comparing genome and interactome similarity of C. acnes strains
To determine whether more closely related strains of C. acnes tended to bind to more similar sets of human proteins (Fig. 3c), we organized n = 174 strain-specific C. acnes proteomes from the bioBakery 3 catalogue57 (as originally derived from UniProt (v201901)49). These proteomes are sets of protein-coding genes that can be mapped to sequenced isolate genomes, a subset of which (n = 40) were also screened here using the BASEHIT methodology. To compare gene content between strains, we first mapped genes to UniRef90 families58, that is, clusters of protein sequences with at least 90% amino acid sequence identity and at least 80% coverage of the longest cluster member (the seed). The 40 tested strains contained 2,172 ± 78 UniRef90 families (mean ± s.d.). We estimated the genomic similarity of each pair of C. acnes strains as the Jaccard overlap score for their respective UniRef90 contents. We similarly quantified the similarity of the interactions of strains with the human proteome as the Jaccard overlap score of their human protein-binding partners (following the procedures used to compute general taxonomic conservation of binding partners introduced above; Fig. 2d). Finally, we applied a Mantel test to quantify the strength of correlation between genomic similarity and binding partner similarity across all pairs of strains (using Spearman correlation to quantify distance similarity and 104 rounds of strain-label permutation to quantify statistical significance).
Phylogenetic distance analysis
We constructed a tree for 108 tested strains with sequenced isolate genomes. Genomes were sourced from bioBakery 3 (ref. 57), having been initially downloaded from NCBI RefSeq and GenBank around January 2019. We systematically (re)extracted and translated open reading frames from these genomes using Prodigal (v2.6.3)59. The translated open reading frame of genomes was provided as input to PhyloPhlAn 3 (ref. 60) for tree reconstruction. We based our tree on the approximately 400 PhyloPhlAn phylogenetic marker genes61 with the PhyloPhlAn 3 software configured to the following modes: (1) high-diversity input, (2) fast tree calculation, and (3) protein supermatrix strategy. All other settings (including settings within these specific modes) were left to their default values. We converted the resulting refined tree to a distance matrix using DendroPy (v4.6.1)62.
We quantified the similarity between the interaction profile of strains as the Jaccard overlap score for their sets of human protein-binding partners (focusing on strains with at least one such binding partner). We quantified the relationship between phylogenetic distance and interaction similarity as the Spearman correlation over 5,565 valid strain pairs. We assessed the statistical significance of the resulting correlation using a Mantel test with 104 rounds of permutation. We performed a similar analysis restricted to pairs of strains with phylogenetic distances below 0.02 amino acid substitutions per amino acid site. This threshold was selected to be roughly analogous to a conspecific relationship, with 879 of 897 pairs of conspecific strains falling below this threshold, alongside only 33 non-conspecific strain pairs. We applied a modified version of the Mantel test in this analysis to account for the use of an incomplete distance matrix. More specifically, any time strain permutation required distance lookup for a pair of strains outside the less than 0.02 threshold, that pair was excluded from the corresponding permuted Spearman correlation. The final number of strain pairs that we assessed was 907.
Whole-genome sequencing of bacterial strains
Staphylococcus species were streaked on tryptic soy agar and incubated at 37 °C aerobically overnight. Single colonies were picked and inoculated into 5 ml tryptic soy broth. Bacterial DNA was extracted from overnight cultures using the DNeasy UltraClean Microbial Kit (Qiagen) according to the manufacturer’s instructions. Sequencing libraries were prepared with the Illumina Nextera XT library preparation kit (Illumina) and barcodes with the Nextera XT Index Kit v2 sets. The sequencing libraries for each strain were pooled and sequenced on a NovaSeq S4 2 × 150 bp by YCGA. Sequences were trimmed and aligned using Trimmomatic 0.36 and scaffold genomes were assembled using SPAdes 3.15.1.
Identifying associations between host protein binding and bacterial genes
To predict the specific microbial gene (or genes) mediating a given strain–human protein interaction, we assessed the overlap between strain genomic content and human binding partner patterns. The process proceeded in several steps. First, we selected the subset of strains with (1) at least one interaction and (2) for which we could obtain sequenced genomes with genes assignable to UniRef90 and UniRef50 (‘UniRef’) families by existing annotations or homology (see ‘Comparing genome and interactome similarity of C. acnes strains’). We then set aside UniRef families that occurred in only one strain, less than 5% of strains or more than 95% of strains (effectively selecting for ‘higher-variance’ genes for downstream association testing). We similarly set aside human proteins that interacted with only one strain and optionally less than 5% or more than 95% of strains. Then, the UniRef families and human proteins were separately grouped according to the strains that they were present in or interacted with, respectively, using a within-group Jaccard similarity threshold of 0.8. The most prevalent member of each group was taken as its ‘representative’. For each pair of UniRef family and human protein representatives, we cross-tabulated the presence or absence of the UniRef and the presence or absence of an interaction with the human protein over strains in our initial annotated set. Each table of counts was then assessed for statistically significant association via Fisher’s exact test, with the nominal P values FDR adjusted by the Benjamini–Hochberg method. In the analysis of C. acnes strains at the UniRef90 level, we identified 130 statistically significant (FDR q < 0.25) pairs of representative UniRef90 families co-occurring with the binding activity for a representative human protein. In the analysis of Staphylococcus strains at the UniRef50 level, we identified 4,657 such interactions.
