

Abstract
Most animals have compound eyes, with tens to thousands of lenses attached rigidly to the exoskeleton. A natural assumption is that all of these species must resort to moving either their head or their body to actively change their visual input. However, classic anatomy has revealed that flies have muscles poised to move their retinas under the stable lenses of each compound eye1,2,3. Here we show that Drosophila use their retinal muscles to smoothly track visual motion, which helps to stabilize the retinal image, and also to perform small saccades when viewing a stationary scene. We show that when the retina moves, visual receptive fields shift accordingly, and that even the smallest retinal saccades activate visual neurons. Using a head-fixed behavioural paradigm, we find that Drosophila perform binocular, vergence movements of their retinas—which could enhance depth perception—when crossing gaps, and impairing the physiology of retinal motor neurons alters gap-crossing trajectories during free behaviour. That flies evolved an ability to actuate their retinas suggests that moving the eye independently of the head is broadly paramount for animals. The similarities of smooth and saccadic movements of the Drosophila retina and the vertebrate eye highlight a notable example of convergent evolution.
Similar content being viewed by others

Main
Eye movements serve many perceptual, cognitive and social functions for primates and other species with single-lens eyes. Most animals, however, have compound eyes that attach rigidly to their heads. Drosophila melanogaster, for example, have compound eyes, and this species serves as an important model for understanding the neural basis of vision. Here we investigate whether Drosophila, somehow, actively move their eyes even when their head is still. We further study what functions such eye movements might serve and how their dynamics compare to those of eye movements in primates and other vertebrates.
In the 1970s, it was discovered that houseflies have a muscle under each compound eye that attaches to the orbital ridge, a thin sheet of cuticle that surrounds the retina and part of the optic lobes1,2. This muscle seemed poised to shift the optical axes of the photoreceptors relative to the lenses of the compound eye1,2,4. ‘Clock spikes’—large, extracellularly recorded action potentials with regular interspike intervals evident in the vicinity of the optic lobes—were shown to reflect the activity of a motor neuron innervating this retinal muscle2. In 1991, a second muscle in houseflies was discovered, suggesting that the retina might move in two dimensions3. The rate of clock spikes is modulated spontaneously and also in response to external stimuli4,5,6,7. In preliminary reports, it has been argued that these spike-rate modulations correlate with movements of photoreceptors3,8,9. In Drosophila, recent work has focused on phototransduction-based movements of photoreceptors10,11,12; thus, despite the initial observations in houseflies, the functions of muscular movements of the fly retina13 remain unclear.
Two muscles move the Drosophila retina
We discovered that Drosophila, like houseflies, have two retinal muscles (Fig. 1a–c and Supplementary Videos 1 and 3). One muscle resembles the musculus orbito-tentoralis of large flies1,2,3. The muscle is attached through a long tendon to the tentorial bar posteriorly and inserts into the antero-medial rim of the orbital ridge (Extended Data Fig. 1). A second muscle, which resembles the musculus orbito-scapalis3 of large flies, inserts more dorsally, into the frontal–medial rim of the orbital ridge. This second muscle originates at the edge of the antennal cup, to which it connects through a short tendon.

a, A frontal view of the Drosophila head showing immunostaining for muscles in red (phalloidin targeting actin) and neurons, including retinal motor neurons, in green (anti-GFP targeting mVenus expression). The musculus orbito-scapalis (MOS), which interconnects the antennal cup to the front of the orbital ridge (which surrounds the retina), is fully visible. The musculus orbito-tentoralis (MOT) is partially visible, at its insertion point to the orbital ridge. b, A horizontal view allows one to fully visualize the musculus orbito-tentoralis (arrow) and its innervating motor neuron. We stained retinal muscles more than 15 times with similar results. c, Schematics of both muscles (red) in coronal (top) and horizontal (bottom) planes. Tendons are shown in blue. The orbital ridge is shown in yellow. Other muscles (proboscis and oesophagus) are shown in brown. d, Part of a retina seen through a water immersion objective. Two images are overlaid. Red image: retina during optogenetic activation of the retinal motor neurons, yielding a maximal shift of photoreceptor tips. Grey image: after turning off the optogenetic light, with the muscles fully relaxed. Whereas the photoreceptors moved with optogenetic activation (red versus grey photoreceptor tips), the lenses, which are in focus on the left (arrow), remained stable. We performed this experiment in four flies, and the images shown represent the maximum movements observed. e, The schematic (left) shows how the deep pseudopupil16 (DPP) is an erect, virtual image of the photoreceptor (PR) tips at the centre of the curvature of the compound eye (adapted from ref. 16, Springer Nature). C, cornea. Right: overlaid image of the deep pseudopupil in one fly during (red) and after (grey) optogenetic activation (as in d). f, Simultaneously tracked centre-of-mass traces of photoreceptor tips, using a water immersion objective visualizing ommatidia at the top of the eye, and the deep pseudopupil, using an air lens aimed at a lower region of the same eye. See Methods for how we converted pseudopupil movements from pixels to degrees. Scale bars, 100 μm (a,b) and 50 μm (d).
As these muscles seem poised to move the Drosophila retina, we visualized the tips of the photoreceptors with a water immersion objective positioned above the eye (Methods)14. We expressed the redshifted channelrhodopsin, CsChrimson15, in the motor neurons innervating the retinal muscles. Using a pulse of red light, we optogenetically drove the retina to an extreme, tensed position (Fig. 1d, red) and then let it relax to an extreme, relaxed position by turning off the light (Fig. 1d, grey). We observed peak-to-peak displacements of the photoreceptors that spanned about three inter-photoreceptor spacings, which corresponds to ~15° in angular space (Fig. 1d).
Submerging the fly eye in water precludes many experiments. We thus decided to track the photoreceptor tips by measuring the position of the deep pseudopupil16—an enlarged, virtual image of photoreceptors that forms at the centre of curvature of the compound eye, which can be visualized with an air lens (Fig. 1e). Specifically, when one places a point source of light abutting the fly, such that light exiting the compound eye is collected by the air lens, the deep pseudopupil appears as seven large, bright dots, arranged in the shape of a single ommatidium’s receptor array. We used 780- or 850-nm light—wavelengths to which the fly’s natural opsins are insensitive17—to visualize the deep pseudopupil without blinding the fly. To verify that movements of the deep pseudopupil accurately reflect photoreceptor movements, we simultaneously tracked the deep pseudopupil (with an air lens) and photoreceptors at the top of the eye (with a water immersion objective) and observed a tight correlation between these two signals (Fig. 1f and Supplementary Video 4). We observed the same maximal angular excursion of ~15° in the deep pseudopupil during optogenetic activation of the retinal muscles as when tracking the positions of the photoreceptors directly (Fig. 1d,e).
Retinal movements shift receptive fields
Drosophila and other dipterans have neural superposition eyes, in which each of the eight photoreceptors in a single ommatidium is in precise optical alignment with specific photoreceptors in neighbouring ommatidia. Signals from all photoreceptors oriented along a common angle in visual space—independently of their ommatidium of origin—are ultimately combined in downstream neurons through a sophisticated neural wiring scheme18. If retinal muscles were to move the optical axes of photoreceptors in different ommatidia by different amounts, this would compromise the fidelity of neural superposition and thus impair the ability of visual neurons to signal effectively. To address this concern, we measured the visual responses of LC14 cells19 (or dorsal cluster neurons20) using whole-cell patch-clamp recordings during optogenetically induced retinal movements. LC14 cells interconnect the two visual lobes19,20. We found that they have ~50°-wide receptive fields along the frontal, vertical midline (Fig. 2a,b). These cells respond strongly to moving bars or spots traversing their receptive fields (Extended Data Fig. 2).

