

Abstract
The transcriptional machinery is thought to dissociate from DNA during replication. Certain proteins, termed epigenetic marks, must be transferred from parent to daughter DNA strands in order to maintain the memory of transcriptional states1,2. These proteins are believed to re-initiate rebuilding of chromatin structure, which ultimately recruits RNA polymerase II (Pol II) to the newly replicated daughter strands. It is believed that Pol II is recruited back to active genes only after chromatin is rebuilt3,4. However, there is little experimental evidence addressing the central questions of when and how Pol II is recruited back to the daughter strands and resumes transcription. Here we show that immediately after passage of the replication fork, Pol II in complex with other general transcription proteins and immature RNA re-associates with active genes on both leading and lagging strands of nascent DNA, and rapidly resumes transcription. This suggests that the transcriptionally active Pol II complex is retained in close proximity to DNA, with a Pol II–PCNA interaction potentially underlying this retention. These findings indicate that the Pol II machinery may not require epigenetic marks to be recruited to the newly synthesized DNA during the transition from DNA replication to resumption of transcription.
Similar content being viewed by others


Main
Maintenance of differential gene expression depends on RNA transcription catalysed by Pol II5. For a cell to retain its lineage from one generation to the next, the transcriptional status at individual genes must be maintained and transmitted during the cell cycle to daughter cells. Retention of transcriptional states is critical during DNA replication and mitosis because chromatin structure may be drastically altered owing to protein displacement by the replication fork2 and by chromatin condensation6, respectively.
Although there is no conclusive experimental evidence, chromatin-based epigenetics assumes that components of the Pol II transcriptional apparatus and immature RNA dissociate from DNA for a significant period of time, and that restoration of chromatin structure (that is, the histone code) is an obligate prerequisite to re-recruiting Pol II to active genes1,4. We interrogated this model and found that active forms of Pol II, the key general transcription factor (GTF) TATA-binding protein (TBP), several elongation factors and immature RNA transcripts, were detected on both strands of nascent DNA immediately following replication. Consistently, new transcription resumes shortly after replication, and may involve completion of synthesis of immature RNAs.
These findings may change the traditional model of epigenetic bookmarking, which assumes dissociation of transcriptional proteins and requires chromatin restoration as a mechanism for the de novo assembly of the Pol II complex at its target genes. On the basis of our results, we suggest that the transcriptionally engaged Pol II complex may be transiently dislodged from DNA but retained at replication forks during the passage of the replication fork through direct protein interactions with PCNA. This may enable the complex to quickly re-associate with its sites on both strands of nascent DNA at active genes and resume transcription, alleviating the need for any additional bookmarking mechanisms during S phase at active genes.
Pol II is bound to short nascent DNA
To determine when Pol II is recruited back to DNA after replication, we measured the recruitment of Pol II to nascent DNA using the chromatin assembly assay7,8 (CAA; Fig. 1a). This assay uses a proximity ligation assay (PLA) to assess whether a chromosomal protein is within 40 nm or less of EdU-labelled nascent DNA. Cells were labelled for 5 min, or for 10 min followed by increasing chase times to assess Pol II accumulation following DNA replication. These experiments were performed on an asynchronous population of cells, so EdU labelling represents firing of different replicons in individual cells. Since hundreds of nuclei are examined in each experiment, the results of CAA reflect association of tested proteins with nascent DNA at every replicating region of the genome7,8.

a, Scheme of CAA. Nascent DNA is pulse-labelled with EdU (red circle) with or without chase and conjugated with biotin (purple circle). PCNA is shown as a red ring and the replisome is shown in blue. b–i, Cells were labelled and chased to detect labelled Pol II (b), Pol II S5P (c), Pol II S2P (d), TBP (e), CDK9 (f), cyclin T1 (g), SPT4 (h) or SPT5 (i). PLA was performed between nascent DNA and the indicated proteins. As negative controls for the PLA reaction, we used EdU-negative nuclei as well as CAA using an antibody to either the transcription protein or anti-biotin and IgG control (Extended Data Fig. 1). Data are mean ± s.d. (n = 3 independent experiments). The number of nuclei examined were: 440 (b), 365 (c), 475 (d), 429 (e), 355 (f), 335 (g), 469 (h) and 536 (i). One-way ANOVA with Tukey’s post hoc test. Scale bars, 10 μm. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Total Pol II was robustly detected on nascent DNA following 5 min of EdU labelling (Fig. 1b). The 5 min-labelled nascent DNA fragments range in length from several hundred base pairs (bp) to approximately 3.7–6.0 kb in euchromatic replication9. The approximately twofold increase in the number of PLA signals at 10 min probably reflects the increased length of the EdU-labelled DNA. Of note, we did not observe CAA signals in cells in which the PLA was performed using either an anti-Pol II antibody or anti-biotin antibody paired with an IgG control (Extended Data Fig. 1).
Further, we performed CAA using antibodies specific to Pol II phosphorylated on Ser5 (Pol II S5P) or Ser2 (Pol II S2P) of its C-terminal domain (CTD), which correspond to transcriptional initiation and elongation, respectively10. The initiating form Pol II S5P and the elongating form Pol II S2P were detected on 5 min EdU-labelled DNA and then increased about twofold within 10 min after replication (Fig. 1c,d).
Treatment of cells for 30 min with the initiation inhibitors THZ-1 (ref. 11) and triptolide12 (TPL) or the elongation inhibitor 5,6-dichloro-1-α-d-ribofuranosylbenzimidazole13 (DRB) did not drastically reduce the number of CAA signals on nascent DNA (Extended Data Fig. 2a), although treatments with THZ-1 and DRB reduced overall levels of phosphorylated Pol II (Extended Data Fig. 2b) (note that DRB also inhibits CDK7 (ref. 14)). This suggests that the detection of phosphorylated Pol II on nascent DNA is probably not the result of a new round of transcription after replication.
Longer treatment with TPL caused near complete loss of signals for Pol II S5P and S2P (Extended Data Fig. 2a), and longer treatment with DRB caused partial loss of Pol II S5P and near complete loss of Pol II S2P on nascent DNA (Extended Data Fig. 2a). The partial loss of Pol II S5P during longer treatment with DRB may either be owing to either non-specific inhibition of CDK7 (ref. 14) or decreased elongating Pol II, since Pol II S5P is not immediately de-phosphorylated once a Pol II molecule moves into elongation15. Notably, neither of these treatments affected the levels of the essential replication component PCNA (Extended Data Fig. 2c).
We observed significant increases of PLA signals for all Pol II isoforms at chase times from 25 to 40 min, reaching their maximum at 40 min after replication, suggesting that by this point Pol II has finished its loading onto nascent DNA (Fig. 1b). These results suggest that both initiating and elongating forms of Pol II associate with DNA within several minutes after passage of the replication fork and are fully loaded onto the new strands by 40 min after replication.
GTFs are detected on short nascent DNA
Detecting Pol II isoforms on nascent DNA shortly after replication raises the question of whether a GTF also associates with short nascent DNA. We assessed this by examining association with nascent DNA of TBP, a representative initiation factor that is the first GTF to bind to the core promoter and also remains associated long after transcription initiates16,17. Results of CAA show that similar to Pol II, TBP is detected on nascent DNA within 5 min of replication and the TBP signal increases after 10 min of labelling as expected (Fig. 1e). This suggests that similar to Pol II, TBP is associated with DNA within minutes of replication. As expected, 4 h of treatment with the elongation inhibitor DRB did not affect the levels of TBP on nascent chromatin (Extended Data Fig. 2d).
In line with detection of both Pol II S5P and S2P shortly after replication, we found that proteins involved in the transition from transcriptional initiation to elongation are also associated with nascent DNA. The P-TEFb complex comprising cyclin T1 and CDK9 is responsible for phosphorylating the CTD of Pol II at serine 2, which is critical for enabling Pol II to escape promoter proximal pausing and move into productive elongation down the gene body18. CAA for cyclin T1 and CDK9 shows association of these proteins following 5 and 10 min of labelling with EdU (Fig. 1f,g).
Similarly, we found that both components of the DRB sensitivity inducing factor (DSIF) complex that is essential for transcriptional elongation and associates with elongating Pol II, SPT4 and SPT5, are also detected on nascent DNA in 5 min after replication (Fig. 1h,i). This adds further support to the presence of the actively elongating Pol II complex on the relatively short nascent DNA. Treatment for 4 h with DRB caused a significant decrease in SPT4 and SPT5 on nascent chromatin (Extended Data Fig. 2e) but not a complete loss of either factor. This is probably owing to the loss of DSIF associated with elongating Pol II S2P, but not DSIF associated with paused Pol II. Thus, as well as Pol II, GTFs are also associated with DNA within minutes after DNA replication.
GTFs and PCNA colocalize on nascent DNA
We developed an approach that we call multiplex CAA to examine whether transcriptional proteins are associated on nascent DNA close to the replisome marker PCNA (Fig. 2a). In this assay, two simultaneous PLA reactions use different pairs of oligonucleotide-tagged antibodies to generate unique amplified DNA regions for each pair of antigens if they are in close proximity, which can then be detected with oligonucleotides labelled with different fluorophores19. To improve the resolution of this analysis, we used stimulated emission depletion (STED) super-resolution microscopy to ensure that the signals of these two PLA reactions indeed overlap.