Bacterial binding to mammalian cells
The R. gnavus strains NWP326 and NWP327 were streaked on GIFU plates and incubated overnight at 37 °C, all while maintaining anaerobic conditions. Single colonies were picked and used to inoculate 3 ml GIFU cultures, which were returned to 37 °C overnight, after which cultures reached an OD of approximately 2.5. Cultures were washed twice with PBS and resuspended in PBS with 20 µM CFSE, then shaken at 230 rpm for 1 h at 37 °C. Bacteria were washed twice with PBS, then resuspended in an equal volume of PBS + 1% BSA such that the OD remained at approximately 2.5. CFSE staining was confirmed by flow cytometry on a Cytoflex (Beckman Coulter). Data were analysed using FlowJo (v10).
Expi293 cells (Thermo Fisher; no additional authentication or mycoplasma testing was performed after purchase) were transfected according to the manufacturer’s instructions. Cells were mock transfected or transfected with a vector expressing full-length human CD7 (OHu26017, GenScript) or CD55 (OHu18890, GenScript). Four or five days after transfection, 1 ml of Expi293 culture was washed once in PBS and resuspended in 1 ml fresh PBS + 1% BSA.
For cell-binding experiments, 10 µl of Expi293 cells and 10 µl of R. gnavus were mixed in a final volume of 100 µl of PBS + 1% BSA. Cell mixtures were incubated at 4 °C for 1 h, then assessed by flow cytometry on a Cytoflex (Beckman Coulter). Data were analysed using FlowJo (v10). Data shown were gated for cell size as in Supplementary Fig. 3a.
Phagocytosis assays
Fusobacterium strains were streaked out on tryptic soy agar + 5% sheep’s blood and incubated at 37 °C under anaerobic conditions. Two days before the phagocytosis assay, colonies were picked to inoculate 7 ml cultures of chopped meat medium (Anaerobe Systems) and returned to 37 °C.
THP-1 cells (American Type Culture Collection; no additional authentication or mycoplasma testing was performed following purchase) were cultured in RPMI with 10% FBS and 1% penicillin–streptomycin, and kept at cell densities between 1 × 105 and 1.5 × 106 cells per millilitre at 37 °C + 5% CO2. To differentiate the THP-1 cells via high cell density, cells were resuspended in RPMI with 10% FBS at 2.5 × 106 cells per millilitre, and 100 µl of the cell suspension (250,000 cells) was plated per well of a tissue culture-treated flat-bottom 96-well plate and incubated at 37 °C + 5% CO2 overnight.
For phagocytosis in cis: Fusobacterium strains were washed once with PBS and resuspended in PBS with 10 µM CFSE at 1 OD ml−1. The bacteria were then shaken at 200 rpm for 1 h at room temperature. The bacteria were then washed three times and resuspended at 1 OD ml−1 in PBS. Particle counts of CFSE+ bacterial cells were performed on a Cytoflex (Beckman Coulter) to calculate multiplicity of infection (MOI). Labelled Fusobacterium strains were resuspended at 1.25 × 107 particles per millilitre in RPMI + 10% FBS, and 100 µl of the bacterial suspension (1.25 × 106 particles, MOI 5:1) was added to replace the media in the THP-1 cells. Labelled bacteria and THP-1 cells were co-incubated at 37 °C + 5% CO2 for 2 h. Cells were washed once in ice-cold PBE and fixed with 4% paraformaldehyde in PBS for 10 min on ice. Cells were washed twice more in PBE and resuspended in 200 µl PBE. Half of the sample (100 µl) was transferred to a new 96-well plate and mixed with 30 µl trypan blue to quench non-internalized CFSE signals. Phagocytosis was assessed by quantifying CFSE+ THP-1 cells on a Cytoflex (Beckman Coulter). Data shown were gated for cell size as in Supplementary Fig. 3b.
