

Abstract
Biosynthesis is an environmentally benign and renewable approach that can be used to produce a broad range of natural and, in some cases, new-to-nature products. However, biology lacks many of the reactions that are available to synthetic chemists, resulting in a narrower scope of accessible products when using biosynthesis rather than synthetic chemistry. A prime example of such chemistry is carbene-transfer reactions1. Although it was recently shown that carbene-transfer reactions can be performed in a cell and used for biosynthesis2,3, carbene donors and unnatural cofactors needed to be added exogenously and transported into cells to effect the desired reactions, precluding cost-effective scale-up of the biosynthesis process with these reactions. Here we report the access to a diazo ester carbene precursor by cellular metabolism and a microbial platform for introducing unnatural carbene-transfer reactions into biosynthesis. The α-diazoester azaserine was produced by expressing a biosynthetic gene cluster in Streptomyces albus. The intracellularly produced azaserine was used as a carbene donor to cyclopropanate another intracellularly produced molecule—styrene. The reaction was catalysed by engineered P450 mutants containing a native cofactor with excellent diastereoselectivity and a moderate yield. Our study establishes a scalable, microbial platform for conducting intracellular abiological carbene-transfer reactions to functionalize a range of natural and new-to-nature products and expands the scope of organic products that can be produced by cellular metabolism.
Similar content being viewed by others
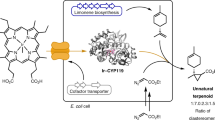

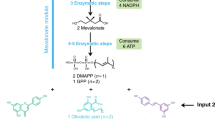
Main
The repertoire of natural enzyme-catalysed reactions lacks many of the common reactions of organic synthesis, including the carbene-transfer reactions4,5,6,7. Although a few natural enzymatic transformations have been proposed to involve carbene intermediates, it has been challenging to repurpose them for unnatural biosynthesis8,9,10,11,12. Recently, metalloenzymes have been engineered to catalyse many abiological carbene-transfer reactions, such as the cyclopropanation of alkenes and insertions of carbene units into N–H and C–H bonds13,14,15. These enzymes exhibit impressive unnatural activity and selectivity (Fig. 1a). Integration of such unnatural carbene-transfer reactions into microbial metabolism would open possibilities to generate new-to-nature products in vivo through biosynthesis. Although abiological carbene-transfer reactions catalysed by small organometallic catalysts or artificial metalloenzymes have been interfaced with natural biosynthesis pathways to generate new products2,3,16, the diazo compound acting as a carbene precursor and the metal cofactors were added to cells exogenously, and this addition limits the scale at which the reactions can be performed economically and the scope of substrates that can be imported into the cell. An intracellular biosynthetic route to the carbene precursor and engineered catalysts would mitigate these limitations and greatly expand the use of enzymatic non-natural carbene-transfer chemistry (Fig. 1b). Here we show that the natural α-diazoester azaserine can be produced by S. albus engineered with a biosynthetic gene cluster and act as the carbene precursor for coupling with intracellularly produced styrene to generate unnatural amino acids containing a cyclopropyl group—which has wide applications in organic synthesis and occurs frequently in medicine development17,18—through a reaction catalysed by an evolved cytochrome P450 mutant with its native cofactor. This study sets the stage for full integration of those carbene-transfer reactions into biosynthesis and broadens the scope and structural diversity of chemicals that can be produced by cellular metabolism.

a, Examples of recently developed carbene-transfer reactions catalysed by engineered cytochrome P450s14. b, Schematic for integrating carbene-transfer reactions into biosynthesis with all components in the reaction produced by microbial cells.
Although there are many different carbene precursors in synthetic chemistry and methods to synthesize them in situ, most of these compounds are not made by living systems7,19,20,21. Diazo compounds, such as the widely used unnatural compound ethyl diazoacetate (EDA)22,23, are common precursors to metal–carbene complexes. There are natural products that contain diazo groups24, and we considered that those natural molecules could be used as carbene precursors that could be produced biosynthetically (Fig. 2a). 6-Diazo-5-oxo-l-norleucine (DON) is a natural diazo compound, and its biosynthetic pathway has been elucidated recently25. DON and its derivatives have been used in carbene-transfer reactions26. However, only N- and O-protected DON (the amino group and carboxyl group in DON were both modified with protecting groups) has been used for intermolecular reactions because the unprotected DON undergoes intramolecular insertion and does not react intermolecularly under the conditions tested26. Although the biosynthetic pathways for some other natural diazo compounds have been reported, their molecular size, instability and/or reactivity may make them unsuitable to act as a carbene precursor24,27,28. However, the size and electronic properties of the natural product azaserine, which was initially identified as a drug for treating cancer29,30, are similar to those of EDA. We therefore reasoned that it could be a suitable reagent for intracellular carbene-transfer reactions.

a, Selected diazo compounds that are found in nature24, except for EDA (black rectangle). b, Azaserine acts as a carbene precursor for cyclopropanation. The extracted ion chromatogram (EIC, [M+H]+, m/z = 250.1074) for target products. Representative traces are shown for two repeated reactions. P1 to P4 are the four diastereomers formed by cyclopropanation of styrene with azaserine (right). The reaction contained 5 mM styrene, 5 mM azaserine, 10 µM Ir(Me)MPIX or no catalyst, 0 or 10 mM Na2S2O4, 5 vol% ethanol and M9-N buffer, and was conducted at 22 °C under aerobic conditions for 18 h. The standard is a mixture of the four chemically synthesized diastereomers. c, Biosynthesis of azaserine in S. albus with the identified azaserine gene cluster in its genome. The EIC ([M+H]+, m/z = 174.0509) for azaserine. Representative traces for two repeated experiments. d, Azaserine production in three different culture media. Data are mean ± s.d. n = 3 biological replicates.
Azaserine was first tested for its ability to act as a carbene precursor for the cyclopropanation of styrene catalysed by the artificial cofactor Ir(Me)MPIX (MPIX for mesoporphyrin IX) and haemin, both of which have been used previously to catalyse cyclopropanation13,31. The reaction products were analysed using liquid chromatography–high-resolution mass spectrometry (LC–HRMS) and compared with the independently synthesized authentic products. The cyclopropanation products were detected, indicating that azaserine can serve as a carbene precursor (Fig. 2b). Although Na2S2O4 was essential for the activity of haemin (Extended Data Fig. 1b), consistent with previous studies13, it was not required for that of Ir(Me)MPIX (Fig. 2b and Extended Data Fig. 1a).