a, Schematic depiction of multiplex CAA with transcription proteins and PCNA. Nascent DNA is EdU-labelled (blue circle) and conjugated to biotin (purple circle). Two PLA reactions are performed in parallel between nascent DNA and a transcription protein (for example, Pol II (red signal)) and between nascent DNA and PCNA (green signal). b, Example distance measurements between multiplex CAA signals with PCNA (green) and Pol II S5P (red). Left, an overlapping pair of signals. Right, a pair of non-overlapping but closely spaced signals. Arrows through the PLA foci are 1 µm in length and correspond to the intensity plots below which depict the intensity for the green (top) and red (bottom) signals. The grey bar within the plot shows the distance measurement between the centres of the foci; the distance is shown in the image. c, Multiplex CAA for Pol II S5P and PCNA. Top, image showing a full nucleus. Bottom, examples of overlap are enlarged and shown as split channels. d, Quantification of multiplex CAA experiments with PCNA; data represent the average percentage of overlaps ± s.d. from n = 3 independent experiments. e, Multiplex CAA for Pol II S2P and PCNA. Top, image showing a full nucleus. Bottom, examples of overlap are enlarged and shown as split channels. f, Multiplex CAA for TBP and PCNA showing enlarged example images of overlap between CAA for TBP and PCNA. g, Multiplex CAA for SPT4 and PCNA showing enlarged example images of overlap between CAA for SPT4 and PCNA. h, Multiplex CAA for SPT5 and PCNA showing enlarged example images of overlap between CAA for SPT5 and PCNA. Scale bars: 2 µm (full nucleus in c,e) and 400 nm (enlarged images in c,e–h). Data shown are representative of three independent experiments.
When measuring the distance between the centres of the PLA foci in multiplex CAA, we found that overlapping signals are on average 136 nm ± 53 nm (mean ± s.d.) from one another. Closely spaced but not overlapping PLA signals are detected when the distance between the centres of signals is greater than 275 nm (see example measurements in Fig. 2b). It is estimated that 100 bp of linear DNA, which is expected at replication forks20, is 34 nm in length21. Therefore, this assay detects transcription proteins on nascent DNA within 400 bp for the overlapping signals (but they may be as close as 244 bp) to about 800 bp for the adjacent signals from the replication protein PCNA.
Multiplex CAA following 10 min of labelling with EdU was performed with PLA between Pol II S5P and EdU (red) and PLA between PCNA and EdU (green) (Fig. 2c). On the basis of the length of DNA synthesized during 10 min of labelling9 (7.4 to 12 kb), and the distances described above, the probability of detecting Pol II overlapping with PCNA (that is, about 400 bp apart) is 5–10%. In line with these estimates, we observed that around 8% of Pol II S5P PLA signals overlap with PCNA PLA signals on nascent DNA (Fig. 2d and Extended Data Fig. 3a). We detected a similar proportion of overlapping signals between Pol II S2P and PCNA on nascent DNA (Fig. 2d,e). We note that in addition to overlapping signals we also detected 15.0 ± 1.68% (mean ± s.d.) of closely spaced signals (Fig. 2b), which reflects association of proteins with PCNA on less than 1 kb of nascent DNA (see above); for simplicity of analysis, we subsequently focused on quantification of the overlapping signals. We expected that longer labelling would decrease the percentage of overlaps since the number of Pol II signals would increase, but the fraction of signals overlapping with PCNA would decrease. In line with this, we observed that the number of overlapping signals for Pol II S5P or S2P decreased from 8% after 10 min of EdU labelling to close to 4% after 20 min of EdU labelling (Extended Data Fig. 3b).
We performed multiplex CAA for TBP and PCNA and found that a large number of signals for these proteins overlap, suggesting that TBP is associated with DNA shortly following replication (Fig. 2d,f). Finally, we found that SPT4 and SPT5 also co-occupy short nascent DNA alongside PCNA (Fig. 2d,g,h). These results indicate that both forms of Pol II and accompanying proteins are associated with nascent DNA within few hundred base pairs of the passed replisome.
Pol II binds to both new strands of DNA
Transcription of active genes should resume on both strands of nascent DNA, and it is not known when transcription proteins re-associate with each strand of nascent DNA. To assess this, we selected a representative protein as a marker for each daughter strand in multiplex CAA experiments. For the leading strand, we chose DNA polymerase ε (Pol ε), the polymerase primarily responsible for leading strand synthesis, and for the lagging strand we selected flap endonuclease 1 (FEN1), which is involved in resolving Okazaki fragments22. PLA between Pol ε and FEN1 detected almost no signals; as a comparison, direct PLA between PCNA and either of these proteins results in a significant number of PLA signals (Extended Data Fig. 3c). These results suggest that Pol ε and FEN1 are not in close proximity to each other and can therefore serve as specific markers for the leading and lagging strands, respectively.
To assess whether transcriptional proteins were bound to the leading and lagging strands, we performed multiplex CAA with antibodies to transcription proteins and Pol ε (Fig. 3a) and FEN1 (Fig. 3d). We found that a large number of CAA signals for both major forms of phosphorylated Pol II, TBP and SPT4 overlap with the CAA signals between Pol ε and EdU (Fig. 3b,c), as well as CAA signals between FEN1 and EdU (Fig. 3e,f). The percentage of overlap for all tested transcription proteins with both Pol ε and FEN1 was approximately 4% (Fig. 2c,f), although a large number of closely spaced signals were also observed. The number of overlapping signals was half that for PCNA, potentially because only half of these proteins can be transferred from the parental strand to each of the daughter strands of nascent DNA. Together, the results of multiplex CAA with Pol ε and FEN1 show that initiating and elongating Pol II, the initiation protein TBP, and the DSIF component SPT4 are all associated with both leading and lagging strands immediately after the passage of the replisome.

a, Schematic depiction of multiplex CAA with transcriptional proteins and Pol ε. Nascent DNA is pulse-labelled for 10 min with EdU (red circle), biotin is conjugated to EdU (purple circle), and the two PLA reactions are performed in parallel. The first is between nascent DNA and a transcriptional protein, such as Pol II, with the generated PLA signal depicted in red. The second is between nascent DNA and the leading strand component Pol ε, with the generated PLA signal depicted in green. STED microscopy was used to analyse proximity or overlap between PLA signals of tested proteins. b, STED images of multiplex CAA for Pol II S5P, Pol II S2P, TBP and SPT4 with Pol ε. Panels show enlarged images of examples of overlap between CAA for each transcription protein (red) and CAA for Pol ε (green). Data shown are representative of three independent experiments. c, Quantification of multiplex CAA experiments with DNA Pol ε. Data are represented as average percentage of overlaps ± s.d. from n = 3 independent experiments. d, Scheme depicting multiplex CAA with transcriptional proteins and FEN1, which is associated with the lagging strand. Experiments were performed as described in a. e, STED images of multiplex CAA for Pol II S5P, Pol II S2P, TBP and SPT4 with FEN1. Panels show enlarged images of examples of overlap between CAA for each transcription protein (red) and CAA for FEN1 (green). Data shown are representative of three independent experiments. f, Quantification of multiplex CAA experiments with FEN1. Data are represented as average percentage of overlaps ± s.d. from n = 3 independent experiments. Scale bars, 400 nm.
Pol II binds to sites on new DNA strands
To understand whether Pol II associates with the same regions of active genes after replication, we performed sequential chromatin immunoprecipitation (re-ChIP) assays with BrdU-labelled nascent DNA7,23 (Extended Data Fig. 4a). BrdU re-ChIP assays following 25 min and 40 min of BrdU labelling detected Pol II at the same upstream and downstream regions of active genes GAPDH, PPIA and TNFAIP3, but not at the repressed gene GFAP, as it was detected by conventional chromatin immunoprecipitation (ChIP) assays (Extended Data Fig. 4b–e). These experiments support the results of the CAA assays and suggest that Pol II associates with the same regions of active genes within at least 25 min following DNA replication.
The BrdU re-ChIP assay7,23 requires about 20 min of DNA labelling. Therefore, to determine whether Pol II is present at specific regions of active genes on DNA immediately after replication, we performed re-ChIP assays first immunoprecipitating for Pol II and then for the replication protein PCNA7. PCNA is quickly removed from DNA following passage of the replication fork (Extended Data Fig. 6b) making it a good biochemical marker for both strands of nascent DNA in re-ChIP assays. In the previous study7, and in the current work, we found that immunoprecipitation with PCNA pulls down DNA fragments of about 100–200 bp (Extended Data Fig. 5a,b, left). Treatment with the single-stranded specific S1 nuclease (Extended Data Fig. 5c) did not change the size of the PCNA-immunoprecipitated fragments (Extended Data Fig. 5a,b, right), suggesting that single-stranded DNA in front of PCNA was not present in the immunoprecipitated material, probably because it is more susceptible to sonication. Thus, this re-ChIP assay determines whether a protein of interest is bound to nascent DNA within 100–200 bp following passage of the replisome, which given the rate of replication corresponds to nascent DNA synthesized within a few seconds after replication.
We found that PCNA was associated with all tested regions, albeit at low levels (Extended Data Fig. 6a). This is expected since PCNA moves across every region of the genome once per cell cycle7. In the re-ChIP experiments with PCNA (Fig. 4a), Pol II was detected at both upstream and downstream replicating regions of the three active genes, but not at a repressed gene (Fig. 4b, bottom). Similarly, re-ChIP assays (Extended Data Fig. 6c) showed that SPT4 is associated with the same nascent upstream and downstream regions of the same three genes, but not at the repressed gene. Since the examined downstream regions (regions 2, 4, 6 and 8; see Extended Data Fig. 4b for primer map) are far enough from the transcription start site (TSS), detection of Pol II and SPT4 by PCNA re-ChIP (Fig. 4b and Extended Data Fig. 5c) precludes that downstream Pol II and SPT4 binding is the result of a new round of transcriptional initiation, but rather suggests that the elongating Pol II complex re-associates with DNA within the gene body within several seconds after the passage of the replication fork. These results imply that Pol II and SPT4 associate with nascent DNA within 200 bp of the replication fork.