For phagocytosis in trans: Fusobacterium strains and THP-1 cells were prepared in the same way as for the phagocytosis in cis experiments. Particle counts for fluorescein-labelled E. coli K12 bioparticles (Invitrogen) and unlabelled Fusobacterium strains were performed on a Cytoflex (Beckman Coulter). Bioparticles and Fusobacterium were resuspended in RPMI + 10% FBS such that 50 µl bioparticles containing 250,000 particles (MOI 1:1) and 50 µl unlabelled Fusobacterium strains containing 500,000 particles (MOI 2:1) were added to THP-1 cells. Bioparticles, Fusobacterium and THP-1 cells were co-incubated at 37 °C + 5% CO2 for 1 h and washed, fixed and quenched as in the previously described experiments. Phagocytosis was assessed by quantifying CFSE+ THP-1 cells on a Cytoflex (Beckman Coulter). Data shown were gated for cell size as in Supplementary Fig. 3b.
Gnotobiotic mouse experiments
Bacteroides strains were streaked out on GIFU agar plates and incubated at 37 °C in an anaerobic chamber. Overnight cultures were inoculated with colonies picked into 5 ml of GIFU broth and returned to 37 °C. Six- to eight-week-old female germ-free C57BL/6 mice were individually gavaged with 200 µl of overnight Bacteroides culture normalized to 1 OD ml−1 and housed for 2 weeks in microisolator cages (ISO72P, Tecniplast).
Gnotobiotic mouse experiments were performed in duplicate (initial RNA sequencing colonization experiments) as a screening procedure, and with five mice per group for follow-up experiments, based on previous experience with similar monocolonization experiments. Sex-matched and age-matched mice were randomized into different treatment groups. No groups requiring blinding were used in this study. Samples from different groups were handled and analysed in parallel in the same way. All mice used in the study were bred in the Palm Lab Gnotobiotic Facility at the Yale School of Medicine. Mice were housed in a controlled temperature (22 °C) and humidity (50%) room under a 12 h–12 h light–dark cycle. All animal protocols were approved by the Yale University Institutional Animal Care and Use Committee (IACUC protocol 11513).
RNA sequencing
The distal 2 cm of tissue of the colon and of the terminal ileum were collected from aforementioned monocolonized mice for RNA sequencing. Tissues were placed in 500 µl RNAprotect (Qiagen) and incubated at 4 °C overnight. The following day, samples were transferred to −80 °C until further processing using the RNeasy Mini Kit (Qiagen) with on-column DNase digestion, according to the manufacturer’s protocol. Sequencing libraries were generated with the Illumina Stranded Total RNA Prep with Ribo-Zero Plus kit (Illumina). The libraries were normalized and pooled for sequencing on a full lane of a NovaSeq S4 2 × 100 bp by YCGA. RNA sequencing results were analysed in PartekFlow 11.0.24.0102.
IgA ELISAs
To quantify faecal IgA, faecal pellets were collected from each mouse after 2 weeks of monocolonization. Half of a pellet was added to 500 µl sterile PBS and homogenized with Lysing Matrix D 1.4-mm zirconium–silicate beads (MP Biomedicals) in a bead beater for 10 s. Samples were centrifuged at 50g for 15 min at 4 °C to discard large debris. The supernatant was collected and centrifuged at 10,000g for 3 min to pellet the bacteria. The supernatant was used as input for protein quantification. Pierce BCA protein assays (Thermo Scientific) were conducted according to the manufacturer’s instructions to quantify total protein in the faecal supernatant and used to normalize IgA ELISA results. Nunc-Immuno Microwell 96-well plates (Thermo Scientific) were coated overnight at 4 °C with 50 µl per well of goat anti-mouse IgA (55478, MP Biomedicals) diluted 1:2,000 in PBS. The plates were washed three times with 200 µl 1× TBS-T (Thermo Scientific) and blocked with 150 µl 3% BSA (Sigma-Aldrich) in PBS for 2 h at room temperature. The plates were washed with TBS-T three times again. Tenfold serial dilutions spanning undiluted to 10,000× were made with samples in PBS and 50 µl was added to each well. A standard curve was established with mouse IgA, κ (553476, BD Biosciences) from 5 µg ml−1 to 2.4 ng ml−1. Samples were then shaken for 2 h at room temperature. The plates were washed with TBS-T four times and then incubated with 50 µl of goat anti-mouse IgA–horseradish peroxidase (A4789, Sigma) diluted 1:5,000 in PBS + 1% BSA for 30 min at room temperature on a shaker. Plates were washed with TBS-T six times, developed with 100 µl Pierce TMB substrate (Thermo Scientific) and stopped with 100 l 2 M H2SO4, as indicated by the manufacturer’s instructions, before reading on a plate reader at 450 nm. IgA ELISA concentrations were calculated based on the standard curve and normalized by total protein content of each sample.