Having confirmed that azaserine can act as a carbene precursor, we searched for the biosynthetic pathway for it. There are two known organisms that produce azaserine naturally—Streptomyces fragilis24 and Glycomyces harbinensis32—and their genome sequences are available. Using comparative genomics, a gene cluster probably encoding the biosynthesis of azaserine (aza), which is similar to those for some natural products containing N–N bonds27,33,34, was identified in both strains (Extended Data Fig. 3a). To verify the prediction, given that the native producers are not genetically tractable, the gene cluster from S. fragilis was cloned and heterologously expressed in other hosts. As the azaserine gene cluster is from S. fragilis, which is a prokaryote, the most commonly used model prokaryote Escherichia coli and a model streptomycete S. albus, which belongs to the same genus as S. fragilis, were selected. As azaserine has antibiotic activity, expression of the pathway in a heterologous host would probably be toxic. To select a suitable host, the toxicity of azaserine towards E. coli and S. albus was tested. Consistent with previous studies35, the growth of E. coli BL21(DE3) was inhibited by 40 mg l−1 of azaserine added exogenously (Extended Data Fig. 2a). However, the growth of S. albus was not inhibited under the tested conditions (Extended Data Fig. 2b). Thus, S. albus was selected as the host. The predicted gene cluster was cloned and integrated into the chromosome of S. albus. Proteomic analysis indicated that 20 out of 23 proteins encoded in the cloned region were expressed (Extended Data Fig. 3b). Under the tested conditions, azaserine was biosynthesized by the engineered S. albus strain, providing functional validation of the predicted gene cluster (Fig. 2c). A detailed analysis of the enzymes encoded by the aza gene cluster and elucidation of the biosynthetic logic of azaserine production through feeding studies and in vitro biochemical characterization are described in a separate study (D.V.C. et al., manuscript in preparation). Under the optimized culture conditions, a titre of 103 mg l−1 was achieved at 48 h when the cells were grown in 1B medium (Fig. 2d). The titre of azaserine decreased with cultivation time in tryptic soy broth (TSB) medium, even after the cells were removed, possibly due to a reaction of azaserine with a component made by the cells when they were cultivated in TSB medium (azaserine was stable in fresh TSB medium and weak alkaline conditions36) (Extended Data Fig. 4a,b).
To incorporate an abiological carbene-transfer reaction into biosynthesis, an appropriately engineered enzyme with the ability to use azaserine as the carbene precursor was required. Several P450s and their mutants (Supplementary Table 1) that were previously reported to catalyse cyclopropanation by carbene transfer3,37,38 were screened for activity towards the reaction of styrene with azaserine by using E. coli expressing their encoding genes. Furthermore, P450-T2, a cytochrome P450 that was recently identified from a metagenomic database originating from a hot-spring collection, was tested because it is a thermophilic enzyme that was reported to bind to l-mimosine39, which is similar in structure to the hydrophilic azaserine. Given that the heterologous host S. albus is an aerobe and the reaction will be introduced into it, the screening process was conducted under aerobic conditions. In the initial screening, E. coli cells expressing the wild-type (WT) P450-T2 and a CYP119 mutant exhibited both significant activity and different diastereoselectivity (dr, the percentage of the major diastereomer versus the sum of all diastereomers in the product) compared with the control expressing red fluorescent protein (RFP) (Fig. 3a). The control formed only a low level of products, possibly produced by the reaction catalysed by the free haem inside the cell. As the activity of P450-T2 was the highest among the enzymes tested in the initial screening, the activity of this enzyme was further confirmed by in vitro reaction with purified protein (Extended Data Fig. 5b). Although the diastereoselectivity from the reaction catalysed by whole cells containing P450-T2 was 87.8%, it was 99.3% when the reaction was catalysed by purified protein (Fig. 3c and Extended Data Fig. 5b). The lower diastereoselectivity achieved by whole cells was possibly due to the interference of free haem inside the cell, which can also catalyse the reaction with low diastereoselectivity (Extended Data Fig. 1b). CYP203A1 from Rhodopseudomonas palustris, a homologue of P450-T239, and mutants of CYP203A1 with modifications to the amino acid acting as the axial ligand to iron (C337H, C337S and C337T) were also tested as catalysts for the reaction of styrene with azaserine. The reactions catalysed by E. coli cells expressing CYP203A1 WT and the C337H mutant formed more products than that from the reactions catalysed by cells expressing RFP. However, the activity of CYP203A1 and its mutant was lower than that of P450-T2 WT (Extended Data Fig. 6). Thus, P450-T2 WT was selected for further engineering.

a, Screening P450s for catalysing the reaction of styrene with azaserine. A list of screened P450s and their mutants is provided in Supplementary Table 1. The EIC ([M+H]+, m/z = 250.1074) for target products of reactions catalysed by E. coli cells expressing different proteins. Representative traces are for two repeated experiments. RFP, E. coli cells expressing RFP as a negative control; Ir(Me)MPIX, trace of the reaction products using Ir(Me)MPIX (with Na2S2O4) as a catalyst for comparison. b, Crystal structure of P450-T2 (Protein Data Bank (PDB): 8FBC). The residues in green are mutation sites in the final evolved mutant. c, Directed evolution of P450-T2 for cyclopropanation of styrene with azaserine. d, P450-T2 WT and P450-T2-5 mutant for insertion of the carbene unit into the sp3 C–H bond of phthalan. C1 and C2 are the two diastereomers formed by the reaction of phthalan with azaserine. The EIC ([M+H]+, m/z = 266.1023) for target products. Representative traces for three biological repeats (left). The reaction conditions were as follows: 5 mM styrene or 10 mM phthalan, 5 mM azaserine, E. coli cells expressing different P450s or RFP (as a control) at a concentration of 30 units based on the optical density at 600 nm (OD600) as catalysts, 5 vol% ethanol and M9-N buffer, conducted at 22 °C under aerobic conditions for 18 h. Ptotal and Ctotal are the sum area for all corresponding diastereomers. In c and d, the numbers on the horizontal axis represent the selected mutants in each round of directed evolution (the specific mutations are shown in Extended Data Table 1). Data are mean ± s.d. n = 3 biological replicates.
To guide the directed evolution, the crystal structure of P450-T2 was determined (Fig. 3b). On the basis of the structure and previous studies, several sites around the haem-binding pocket and on specific loop regions were selected for site-saturation mutagenesis and evaluation of the activity and selectivity of the resulting mutants. The turnover number per cell was used as the figure of merit instead of the turnover number per catalyst because the former value includes any changes in protein production in the cell due to mutations. The first-round mutant (S239V) catalysed the reaction to form 46 times the amount of product as was formed by the WT enzyme (Fig. 3c). This site corresponds to the Thr268 residue in P450 BM3 (Extended Data Fig. 7)—a site that has been reported to be important for increasing the activity of P450 BM3 towards carbene-transfer reactions13. The third-round mutant (S239V, F338H) was about five times more active than the S239V mutant (Fig. 3c). The Phe338 position corresponds to Phe393 in P450 BM3 (Extended Data Fig. 7), the mutation of which was reported to change the reduction potential of the haem and affect its catalytic behaviour40. After five rounds of evolution, the fifth-round mutant (P450-T2-5) catalysed the reaction with high diastereoselectivity (dr > 99%) and a moderate yield (42.7%); this yield is 251 times higher compared with the yield of the reaction catalysed by WT enzyme (Fig. 3c). Aside from catalysing cyclopropanation of styrene with azaserine, P450-T2 WT and P450-T2-5 also catalysed the insertion of the carbene unit from azaserine into the sp3 C–H bond of phthalan. The WT enzyme catalysed the reaction with high diastereoselectivity (dr = 97.2%) (Fig. 3d). The activity of P450-T2-5 was about 220 times higher than that of WT; the diastereoselectivity was high (dr = 85.8%), although slightly lower than that of the WT. At this point, the highest yield for this reaction was 2.5%.