a, Schematic representation of re-ChIP assays. Chromatin was first immunoprecipitated for Pol II, and then subsequently precipitated for either PCNA, FEN1 or Pol ε. As previously demonstrated7, single-stranded DNA fragments ahead of the replication fork are destroyed by sonication; the recovered fragments represent 100–200 bp stretches of nascent DNA (Extended Data Fig. 5). Recovered DNA was analysed by quantitative PCR (qPCR). IP, immunoprecipitation. b, Re-ChIP with PCNA and Pol II. Top, ChIP for either Pol II or IgG at the genes indicated in Extended Data Fig. 4b. Bottom, re-ChIP of samples first immunoprecipitated for Pol II. Following elution, Pol II samples were re-immunoprecipitated for PCNA or IgG. Following DNA purification, samples were analysed by qPCR. c, Re-ChIP with FEN1 and Pol II. Top, ChIP for either Pol II or IgG at the genes indicated in Extended Data Fig. 4b. Bottom, re-ChIP of samples first immunoprecipitated for Pol II. Following elution, Pol II samples were re-immunoprecipitated for FEN1 or IgG. Following DNA purification, samples were analysed by qPCR. d, Re-ChIP with Pol ε and Pol II. Top, ChIP for either Pol II or IgG at the genes indicated in Extended Data Fig. 4b. Bottom, re-ChIP of samples first immunoprecipitated for Pol II. Following elution, Pol II samples were re-immunoprecipitated for Pol ε or IgG. Following DNA purification, samples were analysed by qPCR. b–d, Data are mean values and are representative of two independent experiments.
To confirm these results and to assess whether Pol II re-associates with specific gene regions on the leading and lagging strands of nascent DNA, we developed re-ChIP assays for Pol ε and FEN1. The results of these experiments suggest that immediately after replication, Pol II associates on both strands of nascent DNA with the same upstream and downstream regions of active genes as it was associated on parental DNA (Fig. 4c,d). The results of the re-ChIP assays with PCNA, Pol ε and FEN1 confirm the results of the multiplex CAA assays in that RNA Pol II and other elongational proteins are able to re-associate in a few seconds on both strands of nascent DNA. We suggest that the replisome may only transiently displace components of the general transcriptional and elongation machinery, and that they may be re-directed to both strands of nascent DNA.
RNA is bound to Pol II on nascent DNA
Finding that the elongationally active form of Pol II and the elongation complex DSIF are associated with short nascent DNA raises two related questions. First, regarding the fate of immature RNAs that were being synthesized by Pol II just before replication; and second regarding when transcription resumes on the new strands of DNA after replication. To address these issues, we adapted the PLA-based RNA–DNA interaction assay (RDIA) that detects the proximities of RNA labelled with BrU and nascent DNA labelled with EdU24,25.
First, to assess whether immature transcripts are retained during the passage of the replication machinery, nascent RNAs were labelled with BrU, DNA was labelled with EdU, and then cells were chased for various times. This order of labelling enables us to assess whether transcripts being synthesized before the passage of the replication complex remain in the vicinity of newly replicated DNA. As shown in Fig. 5a, nascent transcripts remain in close proximity to nascent DNA following replication. These RNAs are not the result of a new round of transcription, since inhibition by THZ-1 or DRB did not affect association of the BrU-labelled RNAs with EdU-labelled DNA (Extended Data Fig. 7a). These results imply that immature RNAs synthesized prior to passage of the replication fork are not aborted from the elongational form of Pol II S2P during the passage of the replisome.

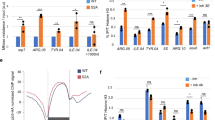
a, Cells were labelled with BrU and EdU, and chased as indicated. b, Scheme depicting multiplex CAA with Pol II and immature RNA. PLA reactions were between nascent DNA and Pol II S2P (red), Pol II S2P and BrU-labelled RNA (green). c, STED images of multiplex CAA described in b. d, Cells were labelled with EdU, chased as indicated, and labelled with BrU. e–g, Co-immunoprecipitation of nuclear extracts with PCNA (e), Pol II S2P (f) and Pol II S5P (g) antibodies. h, GST pull-down of nuclear extracts with GST–PCNA or GST. In e–h, membranes were immunoblotted for indicated proteins. Data shown are representative of three independent experiments. For blot source data, see Supplementary Fig. 1. i, Cells were untreated (UT) or treated with PCNA-I1 for indicated times. j, Cells were untreated or treated for 3 h with PCNA-I1. PCNA-I1 was washed out as indicated. In i,j, CAA was performed for indicated proteins; signal was compared to the untreated condition (bottom). a,d,i,j, Data are mean ± s.d. (n = 3 independent experiments). The number of nuclei examined over 3 independent experiments were: 323 (a); 606 (d); PCNA: 344, S5P: 291, S2P: 340 (i); PCNA: 210, S5P: 192, S2P: 185 (j). One-way ANOVA with Tukey’s post hoc test. k, Proposed model for Pol II transfer from parental to nascent DNA. As DNA polymerase moves head-on towards the Pol II complex, it is displaced from DNA, but maintained at the replisome by interactions between PCNA and Pol II. Pol II rebinds nascent DNA immediately following replication and resumes transcription. NS, not significant. Scale bars: 10 μm (a,d) and 400 nm (c). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To examine this possibility, we used a different version of multiplex CAA, which incorporates analysis of RNA–protein interactions. The first PLA reaction was between nascent DNA and Pol II S2P (red), and the second PLA reaction was between Pol II S2P and immature RNA synthesized prior to replication (green) (Fig. 5b). Given that multiple S2 residues are phosphorylated in the heptad repeats of the CTD of Pol II, several antibodies can independently bind to the same Pol II S2P molecule making this experiment feasible. As shown in Fig. 5c, we observed that about 20% of the BrU Pol II S2P signals (green) (an average of 22 signals in multiple nuclei) overlap with nascent DNA-bound Pol II (red) signals. These results suggest that immature transcripts observed by RDIA (Fig. 5a) are indeed associated with the elongationally engaged Pol II complex bound to nascent DNA.
Rapid resumption of RNA synthesis
To determine the kinetics of resumption of transcription on nascent DNA, cells were pulse-labelled with EdU for 10 min, and after various chase times, nascent RNA was labelled with BrU, followed by PLA. PLA signals were first detected at 25 min of combined labelling with EdU and BrU and their number further increased at subsequent times post-replication. This suggests that transcription resumes at least as early as 25 min following replication, but it is possible that it may resume even earlier (Fig. 5d).
We observed that the number of PLA signals for immature RNA begins to dissipate as early as 20 min after replication (Fig. 5a). Remarkably, the timing of the dissipation of signals for immature RNAs (20 min after replication) (Fig. 5a) and resumption of new transcription on nascent DNA (25 min after replication or earlier) (Fig. 5d) was very close, suggesting that these transcripts may begin to dissociate from DNA potentially after completion of their synthesis. Given that we detected Pol II on very short stretches of nascent DNA in the downstream regions of active genes (Fig. 4), and observed Pol II S2P colocalizing with PCNA (Fig. 2d), Pol ε and FEN1 (Fig. 3b,d), these results may suggest that RNA synthesis resumes on both strands of daughter DNA.
Pol II transiently interacts with PCNA
Our results suggest that Pol II complexes are retained in the vicinity of the replication fork and re-associate with their sites on nascent DNA within seconds after the passage of the replication complex. Mechanistically, this may occur through transient protein–protein interactions between transcription and replication complexes during replisome progression. We examined whether retention of Pol II at the replication fork is mediated by phase condensates. However, in the CAA experiments, 1,6-hexanediol, which disrupts most interactions known to drive the formation of liquid–liquid phase separation26 (LLPS) did not affect association of Pol II with nascent DNA (Extended Data Fig. 7b). Although some other, 1,6-hexanediol-resistant types of LLPS, may also be involved27, it seems unlikely that LLPS has a major role in the retention of Pol II in the vicinity of the replication foci.
Another possibility is direct protein–protein interactions between Pol II and the replisome. We found that PCNA co-immunoprecipitates with several core components of the Pol II complex, Rpb1, Rpb2, Rpb7, Rpb9 and Rpb11, as well as the DSIF components SPT4 and SPT5, suggesting that the entire core Pol II complex and associated factors may interact with the replisome. PCNA-immunoprecipitated material contained S2P and S5P phosphorylated forms of the largest Pol II subunit Rpb1, but not the unphosphorylated Pol II isoform (Fig. 5e). Ethidium bromide did not cause loss of Pol II following anti-PCNA immunoprecipitation (Extended Data Fig. 7c), suggesting that this interaction is DNA-independent28. In reciprocal experiments, we found that Pol II S2P and S5P antibodies each co-immunoprecipitated PCNA together with core components of the Pol II complex (Fig. 5f,g). Further, GST–PCNA fusion protein specifically associated with the hyperphosphorylated forms of Pol II in nuclear extracts (Fig. 5h). This suggests that PCNA, which is associated with both strands of replicating DNA and is known to interact with multiple proteins29,30, is a replisome component that directly interacts with the Pol II complex.