a, We recorded from LC14 neurons on the right side of the brain. b, Example of the receptive field of an LC14 cell (right), as estimated by responses to 100-ms (grey region) flashes on the screen (left). c, We performed whole-cell patch-clamp recordings in rigidly tethered flies. We optogenetically activated the motor neurons (Methods) to induce large retinal shifts. We measured the concomitant retinal position with a camera as well as the Vm in visual neurons in response to a moving black bar (bar width: 9°, bar velocity: 21° s−1). d, An example trace for one fly showing the retinal positions (in the top trace, the left retina is shown in orange and the right retina is shown in blue) and the Vm, starting from a baseline of −72 mV (bottom). Grey and red rectangles respectively indicate trials without and with optogenetic activation. e, Left: the baseline-subtracted Vm for rightward bar motion in one fly. The thin red and black lines show trials with (n = 9 trials) and without (n = 7 trials) optogenetic activation and the thick lines indicate the means. The mean baseline Vm at the start was −72 mV for the black and −70 mV for the red trials. Right: as left, but for leftward bar motion (n = 9 trials with and without optogenetic activation). f, Averaged Vm across trials and for both bar directions. The distance between the red and black vertical lines indicates the shift in the pseudopupil induced by the optogenetic light. g, The same as in f, but for a population of six flies (the light lines are traces for individual flies, aligned to their Vm peaks in trials without optogenetic activation, and the dark lines are averages). In all flies, the baseline Vm (mean during 0.5 s at trial onset) was slightly higher during optogenetic activation than in trials without (mean Vm of −71.1 versus −72.5 mV with standard deviations of 5.5 mV). h, Left: the same as in g, but in which single-fly Vm traces were normalized to their peak value before averaging. Right: normalized Vm data shifted by the measured angular retinal shift (indicated by vertical lines). a.u., arbitrary units.
Using a vertical bar that swept left or right in front of the fly, we measured the horizontal (yaw) position of LC14 receptive fields in the context of different retinal positions (Fig. 2c). To achieve two stable retinal positions, we either bilaterally activated retinal motor neurons optogenetically, or left them unactivated, as the bar swept. The example trace in Fig. 2d shows consistent, depolarizing membrane voltage (Vm) responses to a bar moving across the receptive field, independently of whether the retina was optogenetically repositioned or not. Importantly, however, the trial-averaged Vm traces revealed that this LC14 cell responded to the bar at slightly different positions on the screen during optogenetic activation compared to control trials (Fig. 2e, red versus black). As LC14 cells are insensitive to the direction of visual motion (Extended Data Fig. 2), we could combine trials in which the bar moved to the left and those in which it moved to the right to yield the best possible estimate of the shift in the cell’s receptive field with optogenetic activation (Fig. 2f). All six LC14 cells that we recorded showed a shift in the receptive field in the expected direction. Moreover, when we slid the population-averaged Vm curves with and without optogenetic activation by the mean pseudopupil (that is, retinal image) displacement (5.7°), we observed a precise match of the Vm curves at the two retinal positions (Fig. 2g,h). The fact that retinal movements lead to precise shifts in the position of LC14 receptive fields, rather than degrading their visual responses more generally, indicates that effective neural superposition is preserved at varied retinal positions.
Visually induced retinal movements
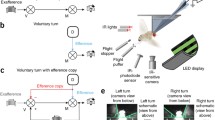
Many animals move their eyes to help stabilize gaze21. We wondered whether retinal movements might serve this function in flies. This would be a role akin to the optokinetic reflex in humans, in which our eyes smoothly move in the direction of a rotating panoramic scene, which acts to minimize visual motion on our retinas. We tethered flies to tungsten pins with their heads rigidly glued to their thorax and placed them at the centre of a panoramic LED display (Fig. 3a). We tracked the position of the pseudopupil of each eye independently. In some experiments, we had the flies perform tethered flight, in which case we simultaneously measured their wing-steering behaviour (Methods).

a, Apparatus to monitor the position of the deep pseudopupil in both eyes alongside the wing-steering behaviour of tethered, flying flies, while visual stimuli are presented on a panoramic LED display. Throughout, the left eye is represented by orange and the right eye is represented by blue. b, Sample traces of the positions of both pseudopupils of one fly as it viewed rightward- and leftward-moving gratings. Data from a quiescent fly are shown on the left and data from a flying fly are shown on the right. Responses in flight are more variable, with varying frequency of nystagmus saccades on a trial-to-trial basis (compare the left and right eyes in flight). c, The plots at the top show the population-averaged two-dimensional (x–y) movements of the pseudopupils for 10 flies in response to full-field rightward- and leftward-moving gratings. The traces below show the horizontal (x) component of the population-averaged pseudopupil movements over time (the grey region indicates when the stimulus is moving). The schematics at the top illustrate the visual stimulus presented. d, Data from the same flies as in c (8–10 flies), but during flight. Wing-steering responses are shown as the difference between the left and right wingbeat amplitudes (L–R WBA) at the bottom, with rightward deflections indicating a rightward steering response. e,f, The same as in c,d, but for unilateral rightward and leftward motion. Data from gratings presented to the fly’s right eye are shown; symmetrical results were obtained when the left eye was stimulated (not shown). g,h, The same as in c,d, but for bilateral back-to-front motion. i,j, The same as in c,d, but for bilateral front-to-back motion. k,l, The same as in c,d, but for up and down motion. Vertical (y) movements are shown in the pseudopupil time series for these panels. The thin lines show single-fly averages (3–10 trials). The thick lines show the population average (n = 8–10 flies).
When we rotated a panoramic square-wave grating around a quiescent, non-flying fly (Fig. 3b, left), we observed smooth, direction-selective, tracking movements of the retina in both eyes. Specifically, when the grating moved to the right, the retina moved smoothly to the left and vice versa. In flight, the same fly showed smooth tracking as well but with interspersed resetting movements in the other direction (that is, counter-saccades; Fig. 3b, right), akin to nystagmus saccades in human optokinetic responses. Responses in flight were more variable, with varying frequency of nystagmus saccades on a trial-to-trial basis (compare the left and right eyes in flight). This fly’s retinal movements were characteristic of the average movements in our population of flies (Fig. 3c,d and Supplementary Videos 5 and 6). Counter-saccades occur at unpredictable times and thus are not easily evident in the population-averaged responses (Extended Data Fig. 3). We note that in flight and in quiescence, we observed a tight correlation between the peak velocity of saccades and their amplitudes (Extended Data Fig. 4a,b), consistent with a power-law relationship, reminiscent of the ‘main sequence’ in human saccades22.
The direction in which the fly retina moves in response to visual motion is opposite to the direction in which the lens of our own eye moves in response to the same visual stimulus, but both movements have the same slowing effect on visual motion. The key point is that when the human eye rotates, both the lens and retina move together, whereas in Drosophila only the retina moves, with the overlying lenses remaining stationary. Because the lenses of the fly (and human) eye form inverted images, the fly retina, moving alone, has to move in a direction opposite to the direction of visual motion to slow down the movement of the image.
Unlike in humans, the optokinetic reflex in Drosophila can operate independently in each eye. When we presented a grating to the right eye alone, the right retina tracked whereas the left retina remained stationary (Fig. 3e,f and Supplementary Video 7) and vice versa (data not shown). Similarly, when we presented back-to-front motion to both eyes, this elicited front-to-back movements of both retinas (that is, divergence; Fig. 3g,h), and when we presented front-to-back visual motion to both eyes, both retinas moved back-to-front (convergence; Fig. 3i,j).
The fact that Drosophila have two retinal muscles per eye (Fig. 1) suggests that they can move their retinas in two dimensions. Indeed, flies also performed a vertical optokinetic response to up and down visual motion (Fig. 3k,l) with peak-to-peak magnitudes that were about 50% the size of horizontal movements.
The mean initial retinal speed in response to gratings moving horizontally at 15° s−1 was about 3° s−1 (Extended Data Fig. 5) and thus too slow to fully cancel the visual motion experienced by the retina. This low gain is consistent with the values observed in mice23 and goldfish24, but not with those observed in primates, for which optokinetic gains can approach 1 (refs. 25,26). It is unlikely that this incomplete cancellation of visual motion is due to muscular constraints, because the retina can move an order of magnitude faster during counter-saccades, with peak velocities exceeding 140° s−1 (Extended Data Fig. 4), and we also observed spontaneous saccades (described later) that exceeded 600° s−1. In unrestrained animals, retinal movements probably work in synergy with head and body movements27,28,29,30,31,32,33 to stabilize the visual image, and thus they may not need to operate with a very high gain.
Recently it was argued that photocontraction34—photoreceptor movements resulting from the subcellular mechanics of rhabdomeric phototransduction—can induce Drosophila photoreceptors to physically move in vivo10. If the retinal movements we observed were due to photocontraction, they should persist in flies with intact photoreceptors but impaired downstream visual processing. We silenced the synaptic output of visual neurons of the lamina (L1–L4)35,36, which are monosynaptically downstream of photoreceptors, by expressing active tetanus toxin light chain in those cells37. Optokinetic responses to visual motion stimuli were entirely abolished after this manipulation (Extended Data Fig. 6), whereas control flies expressing an inactive form of the toxin showed robust retinal movements. These data alongside the bidirectionality of the optokinetic response (which requires the post-photoreceptor calculation of the direction of visual motion), and other results (Extended Data Fig. 7 and Supplementary Discussion), indicate that the retinal movements we describe are not due to photocontraction.