Having shown that the carbene precursor could be generated biosynthetically in bacteria and that it reacts with styrene with a good yield when catalysed by a mutant of P450-T2, we sought to biosynthetically generate the final component of the reaction, styrene. To do so, the biosynthetic pathway for styrene was reconstituted in the azaserine-producing S. albus strain by integrating the required genes into the genome (Fig. 4a). The biosynthesis of styrene had been established in E. coli and Saccharomyces cerevisiae41,42,43, but not in Streptomyces. The pathway consists of two enzymes to convert phenylalanine to styrene—a phenylalanine ammonia lyase, the enzyme catalysing the rate limiting step, and a ferulic acid decarboxylase (FDC). Although it was reported that a prenyltransferase was required to synthesize the cofactor of FDC44, we found that the additional prenyltransferase gene was not required for the activity of FDC, possibly because a homologue of this prenyltransferase (BioCyc database collection, accession ID: XNR_3566) is naturally expressed in S. albus (Extended Data Fig. 8a). Given that styrene is volatile, its production was conducted in sealed tubes possessing sufficient headspace for the oxygen needed to grow S. albus. Styrene was produced in both TSB medium and 1B medium. Feeding the culture with phenylalanine, which is the precursor to styrene, the titre reached a level of 20 mg l−1 (Fig. 4b and Extended Data Fig. 8b).

a, Schematic for producing the final products with the substrates and enzymes made by the cells. b, Styrene biosynthesis in S. albus. 1B medium was supplemented with 0 mM, 2 mM or 4 mM phenylalanine for styrene production. c, Bioproduction of the unnatural cyclopropanes. 1B medium with 4 mM phenylalanine was used to generate the final products. The EIC ([M+H]+, m/z = 250.1074) for target products produced by the styrene- and azaserine-producing strain with or without the P450-T2-5 mutant. Representative traces for three biological replicates. The standard is a chemically synthesized single diastereomer standard of P1. Haemin, trace of the reaction products of azaserine with styrene using haemin as catalyst (with Na2S2O4) for comparison. d, The final product titre optimization. TSB or 1B medium was used for S. albus strains producing azaserine and styrene with 1 or 2 copies of P450-T2-5 to generate the final products. Ptotal is the sum area for all diastereomers. The grey bars indicate the titre (b and d); the white square indicates the dr (d). Data are mean ± s.d. n = 3 biological replicates.
After engineering S. albus to produce both substrates for the cyclopropanation of styrene with azaserine and evolving cytochrome P450-T2 to catalyse the reaction with significantly increased activity, the evolved P450-T2-5 mutant was integrated into the genome of the azaserine- and styrene-producing S. albus strain to construct the final strain, which can biosynthetically produce all of the components for the reaction. The final strain was then cultured to test whether the unnatural cyclopropanes would be produced through the intended biosynthesis. After culturing the strain in 1B medium with 4 mM phenylalanine for 72 h, the unnatural cyclopropanes were detected and confirmed by LC–HRMS and LC coupled with tandem MS (LC–MS/MS) (Fig. 4c and Extended Data Fig. 9a). The results of a time course show that the titre does not increase after 72 h under the current conditions (Extended Data Fig. 9b). Having observed this product, different media were tested, and one more copy of the P450-T2-5 gene was integrated into the chromosome to increase the titre. Under these final conditions, 222 µg l−1 of the cyclopropane product formed with 95% dr (Fig. 4d). Thus, the engineered S. albus was able to biosynthesize all of the reaction components for an abiological carbene-transfer reaction and produce unnatural products without the need for exogenously added carbene precursors and catalysts. Currently, the conversion efficiency for the integrated carbene-transfer reaction and the final product titre are not high, but sufficient for proof of concept. Although we were able to increase the activity of P450-T2 by 251-fold through mutations, we believe the major limitation is still the activity of the P450-T2-5 mutant, which was engineered to catalyse an abiological reaction of two non-native substrates by a non-natural catalytic mechanism.
This study established a platform for introducing abiological carbene-transfer reactions into microbial metabolism by producing the carbene precursor azaserine and showed that abiological carbene-transfer reactions can be fully incorporated into biosynthesis. With all of the components in the carbene-transfer reaction made by the microorganism, the system will be easier and less expensive to scale industrially than if one or more components need to be added to the system. This strategy is not limited to azaserine. There are other natural and potentially unnatural carbene precursors that could be synthesized in vivo. Moreover, many natural and unnatural products, such as limonene, carvone and indoles3,45, have been reported to be substrates for carbene chemistry catalysed by engineered haemoproteins, some of which could be generated biosynthetically as the carbene acceptor, thereby creating new therapeutics, scaffolds for drug discovery and precursors to materials. Furthermore, haemoproteins have been engineered to catalyse carbene insertions into a variety of chemical bonds (C–H, N–H and S–H)14,15 that are common in natural and unnatural compounds. It is also possible that the carbene acceptor can be added exogenously to the medium to react with the azaserine produced in vivo, and the reaction can be catalysed by the engineered enzymes inside the microorganisms. In this case, the product could be collected directly or serve as an intermediate for further enzymatic transformations inside or outside the cell. Finally, it may be possible to incorporate azaserine into a protein as a noncanonical amino acid and then perform the carbene-transfer reaction on the resulting protein or to perform the carbene-transfer reaction of azaserine before its incorporation into a protein, either of which would result in a protein with unnatural amino acids, further broadening the applicable scope of this platform.
Streptomyces was used as the host in this study, and this genus produces many widely used antibiotics and other valuable complex organic chemicals. Modification and diversification of those molecules would generate unnatural medicinal candidates for exploration. Such unnatural biosynthesis is not limited to Streptomyces or even bacteria, as one could imagine introducing those carbene-transfer reactions into other widely used industrial microorganisms, such as Corynebacterium or Saccharomyces. With the increasing number of reactions that were previously possible only through synthetic chemistry being executed by engineered enzymes, the integration of these enzymes into microorganisms and their reactions into biosynthesis should create new opportunities for biomanufacturing.
Methods
Chemical standards
All commercial (ordered from Millipore-Sigma) and chemically synthesized standards have a purity of 95% or higher. Methods for chemical standard synthesis are provided in the Supplementary Information.
In vitro reaction using purified proteins or metal cofactors
In a 2 ml brown gas chromatography (GC) vial, a specific volume of M9-N buffer was transferred to the reaction vial followed by the addition of Na2S2O4 (0.5 M stock in M9-N buffer), azaserine (100 mM stock in M9-N buffer) and styrene (100 mM stock in ethanol). Catalysts, metal cofactor (4 mM stock in DMSO) or purified proteins (100 µM stock in M9-N buffer) were added, resulting in a specific concentration and a final reaction volume of 300 µl. The vials were sealed with caps and shaken at 200 r.p.m. and 22 °C for 18 h. The reaction was quenched by adding 300 µl methanol, vortexed at a speed of 3,000 per min for 10 min and stored at −80 °C before analysis. The samples were thawed, briefly vortexed and centrifuged at 20,000g for 8 min. The supernatant was filtered with a 3 kDa centrifugal filter and analysed using LC–MS. To make the M9-N reaction buffer, salts (47.7 mM Na2HPO4, 22.0 mM KH2PO4, 8.6 mM NaCl) were dissolved in 1 l double-distilled H2O and autoclaved to give a medium with a pH of around 7.4. Solutions of MgSO4 (1 M), CaCl2 (1 M) and glucose (20 w/v%) were added to give a final concentration of 2.0 mM MgSO4, 0.1 mM CaCl2, and 0.4 w/v% glucose.