To examine a potential causal link between PCNA and the detected association of Pol II with short nascent DNA, we used the PCNA inhibitor PCNA-I1, which affects PCNA trimerization31. CAA experiments showed that increasing times of treatment with PCNA-I1 led to a gradual decrease in association of PCNA and to a more profound decrease in association of both Pol II S2P and S5P with nascent DNA (Fig. 5i). Notably, this decrease beginning at 45 min of treatment was before loss of EdU labelling (Extended Data Fig. 7d) suggesting that the decrease is not due to loss of EdU available for CAA. In reciprocal experiments, when complete inhibition of PCNA association with chromatin by PCNA-I1 was reversed by washing out this inhibitor, association of PCNA and, subsequently, association of both phosphorylated Pol II isoforms was restored (Fig. 5j). These results strengthen the results of the protein interaction assays and provide a functional link between PCNA and Pol II at the time of DNA replication.
Discussion
The results presented here suggest a new epigenetic paradigm by introducing a novel mechanism of bookmarking of the active state of genes through DNA replication. The field has long accepted that because Pol II is evicted during mitosis, the bookmarking by transcription factors is necessary32,33. It is a widely held belief that eukaryotic Pol II is evicted during DNA replication, although it remains unknown, and studies in prokaryotes have given differing results34,35,36. Based on this notion, modified histones3,4 are considered to be epigenetic marks. More recently, histone-modifying complexes were also implicated as putative epigenetic marks during S phase7,8,23,37. These theories are based on the assumption that active Pol II complexes dissociate during replication, and that transcription resumes after some time following chromatin rebuilding, which is triggered by presumptive epigenetic marks1,3. Experimental support for this theory is limited to detection of Pol II S5P on nascent chromatin in 30 min post-replication by chromatin occupancy after replication with sequencing (ChOR-seq), which also led to the postulation that this timing reflects restoration of transcription on nascent DNA38. However, labelling with EdU used in ChOR-seq, similar to BrdU re-ChIP shown in Extended Data Fig. 4, limits this assay to 10–20 min after DNA replication, and the only firm conclusion that can be drawn from these results is that Pol II is associated with its target genes within 30 min or less after replication.
The results of our work suggest that active Pol II in complex with RNA and accompanying factors remains in the vicinity of the replication complex. Mechanistically, retention of the Pol II complex near the replication fork may rely on direct protein interactions between PCNA and the Pol II complex, although the details of this interaction remain unknown. Once replication is complete, Pol II quickly binds to both strands of nascent DNA within only few hundred base pairs from the replication fork. Our data also suggest that synthesis of new RNA resumes shortly after replication, and may involve completion of synthesis of immature RNAs that are associated with Pol II (Fig. 5k). Further investigations by methods specifically monitoring directly the transfer of Pol II over the replication fork will be required to finally determine whether and how much old and new Pol II complexes associate with nascent leading and lagging DNA strands.
The possibility of quick rebinding of Pol II to nascent DNA was suggested by spatial colocalization of replication and transcription factories39, although subsequent studies of nuclear factories provided conflicting results showing these two processes may be spatially separated in the nucleus40,41,42. Resumption of synthesis of immature RNAs is supported in the study of common fragile site instability in long human genes43, where it was found that the time of synthesis of very large mRNAs is longer than the entire cell cycle, and therefore synthesis of immature RNAs has to be completed after replication43. However, the putative mechanism of resumption of immature RNA synthesis is unknown and may require identification of a helicase to generate a transcription bubble, such as the XPB helicase component of TFIIH at the promoters44. Whereas XPB does not travel with Pol II during transcription elongation, other helicases—that is, senataxin and RECQ5—have been shown to function at later stages of transcription and to associate with DNA replication. The DNA/RNA helicase senataxin is in a complex that interacts with Pol II and remove R-loops that form during transcription termination. Senataxin also associates with replication forks and facilitates replisome progression by preventing the accumulation of DNA–RNA hybrid when forks encounter nascent transcripts45,46,47. The DNA helicase RECQ5 regulates homologous recombination, co-purifies with PCNA, and is also shown to directly bind to RNA Pol II and to negatively regulate transcription elongation48. It is therefore possible that these helicases may function in the formation of a transcription bubble on nascent DNA within gene bodies. It is also possible that resumption of transcription for some genes may not occur during the S phase, as it was shown that active TSSs of about 400 genes are duplicated only during the G2/M phase49.
In summary, the results of this study suggest a view of the transition between the transcriptional gap phases (G1 to G2) with only a very short interruption of RNA synthesis during replisome passage over a particular genomic region. This presents a new model for the epigenetic transmission of cell state identity. As the transcriptional machinery is only momentarily displaced by the replisome, no additional epigenetic bookmarking by components of chromatin is required during S phase.
Methods
Cell culture and DNA labelling for CAA experiments
H1299 (human non-small cell lung cancer) cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS; Gemini) and 2 mM l-glutamine (Corning). GM22737 (human lymphoblastoid) cells were grown in RPMI supplemented with 10% FBS and 2 mM l-glutamine. Cells were tested and shown to be free of mycoplasma contamination. All shown CAA and RDIA results are for H1299 cells; similar results were obtained in some experiments with GM22737 cells. For CAA and RDIA experiments, cells were trypsinized, plated on chamber slides and grown for two days. Cells were then pulse-labelled with medium containing 10 µM 5-ethynyl-2′-deoxyuridine (EdU, Invitrogen) and then chased with label-free medium. Cells were fixed for 12 min with 3.7% formaldehyde, washed with 1× phosphate buffered saline (PBS), permeabilized for 10 min with 0.25% Triton X-100, and washed with PBS.
Click reaction
In preparation for the click reaction, cells were briefly blocked with 1% bovine serum albumin (BSA). In order to conjugate biotin to EdU, cells were then subjected to click reaction in 0.1× PBS containing (in the following order): copper (ii) sulfate (final concentration 2 mM; Acros Organics), biotin azide (final concentration 5 µM; Invitrogen), and freshly prepared l-ascorbic acid sodium salt (final concentration 10 mM; Acros Organics) for 30 min at room temperature. Samples were washed with PBS and blocked for 30 min with 1× western blocking reagent (Roche) in PBS containing 1.5% donkey serum. Primary antibodies to the protein of interest and to biotin were diluted in 1× western blocking reagent and 0.01% Triton X-100 and incubated with samples overnight at 4 °C.
Chromatin assembly assay and immunostaining
CAA was performed as described7,8. Following overnight incubation with primary antibodies, cells were washed for 20 min with PBS. The PLA (Millipore Sigma) was performed as described by the manufacturer using 15 µl of reagents per spot. The two PLA secondary antibodies, anti-mouse PLUS and anti-rabbit MINUS, were diluted 1:5 in 1× western blocking reagent and added to samples for 1 h at 37 °C. Samples were then washed for 10 min in Duolink II wash buffer A (10 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 0.05% Tween-20). The ligation reaction was performed by diluting the ligation reaction buffer 1:5 in water, adding ligase enzyme (1:40), and incubating samples for 30 min at 37 °C. Samples were washed for 10 min in wash buffer A, and then the amplification reaction was performed by diluting the amplification reaction buffer 1:5 in water and supplementing it with polymerase (1:80) and added to samples for 100 min at 37 °C. Samples were then washed in PBS.
Following PLA, biotin was counterstained with anti-mouse IgG conjugated to Alexa Fluor 488 and anti-biotin conjugated to Alexa Fluor 488 in order to identify replicating nuclei. Samples were washed with PBS and mounted with Vectashield containing DAPI (Vector).
The following antibody dilutions were used for IF and PLA: mouse monoclonal anti-Pol II (Santa Cruz Biotechnology, sc-55492) at 1:50, rabbit polyclonal anti-Pol II PS2 (Bethyl Laboratories, A300-654A) at 1:750, rabbit polyclonal anti-Pol II PS5 (Abcam, ab5131) at 1:10,000, rabbit monoclonal anti-TBP (Cell Signaling Technology, 44059) at 1:1,000, rabbit polyclonal anti-cyclin T1 (Santa Cruz Biotechnology, sc-10750) at 1:100, rabbit polyclonal anti-CDK9 (Santa Cruz Biotechnology, sc-484) at 1:100, rabbit monoclonal anti-SPT4 (Cell Signaling Technology, 64828) at 1:1,000, rabbit polyclonal anti-SPT5 (Millipore, ABE443) at 1:1,000, rabbit polyclonal anti-biotin (Abcam, ab1227) at 1:1,000, mouse monoclonal anti-biotin (Jackson ImmunoResearch, 200-002-211) at 1:1,000, donkey anti-rabbit Alexa Fluor 488 secondary (Jackson ImmunoResearch, 711-545-152), donkey anti-mouse Alexa Fluor 488 secondary (Jackson ImmunoResearch, 715-545-150) and mouse anti-biotin Alexa Fluor 488 secondary (Jackson ImmunoResearch, 200-542-211).
Direct PLA experiments with FEN1, Pol ε, and PCNA were performed similar to CAA, but without EdU labelling and click reaction. PLA reactions were: rabbit anti-FEN1 and mouse anti-Pol ε, rabbit anti-FEN1 and mouse anti-PCNA, and mouse anti-Pol ε and rabbit anti-PCNA. Antibodies used were: rabbit polyclonal anti-FEN1 (generated for this Article) at 1:500, mouse monoclonal anti-PCNA (Cell Signaling Technology, 2586) at 1:1,000, mouse monoclonal anti-DNA Pol ε subunit 3 (Novus Biologicals, H00054107-B02P) at 1:30, and rabbit polyclonal anti-PCNA (this paper) at 1:1,000.