Spontaneous retinal movements
Many animals move their eyes not only in response to external visual motion, but also seemingly spontaneously. We found that this is true in Drosophila as well (for example, Supplementary Video 8). Tracking the deep pseudopupil of a tethered, flying fly in the context of a stationary panoramic grating, we observed that the fly would keep its retina at a stable position for some time, move it rapidly to a new stable position, hold it there, then rapidly move it again, and so on (Fig. 4a, right). The sample fly in Fig. 4a (right) performed fast retinal movements (Methods), or saccades, with amplitudes typically below 1°. Two tethered, flying flies performed either similar or larger saccades in the context of the same panoramic stimulus (Fig. 4b). In darkness, the pseudopupil of the sample fly in Fig. 4a had an x–y position that drifted extensively, and the fly also exhibited larger saccades of up to about 5° (Fig. 4a, left). These data suggest that flying flies need structured visual input to keep their retinas stable.

a, Spontaneous retinal movements in one flying fly viewing a dark screen (left) or a stationary vertical grating (right). Throughout, the left eye is represented by orange and the right eye is represented by blue. Top: x–y plot of pseudopupil movements. Bottom: horizontal (x) component of the pseudopupil movement over time. b, x–y plots of both retinas in two more flies viewing a stationary grating (20 s). c, We isolated moments in which the right eye generated large saccades (left: 160 saccades; right: 147 saccades, thin lines) in the x dimension and averaged the x movements of the right and left eyes around those times for our population of 10 flies (thick black lines), in the context of either a dark screen (left) or a stationary, vertical grating (right).
We quantified the saccades that flying flies performed in the context of a stationary grating and in darkness by detecting large saccades in one eye and plotting these alongside the concomitant retinal movement in the other eye (Fig. 4c). We observed significantly larger saccade amplitudes in flying flies in the context of a dark screen than with vertical gratings (mean ± standard deviation of 2.6° ± 1.6° versus 0.9° ± 0.4°; Fig. 4c), and saccades were also more likely to be synchronized across the two retinas in darkness.
Small saccades activate visual neurons
Spontaneous retinal saccades often have amplitudes below 1°. To examine whether such small movements of the retina have any impact on visual processing, we recorded whole-cell patch-clamp data from the horizontal and vertical system cells: motion-sensitive interneurons in the fly’s lobula plate38,39. We observed direction-selective depolarizations and hyperpolarizations in response to tiny retinal movements (Extended Data Fig. 8), demonstrating that even the smallest retinal saccades are registered by the visual system (see Supplementary Discussion). Voltage responses to saccades were often small, and it is possible that an efference copy is sent to these and other cells to minimize visual responses to retinal saccades (Extended Data Fig. 8c); however, in some flies we observed visual responses with magnitudes approaching the cell’s full dynamic range as estimated by responses to drifting gratings (Extended Data Fig. 8b), arguing that microsaccades produce potent sensory drive in the visual system.
Retinal movements in gap crossing
Having shown that retinal movements affect the physiology of visual neurons both by shifting receptive fields and by activating motion-sensitive neurons, we investigated how retinal movements might affect fly behaviour (beyond improving image stability).
Past work has argued that flies visually assess the length of a gap in deciding whether and how to cross it40,41. Specifically, if a gap is deemed crossable, Drosophila perform a set of leg-reaching movements to contact the other side, ultimately pulling themselves over; if a gap is uncrossable, flies are less likely to attempt to cross the gap at all40. Motion parallax has been suggested as one depth-estimating mechanism that flies might use in assessing depth during this task40. We wondered whether Drosophila might perform active retinal movements during gap crossing. We were particularly curious whether flies performed active vergence movements as they crossed the gap. As flies have a binocular overlap zone of about ~15° in the frontal visual field, such movements, in principle, could help flies to assess depth through either binocular triangulation or binocular ruler mechanisms2,42 (see Supplementary Discussion). Retinal movements during gap crossing could serve other roles as well.
As we cannot yet measure retinal movements in freely moving flies, we developed a head-fixed gap-crossing paradigm for Drosophila. Pin-tethered flies walked on a wheel that rotated along one axis43. The wheel included two, 2.5-mm-wide gaps, 180° apart (Fig. 5a). One gap had horizontal stripes on the walls and the other had vertical stripes (Methods); data associated with the two gaps have been combined because we noticed no consistent difference in the flies’ behaviour across them. We tracked the position of the wheel (Methods) as the flies walked with lights on for 15 min, in complete darkness for 15 min, and with the lights on again for 15 min. On average, flies crossed a gap in the forward direction every 51 s with the lights on and every 67 s in darkness, with considerable variability across individuals; that flies crossed gaps at a lower rate in darkness—alongside control experiments that showed no measurable optokinetic responses to a physically moving grating in darkness—indicates that the flies could not see during the period that the lights were off (Extended Data Fig. 9). Whereas freely walking flies are unlikely to cross a gap that they cannot see, pin-tethered flies do so extensively, perhaps because their only navigational alternative, being rigidly tethered, is to walk backwards.

a, Tethered flies walked on a wheel with two 2.5-mm gaps that had horizontal or vertical stripes on the walls. We tracked wheel and retinal positions. b, Retina (top) and wheel (bottom) positions in an example fly with particularly large vergence movements. The dashed horizontal lines show the threshold for crossing detection; the vertical lines indicate crossing in the forward direction (Methods). c, Gap-crossing-triggered averages for a single fly revealed consistent convergent retinal movements. Top: the sign-inverted product of the shifts of the left and right eyes as the vergence metric. d, The same as in c, but for a population of 23 flies. e, Quantification of the time traces in d. The wheel and retina position values were calculated as the difference of the means in 3-s windows starting 5 s before and 2 s after a cross; the vergence metric value was calculated as the difference between the means in 1-s windows starting 2.5 s before and 0.5 s before a cross. Each data point represents a single fly and the mean ± s.e.m is shown. These distributions are significantly different from zero (two-sided t-test, P values ranging from 1.5 × 10−7 to 0.045, after Bonferroni correction for nine tests), except the vergence measure in darkness (P = 0.075). f, We expressed Kir2.1 in two split-GAL4 lines targeting retinal motor neurons. The schematic (top) shows the retinal motor neurons, and the micrographs (bottom) show immunostaining of split-GAL4 lines driving CsChrimson–tdTomato (examples chosen to be high intensity bilaterally, see Extended Data Fig. 10). Scale bars, 100 μm. g, Image of a fly crossing a gap. h, x–y trajectories of crossings in two example flies. i, For each gap-crossing event, we measured the lowest y value during a crossing event (Methods). The data points show the median of these y values across crossing events for each fly (left to right, n = 36, 36, 37 and 34 flies). The black bars show the mean ± s.e.m. across populations of flies for two split-GAL4 lines. Control and experimental distributions differed significantly as indicated by the asterisks (two-sided Welch test, P = 0.005 for R44A07-AD;R13D09-DBD and P = 0.008 for R14B04-AD;R13D09-DBD). j, Optokinetic responses of both eyes in control (black; n = 6 flies for each genotype) and silenced (red; n = 6 flies for R44A07-AD;R13D09-DBD and n = 7 flies for R14B04-AD;R13D09-DBD) flies for both lines.
An example fly with particularly large retinal movements consistently exhibited convergent pseudopupil movements (that is, divergent movements of the optical axis across the two eyes) when crossing gaps (Fig. 5b, vertical grey lines, and Supplementary Videos 9–11). This observation held when we averaged retinal movements across all gap-crossing events for this fly (Fig. 5c) and for a population of 23 flies (Fig. 5d,e). We observed similar vergence movements with the lights on and in darkness, albeit with more variability in darkness. That tethered flies made vergence retinal movements when crossing gaps in darkness indicates that these movements reflect an active vision strategy rather than sensory responses to visual features of the gap. Although moving the retina to improve visual perception is futile in darkness, it is still attempted, probably as a reflex, much like a human would probably move their eyes in reflexive ways if forced to cross an obstacle in darkness.