Reaction using E. coli whole cells
Plasmids containing the genes encoding the desired enzymes were transformed into BL21(DE3) competent cells (NEB). Selection plates were incubated at 30 °C overnight. Three colonies for each strain were inoculated into 3 ml Terrific Broth (TB) medium containing 50 µg ml−1 kanamycin and grown at 30 °C with shaking (200 r.p.m.) overnight. In a 50 ml glass tube, 50 µl overnight culture was inoculated into 5 ml fresh TB medium containing 50 µg ml−1 kanamycin and grown at 37 °C with shaking at 200 r.p.m.. When the optical density at 600 nm (OD600) reached approximately 0.6, the culture was cooled to 22 °C with shaking (200 r.p.m.) for 30 min. IPTG and 5-aminolevulinic acid were added to final concentrations of 0.2 mM and 0.5 mM, respectively. The cultures were grown at 22 °C with 200 r.p.m. shaking for 18 h. After measuring the OD600, 9 OD600 units of cells were collected by centrifugation at 10,000g for 1 min and resuspended in 270 µl M9-N buffer. The cell suspension was transferred to a 2 ml brown GC vial. Azaserine (100 mM stock in M9-N buffer) and styrene (100 mM stock in ethanol) or phthalan (200 mM stock in ethanol) were added resulting in specific final concentrations. The final reaction volume was 300 μl, and the final cell concentration was 30 OD600. The vials were sealed with screw caps and shaken at 200 r.p.m. and 22 °C for 18 h. The reaction was quenched by adding 300 µl methanol, vortexed at speed of 3,000 per min for 10 min and stored at −80 °C before the analysis. Before the analysis, the samples were thawed, briefly vortexed and centrifuged at 20,000g for 8 min. The supernatant was filtered through a 3 kDa centrifugal filter and analysed using LC–MS.
Azaserine toxicity
For E. coli, three single colonies were inoculated into Luria–Bertani (LB) medium and grown overnight at 30 °C with shaking at 200 r.p.m. The culture was diluted 1:100 into 5 ml of fresh LB medium with different concentrations of azaserine in 50 ml glass tubes and grown at 30 °C with shaking at 200 r.p.m. The cell density was analysed at 24 h and 48 h.
For S. albus, 20 µl spores were inoculated into 50 ml glass tubes containing 3 ml TSB medium with 4–6 glass beads of around 5 mm diameter and grown at 30 °C under 200 r.p.m. shaking for 48 h. The culture was then diluted 1:50 into 5 ml of fresh TSB medium with different concentrations of azaserine in 50 ml glass tubes containing 4–6 glass beads. The cells were grown at 30 °C and 200 r.p.m. shaking. At 24 and 48 h after inoculation, 1 ml samples of the cultures were collected and stored at −80 °C. Biomass was quantified using the methylene blue method46.
Identification of the biosynthetic gene cluster for azaserine
Comparative genomics was used to identify the azaserine biosynthetic gene cluster (aza BGC). A database was assembled with the genomes of 7,763 bacterial strains retrieved from the GenBank and 1,913 BGC entries from the MiBIG database. This database included the genomes of S. fragilis NBRC 12862 (also known as strain PD 04926) and G. harbinensis CGMCC 4.3516, as well as the biosynthetic gene clusters for the triacsins from Kitasatospora aureofaciens (MIBIG: BGC0001983), s56-p1 from Streptomyces sp. SoC090715LN (BGC0001764), pyrazomycin from Streptomyces candidus (BGC0001985) and actinopyridazinone from Streptomyces sp. MSD090630SC-05. All of these DNA sequences were annotated using RAST47, and their amino acid sequences and tabular feature files were used as an input for the CORASON pipeline48. The pipeline was modified to improve the command line interface, allow processing of large genome datasets, eliminate dependencies, increase the speed of the analysis and improve phylogenetic analysis by incorporation of maximum-likelihood phylogenetic calculations with model selection using IQtree49. The modified code is freely available at GitHub (https://github.com/WeMakeMolecules/myCORASON). The genome mining was performed using tri17 (protein ID: QCT05750.1) and tri28 (protein ID: QCT05761.1) as queries, with a cut-off e value of 1 × 10−12 and a bit score cut-off of 200. Orthologous BGCs were called only if homologues of tri28 or tri17 were found within a gene neighbourhood that included at least one more orthologue from the triacsin BGCs. The gene neighbourhoods were defined as 15 genes upstream and 15 genes downstream of the tri17 or tri28 homologues.
Protein expression and purification
For protein expression, a single colony of E. coli BL21(DE3) containing a plasmid encoding P450-T2 WT or mutant was grown overnight in 30 ml of TB medium with 50 µg ml−1 kanamycin at 37 °C with 200 r.p.m. shaking. The culture was used to seed (1:100 dilution) 500 ml TB medium containing 50 µg ml−1 kanamycin in a 2 l non-baffled flask. The cells were grown at 37 °C with 200 r.p.m. shaking to OD600 = 0.7 before moving to 22 °C with 200 r.p.m. shaking. After 30 min, IPTG and 5-aminolevulinic acid were added to final concentrations of 0.25 mM and 0.5 mM, respectively. The cultures were grown at 22 °C with 200 r.p.m. shaking for 18–20 h before collecting the cells by centrifugation at 4,000g for 30 min. The pellets were weighed (~5 g) and stored at −80 °C.
For protein purification, cells collected above were resuspended in 50 ml lysis buffer (25 mM Tris-HCl, pH 8.0, 100 mM NaCl, 30 mM imidazole, 0.1 mg ml−1 lysozyme, 0.05 mg ml−1 DNase, 1 mM MgCl2 and 5 mg haemin) and mixed by vortexing for 30 min. The lysate soluble fraction was obtained by further sonication, high-speed centrifugation at 37,000g for 30 min and filtering through a 0.45 µm filter. The lysate was then subjected to a 5 ml Ni-NTA column, washed with 10 column volumes of Ni-NTA wash buffer (25 mM Tris-HCl, pH 8.0, 100 mM NaCl, 10 mM imidazole) at a flow rate of 3 ml min−1 and eluted with an imidazole gradient (10 mM–300 mM in 20 min). The eluted fractions were assessed by SDS–PAGE and fractions with the target protein were further subjected to a 5 ml Q-trap column. The protein was washed with 5 column volumes of Q-trap wash buffer (25 mM Tris-HCl, pH 8.0) at a flow rate of 3 ml min−1 and eluted with a NaCl gradient (0 mM–500 mM in 20 min). Elution fractions were assessed by SDS–PAGE and selected fractions were dialysed (1:100, v:v) at 4 °C overnight in storage buffer (25 mM Tris-HCl, pH 8.0, 200 mM NaCl, 10% glycerol). Finally, the protein concentration was determined using the NanoDrop, followed by liquid N2 flash-cooling and storage at −80 °C.
Crystallization, X-ray data collection and structure determination
P450-T2 was concentrated to 10 mg ml−1. Crystals of P450-T2 were found in 0.2 M ammonium sulfate and 20% PEG 3,350. The crystal of P450-T2 was placed in a reservoir solution containing 20% (v/v) glycerol, then flash-cooled in liquid nitrogen. The X-ray diffraction dataset for P450-T2 was collected at the Berkeley Center for Structural Biology beamline 8.2.1 at the Advanced Light Source at Lawrence Berkeley National Laboratory. The diffraction data were collected using a wavelength of 1.000 Å and a temperature of 100 K. The dataset was processed using the Xia2 (ref. 50). The crystal structure of P450-T2 was solved by molecular replacement with PHASER51 using a model generated by AlphaFold52. The atomic positions obtained from the molecular replacement were used to initiate refinement with the Phenix suite53. Structure refinement was performed using the phenix.refine program54. Manual rebuilding was performed with COOT55. Root-mean-square deviations from ideal geometries for bond lengths, bond angles and dihedral angles were calculated using Phenix54. The stereochemical quality of the final model of P450-T2 was assessed using MOLPROBITY56. The Ramachandra plot showed 97.4% of residues are present in favoured regions, 2.6% of residues are present in allowed regions and no residues are outliers. A summary of the crystal parameters, data collection and refinement statistics is provided in Supplementary Table 3.