RNA–DNA interaction assay
To assess resumption of transcription, cells were labelled for 10 min with 10 µM EdU and chased as indicated. Following the chase, nascent RNA was labelled for 15 min with 5 mM 5-bromouridine (BrU; Sigma Aldrich, 850187). To assess the stability of transcripts, RNA was labelled for 30 min with 5 mM BrU, DNA was labelled for 10 min with 10 µM EdU, and cells were chased in label-free medium. Following labelling, samples were fixed and permeabilized as described above. For RDIA experiments, the click reaction was performed twice for 15 min each, and samples were subsequently washed and blocked as above. Samples were incubated at 4 °C overnight in primary antibodies, and PLA was performed the following day as described above. Antibodies for RDIA were rabbit polyclonal anti-biotin (Abcam, ab1227) at 1:1,000 and mouse monoclonal anti-BrdU (clone: MoBu-1; Santa Cruz Biotechnology, sc-51514) at 1:50. The full protocol is in refs. 24,25.
Inhibitors
H1299 cells were incubated in medium containing either 0.4 µM THZ-1 (Selleckchem, S7549), 2 µM TPL (Tocris, 3253), or 200 µM DRB (Sigma Aldrich, D1916), or left untreated as a control. Following treatment cells were either collected and western blotted for Pol II S5P and Pol II S2P, or were labelled for 10 min with 10 µM EdU in the presence of inhibitors, fixed, and assessed by CAA, as described above. For RDIA experiments, cells were labelled with 5 mM BrU and then incubated in medium containing 10 µM EdU and either 0.4 µM THZ-1, or 200 µM DRB, or left untreated as a control. Following EdU labelling, cells were fixed and RDIA was performed as described above.
For 1,6-hexanediol experiments, a 20% v/v solution of 1,6-hexanediol (Sigma Aldrich, 240117) was prepared in cell culture medium and then further diluted to either 2% or 3%. Cells were incubated for 15 min in the medium containing 1,6-hexanediol and 10 µM EdU for 15 min, then were fixed and assessed by CAA as described above.
For PCNA-I1 (Cayman Chemical, 20454) experiments, cells were treated at 3 µM for the indicated times, incubated for 10 min in the medium containing PCNA-I1 and 10 µM EdU, and then fixed and assessed by CAA as described above.
Multiplex CAA
Generation of oligonucleotide-tagged secondary antibodies was carried out following manufacturer’s instructions (Sigma Aldrich). For all experiments, donkey anti-rabbit IgG and donkey ant-goat IgG were conjugated to oligo A and oligo B, respectively, which generates a red PLA signal. For experiments assessing proximity of biotin, Pol II S2P, and BrU, donkey anti-rabbit IgG and donkey anti-mouse IgG were conjugated to oligo H and oligo I, respectively, which generate a PLA signal in far red. For experiments assessing proximity of two proteins with biotin, donkey anti-goat IgG and donkey anti-mouse IgG were conjugated to oligo H and oligo I, respectively.
For multiplex CAA with two proteins, cells were pulse-labelled with 75 µM EdU. For multiplex CAA with nascent RNA and DNA, RNA was labelled for 30 min with 5 mM BrU and then DNA was labelled for 10 min with 75 µM EdU. Cells were then fixed, permeabilized, conjugated with biotin and blocked as described above for CAA. Primary antibodies were added and incubated overnight at 4 °C. Secondary antibodies diluted 1:100 in 1× western blocking reagent were added to samples and incubated for 1 h at 37 °C. Samples were washed in Duolink II wash buffer A. The ligation reaction was then prepared by diluting the ligation reaction buffer 1:5 in water and adding ligase enzyme (1:20), and incubating samples for 30 min at 37 °C. Samples were washed for 10 min in wash buffer A, and then the amplification reaction was prepared by diluting the amplification reaction buffer 1:5 in water and supplementing it with polymerase (1:40) and added to samples for 100 min at 37 °C. Samples were washed for 10 min in wash buffer A, and then the detection reagent was prepared by diluting the dilution reaction buffer 1:5 in water. The detection solution was added to the samples and they were incubated for 100 min at 37 °C. Samples were washed with PBS. Samples were then stained with anti-goat IgG conjugated to Alexa Fluor 488 and anti-biotin conjugated to Alexa Fluor 488 to identify replicating nuclei. Slides were mounted in ProLong Glass Antifade Mountant (Invitrogen) and covered with a no. 1.5 coverslip in preparation for STED imaging.
Primary antibody dilutions for transcriptional proteins were the same as for regular CAA. Additional antibodies for multiplex CAA were: goat polyclonal anti-biotin (Vector Laboratories, SP-3000) at 1:1,000, mouse monoclonal anti-PCNA (Cell Signaling Technology, 2586) at 1:1,000, mouse monoclonal anti-Pol ε subunit 3 (Novus Biologicals, H00054107-B02P) at 1:30, mouse monoclonal anti-FEN1 (Novus Biologicals, NB100-150) at 1:30, and anti-goat Alexa Fluor 488 secondary (Jackson ImmunoResearch, 200-542-211).
EdU immunofluorescence quantification
To assess EdU labelling, images were captured at the same exposure time. Then, fluorescence intensity of EdU labelling was quantified using ImageJ50. Nuclei were traced and then integrated density in pixels was reported. Additionally, background fluorescence from outside of cells was measured. Corrected total cell fluorescence (CTCF) was calculated using the formula CTCF = integrated density − (area of the nucleus × mean fluorescence of the background) as previously described51. CTCF was is reported as mean ± standard deviation.
Microscopy
CAA and RDIA images were acquired using an Olympus BX51 microscope equipped with a ×100 objective. The microscope is mounted with a RETIGA 3000 camera and connecting to a computer running Q-Capture Pro7 software.
STED images were acquired using a Leica TCS SP8 STED 3X confocal/super-resolution microscope equipped with a ×100 STED objective. Images were captured using HyD detectors. Far red PLA (excitation at 646 nm) and red PLA (excitation at 593 nm) were both depleted using the 775 nm STED depletion laser with the following settings: gating at 0.5 to 6 ms, pinhole of 0.75 Airy units, and 50% gain. Images were acquired using three times line averaging, an imaging speed of 400 Hz, and an image format of 2,048 by 2,048. Images were subsequently deconvoluted using Leica’s Lightning software.
ChIP
Approximately 5 million GM22737 cells were used per ChIP experiment. Cells were crosslinked in 1% formaldehyde (16% methanol-free stock, Fisher) for 10 min while rocking at room temperature. Following crosslinking, samples were quenched by adding glycine to a final concentration of 0.18 M and rocking for five minutes at room temperature. Cells were collected by centrifugation at 1,000 rpm for 5 min at 4 °C, and then washed 3 times with cold PBS. Nuclei were then isolated by resuspending in nuclear extraction buffer A (20 mM HEPES-KOH (pH 7.5), 10 mM EDTA, 0.25% Triton X-100, and protease inhibitors (Roche)), rocked for 10 min at 4 °C, and pelleted at 1,300g for 10 min at 4 °C. The pellet was resuspended in nuclear extraction buffer B (50 mM HEPES-KOH (pH 7.5), 150 mM NaCl, 1 mM EDTA, and protease inhibitors), rocked for 10 min at 4 °C, and then nuclei were collected by centrifugation for 10 min at 1,300g at 4 °C. Nuclei were resuspended in sonication buffer (10 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-lauroylsarcosine, 0.1% SDS, and protease inhibitors) and allowed to incubate on ice for 10 min. Samples were then transferred to sonication tubes and sonicated until the average fragment size was less than 500 bp. Triton X-100 was added to a final concentration of 1% and samples were spun at max speed for 10 min at 4 °C. The supernatant was collected, 5% was stored as input material, and the rest was applied to Protein A/G magnetic beads (MedChemExpress) pre-bound to antibodies and allowed to incubate overnight at 4 °C with rocking.
The following day the beads were washed with 1 ml of each of the following chilled buffers: low salt immune complex wash buffer (20 mM Tris-HCl (pH 8.1), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), LiCl immune complex wash buffer (10 mM Tris-HCl (pH 8.1), 250 mM LiCl, 1 mM EDTA, 1% NP-40, 1% deoxycholate), and twice with TE (10 mM Tris-HCl (pH 8.0), 1 mM EDTA). Following washes, material was eluted by rocking for 15 min at room temperature in 250 µl of elution buffer (0.1 M sodium bicarbonate, 1% SDS). This was repeated and the two eluates were combined for a final volume of 500 µl. Following addition of 10 µl 5 M NaCl, 10 µl 0.5 M EDTA, and 20 µl 1 M Tris-HCl (pH 6.8), crosslinking was reversed by incubating at 65 °C for 16 h in the presence of 2 µg proteinase K. DNA was then purified by phenol/chloroform extraction and ethanol precipitation, and analysed by real-time PCR.