If retinal movements contribute to gap crossing, then impairing their dynamics might be expected to alter gap-crossing trajectories. Using two different split-GAL4 lines, we muted electrical signalling in retinal motor neurons by expressing in them a modified-mouse Kir2.1 ion channel; genetic-background-matched controls expressed a mutated, non-conducting form of this channel43. We tracked freely walking flies crossing 3.5-mm-long gaps. Gaps of this length are challenging but crossable in free behaviour40 (Fig. 5g,h). Control flies predominantly crossed the gap near the top, whereas experimental flies were more likely to walk down the near wall before crossing, yielding a statistically lower mean crossing height (y) for both experimental genotypes (y = −1.4 ± 0.1 mm, mean ± s.e.m., in silenced flies compared to −1.0 ± 0.1 mm in control flies in one line, and y = −1.5 ± 0.2 mm versus y = −1.0 ± 0.1 mm in the second line; P < 0.01 for both split-GAL4 lines, Welch test; Fig. 5i). A quantitative phenotype—that is, 40–50% lower y values—is consistent with the fact that expression of Kir2.1 yielded only a partial, about 33–35%, impairment to the magnitude of optokinetic retinal movements in both genotypes (Fig. 5j). Improved split-GAL4 lines (should they be possible to generate) that target the set of motor neurons more comprehensively and with higher transgene expression levels should allow one to test the effect of more complete silencing of retinal movements on behaviour in the future. Regardless, these data demonstrate that normal physiology of the retinal motor system is needed for flies to cross challenging gaps in a canonical fashion.
Discussion
Retinal optokinetic responses (Fig. 3) and spontaneous saccades (Fig. 4) in Drosophila conspicuously resemble human eye movements made in similar contexts. That the dynamics of an insect retina, actuated by two muscles, shows similarities to those of the vertebrate eye, actuated by six muscles of different origin, reveals a remarkable example of homoplasy in animal vision. Drosophila compound eyes have no equivalent of an area centralis or fovea, and many primate saccades are thought to be related to the act of foveation. Instead of foveation, spontaneous retinal saccades in flies could help to refresh the visual image in the face of receptor adaptation, and we speculate that they may also improve the ability of flies to perceive fine spatial features by dynamically realigning the photoreceptors in relation to those features. Retinal movements, in principle, could also contribute to other functions in Drosophila such as spatial attention and visual object recognition, alongside providing a potential proxy measure for whether flies are awake, asleep or experiencing varying levels of arousal.
We found that the flies reliably perform vergence retinal movements when crossing gaps (Fig. 5). One function these movements might serve is to estimate the length of the gap through a binocular ruler mechanism42 (see Supplementary Discussion). Notably, LC14 neurons (Fig. 2) seem particularly well suited for implementing such a function11,19,20,44 because: they have front-facing receptive fields in the binocular overlap region; they are particularly sensitive to visual features such as edges or small objects; and they have axons that interconnect the two visual lobes, providing an anatomical substrate for a binocular comparison. The advanced experimental toolkit in Drosophila should allow us to test this idea, and many related ideas on the role of retinal movements, in the coming years. More generally, the discovery of retinal movements in Drosophila—an important genetic model organism—should allow experimenters to develop a deep understanding of how active vision works in one species10,11,12,45,46,47,48,49,50, serving as an example for many others in the future.
Methods
Immunohistochemistry and imaging of retinal muscles and motor neurons
To visualize retinal muscles alongside motor neurons in intact, or semi-intact, fly heads (Fig. 1a,b), we expressed mVenus in a split-GAL4 line51 labelling retinal motor neurons (w+;Mi{Trojan-GAL4DBD.2}VGlut[MI04979-TG4DBD.2]/VT012411-p65ADZp;CsChrimson::mVenus/CsChrimson::mVenus) and performed the following protocol. Flies were anaesthetized with CO2 and briefly washed with 70% ethanol. Heads were isolated. Antennae, maxillary palps and the labrum were removed under 2% paraformaldehyde in phosphate-buffered saline (PBS) with 0.1% Triton X-100. Heads were fixed in this solution overnight at 4 °C. After washing in PBS containing 1% Triton X-100, the samples were embedded in 7% agarose and sectioned on a Leica vibratome (VT1000s) either horizontally or coronally, at 0.2 mm. The slices or whole heads were incubated in PBS with 1% Triton X-100, 0.5% dimethylsulfoxide and aescin (0.05 mg ml−1, Sigma-Aldrich, E1378) containing Texas Red-X phalloidin (1:50, Life Technologies no. T7471), anti-GFP rabbit polyclonal antibodies (1:1,000, Thermo Fisher, no. A10262) and a chitin-binding dye Calcofluor white (0.1 mg ml−1, Sigma-Aldrich no. F3543-1G) at room temperature with agitation for 2 days (slices) or 5 days (whole mounts). Long incubation times and the presence of surfactants ensured better penetration of phalloidin into the muscles. After a series of four approximately 1-h-long washes in PBS containing surfactants, the sections were incubated for another 24 h in the above buffer containing secondary antibodies (1:1,000, goat anti-rabbit, Thermo Fisher no. A32731). The samples were then washed in PBS with 1% Triton and fixed for 4 h in 2% paraformaldehyde to reduce leaching of bound phalloidin from muscles during the subsequent ethanol dehydration step. To avoid artefacts caused by osmotic shrinkage of soft tissue, samples were gradually dehydrated in glycerol (2 to 80%) and then ethanol (20 to 100%)52 and mounted in methyl salicylate for imaging.
Serial optical sections were obtained at 1-µm intervals on a Zeiss 710 confocal microscope with a LD-LCI 25×, 0.8 NA objective. Calcofluor white, anti-rabbit Alexa Fluor 488 antibodies and Texas Red phalloidin-treated samples were imaged using a 405-, 488- and 594-nm laser, respectively. As the blue–green emission of Calcofluor bleeds into the green channel, we applied a sequential scanning approach in which we excited only one fluorophore at a time. Images were processed in Fiji (http://fiji.sc/), Icy (http://icy.bioimageanalysis.org/) and Photoshop (Adobe Systems).
To visualize the structures of the orbital ridge (Extended Data Fig. 2), we followed the following protocol. Flies were frozen at −20 °C and decapitated in PBS with 0.1% Triton X-100. Heads were embedded in 7% agarose and sectioned on a vibratome at 150 µm. The soft tissue was digested by overnight incubation in a mixture of hyaluronidase and trypsin at 0.5 mg ml−1 in PBS at 37 °C. Simultaneously, cuticle was stained with the chitin-binding dyes Congo red (0.25 mg ml−1, Sigma-Aldrich no. C6767) and Calcofluor white (0.1 mg ml−1, Sigma-Aldrich no. F3543). The sections were washed, dehydrated and embedded in methyl salicylate. Cuticle treated with Congo red and Calcofluor white was imaged using 405- and 560-nm laser lines, respectively.
Immunohistochemistry and imaging of the L1–L4 and motor neuron split-GAL4 lines in the brain
We dissected the brains out of female flies to visualize neuronal GFP expression in a split-GAL4 line (R34G07-p65.AD;R9B08-GAL4.DBD) that targets L1–L4 neurons35,36 (Extended Data Fig. 6). Brains were dissected, fixed in 1% paraformaldehyde (PFA; Electron Microscopy Sciences no. 15713) in S2 medium (Sigma-Aldrich no. S0146) and nutated overnight at 4 °C. We next blocked specimens in 5% normal goat serum (NGS) in PBT (TritonX-100 in PBS) for 1.5 h and incubated at least overnight in a primary antibody solution containing 1:30 mouse nc82 (anti-Brp; Developmental Studies Hybridoma Bank no. nc82-s) and 1:1,000 chicken anti-GFP (Rockland no. 600-901-215). Finally, samples were incubated in a secondary antibody solution containing 1:800 goat anti-chicken Alexa Fluor 488 (Life Technologies (Invitrogen) no. A11039) and 1:400 goat anti-mouse Alexa Fluor 594 (Life Technologies (Invitrogen) no. A11032), for 3–4 days. Samples were washed in PBT three times after each step. We mounted the brains on slides with the antennal lobe down in the case of L1–L4 and with the antennal lobe up for visualizing the motor neuron. We immersed the brains in VectaShield (Vector Labs), added a coverslip, and took z stacks with a confocal microscope (Zeiss LSM780) and a Plan-Apochromat 20×, 0.8 NA objective. Image processing and analysis were performed using Fiji software53.