Site-saturation mutagenesis
The plasmid pBbE7k57 was used as the vector for cloning and expression in E. coli and digested by NdeI and BamHI. In total, 19 individual PCR reactions (20 µl for each) were performed for each screened residue site with primers containing specific mutations for saturation mutagenesis. A total of 19 PCR reactions were divided into three groups, each group with 6–7 PCR products. Aliquots of 8 µl from individual PCR reactions from the same group were taken and mixed together, resulting in three PCR product mixtures. Then, 5 µl of DpnI was added to the mixtures, and the samples were incubated at 37 °C for 3 h. All of the fragments were purified by gel electrophoresis. Purified DNA fragments were ligated by Gibson assembly. Then, 2 µl of the assembly products was added to 50 µl BL21(DE3) competent cells (NEB) for each group. In total, 24 colonies were picked from the transformation plate for each group and patched to two parallel LB agar plates containing 50 µg ml−1 kanamycin. One plate was sent for sequencing and the other was kept for inoculation.
Strain construction
The S. albus J1074 strains containing the exogenous genes were constructed according to a previously described protocol58 with minor modifications as described below. When spectinomycin resistance was used for selection, 100 µg ml−1 spectinomycin was used for E. coli selection and 200 µg ml−1 for S. albus selection. Single S. albus colonies from conjugation plates were inoculated into 2 ml TSB medium containing 200 µg ml−1 spectinomycin (or 50 µg ml−1 apramycin) and 25 µg ml−1 nalidixic acid in 24-well plates (about 4 glass beads with 5 mm diameter were added to each well), and the plates were incubated at 30 °C with 200 r.p.m. shaking for 3 days. Candidates with the correct integration were screened and verified by PCR or whole-cell reaction (using cells as catalysts to convert phenylalanine to styrene for verifying strains constructed to produce styrene). For the whole-cell conversion, 1 ml culture was centrifuged at 20,000g for 2 min. The supernatant was discarded, and the pellets were washed with 500 µl M9-N buffer, resuspended in 300 µl M9-N buffer and transferred to 2 ml GC vials. Phenylalanine was added to a final concentration of 5 mM (15 µl 0.1 M phenylalanine stock), the vials were capped and shaken at 200 r.p.m. at 30 °C for 24 h. Next, 600 µl hexane containing 80 µg ml−1 beta-pinene was added. The vials were vortexed at a speed of 3,000 per min for 5 min. The samples were transferred to a 1.7 ml tube and centrifuged at 20,000g for 8 min. The organic layer (200 µl) was transferred to a vial for styrene analysis by GC–MS. Strains with positive PCR results or styrene production were stored at −80 °C.
Media and S. albus cultivation
For azaserine production, spores (30 µl) of S. albus J1074 with the azaserine gene cluster integrated into its genome were inoculated into 4 ml TSB medium in 50 ml glass tubes with approximately 10 glass beads of 5 mm diameter. The cells were grown at 30 °C with 200 r.p.m. shaking for 48 h. The cells were next diluted 1:50 into 30 ml various media in 250 ml non-baffled flasks with eight-layer cotton gauze caps and grown at 28 °C with 200 r.p.m. shaking. At several timepoints, samples of 500 µl were removed and stored at −80 °C. For analysis of azaserine titres, the samples were thawed, 500 µl of methanol was added, and the mixture was vortexed at 3,000 per min for 10 min and then centrifuged at 20,000g for 8 min. The supernatant was filtered through a 3 kDa centrifugal filter and analysed using LC–MS.
For styrene production, 60 µl of mycelium stock (stored at −80 °C) was inoculated into 3 ml (1:50) TSB medium with 200 µg ml−1 spectinomycin and 25 µg ml−1 nalidixic acid in 50 ml glass tubes containing approximately 10 glass beads of 5 mm diameter. The cultures were grown at 30 °C with 200 r.p.m. shaking for 72 h. The cells were next diluted 1:50 into 5 ml fresh medium (containing 200 µg ml−1 spectinomycin and 25 µg ml−1 nalidixic acid) with various concentrations of phenylalanine in 50 ml conical tubes containing 6 glass beads of 5 mm diameter, which were sealed, incubated at 28 °C with 200 r.p.m. shaking. At the specific timepoints, 500 µl samples were taken and extracted with 500 µl hexane containing 80 ng µl−1 beta-pinene, vortexed at 3,000 per min for 10 min and centrifuged at 20,000g for 8 min. The organic layer (200 µl) was transferred to a vial for GC–MS analysis.
For generation of the product by styrene and azaserine cross-coupling, the cells were cultured as described above for styrene production, except that 0.5 mM 5-aminolevulinic acid was added to the growth medium. The samples were analysed using LC–MS as described above for azaserine. MYI medium (1 l) consisted of 4 g yeast extract, 10 g malt extract and 4 g glucose, with double-distilled H2O added to 1 l, autoclaved at 121 °C for 20 min. 1B medium (1 l) (modified from an expired patent59) was as follows: 10 g glucose, 10 g soybean meal, 5 g casein lysate, 5 g yeast extract, 0.5 g sodium chloride and 2.5 g ammonium nitrate with double-distilled H2O added to 1 l, adjusted pH to 7.5. A total of 1 g of CaCO3 was added to 1 l of the medium before autoclaving at 121 °C for 20 min.
Plasmid construction
All plasmids (Supplementary Table 2) were constructed using Gibson assembly or restriction enzyme cloning. The DNA products were transformed into E. coli DH5α competent cells, plated onto LB agar plates with the corresponding antibiotics (100 µg ml−1 spectinomycin, 50 µg ml−1 kanamycin or 50 µg ml−1 apramycin). For cloning fragments larger than 15 kb, the transformed cells were incubated at 30 °C, whether on agar plates or the subsequent liquid cultures. All other transformed cells were incubated at 37 °C. Mini-prepped plasmids were digested with restriction enzymes. Candidates with the correct digestion patterns were sent for sequencing. To clone the azaserine gene cluster, S. fragilis genomic DNA was used as a template for amplification of the gene fragment. All other exogenous genes that were to be expressed in S. albus were codon-optimized for S. albus and ordered from IDT or GenScript. The conjugation plasmids used for integrating genes into S. albus contained the p15A origin.