Re-ChIP
BrdU re-ChIP assays were performed as previously described7,23 using approximately 20 million cells per experiment. Cells were labelled with 200 µM BrdU, crosslinked, and ChIP was performed as described above. Following DNA purification, DNA pellets were resuspended in 480 µl of TE and 5% of material was saved to serve as input for the second immunoprecipitation. Then, 100 µg salmon sperm DNA (Invitrogen) was added, samples were boiled for 5 min and then were cooled on ice for 2 min. Then, 50 µl of 10× adjusting buffer (110 mM sodium phosphate buffer (pH 7.0), 1.52 M NaCl, 0.55% Triton X-100), 10 µl of 10% BSA (Sigma), and 2 µg of anti-BrdU antibody were added, and samples were rocked for 20 min at room temperature. Following incubation with anti-BrdU antibody, 35 µg of rabbit anti-mouse IgG was added to each sample, and allowed to incubate for 20 min at room temperature with rocking. Antibody–DNA complexes were then pelleted by centrifugation at maximum speed for 15 min at 4 °C. The pellet was washed 3 times with 1× adjusting buffer (11 mM sodium phosphate buffer (pH 7.0), 152 mM NaCl, 0.055% Triton X-100), and then resuspended in 0.5 ml of re-ChIP lysis buffer (50 mM Tris-HCl (pH 8.0), 10 mM EDTA, 0.5% SDS, 2 µg proteinase K) and incubated for 8 h at 55 °C. DNA was purified by phenol/chloroform extraction and ethanol precipitation, and then used for qPCR.
PCNA re-ChIP assays were performed similar to previously described7 using approximately 75 million cells per experiment. Cells were crosslinked and immunoprecipitated as described above using anti-Pol II, anti-SPT4, or anti-IgG as a negative control. Protein-DNA complexes were eluted in 35 µl TE supplemented with 50 mM DTT and 0.125% SDS at 37 °C for 25 min. The elution was repeated and the two eluates were combined (total volume 70 µl) and diluted 15-fold in re-ChIP dilution buffer (15 mM Tris-HCl (pH 8.1), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100), supplemented with 10 µg salmon sperm DNA and 0.2% BSA. Five per cent of material was saved as ChIP material, and the rest was divided in half and incubated overnight at 4 °C with either biotinylated anti-PCNA, biotinylated FEN1, biotinylated Pol ε, or biotinylated-IgG. (Antibodies were biotinylated with Pierce Antibody Biotinylation Kit for IP according to manufacturer’s instructions.) The following day, complexes were captured by incubation with streptavidin magnetic beads (Pierce) for 2 h at room temperature. Complexes were washed, eluted, and reverse-crosslinked as described above for ChIP. DNA was purified by phenol/chloroform extraction and ethanol precipitation, and analysed by qPCR. Re-ChIP experiments were performed in biological duplicate.
ChIP and re-ChIP experiments were performed using the following antibodies: rabbit monoclonal anti-Rbp1 NTD (Cell Signaling Technology, 14958), rabbit monoclonal anti-SPT4 (Cell Signaling Technology, 64828), mouse monoclonal anti-BrdU (BD Bioscience, 555627), rabbit polyclonal anti-mouse IgG (Jackson ImmunoResearch, 315-005-003), rabbit polyclonal anti-PCNA (this paper), rabbit polyclonal anti-FEN1 (this paper), mouse monoclonal anti-Pol ε (Millipore, MABE966), and rabbit IgG, whole molecule (Jackson ImmunoResearch, 011-000-003).
Sizing and S1 nuclease treatment of PCNA-precipitated fragments
DNA purified after PCNA ChIP was incubated in the presence of 1 U of S1 nuclease (Invitrogen) for 10 min at 37 °C. In order to ensure that all single-stranded DNA was degraded, S1 treatment was done in the presence of the 25 pg of single-stranded viral M13mp18 (New England Biolabs) which was then assessed by qPCR to determine if digestion was complete (see Extended Data Fig. 5c). Following digestion, samples were incubated for 5 min at 80 °C in the presence of 10 mM EDTA to inactivate the S1 nuclease. DNA was purified by phenol/chloroform extraction and ethanol precipitation, and analysed by qPCR.
Generation of PCNA and FEN1 antibodies
PCNA rabbit polyclonal antibodies were raised to amino acids 10–193 of the human protein. FEN1 rabbit polyclonal antibodies were raised to amino acids 1–301 of the Drosophila protein. See Extended Data Fig. 6d for western blot validation of these antibodies.
Co-immunoprecipitation
GM22737 cells were collected by centrifugation at 1,000 rpm for 5 min at 4 °C, and then washed 3 times with cold PBS. The cell pellet was resuspended in nuclear extraction buffer A (20 mM HEPES-KOH (pH 7.5), 10 mM EDTA, 0.25% Triton X-100, protease inhibitors, and phosphatase inhibitors (Roche)), and incubated on ice for 10 min. Cells were then lysed by Dounce homogenization using pestle B for 15 passes, and nuclei were collected by centrifuging the lysate at 3,500 rpm for 10 min at 4 °C. The nuclear pellet was resuspended in IP lysis buffer (20 mM HEPES, 1 mM EDTA, 300 mM NaCl, 0.1% NP-40, 5% glycerol protease inhibitors, and phosphatase inhibitors) and incubated on ice for 10 min followed by sonication to lyse nuclei. Following sonication, samples were centrifuged at max speed for 10 min 4 °C to pellet debris. The supernatant was diluted 1:1 in IP lysis buffer without NaCl, such that the final IP buffer concentration was 20 mM HEPES, 1 mM EDTA, 150 mM NaCl, 0.1% NP-40, 5% glycerol, protease inhibitors, and phosphatase inhibitors. Input was saved from this material, and the rest was incubated overnight at 4 °C with rocking with either with anti-PCNA, anti-Pol II S5P, anti-Pol S2P, or IgG.
The next day, samples were incubated with magnetic protein A/G beads (MedChemExpress) for 45 min at 4 °C with rocking in order to immunoprecipitate complexes. The immunoprecipitated material was washed with chilled buffers as follows: twice with IP buffer (20 mM HEPES, 1 mM EDTA, 150 mM NaCl, 0.1% NP-40, 5% glycerol), once with high salt IP wash buffer (20 mM HEPES, 1 mM EDTA, 300 mM NaCl, 0.75% NP-40, 5% glycerol), once with IP wash buffer, and finally once with PBS. Complexes were eluted by boiling in Laemmli buffer and analysed by SDS–PAGE and western blotting.
For experiments using ethidium bromide, nuclear extracts were treated for 30 min on ice with 100 µg/ml ethidium bromide. Samples were then centrifuged for 5 min at max speed, the supernatant was transferred to a new tube, antibody was added, and samples were rocked overnight at 4 °C. Samples were immunoprecipitated with protein A/G and then washed as described above, except that all wash buffers contained 100 µg/ml ethidium bromide. Proteins were eluted by boiling in Laemmli buffer and analysed by SDS–PAGE and western blotting.
GST pull-down
GST and GST–PCNA were expressed in BL21 cells and purified as described by manufacturer (GenScript). Nuclear lysates were prepared as described for co-immunoprecipitation. Lysates pre-cleared by incubation with glutathione resin at 4 °C for 1 h with rocking. Pre-cleared lysates were then incubated with 30 µg of either GST or GST–PCNA overnight at 4 °C with rocking. The next day, glutathione resin was blocked by rocking for 2 h at 4 °C in IP buffer containing 0.5% BSA. The resin was then incubated with GST or GST–PCNA containing lysates for 30 min at 4 °C in order to capture GST. The resin was washed as described for co-immunoprecipitation experiments. Complexes were eluted by boiling in Laemmli buffer and analysed by SDS–PAGE and western blotting.
Western blotting
Samples were electrophoresed on a 4–20% gradient gel (Bio-Rad) and then wet transferred onto PVDF membrane (Millipore). The membranes were blocked for 30 min with 10% milk in Tris-buffered saline containing Tween-20 (TBS-T; 10 mM Tris, pH 8.0, 150 mM NaCl, 0.2% Tween-20). The primary antibodies were diluted in 5% milk in TBS-T and incubated overnight at 4 °C with rocking. The following day the membranes were washed thrice with 1% milk in TBS-T and then incubated in secondary antibody for 1 h at room temperature with rocking. The membranes were washed thrice in TBS-T and then detected using SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and imaged on a ChemiDoc MP Imaging System (Bio-Rad).
The following antibody dilutions were used for western blotting: rabbit monoclonal anti-Rbp1 NTD (Cell Signaling Technology, 14958) at 1:1,000, rabbit polyclonal anti-Pol II phospho S2 (Bethyl Laboratories, A300-654A) at 1:3,000, rabbit polyclonal anti-Pol II phospho S5 (Abcam, ab5131) at 1:3,000, rabbit polyclonal anti-Rpb2 (Active Motif, 61558) at 1:250, mouse monoclonal anti-POLR2G (Rpb7; Santa Cruz, sc-398213), mouse monoclonal anti-POLR2I (Rpb9; Santa Cruz, sc-398049), mouse monoclonal anti-POLR2J1/2/3 (Rpb11; Santa Cruz, sc-514129), mouse monoclonal anti-PCNA (Cell Signaling Technology, 2586) at 1:3,000, rabbit monoclonal anti-SPT4 (Cell Signaling Technology, 64828) at 1:1,000, rabbit polyclonal anti-SPT5 (Milipore, ABE443) at 1:1,000, rabbit monoclonal anti-histone H3 (Cell Signaling, 4499) at 1:2,000, anti-rabbit IgG HRP-linked (Cell Signaling, 7074), and anti-mouse IgG HRP-linked (Cell Signaling, 7076).
Statistics and reproducibility
Prism software was used for graphing and statistical analysis. Significance throughout the paper is indicated as: NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
For CAA and RDIA experiments, slides were first examined to make sure that PLA signals were detected only in the EdU-labelled nuclei. Next, all EdU-labelled nuclei were examined to make sure that they contain approximately the same number of PLA signals. Finally, PLA signals were counted and used for statistical analysis from three biological replicates. Statistical significance was assessed using a one-way ANOVA with Tukey’s post hoc analysis or by Student’s two-way, unpaired t-test.