Statistics and reproducibility
All strains were run in triplicate during BASEHIT screening. Initial BASEHIT optimization experiments indicated that triplicate data were sufficient to identify outlier (non-concordant) results. The full strain collection screen was run once. Flow cytometry validation of BASEHIT results by recombinant protein staining were performed 2–5 times on new cultures of bacteria to confirm consistency of the phenotype, and all replication attempts were successful (Figs. 1c and 4c,d). Experiments testing bacterial binding to mammalian cells were performed four times independently, and all replication attempts were successful (Fig. 5b and Extended Data Fig. 10a). Phagocytosis experiments were performed three times independently, and all replication attempts were successful (Fig. 5d,e and Extended Data Fig. 10b). Data shown are representative for the experiments. RNA sequencing experiments were performed once with two mice per group (Fig. 5g,h). Two independent faecal IgA experiments were performed and successfully replicated (Fig. 5i).
In vitro experiments were performed in triplicate or quadruplicate based on previous experience with similar experiments. Gnotobiotic mouse experiments were performed in duplicate (initial RNA sequencing colonization experiments) as a screening procedure, and with five mice per group for follow-up experiments, based on previous experience with similar monocolonization experiments. Sex-matched and age-matched mice were randomized into different treatment groups.
GraphPad Prism (v9 and v10) and R (v4.1.0) were used for statistical tests and figure generation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting this study are included in the paper and its associated supplementary tables or deposited in publicly available databases. Source Data is available for all figures (Figs. 1–5 and Extended Data Figs. 1–10). Raw BASEHIT sequence data were deposited and are available at the NCBI Sequence Read Archive with the BioProject identifier: PRJNA1039280. Mapped barcode data have been deposited and are available at Zenodo (https://doi.org/10.5281/zenodo.10606150)51. RNA sequencing data and whole-genome sequences for Staphylococcus strains were also deposited and can be found at PRJNA1039280. Public databases used: bioBakery 3 (https://github.com/biobakery), Species Genome Bin (http://segatalab.cibio.unitn.it/data/Pasolli_et_al.html), ProTraits (http://protraits.irb.hr/), UniProt (https://www.uniprot.org/), Gene Ontology (https://geneontology.org/), proteins physical properties55 and the Human Protein Atlas (https://www.proteinatlas.org). Source data are provided with this paper.
Code availability
The custom code for the analysis of BASEHIT data has been deposited and is available at Zenodo (https://doi.org/10.5281/zenodo.10606150)51.
References
Ruff, W. E., Greiling, T. M. & Kriegel, M. A. Host–microbiota interactions in immune-mediated diseases. Nat. Rev. Microbiol. 18, 521–538 (2020).
Fan, Y. & Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 19, 55–71 (2021).
Fischbach, M. A. Microbiome: focus on causation and mechanism. Cell 174, 785–790 (2018).
Niemann, H. H., Schubert, W. D. & Heinz, D. W. Adhesins and invasins of pathogenic bacteria: a structural view. Microbes Infect. 6, 101–112 (2004).
Poole, J., Day, C. J., von Itzstein, M., Paton, J. C. & Jennings, M. P. Glycointeractions in bacterial pathogenesis. Nat. Rev. Microbiol. 16, 440–452 (2018).
Chatterjee, S., Basak, A. J., Nair, A. V., Duraivelan, K. & Samanta, D. Immunoglobulin-fold containing bacterial adhesins: molecular and structural perspectives in host tissue colonization and infection. FEMS Microbiol. Lett. 368, fnaa220 (2021).
Foster, T. J., Geoghegan, J. A., Ganesh, V. K. & Hook, M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 12, 49–62 (2014).
Langley, R., Patel, D., Jackson, N., Clow, F. & Fraser, J. D. Staphylococcal superantigen super-domains in immune evasion. Crit. Rev. Immunol. 30, 149–165 (2010).
Rooijakkers, S. H. & van Strijp, J. A. Bacterial complement evasion. Mol. Immunol. 44, 23–32 (2007).
Okumura, R. et al. Lypd8 promotes the segregation of flagellated microbiota and colonic epithelia. Nature 532, 117–121 (2016).
Gur, C. et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42, 344–355 (2015).
Walch, P. et al. Global mapping of Salmonella enterica–host protein–protein interactions during infection. Cell Host Microbe 29, 1316–1332.e12 (2021).
Penn, B. H. et al. An Mtb–human protein–protein interaction map identifies a switch between host antiviral and antibacterial responses. Mol. Cell 71, 637–648.e5 (2018).
Schweppe, D. K. et al. Host–microbe protein interactions during bacterial infection. Chem. Biol. 22, 1521–1530 (2015).