GC–MS and LC–MS analyses
Styrene was analysed using an Agilent Technologies GC–MS system equipped with an HP-5 column (Agilent). The temperature in the GC oven was increased from 50 °C to 110 °C at a rate of 10 °C min−1. The solvent delay was set at 3 min. Single ions (m/z = 78, 93, 104, 121) were detected. The other products were analysed using high-performance LC–MS (HPLC–MS) using the Agilent 1260 infinity II system with mass selective detector iQ (MSD iQ) or an Agilent Technologies 6520 with quadrupole time-of-flight mass spectrometer (Q-TOF-MS). The HPLC autosampler and column compartment were maintained at 6 °C and 25 °C, respectively. An Agilent Technologies ZORBAX Eclipse Plus C18 (3.5 µm, 4.6 mm × 150 mm) column was used for azaserine analysis. A Millipore-Sigma Astec CYCLOBOND I 2000 HP-RSP Chiral HPLC (5 µm, 25 cm × 4.6 mm) column was used to analyse the reaction products of azaserine with styrene (aza–sty) or with phthalan (aza–phtha). For aza–sty analysis, the mobile phase comprised 10 mM ammonium formate in water with 50 µl formic acid added per litre (solvent A) and 0.1% (v/v) formic acid in acetonitrile (solvent B). For aza–sty separation and analysis, the column was eluted with 14% B from 0–32 min at a flow rate of 0.35 ml min−1 for 0–23 min, increased to 0.8 ml min−1 over 0.2 min and held at 0.8 ml min−1 for 23.2–32 min, and then flushed for the next analysis. For aza–phtha analysis, the column was eluted with a linearly increasing concentration of B from 13% to 13.7% over 40 min at a flow rate of 0.2 ml min−1 and then flushed for the next analysis.
Electrospray ionization was performed in positive-ion mode with a capillary voltage of 3,500 V, for [M+H]+ ions. Drying gas temperature and flow rate were maintained at 340 °C and 11 l min−1, respectively, and the nebulizer gas was set to 30 lb in−2 (1 pound per square inch is equivalent to 6.894757 kPa). Fragmentor, skimmer and OCT 1 RF Vpp voltages were set to 120 V, 60 V and 250 V, respectively. Targeted MS/MS was performed with a 250.107384 precursor m/z ion selection in the quadrupole at the analyte’s retention time using a delta retention time window of 2.5 min, an isolation width of ~1.3 m/z and a collision energy of 20 eV for collision-induced dissociation. The resulting fragment ions were analysed by the TOF mass analyser.
Proteomics method
Azaserine-producing S. albus strains were grown in TSB medium. After 24 h of growth at 30 °C, the samples were collected for proteomic analysis. Protein was extracted and tryptic peptides were prepared according to established proteomic sample preparation procedures. In brief, cell pellets were resuspended in Qiagen P2 Lysis Buffer (Qiagen, 19052) to promote cell lysis. Proteins were precipitated by addition of 1 mM NaCl and 4 volumes of acetone, followed by two additional washes with 80% acetone in water. The recovered protein pellet was homogenized in 100 mM ammonium bicarbonate in 20% methanol. The protein concentration was determined by the DC protein assay (BioRad). Protein reduction was accomplished using 5 mM Tris 2-(carboxyethyl)phosphine for 30 min at room temperature, and alkylation was performed with 10 mM iodoacetamide (final concentration) for 30 min at room temperature in the dark. Overnight digestion with trypsin was accomplished with a 1:50 trypsin:total protein ratio. The resulting peptide samples were analysed on the Agilent 1290 UHPLC system coupled to a Thermo Fisher Scientific Orbitrap Exploris 480 mass spectrometer for discovery proteomics60. In brief, the peptide samples were loaded onto an Ascentis ES-C18 Column (Sigma-Aldrich) and separated by a LC gradient previously described60. Eluting peptides were introduced to the mass spectrometer operating in positive-ion mode and were measured in data-independent acquisition (DIA) mode with a duty cycle of 3 survey scans from 380 m/z to 985 m/z and 45 MS2 scans with precursor isolation width of 13.5 m/z to cover the mass range. DIA raw data files were analysed using the integrated software suite DIA-NN61. The database used in the DIA-NN search (library-free mode) is the latest UniProt S. albus proteome FASTA sequence plus the protein sequences of the identified azaserine gene cluster and common proteomic contaminants. DIA-NN determines mass tolerances automatically based on first-pass analysis of the samples with automated determination of optimal mass accuracies. The retention time extraction window was determined individually for all MS runs analysed using the automated optimization procedure implemented in DIA-NN. Protein inference was enabled, and the quantification strategy was set to Robust LC = High Accuracy. Output main DIA-NN reports were filtered with a global false-discovery rate of 0.01 on both the precursor level and the protein group level. The Top3 method, which is the average MS signal response of the three most intense tryptic peptides of each identified proteins, was used to plot the quantity of the azaserine gene cluster proteins in the samples62.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The DNA sequences of plasmids used in this study have been deposited in the public version of the JBEI registry (http://public-registry.jbei.org). Accession codes are provided in Supplementary Table 2 (Part ID). The sequences and annotation of the aza BGC is available at GenBank (NZ_BEVZ01000003.1) spanning bases 82400–111549. The determined structure has been deposited in the PDB (8FBC). The MS proteomics data have been deposited at the ProteomeXchange Consortium through the PRIDE63 partner repository under the dataset identifier PXD037509. Source data are provided with this paper.
Change history
21 December 2023
In the version of the article originally published, the panel label ‘d’ was missing in Fig. 4. The error has been corrected in the HTML and PDF versions of the article.
References
Arnold, F. H. Directed evolution: bringing new chemistry to life. Angew. Chem. Int. Ed. Engl. 57, 4143–4148 (2018).
Wallace, S. & Balskus, E. P. Interfacing microbial styrene production with a biocompatible cyclopropanation reaction. Angew. Chem. Int. Ed. Engl. 54, 7106–7109 (2015).
Huang, J. et al. Unnatural biosynthesis by an engineered microorganism with heterologously expressed natural enzymes and an artificial metalloenzyme. Nat. Chem. 13, 1186–1191 (2021).
Davies, H. M. L. & Manning, J. R. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 451, 417–424 (2008).
Doyle, M. P., Duffy, R., Ratnikov, M. & Zhou, L. Catalytic carbene insertion into C-H bonds. Chem. Rev. 110, 704–724 (2010).
Ford, A. et al. Modern organic synthesis with α-diazocarbonyl compounds. Chem. Rev. 115, 9981–10080 (2015).
Zhu, D., Chen, L. F., Fan, H. L., Yao, Q. L. & Zhu, S. F. Recent progress on donor and donor-donor carbenes. Chem. Soc. Rev. 49, 908–950 (2020).
Kluger, R. Thiamin diphosphate: a mechanistic update on enzymic and nonenzymic catalysis of decarboxylation. Chem. Rev. 87, 863–876 (1987).
Lee, J. K. & Houk, K. N. A proficient enzyme revisited: the predicted mechanism for orotidine monophosphate decarboxylase. Science 276, 942–945 (1997).
Meyer, D., Neumann, P., Ficner, R. & Tittmann, K. Observation of a stable carbene at the active site of a thiamin enzyme. Nat. Chem. Biol. 9, 488–490 (2013).
Cochrane, A. R. et al. The natural product lepidiline A as an N-heterocyclic carbene ligand precursor in complexes of the type [Ir(cod)(NHC)PPh3)]X: synthesis, characterisation, and application in hydrogen isotope exchange catalysis. Catalysts 10, 161 (2020).
Cheng, R. H. et al. Implications for an imidazole-2-yl carbene intermediate in the rhodanase-catalyzed C-S bond formation reaction of anaerobic ergothioneine biosynthesis. ACS Catal. 11, 3319–3334 (2021).
Coelho, P. S., Brustad, E. M., Kannan, A. & Arnold, F. H. Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science 339, 307–310 (2013).
Brandenberg, O. F., Fasan, R. & Arnold, F. H. Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr. Opin. Biotechnol. 47, 102–111 (2017).