Multiplex CAA experiments were quantified by counting the number of overlaps compared to the total number of PLA foci for the given transcription protein from at least three biological replicates. Distances between the centres of the foci were measured using the line profiles tool in the LasX software. All data are presented as mean ± s.d.
Experiments analysed by western blotting were performed in biological triplicate.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available from the corresponding author on reasonable request.
References
Steffen, P. A. & Ringrose, L. What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat. Rev. Mol. Cell Biol. 15, 340–356 (2014).
Bertoli, C., Skotheim, J. M. & de Bruin, R. A. M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 14, 518–528 (2013).
Stewart-Morgan, K. R., Petryk, N. & Groth, A. Chromatin replication and epigenetic cell memory. Nat. Cell Biol. 22, 361–371 (2020).
Almouzni, G. & Cedar, H. Maintenance of epigenetic information. Cold Spring Harb. Perspect. Biol. 8, a019372 (2016).
Li, B., Carey, M. & Workman, J. L. The role of chromatin during transcription. Cell 128, 707–719 (2007).
Kadauke, S. & Blobel, G. A. Mitotic bookmarking by transcription factors. Epigenetics Chromatin 6, 6 (2013).
Petruk, S. et al. TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell 150, 922–933 (2012).
Petruk, S., Black, K. L., Kovermann, S. K., Brock, H. W. & Mazo, A. Stepwise histone modifications are mediated by multiple enzymes that rapidly associate with nascent DNA during replication. Nat. Commun. 4, 2841 (2013).
Herrick, J. & Bensimon, A. Global regulation of genome duplication in eukaryotes: an overview from the epifluorescence microscope. Chromosoma 117, 243–260 (2008).
Buratowski, S. Progression through the RNA polymerase II CTD cycle. Mol. Cell 36, 541–546 (2009).
Kwiatkowski, N. et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620 (2014).
Titov, D. V. et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 7, 182–188 (2011).
Wada, T. et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 12, 343–356 (1998).
Mancebo, H. S. Y. et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 11, 2633–2644 (1997).
Adelman, K. & Lis, J. T. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat. Rev. Genet. 13, 720–731 (2012).
Shandilya, J. & Roberts, S. G. E. The transcription cycle in eukaryotes: From productive initiation to RNA polymerase II recycling. Biochim. Biophys. Acta - Gene Regul. Mech. 1819, 391–400 (2012).
Yudkovsky, N., Ranish, J. A. & Hahn, S. A transcription reinitiation intermediate that is stabilized by activator. Nature 408, 225–229 (2000).
Jonkers, I. & Lis, J. T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 16, 167–177 (2015).
Leuchowius, K.-J. et al. Parallel visualization of multiple protein complexes in individual cells in tumor tissue. Mol. Cell. Proteomics 12, 1563–1571 (2013).
McKnight, S. L. & Miller, O. L. Electron microscopic analysis of chromatin replication in the cellular blastoderm Drosophila melanogaster embryo. Cell 12, 795–804 (1977).
Franklin, R. E. & Gosling, R. G. Molecular configuration in sodium thymonucleate. Nature 171, 740–741 (1953).
Burgers, P. M. J. & Kunkel, T. A. Eukaryotic DNA replication fork. Annu. Rev. Biochem. 86, 417–438 (2017).
Francis, N. J., Follmer, N. E., Simon, M. D., Aghia, G. & Butler, J. D. Polycomb proteins remain bound to chromatin and DNA during DNA replication in vitro. Cell 137, 110–122 (2009).
Petruk, S., Fenstermaker, T. K., Black, K. L., Brock, H. W. & Mazo, A. Detection of RNA–DNA association by a proximity ligation-based method. Sci. Rep. 6, 27313 (2016).
Fenstermaker, T. K., Sun, G., Mazo, A. & Petruk, S. A proximity ligation-based method to detect RNA–DNA association. Methods Mol. Biol. 2008, 121–129 (2019).
Geiger, F. et al. Liquid–liquid phase separation underpins the formation of replication factories in rotaviruses. EMBO J. 40, e107711 (2021).
Parker, M. W., Kao, J. A., Huang, A., Berger, J. M. & Botchan, M. R. Molecular determinants of phase separation for Drosophila DNA replication licensing factors. eLife 10, e70535 (2021).
Lai, J. S. & Herr, W. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl Acad. Sci. USA 89, 6958–6962 (1992).
Moldovan, G.-L. L., Pfander, B. & Jentsch, S. PCNA, the maestro of the replication fork. Cell 129, 665–679 (2007).
González-Magaña, A. & Blanco, F. J. Human PCNA structure, function and interactions. Biomolecules 10, 570 (2020).
Tan, Z. et al. Small-molecule targeting of proliferating cell nuclear antigen chromatin association inhibits tumor cell growth. Mol. Pharmacol. 81, 811–819 (2012).
Ito, K. & Zaret, K. S. Maintaining transcriptional specificity through mitosis. Annu. Rev. Genomics Hum. Genet. 23, 53–71 (2022).
Bellec, M. et al. The control of transcriptional memory by stable mitotic bookmarking. Nat. Commun. 13, 1176 (2022).
Liu, B. & Alberts, B. M. Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science 267, 1131–1137 (1995).
Liu, B., Wong, M. L., Tinker, R. L., Geiduschek, E. P. & Alberts, B. M. The DNA replication fork can pass RNA polymerase without displacing the nascent transcript. Nature 366, 33–39 (1993).
Pomerantz, R. T. & O’Donnell, M. What happens when replication and transcription complexes collide? Cell Cycle 9, 2537–2543 (2010).
Leung, K. H. T., Abou El Hassan, M. & Bremner, R. A rapid and efficient method to purify proteins at replication forks under native conditions. Biotechniques 55, 204–206 (2013).
Stewart-Morgan, K. R., Reverón-Gómez, N. & Groth, A. Transcription restart establishes chromatin accessibility after DNA replication. Mol. Cell 75, 284–297.e6 (2019).
Hassan, A. B., Errington, R. J., White, N. S., Jackson, D. A. & Cook, P. R. Replication and transcription sites are colocalized in human cells. J. Cell Sci. 107, 425–434 (1994).
Malyavantham, K. S., Bhattacharya, S. & Berezney, R. The architecture of functional neighborhoods within the mammalian cell nucleus. Adv. Enzyme Regul. 50, 126–134 (2010).
Wansink, D. G. et al. RNA polymerase II transcription is concentrated outside replication domains throughout S-phase. J. Cell Sci. 107, 1449–1456 (1994).
Wei, X. et al. Segregation of transcription and replication sites into higher order domains. Science 281, 1502–1506 (1998).
Helmrich, A., Ballarino, M. & Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44, 966–977 (2011).
Rimel, J. K. & Taatjes, D. J. The essential and multifunctional TFIIH complex. Protein Sci. 27, 1018–1037 (2018).
Alzu, A. et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 151, 835–846 (2012).
Groh, M., Albulescu, L. O., Cristini, A. & Gromak, N. Senataxin: genome guardian at the interface of transcription and neurodegeneration. J. Mol. Biol. 429, 3181–3195 (2017).
Hasanova, Z., Klapstova, V., Porrua, O., Stefl, R. & Sebesta, M. Human senataxin is a bona fide R-loop resolving enzyme and transcription termination factor. Nucleic Acids Res. 51, 2818–2837 (2023).
Andrs, M., Hasanova, Z., Oravetzova, A., Dobrovolna, J. & Janscak, P. RECQ5: A mysterious helicase at the interface of DNA replication and transcription. Genes 11, 232 (2020).
Wang, J. et al. Persistence of RNA transcription during DNA replication delays duplication of transcription start sites until G2/M. Cell Rep. 34, 108759 (2021).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
McCloy, R. A. et al. Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle 13, 1400–1412 (2014).
Acknowledgements
The authors thank B. Calabretta and S. McMahon for feedback on experiments and the manuscript. This work was supported by the following grants: NIH R01GM075141 to A.M.; T.K.F. was supported by NIH F31GM128300 and NIH training grant T32GM100836. Leica SP8 STED microscope imaging was supported by the Bioimaging Shared Resource of the Sidney Kimmel Cancer Center (NCI 5 P30 CA-56036).
Author information
Authors and Affiliations
Contributions
T.K.F., S.P., H.W.B. and A.M. conceptualized the study. T.K.F. performed experiments presented in Figs. 1–5 and Extended Data Figs. 1–4, 6 and 7. S.P. performed experiments in Extended Data Fig. 5 and performed preliminary experiments pertaining to the results in Figs. 1 and 5 and Extended Data Fig. 4. S.K.K performed preliminary experiments pertaining to the results in Fig. 1. T.K.F. and S.P. designed and developed the RDIA assay. T.K.F. developed the multiplex CAA assay. T.K.F. wrote the manuscript with input from S.P., H.W.B. and A.M. S.P., H.W.B., T.K.F. and A.M. acquired the funding. A.M. provided overall supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Martijn Luijsterburg, Sheila Teves and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 CAA reactions with IgG control do not generate PLA signals.
Cells were labelled for 10 min and PLA was performed using the indicated antibodies and an IgG control. Red, PLA, Green, EdU, Blue, DAPI. Scale bar = 10 μm. Data shown are representative of 3 independent experiments.