Weimer, B. C., Chen, P., Desai, P. T., Chen, D. & Shah, J. Whole cell cross-linking to discover host–microbe protein cognate receptor/ligand pairs. Front. Microbiol. 9, 1585 (2018).
Nicod, C., Banaei-Esfahani, A. & Collins, B. C. Elucidation of host–pathogen protein–protein interactions to uncover mechanisms of host cell rewiring. Curr. Opin. Microbiol. 39, 7–15 (2017).
Martinez-Martin, N. Technologies for proteome-wide discovery of extracellular host–pathogen interactions. J. Immunol. Res. 2017, 2197615 (2017).
Wood, L. & Wright, G. J. Approaches to identify extracellular receptor–ligand interactions. Curr. Opin. Struct. Biol. 56, 28–36 (2019).
Wang, E. Y. et al. High-throughput identification of autoantibodies that target the human exoproteome. Cell Rep. Methods 2, 100172 (2022).
Korotkova, N. et al. A subfamily of Dr adhesins of Escherichia coli bind independently to decay-accelerating factor and the N-domain of carcinoembryonic antigen. J. Biol. Chem. 281, 29120–29130 (2006).
Berger, C. N., Billker, O., Meyer, T. F., Servin, A. L. & Kansau, I. Differential recognition of members of the carcinoembryonic antigen family by Afa/Dr adhesins of diffusely adhering Escherichia coli (Afa/Dr DAEC). Mol. Microbiol. 52, 963–983 (2004).
Garrett, W. S. et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8, 292–300 (2010).
Brbic, M. et al. The landscape of microbial phenotypic traits and associated genes. Nucleic Acids Res. 44, 10074–10090 (2016).
Jung, P. et al. Isolation and in vitro expansion of human colonic stem cells. Nat. Med. 17, 1225–1227 (2011).
Lee, S. M. et al. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 501, 426–429 (2013).
Van Rossum, T., Ferretti, P., Maistrenko, O. M. & Bork, P. Diversity within species: interpreting strains in microbiomes. Nat. Rev. Microbiol. 18, 491–506 (2020).
Crost, E. H. et al. Utilisation of mucin glycans by the human gut symbiont Ruminococcus gnavus is strain-dependent. PLoS ONE 8, e76341 (2013).
Hall, A. B. et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 9, 103 (2017).
Kostic, A. D. et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22, 292–298 (2012).
Castellarin, M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22, 299–306 (2012).
Kostic, A. D. et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor–immune microenvironment. Cell Host Microbe 14, 207–215 (2013).
Gur, C. et al. Fusobacterium nucleatum supresses anti-tumor immunity by activating CEACAM1. Oncoimmunology 8, e1581531 (2019).
Abed, J. et al. Colon cancer-associated Fusobacterium nucleatum may originate from the oral cavity and reach colon tumors via the circulatory system. Front. Cell. Infect. Microbiol. 10, 400 (2020).
Parhi, L. et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat. Commun. 11, 3259 (2020).
Matsui, S. et al. Human Fat2 is localized at immature adherens junctions in epidermal keratinocytes. J. Dermatol. Sci. 48, 233–236 (2007).
Jonca, N. et al. Corneodesmosomes and corneodesmosin: from the stratum corneum cohesion to the pathophysiology of genodermatoses. Eur. J. Dermatol. 21, 35–42 (2011).
Johnson, N. C. XG: the forgotten blood group system. Immunohematology 27, 68–71 (2011).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Bourhis, E. et al. Wnt antagonists bind through a short peptide to the first β-propeller domain of LRP5/6. Structure 19, 1433–1442 (2011).
Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 13, 513–532 (2014).
Anastas, J. N. & Moon, R. T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 13, 11–26 (2013).
Carvalheiro, T. et al. Leukocyte associated immunoglobulin like receptor 1 regulation and function on monocytes and dendritic cells during inflammation. Front. Immunol. 11, 1793 (2020).
Weiskopf, K. et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science 341, 88–91 (2013).
Blondel, C. J. et al. CRISPR/Cas9 screens reveal requirements for host cell sulfation and fucosylation in bacterial type III secretion system-mediated cytotoxicity. Cell Host Microbe 20, 226–237 (2016).
Sauer, M. M. et al. Catch-bond mechanism of the bacterial adhesin FimH. Nat. Commun. 7, 10738 (2016).
Adrian, J., Bonsignore, P., Hammer, S., Frickey, T. & Hauck, C. R. Adaptation to host-specific bacterial pathogens drives rapid evolution of a human innate immune receptor. Curr. Biol. 29, 616–630.e5 (2019).
Baker, E. P. et al. Evolution of host–microbe cell adherence by receptor domain shuffling. eLife 11, e73330 (2022).
Xiang, H. et al. Crystal structures reveal the multi-ligand binding mechanism of Staphylococcus aureus ClfB. PLoS Pathog. 8, e1002751 (2012).