To visualize the retinal motor neurons in the two split-GAL4 lines shown in Fig. 5 and Extended Data Fig. 10a,b, we used particularly long incubation times and antibody concentrations, as follows. Brains were dissected in chilled PBS and placed in 4% paraformaldehyde (by mass) mixed in 0.5% PBT for 20 min at room temperature. Samples were washed three times in PBT for 20 min at room temperature, and then were placed in alternating vacuum for 3 min and ambient pressure for 1 min five times at room temperature. Primary antibodies in PBT (1:100 rabbit anti-DsRed (Living Colors), 1:7 mouse hybridoma nc82) were added to the samples, which were then incubated for 4 days at 4 °C. Samples were then washed three times at room temperature in PBT for 20 min before application of secondary in PBT (1:100 goat anti-rabbit Alexa Fluor 594, 1:100 goat anti-mouse Alexa Fluor 488) and incubated at 4 °C for 4 days. Samples were washed again in PBT, three times at room temperature for 20 min, before being pipetted onto glass slides between two glass coverslips adhered to the slide with nail polish, and nearly all PBT was removed through capillary action using a Kimwipe. A third coverslip was placed above the samples, supported by the other two coverslips (forming a bridge under which the samples sat), and adhered to the side coverslips using nail polish. FocusClear was slowly pipetted under the coverslip bridge through one of the two unsealed ends while the samples were watched to ensure they remained adhered to the glass slide. After the channel was completely filled, the remaining two ends of the top coverslip were sealed with nail polish and the slide was stored at 4 °C until imaging. Slides were imaged on a Zeiss LSM instrument, with the Alexa Fluor 594 excited using an SDSS 561-10 solid-state laser and the Alexa Fluor 488 excited using an argon lamp through a 25× immersion objective (Zeiss LCI Plan-Neofluar 25×, 0.8 NA) and Immersol G immersion oil with z-step sizes of 1–1.8 μm. No correction was used for the depth of image collection plane.
We checked expression in Kir2.1 flies used for behaviour (10XUAS-IVS-Kir2.1-T2A-tdTomato expressed in two split-GAL4 lines) using the following steps. One-week-old flies were anaesthetized on ice and soon thereafter placed in a glass staining cup with 80% ethanol. After 30 s, flies were placed in a staining cup with 1× PBS. Flies were dissected under the microscope in PBS and brains were stored in a PCR cap with 1:4 solution of 4% PFA (Sigma) and 0.1% PBT until final fixation. Brains were then fixed in 4% PFA and a drop of 0.1% PBT for 60 min at room temperature. After fixation, PFA solution was removed, and brains were washed with 0.1% PBT three times for 20 min at room temperature. The PBT was removed and replaced by 3% NGS in 0.1% PBT for 30 min at room temperature. The first antibody mix was incubated with Living Colors anti-DsRed polyclonal AB (rabbit; Clontech; 1:200), anti-bruchpilot (nc82; mouse; DSHB; 1:200) and 3% NGS in 0.1% PBT for 48 h at 4 °C in darkness. Brains were washed with 0.1% PBT for 5 s and then three times for 20 min at 4 °C in darkness. Brains were incubated in second antibody mix with goat anti-rabbit Alexa Fluor 568 (Invitrogen; 1:200), goat anti-mouse Alexa Fluor 633 (Invitrogen; 1:200) and 3% NGS in 0.1% PBT for 24 h at 4 °C in darkness. Brains were washed with 0.1% PBT for 5 s, followed by washing three times for 20 min at 4 °C in darkness. After washing with 0.1% PBT for 1 h at 4 °C in darkness, brains were mounted on a glass slide with VectaShield. Imaging was carried out at a Leica SP8 confocal microscope with a 20× microscope lens.
Fly genetics and handling
We studied female D. melanogaster (or Drosophila suzukii for Extended Data Fig. 7), reared in 25 °C incubators with a 12-h light–dark cycle. In electrophysiology experiments, we used flies that were 36–60 h post-eclosion; in ball-walking and flight experiments, we generally used flies 2–5 days post-eclosion. For the tethered and free-behaviour gap-crossing experiments in Fig. 5, we used 3–6-day-old flies. Flies were raised on standard cornmeal agar medium. Unless specified otherwise, we used wild-type Canton-S flies for flight and walking experiments. The investigators were not blind to the flies’ genotypes, but data collection and analysis were carried out in an automated fashion.
All flies used in any behavioural experiments carried at least one copy of the wild-type white gene. For silencing of L1–L4 visual neurons, we expressed the tetanus toxin light chain37 in R34G07-p65.AD;R9B08-GAL4.DBD and expressed an inactive form of tetanus toxin light chain as a control. For electrophysiological recordings and optogenetic activation (Fig. 2), we used flies with the following genotype: w+;R39H04-lexA/lexAopmyrGFP;R44A07-GAL4/20xUAS-CsChrimson::mVenus. Flies again were raised on retinal-poor food (standard cornmeal medium) and transferred to retinal enriched food after eclosion (0.4 mM all-trans retinal, CAS no. 116-31-4, added to our standard cornmeal food). Flies were kept in light-shielded boxes to minimize CsChrimson activation during development and as adults. For optogenetic activation in Extended Data Fig. 1, we drove 20xUAS-CsChrimson::mVenus in R44A07-AD;R13D09-DBD. To guide recordings in Extended Data Fig. 2, we drove mCD8GFP in VT037804-GAL4. In Extended Data Fig. 8, we targeted horizontal system (HS) and vertical system (VS) cells by driving eGFP in two GAL4 lines: w+;UAS-2xeGFP/+;GMR81G07-GAL4 and w+;UAS-2xeGFP/tshirt-GAL80;VT058487-GAL4/+.
To impair retinal movements (Fig. 5), we expressed a mouse Kir2.1 ion channel (w+/w−;10XUAS-IVS-Kir2.1-T2A-tdTomato, tubP-Gal80[ts]) in retinal motor neurons or a mutated Kir2.1 channel, which is non-conducting, as a control (w+/w−;10XUAS-IVS-Kir2.1Mut-T2A-tdTomato; tubP-GAL80[ts]). We used two split-GAL4 driver lines for retinal motor neurons, which we generated: R44A07-AD;R13D09-DBD and R14B04-AD;R13D09-DBD.
Flies were anaesthetized on a Peltier stage at ~4 °C and were glued to a custom stage49 for electrophysiological experiments (Fig. 2 and Extended Data Figs. 2 and 8) or when we needed to image photoreceptor movements with a water immersion objective (Fig. 1f). We used a blue-light-activated glue (Bondic). For behavioural flight experiments, we also anaesthetized flies on a stage at about 4 °C, and then we glued them to a tungsten pin. In all experiments (except the free behaviour in Fig. 5), the head was rigidly tethered to the thorax, or to the fly holder. For electrophysiology experiments, we also glued the proboscis in place.
Recording the movements of the deep pseudopupil and photoreceptor tips directly
We imaged the deep pseudopupil with several different air objective and camera combinations (see below). We used fview54 to capture videos of the deep pseudopupil for either offline tracking or in combination with custom-written real-time tracking software (see below).
For the simultaneous imaging of the deep pseudopupil using an air lens and of photoreceptors, directly, using a water immersion objective (Fig. 1f), we used two externally triggered Chameleon3 cameras (CM3-U3-13Y3M, FLIR) one fitted with a 4× Infinistix air lens (Infinity Photo-Optical) and the other camera recording the photoreceptors through a 40×, 0.8 NA (Olympus) water dipping microscope objective. The fly head was glued onto a custom-made holder designed for electrophysiological recordings in flight49. The fly head was tilted to make the dorsal part of the eye visible in the 40× water dipping micrograph while the ventral part of the eye was visible to the 4× air lens. The fly performed tethered flight during these simultaneous measurements, but we did not measure its steering behaviour.
For Fig. 1d, in which we measured photoreceptor movements triggered by optogenetic activation of motor neurons, we used a CoolSnap EZ camera (Photometrics) visualizing the photoreceptors through a 40×, 0.8 NA (Olympus) water dipping objective. Using a 90:10 beamsplitter (Thorlabs, BSX10R), we sent focused red light (660 nm, Thorlabs M660L4-C1) down on the fly head, to activate CsChrimson, through the epifluorescence port of the microscope, while visualizing the position of the photoreceptors in the camera. In this experiment, the fly was tethered to a metal pin and fully immersed in water, to allow us to rotate the fly under the microscope and obtain the highest possible quality images of the photoreceptors around the midline of the eye. When we performed our initial optogenetic activation of the muscles in the context of imaging the deep pseudopupil (Fig. 1e), we used a Chameleon3 (CM3-U3-13Y3M, FLIR) camera with a 6× Infinistix lens (Infinity Photo-Optical). We adjusted the red-light intensity in these experiments upward until the photoreceptor or deep pseudopupil excursions stopped growing in size.