Liu, Z. & Arnold, F. H. New-to-nature chemistry from old protein machinery: carbene and nitrene transferases. Curr. Opin. Biotechnol. 69, 43–51 (2021).
Wallace, S. & Balskus, E. P. Designer micelles accelerate flux through engineered metabolism in E. coli and support biocompatible chemistry. Angew. Chem. Int. Ed. Engl. 55, 6023–6027 (2016).
Wong, H. N. C. et al. Use of cyclopropanes and their derivatives in organic synthesis. Chem. Rev. 89, 165–198 (1989).
Talele, T. T. The “cyclopropyl fragment” is a versatile player that frequently appears in preclinical/clinical drug molecules. J. Med. Chem. 59, 8712–8756 (2016).
Fulton, J. R., Aggarwal, V. K. & de Vicente, J. The use of tosylhydrazone salts as a safe alternative for handling diazo compounds and their applications in organic synthesis. Eur. J. Org. Chem. 2005, 1479–1492 (2005).
Morandi, B. & Carreira, E. M. Iron-catalyzed cyclopropanation in 6 M KOH with in situ generation of diazomethane. Science 335, 1471–1474 (2012).
Zhang, L. M., DeMuynck, B. M., Paneque, A. N., Rutherford, J. E. & Nagib, D. A. Carbene reactivity from alkyl and aryl aldehydes. Science 377, 649–654 (2022).
Xia, Y., Qiu, D. & Wang, J. B. Transition-metal-catalyzed cross-couplings through carbene migratory insertion. Chem. Rev. 117, 13810–13889 (2017).
Damiano, C., Sonzini, P. & Gallo, E. Iron catalysts with N-ligands for carbene transfer of diazo reagents. Chem. Soc. Rev. 49, 4867–4905 (2020).
Nawrat, C. C. & Moody, C. J. Natural products containing a diazo group. Nat. Prod. Rep. 28, 1426–1444 (2011).
Kawai, S. et al. Complete biosynthetic pathway of alazopeptin, a tripeptide consisting of two molecules of 6-diazo-5-oxo-l-norleucine and one molecule of alanine. Angew. Chem. Int. Ed. 60, 10319–10325 (2021).
Le Maux, P., Nicolas, I., Chevance, S. & Simonneaux, G. Chemical reactivity of 6-diazo-5-oxo-l-norleucine (DON) catalyzed by metalloporphyrins (Fe,Ru). Tetrahedron 66, 4462–4468 (2010).
Sugai, Y., Katsuyama, Y. & Ohnishi, Y. A nitrous acid biosynthetic pathway for diazo group formation in bacteria. Nat. Chem. Biol. 12, 73–75 (2016).
Ma, G. L. et al. Biosynthesis of tasikamides via pathway coupling and diazonium-mediated hydrazone formation. J. Am. Chem. Soc. 144, 1622–1633 (2022).
Bartz, Q. R. et al. Isolation and characterization of azaserine. Nature 173, 72–73 (1954).
Stock, C. C., Clarke, D. A., Reilly, H. C., Rhoads, C. P. & Buckley, S. M. Azaserine, a new tumour-inhibitory substance: studies with crocker mouse sarcoma 180. Nature 173, 71–72 (1954).
Key, H. M. et al. Beyond iron: iridium-containing P450 enzymes for selective cyclopropanations of structurally diverse alkenes. ACS Cent. Sci. 3, 302–308 (2017).
Lee, M. D. et al. New antitumor antibiotic, LL-D05139β. Fermentation, isolation, structure determination and biological activities. J. Antibiot. 40, 1657–1663 (1987).
Matsuda, K. et al. Genome mining of amino group carrier protein-mediated machinery: discovery and biosynthetic characterization of a natural product with unique hydrazone unit. ACS Chem. Biol. 12, 124–131 (2017).
Twigg, F. F. et al. Identifying the biosynthetic gene cluster for triacsins with an N-hydroxytriazene moiety. ChemBioChem 20, 1145–1149 (2019).
Williams, M. V. & Tritz, G. J. Studies on the modes of action of azaserine inhibition of Escherichia coli. Potentiation of phenylalanine reversal. J. Antimicrob. Chemother. 3, 65–77 (1977).
Geisen, S. M. et al. Direct alkylation of deoxyguanosine by azaserine leads to O6-carboxymethyldeoxyguanosine. Chem. Res. Toxicol. 34, 1518–1529 (2021).
Gober, J. G. et al. P450-mediated non-natural cyclopropanation of dehydroalanine-containing thiopeptides. ACS Chem. Biol. 12, 1726–1731 (2017).
Zhang, R. J. K. et al. Enzymatic assembly of carbon-carbon bonds via iron-catalysed sp3 C-H functionalization. Nature 565, 67–72 (2019).
Nguyen, K. T. et al. Characterization of a thermophilic cytochrome P450 of the CYP203A subfamily from Binh Chau hot spring in Vietnam. FEBS Open Bio 11, 124–132 (2021).
Ost, T. W. B. et al. Oxygen activation and electron transfer in flavocytochrome P450BM3. J. Am. Chem. Soc. 125, 15010–15020 (2003).
McKenna, R. & Nielsen, D. R. Styrene biosynthesis from glucose by engineered E. coli. Metab. Eng. 13, 544–554 (2011).
McKenna, R., Thompson, B., Pugh, S. & Nielsen, D. R. Rational and combinatorial approaches to engineering styrene production by Saccharomyces cerevisiae. Microb. Cell Fact. 13, 123 (2014).
Lee, K., Bang, H. B., Lee, Y. H. & Jeong, K. J. Enhanced production of styrene by engineered Escherichia coli and in situ product recovery (ISPR) with an organic solvent. Microb. Cell Fact. 18, 79 (2019).
Lin, F. M., Ferguson, K. L., Boyer, D. R., Lin, X. X. N. N. & Marsh, E. N. G. Isofunctional enzymes PAD1 and UbiX catalyze formation of a novel cofactor required by ferulic acid decarboxylase and 4-hydroxy-3-polyprenylbenzoic acid decarboxylase. ACS Chem. Biol. 10, 1137–1144 (2015).
Brandenberg, O. F., Chen, K. & Arnold, F. H. Directed evolution of a cytochrome P450 carbene transferase for selective functionalization of cyclic compounds. J. Am. Chem. Soc. 141, 8989–8995 (2019).
Fischer, M. & Sawers, R. G. A universally applicable and rapid method for measuring the growth of streptomyces and other filamentous microorganisms by methylene blue adsorption-desorption. Appl. Environ. Microbiol. 79, 4499–4502 (2013).
Aziz, R. K. et al. The RAST server: rapid annotations using subsystems technology. BMC Genom. 9, 75 (2008).
Navarro-Munoz, J. C. et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 16, 60–68 (2020).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Winter, G., Lobley, C. M. C. & Prince, S. M. Decision making in xia2. Acta Crystallogr. D 69, 1260–1273 (2013).
Mccoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 68, 352–367 (2012).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Lee, T. S. et al. BglBrick vectors and datasheets: a synthetic biology platform for gene expression. J. Biol. Eng. 5, 12 (2011).
Yuzawa, S. et al. Short-chain ketone production by engineered polyketide synthases in Streptomyces albus. Nat. Commun. 9, 4569 (2018).
Ehrlich, J. et al. Process for producing azaserine. US patent 2996435A (1961).