UniProt, C. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489 (2021).
Carpenter, B. et al. Stan: a probabilistic programming language. J. Stat. Softw. 76, 1–32 (2017).
andrewGhazi/basehitmodel: basehitmodel-0.1.0. Zenodo https://doi.org/10.5281/zenodo.10606151 (2024).
Pasolli, E. et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649–662.e20 (2019).
The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still going strong. Nucleic Acids Res. 47, D330–D338 (2019).
Zhou, X., Kao, M. C. & Wong, W. H. Transitive functional annotation by shortest-path analysis of gene expression data. Proc. Natl Acad. Sci. USA 99, 12783–12788 (2002).
Wang, T. & Tang, H. The physical characteristics of human proteins in different biological functions. PLoS ONE 12, e0176234 (2017).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate — a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995).
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 10, e65088 (2021).
Suzek, B. E. et al. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Asnicar, F. et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 11, 2500 (2020).
Segata, N., Bornigen, D., Morgan, X. C. & Huttenhower, C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4, 2304 (2013).
Sukumaran, J. & Holder, M. T. DendroPy: a Python library for phylogenetic computing. Bioinformatics 26, 1569–1571 (2010).
Acknowledgements
We thank all members of the Palm, Ring and Huttenhower laboratories for helpful advice and assistance. This work was supported by a grant from the Leona M. and Henry B. Helmsley Charitable Trust (3083 to N.W.P. and A.M.R.). N.W.P. is additionally supported by an NIH Director’s New Innovator Award (DP2DK125119), the NIA and NIGMS (R01AG068863 and RM1GM141649), a Pew Scholar Award, the Chan Zuckerberg Initiative, Aligning Science Across Parkinson’s, F. Hoffmann-La Roche Ltd, and gifts from the Mathers Family Foundation and Ludwig Family Foundation. A.M.R. is additionally supported by an NIH Director’s Early Independence Award (DP5OD023088), a Pew-Stewart Scholar award, and gifts from the Mathers Family Foundation, the Ludwig Family Foundation and the Robert T. McCluskey Foundation. C.E.R. and N.D.S. were supported by the National Science Foundation Graduate Research Fellowship Program. The computations in this paper were run in part on the FASRC Cannon cluster supported by the FAS Division of Science Research Computing Group at Harvard University. Illustrations in Figs. 1a, 4a and 5a,c were generated with BioRender (https://biorender.com).
Author information
Authors and Affiliations
Contributions
C.E.R., N.D.S., N.W.P. and A.M.R. designed the study. C.E.R. and N.D.S. established the BASEHIT platform and performed BASEHIT screens. C.E.R., Y.D., S.F. and A.M.R. created the exoprotein yeast display library. A.R.G. developed the BASEHIT statistical model and performed associated analysis. E.A.F. performed the global network and phylogenetic analysis. N.D.S. and C.E.R. performed all other analyses. C.E.R., N.D.S., A.A.B. and Y.C. acquired and grew bacteria for BASEHIT screens. C.E.R., N.D.S. B.D.-L., J.A.G.-H., J.D.H. and T.A.R. contributed essential reagents for and performed orthogonal validations. N.D.S., B.D.-L. and J.A.G.-H. performed the in vitro functional experiments. N.D.S., Y.Y., M.T.N. and D.S. assessed potential phenotypes and performed the in vivo experiments. Y.Y. performed the whole-genome sequencing of Staphylococcus strains. C.G. and J.O. contributed Staphylococcus strains. A.L.M. assisted with the gnotobiotic mouse experiments. C.H., A.M.R. and N.W.P. supervised the study. C.E.R., N.D.S., A.R.G., E.A.F., C.H., A.M.R. and N.W.P. wrote the paper with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
C.E.R., N.W.P. and A.M.R. are inventors of patents related to the BASEHIT technology and specific host–microorganism interactions discovered through BASEHIT. N.W.P. is a co-founder of Artizan Biosciences and Design Pharmaceuticals. All other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Mikhail Savitski and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Yeast exoproteome library composition and diversity and bacterial strain collection composition and diversity.
a, Extracellular protein sequences are curated and cloned into a standardized backbone featuring a C-terminal epitope tag. Proper display is confirmed via epitope tag staining, as well as binding by confirmation-specific antibodies or endogenous ligands for a subset of proteins. b, Schematic of expression construct used in the yeast display library. c, Proportion of the human exoproteome represented in the yeast display library. d, Each protein is represented by multiple barcodes, with a median of 20 barcodes per protein. Boxplot shows median, IQR, and whiskers extend to 1.5x IQR for n = 3,406 epitopes from 3,336 proteins in the library. e, Tissue expression (defined as Normalized Expression (NX) > 10 in the Human Protein Atlas) of proteins in the library, grouped by barrier, immune, and sterile tissues. f, Percentage of proteins in the library belonging to highly represented protein families. g, Number of strains from indicated genera, showing all genera with 9 or more strains. h, Number of strains from indicated species, showing all species with 5 or more strains. i, Number of strains from different body sites, showing all body sites with 5 or more strains.