To measure deep pseudopupil and flight steering responses to visual stimuli (Fig. 3), we used two Prosilica GE680 cameras (Allied Vision Technologies), one for each eye, each imaging with a 6× Infinistix lens (Infinity Photo-Optical). To illuminate the pseudopupil, we placed an 850-nm fibre optic abutting the fly’s head from behind. To get crisp images of the pseudopupil, it was crucial to move the light guide as close as possible to the fly head to maximize light that scattered in the head capsule and out of the eyes in comparison to stray light hitting the eyes from the outside. Synchronized videos from the two cameras were recorded at 100 Hz, and the movement of the deep pseudopupil was tracked offline using a custom-written algorithm (see below).
In electrophysiology experiments in Fig. 2, we recorded both retinas using a Chameleon3 cameras and InfiniStix lenses (3×, 44 mm, Infinity Photo-Optical) at 60 Hz. To activate the motor neurons expressing ChrimsonCS, we used an achromatic lens pair (Thorlabs MAP10100100-A) to focus a dot of red light between the eyes (660 nm LED, Thorlabs M660FPI, light guide: M15L01). We filtered this wavelength using bandpass filters in the cameras (725/50 nm, Edmund Optics no. 86-943). For the HS and VS recordings in Extended Data Fig. 8, we collected videos of the left eye’s deep pseudopupil at 200 Hz using a Prosilica GE680 camera and a 4× InfiniStix lens. We illuminated the deep pseudopupil in these experiments by shining 735-nm light (Thorlabs M735L3-C1) focused onto the head through the 40× water dipping objective.
In ball-walking experiments (Extended Data Fig. 7), we measured retinal movements and walking behaviour by placing flies on a small airborne ball (see below) and using two Chameleon3 (CM3-U3-13Y3M, FLIR) cameras with 6× Infinistix lenses (Infinity Photo-Optical) to visualize the deep pseudopupils of each eye. The light guide (200 µm, Thorlabs M87L01) used to illuminate the deep pseudopupil was placed slightly above and behind the head. It was connected to an 850-nm LED (Thorlabs M850F2).
In tethered gap-crossing experiments (Fig. 5 and Extended Data Fig. 9d), we visualized the movements of the deep pseudopupil with 6× Infinistix lenses (Infinity Photo-Optical), one per eye. In Extended Data Fig. 9a–c, we tracked only the pseudopupil of the right eye. We illuminated the head with an 850-nm LED (Thorlabs M850F2) coupled to a light guide (105 µm, Thorlabs M15L01) that delivered light to a matched achromatic pair of lenses (MAP10100100-A, Thorlabs), which focused the infrared light to a small spot on the head from slightly above and behind the fly.
Flight and walking behaviour on a ball or wheel
In experiments with tethered, flying flies (Fig. 3), we measured the right and left wing-stroke amplitudes using a wingbeat analyser (JFI Electronics Laboratory, University of Chicago, Chicago, IL)55 and took the difference between these two signals as the fly’s steering response. Specifically, flies were illuminated with an infrared diode (880 nm wavelength) from above, so that the shadows of the flapping wings can be recorded with two photodiodes placed below the flies. The signal from the photodiodes oscillates in synchrony with the wings flapping forward and backward, with the maximum (inverted) photodiode signal on each cycle indicating, quantitatively, the maximum forward wing-stroke angle achieved on that stroke, otherwise known as the WBA. The difference between the left and right WBA (L–R WBA) is correlated with yaw torque33 and thus provides a good measure of the direction and strength in which the fly is attempting to steer. We used a second LED at a slightly different wavelength (850 nm), to illuminate the fly head for visualizing the deep pseudopupil. We placed a bandpass filter (880 ± 5 nm, Edmund Optics, NT43–150) over the photodiodes that record the wing movements to prevent any impact of the 850-nm light on wing tracking. We recorded the L–R WBAs, camera triggers and voltages encoding the visual stimuli on our panoramic LED display at 1 kHz using a Digidata 1440a (Molecular Devices). At the beginning and end of each experiment, we paused the camera triggers and used these pauses to precisely align the pseudopupil estimates on each frame to the L–R WBA and voltage signals of the visual stimulus that were simultaneously recorded.
We developed a tethered gap-crossing paradigm (Fig. 5a–e and Extended Data Fig. 9). We 3D printed fly-sized wheels with a Projet MJP 3600 series 3D printer (30 µm resolution) using VisiJet M3 Crystal material. We press-fitted a precision shaft (Swissjewel, N-1D) through the middle of the wheel and placed that shaft between two spring-loaded jewel bearings (Swissjewel, VS-30), which allowed the wheel to rotate with low friction. The wheels had a 5-mm-wide walking surface and a radius of 4 mm. Each wheel weighed ~89 mg, with a rotational inertia around the central axis of 0.87 g mm−2. The wheel had two equally sized gaps, 180° apart. We spray painted the wheel black and we painted white, horizontal stripes on the walls of one gap and white, vertical stripes on the walls of the other gap. We 3D printed the stripes to be slightly protruding, which allowed us to paint accurate stripes with a fine-point Sharpie. The vertical stripes had a 1.4 mm wavelength (0.7 mm white, 0.7 mm dark) and the horizontal stripes had a 1 mm wavelength (0.5 mm white and 0.5 mm dark). Both of these periodicities corresponded to a grating with an optical wavelength of ~20° for a fly standing on the far edge of the gap. Our algorithm for analysing the gap-crossing wheel data is described later in the Methods.
In experiments with tethered flies walking on a spherical ball (Extended Data Figs. 7), we tracked the flies’ ball-walking behaviour as reported previously56. Specifically, the flies walked on a 6.35-mm (1/4-inch)-diameter ball carved by hand from Last-A-Foam FR-4618 (General Plastics) using a custom-machined steel hemispherical cutter57. The ball was placed at the top of an aluminium post with a 6.75-mm (17/64-inch)-diameter hemispherical cavity machined into the top58. Air was flowed into a 1 -mm hole at the bottom of the hemispherical cavity at the minimum rate to support the ball floating freely. The ball was marked with irregular black spots and tracked using FicTrac, which uses a map of the ball’s black spot pattern to calculate its 3D angular position on each frame59. The software we used to display visual stimuli (see below) and FicTrac ran on separate computers—one Windows machine and one Linux machine—and we therefore used network time protocol to synchronize the system clocks between the two computers to below 2 ms. In this way, the timestamps from the data files for the ball position and the visual stimulus could be accurately compared. The Linux computer was set up as the network time protocol server and the Windows computer was configured to set its system clock from this server over the local network.
Visual stimuli
For experiments in tethered, flying or quiescent flies (Figs. 3 and 4), we presented visual stimuli using a cylindrical green LED (570 nm) visual display that extended 360° in azimuth and 45° in elevation (IORodeo)60. We removed LEDs covering 30° of azimuthal space centred around 60° to the fly’s left and 60° to the fly’s right, which allowed us to bring two cameras close to the fly’s head, one for imaging the deep pseudopupil of the left eye and the other for imaging the deep pseudopupil of the right eye (Fig. 3a). Each LED pixel subtended 1.875° around the yaw axis. In one set of experiments (Fig. 3), we presented vertical gratings (15° wavelength) that moved with a temporal frequency of 1 Hz. The gratings were visible either panoramically or just in one hemisphere (with mean luminance on the other side). We also presented expansion stimuli (for which the gratings moved front-to-back on both sides) or contraction stimuli (for which the gratings moved back-to-front on both sides) as well as a horizontal grating that moved up or down. For all stimuli aside from the panoramic gratings, the frontal ±30° of the arena was kept at mean luminance (that is, without a moving stimulus) to ensure no motion stimulation of the contralateral eye. We used green-scale dithering with three (vertical grating) or two (horizontal grating) intensity levels for each LED pixel to allow for edges in visual patterns to appear to move more smoothly across frames. Trials were 5-s long, and each stimulus was presented with two motion directions (left–right or up–down), repeating all of the stimuli ten times for each fly. Retinal movements in response to grating motion were recorded in the same cohort of 10 flies in both non-flight and during tethered flight. For some conditions, only eight or nine flies had enough valid trials in tethered flight. Using the same experimental setup, we also presented stationary visual environments to quiescent and flying flies and recorded eye movements that occur spontaneously (that is, with no moving stimulus to elicit them; Fig. 4). Each stationary panorama—dark screen, bright screen, vertical grating, horizontal grating—was presented five times for 30 s each time. In electrophysiological experiments, we used a cylindrical green LED display that covered 216° in azimuth and 76° in elevation (IORodeo)60. For the experiments in Extended Data Fig. 8, we presented the same stationary stimuli just described for 15 s—dark screen, bright screen, vertical grating, horizontal grating—as well as horizontal and vertical grating motion (1 Hz temporal frequency, for 5 s) to assess the motion-direction preferences of the recorded cells.