Chen, Y., Gin, J. & Petzold, C. Discovery proteomic (DIA) LC-MS/MS data acquisition and analysis V.2. Protocols.io https://doi.org/10.17504/protocols.io.e6nvwk1z7vmk/v2 (2022).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Ahrne, E., Molzahn, L., Glatter, T. & Schmidt, A. Critical assessment of proteome-wide label-free absolute abundance estimation strategies. Proteomics 13, 2567–2578 (2013).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Madeira, F. et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50, W276–W279 (2022).
Acknowledgements
This work was supported by Joint BioEnergy Institute (https://www.jbei.org), which is supported by the DOE, Office of Science, Office of Biological and Environmental Research under contract DE-AC02-05CH11231 and National Science Foundation grant 2027943. We thank H. Celik and the staff at UC Berkeley’s NMR facility in the College of Chemistry (CoC-NMR) for spectroscopy assistance. Instruments in the CoC-NMR are supported in part by NIH S10OD024998.
Author information
Authors and Affiliations
Contributions
J.H. and J.D.K. conceived the project. J.H. and J.D.K. wrote the manuscript with input from all of the other authors. J.D.K., A.M., J.F.H. and D.S.C. provided project guidance. A.Q. and K.D. synthesized the chemical standards. J.H., P.C.-M. and D.V.C. identified the gene cluster. J.H.P. determined the crystal structure. Q.D. purified the proteins. J.H., R.K. and E.E.K.B. performed MS. Y.C. performed proteomics analysis. J.H. conducted the other experiments. E.P.B., P.D.A., T.R.N. and C.J.P. provided resources. All of the authors contributed to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.D.K. has a financial interest in Amyris, Demetrix, Maple Bio, Lygos, Napigen, Berkeley Yeast, Zero Acre Farms, Ansa Biotechnologies, Apertor Pharmaceuticals, ResVit Bio and Cyklos Materials. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Frances Arnold, Nicholas Turner and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Effect of Na2S2O4 on the activity of haemin and Ir(Me)MPIX for the reaction of styrene with azaserine.
a, Addition of Na2S2O4 decreased the reaction yield when using Ir(Me)MPIX as catalyst. TON, turn-over number. Reaction conditions are described in Fig. 2 legend. Data are mean value for 2 reaction replicates. b, Na2S2O4 is necessary for the activity of haemin toward the reaction. EIC ([M+H]+, m/z 250.1074) for target products. The traces are representative of two reaction replicates. The reaction contained 5 mM styrene, 5 mM azaserine, 10 µM haemin or no catalyst, 0 or 10 mM Na2S2O4, 5 vol% ethanol, and M9-N buffer and was conducted at 22 °C under aerobic conditions for 18 h. Standard, chemically synthesized authentic standard mixture of the four diastereomers.
Extended Data Fig. 2 Azaserine toxicity on E. coli and S. albus.
a, Azaserine is toxic to E. coli BL21(DE3). b, Azaserine does not affect the growth of S. albus under tested concentrations. Biomass was normalized to that of culture without addition of azaserine. Data are mean ± s.d.; n = 3 biological replicates.
Extended Data Fig. 3 Expression of azaserine gene cluster from S. fragilis in S. albus.
a, Bioinformatic annotation of the azaserine gene cluster and comparison with the biosynthetic gene clusters for some natural N–N bond-containing compounds. b, Proteomic analysis of the azaserine gene cluster when expressed in S. albus. Data are mean ± s.d.; n = 3 biological replicates.
Extended Data Fig. 4 Differences in azaserine degradation in various media.
a, Azaserine titre continues to decrease after removal of S. albus cells. The azaserine-producing S. albus was grown in TSB medium. After 24 h, the cells were removed from culture broth using a 0.22-µm sterile filter, the filtrate was incubated at 30 °C (labelled as 0 h), and the azaserine concentration was monitored at different time points. b, Azaserine is stable in fresh TSB medium of normal pH 7.3 or adjusted pH 8.4. Equal volume of azaserine stock was added to a final concentration of about 35 mg/L at 0 h. Data are mean ± s.d.; n = 3 biological (a) or technical (b) replicates.
Extended Data Fig. 5 Purified P450-T2 WT and mutant catalysing the reaction of styrene with azaserine in vitro.
a, Coomassie Blue stained SDS-PAGE gel of purified protein P450-T2 WT (left) and P450-T2-5 mutant (right). Each lane is a sample from fractions collected during ion exchange purification. b, Purified P450-T2 WT and P450-T2-5 mutant proteins catalyse the reaction in vitro with high diastereoselectivity. 5th, P450-T2-5 mutant. Reaction conditions: 5 mM styrene, 5 mM azaserine, 10 µM enzyme, 10 mM Na2S2O4, 5 vol% ethanol, M9-N buffer, conducted at 22 °C under aerobic condition for 18 h. Ptotal, sum area for all diastereomers. Grey bars indicate the dr. Data are mean ± s.d.; n = 3 reaction replicates.
Extended Data Fig. 6 CYP203A1 WT and axial ligand mutants for the reaction of styrene with azaserine.
EIC ([M+H]+, m/z 250.1074) for target products. Representative traces for two repeated experiments. The reactions contained 5 mM styrene, 5 mM azaserine, E. coli cells with concentration of 30 OD600 as catalyst, 5 vol% ethanol, and M9-N buffer and were conducted at 22 °C under aerobic conditions for 18 h.
Extended Data Fig. 7 Conserved amino acids selected for saturation mutagenesis.
The residue labelled in blue is the haem ligand cysteine in those P450s. Residues labelled in red are two conserved residues previously reported to affect the catalytic behaviour in P450 BM3. Sequences were aligned with Clustal Omega64.
Extended Data Fig. 8 Exploration of the conditions for styrene biosynthesis in S. albus.
a, Heterologously expressing PAL2 and FDC1 are sufficient to generate styrene. pAZA121 and pAZA138 are two integration plasmids used to introduce the styrene pathway into S. albus. b, Production of styrene by engineered S. albus grown in TSB medium supplemented with additional 0 mM, 2 mM or 4 mM Phe. Data are mean ± s.d.; n = 3 biological samples.
Extended Data Fig. 9 Characterization and time course of accumulation of the final products.
a, MS/MS (20 eV) spectra of the P1 standard (red) and the biosynthesized major product (black). b, Biosynthesis of final products during the 96-h fermentation process. The 24-h data are not presented because small quantities of product were observed (<10 µg/L), and thus the titre could not be accurately calculated. 1B medium with 4 mM Phe was used to generate the final products. Data are mean ± s.d.; n = 3 biological samples.
Supplementary information
Supplementary Information
Supplementary Tables 1–3 (DNA and proteins sequences, synthesis of compounds), Supplementary Figs. 1–38 and Supplementary References.
Rights and permissions
About this article

Cite this article
Huang, J., Quest, A., Cruz-Morales, P. et al. Complete integration of carbene-transfer chemistry into biosynthesis. Nature 617, 403–408 (2023). https://doi.org/10.1038/s41586-023-06027-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-06027-2
- Springer Nature Limited
This article is cited by
-
Expanding chemistry through in vitro and in vivo biocatalysis
Nature (2024)
-
Ultrafast photoinduced C-H bond formation from two small inorganic molecules
Nature Communications (2024)
-
De novo engineering of programmable and multi-functional biomolecular condensates for controlled biosynthesis
Nature Communications (2024)
-
Carbene chemistry for unnatural biosynthesis
Science China Life Sciences (2024)