Extended Data Fig. 2 BASEHIT optimization with AIEC identifies conditions that yield selectivity and specificity and are broadly specific across diverse known host-microbe interactions.
a, Enrichment of CD55 and CEACAM1 by AIEC using different bead:cell ratios. Enrichment is defined as the fold change in frequency of reads for the indicated protein in the post-selection library relative to the pre-selection library. Enrichment of both CD55 and CEACAM1 decreases with increasing cell:bead ratio. b, Enrichment of CD55 and CEACAM1 by AIEC labelled with variable concentrations of sulfo-NHS-biotin reagent. Increasing or decreasing concentrations of biotin decrease enrichment of CD55 and CEACAM1. c, Enrichment of CD55 and CEACAM1 by various E. coli strains with or without expression of Dr-family adhesins as indicated. CD55 and CEACAM1 are specifically enriched by the Dr-adhesin containing AIEC strain. d, Exoproteome-wide host exoprotein binding pattern of AIEC determined by BASEHIT. CD55 and CEACAM1 are enriched substantially more than any other protein. Data in a, b represent the mean ± s.d., from n = 3 independent samples. e, Diverse bacterial strains with previously described interactions with human exoproteins were screened by BASEHIT and assessed for enrichment. Interactions that were successfully detected by BASEHIT are shown as filled circles, while interactions that BASEHIT failed to detect are shown as empty circles. The overall rate of detection of previously reported interactions (54%) is shown in the pie chart on the right.
Extended Data Fig. 3 Impacts of biotinylation and bacterial cell density on the detection of interactions via BASEHIT.
a, Four bacterial strains with differing interaction profiles were grown and labeled with a titration of biotin ranging from 50 nM to 500 µM and then screened by BASEHIT. The enrichments of each protein hit are shown across all conditions, along with the enrichments of two predicted inert proteins — the coronaviral spike protein 229E-S1, and the arylsulfatase ARSA, which serve as internal negative controls. The biotin concentration used for labelling in our large-scale screen (5 µM) is highlighted in teal. Across all tested interactions, 5 µM biotin exhibited enrichments within two-fold of the “optimal” condition, and no appreciable enrichment of inert proteins was observed under any conditions. Data represent the mean ± s.d. from n = 3 independent experiments. b, Five strains were screened via BASEHIT at bacterial amounts ranging from 50 µL of 0.25 OD/mL to 10 OD/mL per well. The enrichments of hits identified in the BASEHIT screen, as well as the predicted inert proteins 229E-S1 and ARSA. The density used in our large-scale BASEHIT screen, 5 OD/mL, is highlighted in each graph. Across all tested interactions, an input of 50 µL of 5 OD/mL provided enrichment within two-fold of the “optimal” condition, and no appreciable enrichment of inert proteins was observed under any conditions. The density of bacterial particles was determined via volumetric counts for 97 strains used in our large-scale BASEHIT screen (all strains were at ~5 OD/mL). The five strains selected approximated the lower and upper bounds of particle density (~1 × 107 to ~3 × 108 particles/mL).
Extended Data Fig. 4 Modelling and scoring procedure metrics.
a, A histogram of the protein barcode representation in the input library. The wide spread on the log10 x-axis indicates a high degree of variability. The model accounts for this by using barcode input concentration as an offset term. Each tick mark across the x-axis below the histogram represents a protein. b, A Venn diagram showing interaction counts that pass each of the three hit-calling thresholds for the standard threshold set (95% interval excludes zero, estimated effect size > 0.5, and concordance score > 0.75). c, A plot of normalized counts demonstrating the utility of the concordance threshold. Both interactions shown have about the same interaction score (around 1.9) and similarly variable inputs in the Pre library (top panels), but the concordance between normalized output counts (bottom panels) in the TFF2:HM645 interaction is much higher than in SLC6A9:HM1171. Grey cells represent zero counts. d, A histogram of concordance scores for all interactions in the assay. Dashed vertical lines indicate the stringent and standard thresholds. e, Saturation curves from repeated rarefaction analysis. Given that both sets of thresholds have roughly plateaued, we can conclude that we have identified most of the interactions that are detectable under the experimental conditions. f, Comparison of the results of an initial run of the scoring method against five repeated runs where the standard deviation of the normal prior on interaction scores varied from 0.075 to 0.3. Each dot represents the score of a particular interaction. Only interactions that were a hit in at least one run are shown. The middle panel uses the same value as the initial run, showing the extent of Monte Carlo error. As expected, the rank and relative magnitude of scores are highly consistent between runs, while narrower priors lead to lower scores and fewer hits and wider priors lead to higher scores and more hits. The two distinct groups of interactions visible in the panels with wide priors represent subpopulations of interactions that are either more or less amenable to the zero-inflation component of the model.