In tethered, ball-walking flies (Extended Data Fig. 7), we presented visual stimuli with a projector that allowed us to accurately present gratings with a significantly smaller wavelength (as small as 1°) than those we could present on our LED display. Specifically, visual stimuli were displayed with a TI DLP LightCrafter 4500 projector, reflected off a flat mirror (https://firstsurfacemirror.com), onto a conical screen made out of white card stock supported by an acrylic frame. The screen covered 360° in azimuth and 90° in elevation, with about 20° of the screen blocked from view, directly behind the fly, by a camera apparatus used to track the ball. Visual stimuli were generated using ViRMEn, an open-source, Matlab-based virtual reality platform61. We created virtual striped cylinders in the ViRMEn virtual world and presented these to the fly. To measure the flies’ spatial acuity (Extended Data Fig. 7), we created virtual cylinders textured with a repeating sinusoidal grating of the desired spatial wavelength. The cylinder gently faded to grey at the top and bottom of the conical screen, along a Gaussian profile, to minimize the presence of sharp edges. We then rotated this virtual cylinder around the fly, and we measured the resulting retinal optokinetic responses and walking optomotor responses.
Electrophysiology
We recorded whole-cell current-clamp measurements from LC14 cells (that is, dorsal cluster neurons; Fig. 2 and Extended Data Fig. 2) and from HS and VS cells (Extended Data Fig. 8), using methods described previously27,62. We recorded from LC14 cells on the right side of the brain and tracked the retinas of both eyes during those experiments; we recorded from HS and VS cells on the left side of the brain and tracked the left retina alone during those experiments. Specifically, we affixed flies to a custom stage that allows for patch-clamp recording in tethered, flying flies49. We dissected the cuticle, muscles and trachea above the brain and perfused the tissue with extracellular solution (275–285 mOSM) that contained 103 mM NaCl, 3 mM KCl, 5 mM N-tris(hydroxymethyl) methyl-2-aminoethanesulfonic acid, 10 mM trehalose, 10 mM glucose, 2 mM sucrose, 26 mM NaHCO3, 1 mM NaH2PO4, 1.5 mM CaCl2 and 4 mM MgCl2. We bubbled this solution with 95% O2/5% CO2 to achieve a pH of 7.3. As described previously49, we used locally applied collagenase IV in extracellular solution (at a maximum concentration of 0.5 mg ml−1) and mechanical pressure to break through the neurilemma and perineurial glial sheath, providing access to the HS and VS cell bodies. Throughout data collection, the extracellular bath was held at 25–26 °C, slightly higher than room temperature, because this temperature seemed to promote an increased rate of retinal movements. Pipette resistances ranged from 4.5 to 8 MΩ. For recordings from LC14 cells in Fig. 2, we used forceps to remove the neurilemma and perineurial glia, and did not use collagenase IV. Pipettes were filled with intracellular solution that contained 140 mM K-aspartate, 1 mM KCl, 10 mM HEPES, 1 mM EGTA, 0.5 mM Na3GTP, 4 mM MgATP, 0.02 mM Alexa Fluor 568–hydrazide-Na and 13 mM biocytin hydrazide. The Vm was amplified (MultiClamp 700B, Molecular Devices), digitized at 10 kHz (Digidata 1440a, Molecular Devices) and saved to a computer (pClamp software suite, Molecular Devices). Voltage measurements were corrected for a 13-mV junction potential.
Tracking the deep pseudopupil
We tracked the deep pseudopupil movements either offline or, in later experiments, online (see below). We tracked the deep pseudopupil offline using a custom algorithm written in Python 2.7 using OpenCV (https://opencv.org). We defined a region of interest in the image that included all potential pseudopupil positions but excluded other bright cuticular features, mainly near the edge of the eye. We smoothed the image to minimize pixel noise, sometimes enhanced local contrast, binarized the image and subsequently calculated the contours of the resultant blob, saving the centroid position of this blob as the x–y position of the deep pseudopupil in pixel coordinates. The parameters for filtering, contrast enhancement and thresholding were adjusted for each eye and experiment because the exact nature of the illumination changed between flies. All parameters, however, were held constant for all trials for a given experiment.
For ball and wheel experiments, as well as Fig. 2, the deep pseudopupils were tracked in real time using a custom application written in C#, available at https://github.com/MaimonLab/EyeTrackerForm. In brief, images were captured from FLIR Chameleon3 cameras (CM3-U3-13Y3M-CS) using the FLIR Spinnaker API. A rectangular region of interest was selected for each eye at the beginning of each experiment. This region of interest was thresholded, smoothed using a Gaussian filter, and the largest contour not touching an edge was chosen as the deep pseudopupil. The coordinates of the centroid of this contour, along with the image timestamp from the camera, were recorded into a csv file, and this was carried out for each frame of the incoming video stream.
To convert how many pixels the pseudopupil moved on the camera image to an estimate of how many degrees of visual space the photoreceptors’ field of view moved, we performed the following transformation with Fiji (https://imagej.net/Fiji). Using a single image frame of each fly eye, we measured the distance in pixels between the midpoints of the outermost photoreceptors (with one photoreceptor between them). Assuming that D. melanogaster have an inter-ommatidial angle of about 5° (refs. 63,64), this distance should reflect approximately 10° of angular space. For the conversion of D. suzukii retinal movements from pixel units to degrees, we assumed an inter-ommatidial angle of 3.25° on the basis of the sign inversion of the responses at a wavelength of approximately 6.5° (Extended Data Fig. 7d). In ball- and wheel-walking experiments, we calibrated the pixel movements with an averaged pseudopupil size.
Analysing shifts in receptive fields due to retinal movements
In Fig. 2 we recorded from six LC14 neurons, while optogenetically activating retinal motor neurons. We tracked both retinas but analysed only the movement of the right retina as we were recording in the right visual hemisphere. The cameras viewed the deep pseudopupil from a rear, downward position (owing to spatial constraints), and the cameras were in this case not aligned to the visual stimulus. To estimate the horizontal shifts of the deep pseudopupil relative to the visual stimulus, we inclined the screen to the head of the fly to align so that the vertical bars on the screen move orthogonal to the horizontal ‘h-row’ of the ommatidial array (Figs. 4 and S3 in ref. 27). We then rotated the tracking vectors to obtain the deep pseudopupil movements along the h-row. Each fly was shown several repetitions of a vertical bar moving clockwise and anticlockwise, both with and without optogenetic lights on. Rarely, we observed unexplained depolarizations that recovered after several seconds, and we thus removed trials in which the Vm was more than 10 mV higher than the overall mean Vm (2 out of 170 total trials across 6 flies; note that both excluded trials were from 1 fly). We then calculated the average, baseline-subtracted membrane voltage for each fly across all trials for both motion directions with and without optogenetic activation. To compare these single-fly averages across the population, we shifted the curves to align the peaks of the receptive fields for no optogenetic activation. The peak was determined for low-pass-filtered data (Gaussian filter, s = 10 ms), but the unfiltered curves are plotted. This allowed us to calculate mean receptive fields across the population despite slightly varying peak response in yaw. We calculated the mean angular shift of the right deep pseudopupil for each fly while the stripe was in the ipsilateral half of the screen, including a 25° wedge into the contralateral hemifield; this region was certain to include any LC14 receptive field we recorded. Finally, we normalized the receptive fields of each fly to their peak (see above), and shifted the mean receptive fields by the mean angular separation of the deep pseudopupils in the two conditions. Data were analysed and plotted on a 1,000 Hz common time base.
Analysing optokinetic responses in flight and quiescence
In plots showing the population responses of flying or quiescent flies to visual stimuli (Figs. 3 and Extended Data Fig. 6), we calculated the mean for each fly over all valid stimuli presentations, with invalid trials being those in which the retina was poorly tracked, or, in flight conditions, when the fly stopped flying. Data were upsampled and plotted on a 1,000 Hz time base used for alignment to the visual stimulus. The right and left WBA signals were low-pass filtered using a Butterworth filter (order = 8; cutoff frequency = 10 Hz) applied bidirectionally to not introduce a time shift. All traces in Figs. 3 and Extended Data Fig. 6 were baseline corrected by subtracting the mean signal in a 1.5-s window before the onset of visual motion from the entire trace. We convolved the retinal position trace with a Gaussian filter (s = 4 ms) and then took its derivative. In this filtered velocity trace, we isolated retinal saccades by finding local minima and maxima whose magnitude exceeded two standard deviations from the mean velocity during that trial (Extended Data Fig. 3). In Extended Data Fig. 3, we triggered on saccades thus detected in the right retina and plotted concomitant movements of the left retina, as well as the difference between the left and right wingbeat amplitude (L–R WBA), when flies were flying. For the amplitude versus velocity plots (Extended Data Fig. 4), we estimated the amplitude of isolated saccades by subtracting the maximum and minimum retinal position observed in a 200-ms time window surrounding the peak velocity. We used the peak velocity in the same time window for the saccade’s velocity. We estimated the initial speed of retinal responses from the mean over all trials for each fly in a 100 ms time window starting 50 ms after the onset of visual motion (Extended Data Fig. 5) for three conditions (full-field rightward, rightward motion in the right hemisphere and bilateral front-to-back motion) in flight and quiescence.