Extended Data Fig. 2 Detection of RNA Pol II on nascent DNA is not the result of a new round of transcription.
a, Cells were treated for 30 min 2 h, or 4 h with the indicated inhibitors or left untreated as a control and then labelled with EdU for 10 min in the presence of the inhibitors and CAA was performed for Pol II S5P (left) or Pol II S2P (right). Data are presented as mean values ± s.d. (n = 3 independent experiments). For each of the following, the number of nuclei examined over 3 independent experiments are: 112 (S5P top), 153 (S5P bottom), 70 (S2P top), and 139 (S2P bottom). Statistical significance was determined by one-way ANOVA and p-values shown were determined with Tukey’s post-hoc. ns = not significant, **P < 0.01, ****P < 0.0001. b, Cells were treated for 30 min with either THZ-1 or DRB or left untreated as a control and then blotted for the indicated proteins. Data shown are representative of 3 independent experiments. For blot source data, see Supplementary Fig. 1. c, Cells were treated with either TPL or DRB or left untreated as a control and then labelled with EdU for 10 min in the presence of the inhibitors, and CAA was performed for PCNA. Data are presented as mean values ± s.d. (n = 3 independent experiments), and 117 nuclei were examined over 3 independent experiments, and statistical significance was determined by one-way ANOVA. ns = not significant. d, Cells were treated for 4 h with DRB or left untreated as a control and then labelled with EdU for 10 min in the presence of the inhibitors and CAA was performed for TBP. Data are presented as mean values ± s.d. (n = 3 independent experiments), 77 nuclei were examined over 3 independent experiments, and statistical significance was determined by two-way, unpaired t-test. ns = not significant. e, Cells were treated for 4 h with DRB or left untreated as a control and then labelled with EdU for 10 min in the presence of the inhibitors and CAA was performed for either SPT4 (top) or SPT5 (bottom). Data are presented as mean values ± s.d. (n = 3 independent experiments), and 77 (SPT4) or 88 (SPT5) nuclei were examined over 3 independent experiments. Statistical significance was determined by two-way, unpaired t-test. ****P < 0.0001.
Extended Data Fig. 3 Multiplex CAA detects proteins close to each other on labelled DNA.
a, Schematic representation of multiplex CAA illustrating that while 7.4 to 12 kb of DNA is labeled in 10 min, overlapping PLA signals are on average within 400 bp of one another. b, Quantification of the percent of overlaps following 10 min and 20 min EdU labelling for multiplex CAA with PCNA and either Pol II S5P (top) or Pol II S2P (bottom). Data are presented as mean values ± s.d. from n = 5 independent samples. Statistical significance was determined by a two-way, unpaired t-test. **P < 0.01, ****P < 0.0001. c, Direct PLA between replication proteins: Pol ε and PCNA, Pol ε and FEN1, and FEN1 and PCNA. Data are presented as mean values ± s.d. (n = 3 independent experiments), and 152 nuclei were examined over 3 independent experiments. Statistical significance was determined by one-way ANOVA and p-values shown were determined with Tukey’s post-hoc. ****P < 0.0001.
Extended Data Fig. 4 RNA polymerase II associates with target genes on nascent DNA.
a, Schematic representation of BrdU re-ChIP. DNA was labelled with BrdU (red circle) and then immunoprecipitated with anti-Pol II antibody. Following reverse crosslinking, DNA was denatured and immunoprecipitated with anti-BrdU antibody. Recovered DNA was analyzed by qPCR. Red ring is PCNA, replisome is in blue. b, Schematic representing locations of qPCR products within each analysed gene. Bent arrow represents the TSS and pA indicates the polyadenylation site. For each gene two sets of primers were designed: one close to the TSS and one farther downstream in the gene. c, Pol II ChIP. ChIP was performed for Pol II or IgG and subsequently qPCR was performed at upstream and downstream regions the active genes GAPDH, PPIA, and TNFAIP3 and at the repressed gene GFAP. d, BrdU re-ChIP following 25 min BrdU labelling. DNA was labelled with BrdU for 25 min or unlabelled as a control. ChIP was performed for Pol II or IgG. Following purification of DNA, Pol II ChIP samples were subjected to a second round of immunoprecipitation for BrdU. e, BrdU re-ChIP following 40 min BrdU labelling. DNA was labelled with BrdU for 40 min or unlabelled as a control. ChIP was performed for Pol II or IgG. Following purification of DNA, Pol II ChIP samples were subjected to a second round of immunoprecipitation for BrdU. Numbering represents primer sets shown in b. Data in c–e are presented as mean values and are representative of 2 independent experiments.
Extended Data Fig. 5 PCNA immunoprecipitates short fragments of nascent DNA.
a, Top: Primer map showing forward primer (at position zero) and reverse primers at increasing distances within the TNFAIP3 gene. Bottom: The percent of input for PCNA immunoprecipitated DNA either without S1 nuclease (left) or following digestion with 1 U S1 nuclease (right). b, Top: Primer map showing forward primer (at position zero) and reverse primers at increasing distances within the GFAP gene. Bottom: The percent of input for PCNA immunoprecipitated DNA either without S1 nuclease (left) or following digestion with 1 U S1 nuclease (right). c, m13mp18 ssDNA was treated with 1 U S1 nuclease for 10 min in order to ensure that single stranded DNA was completely digested under treatment conditions. Data in a–c are presented as mean values and are representative of 2 independent experiments.
Extended Data Fig. 6 PCNA is a mark of nascent chromatin throughout the genome.
a, PCNA ChIP. Cells were immunoprecipitated with an anti-PCNA antibody or with IgG and analysed by qPCR at upstream and downstream regions the active genes GAPDH, PPIA, and TNFAIP3 and at the repressed gene GFAP. Numbering represents primer sets shown in Extended Data Fig. 4b. Data are presented as mean values and are representative of 2 independent experiments. b, Cells were labelled for 5 min with EdU, chased for the indicated times, and then CAA was performed for PCNA. Data are presented as mean values ± s.d. (n = 3 independent experiments), and 208 nuclei were examined over 3 independent experiments. Statistical significance was determined by one-way ANOVA and p-values shown were determined with Tukey’s post-hoc. ns = not significant, ****P < 0.0001. c, Sequential ChIP with PCNA and SPT4. Upper panel: ChIP was performed for SPT4 or IgG and then assessed by PCR at the same genes indicated in Extended Data Fig. 4b. Lower panel: re-ChIP of samples first immunoprecipitated for SPT4. Following elution, samples were re-immunoprecipitated for PCNA or IgG. Following DNA purification, samples were analyzed by qPCR. Data are presented as mean values and are representative of 2 independent experiments. d, Western blotting validation of PCNA and FEN1 polyclonal antibodies generated in this study. Data shown are representative of 3 independent experiments. For blot source data, see Supplementary Fig. 1.
Extended Data Fig. 7 Pol II transfer to nascent DNA is likely not the result of LLPS and the interaction between PCNA and Pol II is DNA-independent.
a, Cells were labelled for 30 min with BrU and then labelled for 10 min with EdU in the presence of either THZ-1 or DRB or left untreated as a control. Subsequently, RDIA was performed to assess immature RNA retention post-replication. Data are presented as mean values ± s.d. (n = 3 independent experiments), and 84 nuclei were examined over 3 independent experiments. Statistical significance was determined by one-way ANOVA. ns = not significant. b, Cells were labelled for 15 min with EdU either in the presence of 2% hexanediol, 3% hexanediol, or without hexanediol as a control, then CAA was performed for Pol II S5P (top) or Pol II S2P (bottom). Data are presented as mean values ± s.d. (n = 3 independent experiments). 50 nuclei were examined over 3 independent experiments. Statistical significance was determined by one-way ANOVA. ns = not significant. c, Nuclear extracts were either treated or untreated with ethidium bromide prior to immunoprecipitation with anti-PCNA antibody and subsequent western blotting. Data shown are representative of 3 independent experiments. For blot source data, see Supplementary Fig. 1. d, Top: Cells were untreated or treated with 3 µM PCNA-I1 for the indicated times and then EdU labelled and fixed. Bottom: Cells were untreated or treated with 3 µM PCNA-I1 for 3 h and then the inhibitor was washed out for the indicated times. Cells were labelled with EdU and fixed. In both experiments, following fixation, biotin was conjugated to EdU using click chemistry and immunostained in order to assess DNA replication. Corrected cell total fluorescence (CTCF) was quantified as described in the methods. Data are presented as mean values ± s.d. (n = 3 independent experiments), and 140 (top) or 94 (bottom) nuclei were examined over 3 independent experiments. Statistical significance was determined by one-way ANOVA and p-values shown were determined by Tukey’s post-hoc. ns = not significant, *P < 0.05, **P < 0.01, ****P < 0.0001.
Supplementary information
Supplementary Information
This file contains Supplemental Table 1: List of PCR primers used in this study; and Supplementary Fig. 1: Source western blot images.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fenstermaker, T.K., Petruk, S., Kovermann, S.K. et al. RNA polymerase II associates with active genes during DNA replication. Nature 620, 426–433 (2023). https://doi.org/10.1038/s41586-023-06341-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-06341-9
- Springer Nature Limited
This article is cited by
-
Genome-wide identification of replication fork stalling/pausing sites and the interplay between RNA Pol II transcription and DNA replication progression
Genome Biology (2024)
-
RNA polymerase II promotes the organization of chromatin following DNA replication
EMBO Reports (2024)
-
The BAF chromatin remodeler synergizes with RNA polymerase II and transcription factors to evict nucleosomes
Nature Genetics (2024)