Extended Data Fig. 5 Proteins from multiple tissues bind bacteria with a power-log distribution, and bacteria from different tissues or phyla show similar distributions of host protein binding.
a, Plot of number of bacterial strains bound (interaction called as a hit) for proteins expressed in multiple host tissues. Tissues expression is defined as Human Protein Atlas normalized expression NX > 10. b, Plot of number of proteins bound (interaction called as a hit) for all bacteria with hits as well as for all bacteria including non-binders. c, Same plot as b but depicting strains isolated from specific tissues. Maximum and mean reported for bacteria with one or more hits. d, Same plot as b but depicting strains from indicated phyla. Maximum and mean reported for bacteria with one or more hits.
Extended Data Fig. 6 Biophysical properties are significantly different between interacting and non-interacting proteins.
Proteins which bound at least one bacterial strain (“Targets”) are compared with “Non-targets” for various biophysical properties as indicated. FDR shown is for a two-tailed Wilcoxon Rank-Sum test. Box plots show median, IQR, and whiskers extending to 1.5x IQR, for n = 631 “Targets” and n = 2,705 “Non-targets”.
Extended Data Fig. 7 Relationships between similarity in strains’ interaction profiles and their phylogenetic distance.
a, We computed a phylogenetic tree over 108 genomes of tested strains based on ~ 400 broadly distributed protein families. We compared distances in this tree with similarity of strains’ interaction profiles using Spearman correlation (n = 5,565 strain pairs). Phylogenetic distance is expressed in units of amino acid substitutions per amino acid site. Interaction similarity was measured as the Jaccard overlap score between strains’ sets of human protein binding partners (ignoring strains with no binding partners). b, We separately considered the subset of n = 907 strain pairs with phylogenetic distance <0.02 substitutions per site, which was largely synonymous with a conspecific relationship in taxonomy. In both regimes, interaction similarity and phylogenetic distance were strongly and significantly negatively correlated. In both cases a two-tailed Mantel test with 104 permutations with FDR adjustments was performed.
Extended Data Fig. 8 Superbinder Staphylococcus show highly overlapping sub-networks.
a, Network of 7S. pasteuri and 8 other Staphylococcus superbinders, highlighted in green and orange respectively. The 5 proteins bound by the most strains are labeled. b, Overlap in interaction profiles across strains. Proteins are binned according to whether they are bound by more than half of the S. pasteuri strains (“Pasteuri core”), or by multiple or only one superbinder strains (“Multiple” and “Unique”, respectively). c, Top proteins bound by multiple superbinders. Overall interaction profiles of proteins bound by 7 or more superbinder strains are colored according to the strains they recognize, including all other Staphylococcus strains as well as non-Staphylococcus strains. d, Interactions for skin-expressed proteins CDSN, FAT2, and XG for all 519 bacterial strains organized by tissue of origin. Dashed red line at 0.5 represents hit threshold.
Extended Data Fig. 9 Phylogenetic specificity of interactions with tissue-specific proteins across all tested strains.
The interaction scores for all 519 tested strains are shown for the indicated proteins, which are highlighted in Fig. 4a. Strains are colored by phylum, and all scores above the hit threshold line at 0.5 are indicated and labeled with the genus of the strain. Parentheses indicate the frequency of hits within a genus.
Extended Data Fig. 10 Ruminococcus gnavus and Fusobacterium strains influence host cell binding and function.
a, Representative flow cytometry plots of CD7-binding and non-binding R. gnavus strains labelling mock, CD7-, and CD55-expressing EXPI293 cells as shown in Fig. 5b. b, Representative flow cytometry plots of THP-1 phagocytosis of CFSE-labelled Fusobacterium spp. and of fluorescein-labelled E. coli K12 BioParticles incubated with unlabelled Fusobacterium spp. from Fig. 5d,e.
Supplementary information
Source data
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sonnert, N.D., Rosen, C.E., Ghazi, A.R. et al. A host–microbiota interactome reveals extensive transkingdom connectivity. Nature 628, 171–179 (2024). https://doi.org/10.1038/s41586-024-07162-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07162-0
- Springer Nature Limited
This article is cited by
-
Semi-supervised meta-learning elucidates understudied molecular interactions
Communications Biology (2024)