For Extended Data Fig. 6, we calculated an optomotor index as a measure of how strongly flies respond to visual motion. This index was calculated by subtracting the mean response strength during visual motion in both motion directions from the mean signal in a 1.5-s window before the onset of visual motion, and then subtracting these two values from each other. To test whether the difference in the optomotor index of silenced and control flies was significant, we used a two-sided t-test for the means of two independent samples. The differences in the optomotor index for retinal movements between the two genotypes in non-flight and flight were significant with P < 0.0001 in both cases. The difference in the optomotor index of the wing movements was also significant (P < 0.00001).
Analysing retinal movements in stationary environments
In Fig. 4, to select saccades we found local maxima in the retinal velocity trace (filtered as above) that exceeded three standard deviations of the mean on that trial. We show smoothed saccades (Gaussian filter, s.d. = 4 ms) from 10 flies triggered on retinal movements in the right eye.
Analysing electrophysiological responses to spontaneous retinal movements
In Extended Data Fig. 8, to test whether HS and VS cells activate with eye movements, we simultaneously recorded their Vm alongside the fly’s retinal movements in the context of varying stationary scenes, presented for 15 s each (see above). At the end of each recording, we also measured the Vm response of the cell to moving gratings (1 Hz temporal frequency), presented for multiple 5-s presentations, in both the preferred direction (leftward for HS cells and downward for VS cells) and in the opposite, or null, direction (for example, Extended Data Fig. 8a). We recorded from seven VS cells, which respond best to vertical motion, and five HS cells, which respond best to horizontal motion. In each experiment, we presented a uniformly lit arena, darkness, a stationary vertical grating and a stationary horizontal grating (both at 15° wavelength). We placed a shroud over the rig, such that when the visual display was turned off (darkness), there should have been very little light reaching the fly’s eyes. For VS cells, we focused our analysis on vertical retinal movements, and for HS cells, we focused on horizontal movements (because movements along the other axes are expected to create visual motion sensed less effectively by the cells). We analysed the Vm and retinal movement data on a 1,000-Hz common time base. Retinal position traces were filtered using a Butterworth filter (order = 8, cutoff frequency = 100 Hz) applied bidirectionally. We selected up- and downward retinal movements for VS cells and right- and leftward retinal movements for HS cells, by finding local maxima or minima of the derivative of the smoothed (Gaussian, s = 50 ms) retinal position trace. We analysed those retinal movements (that is, probably saccades) in which the retinal position change (without filtering) exceeded 0.15° in a window from 200 ms before to 300 ms after the local minimum or maximum was found. We used this requirement rather than the minimum deviation from the mean used for the behavioural data, because the retinal movements varied largely from fly to fly during electrophysiological recordings, and we wished to compare retinal movements of a similar amplitude across individuals. We did not analyse the first 1.5 s after the onset of a stationary stimulus to avoid any stimulus-flash-driven saccades or flash Vm responses from the neuron. The number of saccades per fly varied depending on the quality of the preparation and was typically 10–50 total saccades in each direction per fly and visual condition, with the smallest number of saccades being nine for one fly in the grating condition. For each fly and stimulus condition, we calculated the average pseudopupil and Vm time course and then calculated the pooled time course of all cells (n = 12 cells) for darkness, a uniformly lit arena, and the stationary grating orientation with bars oriented orthogonally to the preferred-null axis of the cell (that is, vertical gratings for HS cells and horizontal gratings for VS cells). We tested whether the small Vm fluctuation observed in darkness was significant by comparing single-fly Vm averages in a 20-ms window around the onset of a saccade compared to the averages in a 50-ms window starting 100 ms before the saccade. The differences between those two values were significant for both eye movement directions (two-sided Wilcoxon signed rank test, P = 0.0096 for downward or leftward retinal movements and P = 0.005 for upward or rightward movements). To test for significance of the visual responses, we compared the averages in a 50-ms window around the peak response to the average in a 50-ms window at the onset of the saccade. The differences were significant for both retinal movement directions (two-sided Wilcoxon signed rank test, P = 0.01 for preferred movements, and P = 0.028 for null direction responses).
Analysing tethered gap-crossing data
To track the gap-crossing wheel’s angle over time, we added two, prominent, 3D-printed marks (one diamond and one line) to the mid-point of the wheel’s spokes, and we painted these markers white. We trained a DeepLabCut (version 2.2.0.6) model, with default parameters, on 160 frames in which we labelled the x–y position of these two markers, from 8 videos for a total of 1,030,000 iterations, yielding a test error of 8.64 pixels (image size 1,280 × 1,024 pixels). The mature neural network could take in images of the wheel and output the x–y position of the markers. We wrote an algorithm in Python that used the x–y positions of these two markers to estimate the wheel’s angle on each frame. On any given frame, one marker (the diamond or the line) could be occluded by the base that held the jewel bearings; however, at least one marker was always visible, making the positional tracking of the wheel reliable, as determined by visual inspection of many videos. We tracked the wheel at 150 Hz using a Chameleon3 (CM3-U3-13Y3M, FLIR) with a Computar Macro Zoom Lens (COMLM3XMP, B&H Photo).
We imaged, at 70–100 Hz, the left and right pseudopupils, each with a Chameleon3 (CM3-U3-13Y3M, FLIR) affixed with a 6× Infinistix lens (118600, Infinity Photo-Optical). After data acquisition, we created a common 100 Hz time base to which we aligned the pseudopupil and wheel data, using linear interpolation as needed. We isolated moments when the fly crossed a gap in the forward direction by defining two angular thresholds, one for each gap, and requiring the wheel to have rotated across the relevant threshold. To define a genuine gap-crossing event, we also required that the mean wheel position stay >0.1 rad (~5.7°) above this threshold in the 2 s after gap crossing, to avoid annotating unsuccessful lunges or reaches as crossing events (that is, those in which the fly quickly returns to the its pre-crossing location after crossing the threshold briefly).
We found all distributions of fly averaged retinal movements and the corresponding vergence metric (Fig. 5e) to be significantly different from zero (two-sided t-test, after Bonferroni correction for nine comparisons), except the vergence metric in darkness. We found the following P values: P = 0.015, 0.075 and 0.045 for the vergence metric in the first lights-on period, in darkness and the second lights-on period, respectively; P = 2.5 × 10−6, 0.002 and 2.4 × 10−5 for the right eye, and P = 1.5 × 10−7, 9.2 × 10−4 and 3.9 × 10−4 for the left eye in the same experimental conditions as above.
Analysing free-behaviour gap-crossing data
To test for the ability of flies to cross gaps in free behaviour (Fig. 5f–j), we used a 3D-printed ‘catwalk’ (40 mm long, 10 mm high and 5 mm wide) made of black LCD resin, which had a 3.5-mm-long and 7-mm-deep gap in the middle. We placed the catwalk on a circular black acrylic platform, which was 50 mm in diameter and surrounded by water (to prevent escape). The platform was also surrounded by a white cardboard cylinder to provide a uniform visual background. The arena was evenly illuminated by two overhead white LED lights (HETP, OO7). We cut a hole in the cardboard to allow us to film the catwalk from the side at 50 Hz (camera BFS-U3-16S2M-CS, Teledyne FLIR; lens Fujinon HF12XA-5M, FUJIFILM). At least 24 h before each trial, we cut the flies’ wings under CO2-induced anaesthesia. We also starved flies of food, but not water, for at least 4 h before a trial. We alternated running experimental and control flies in consecutive trials. In each trial, we placed a single fly on the circular platform, and after her first voluntary visit to the catwalk, the fly was given 10 min to freely explore. We used DeepLabCut (version 2.2.0.6) to track the position of the fly’s abdominal tip as she traversed the gap. Before video analysis, the DeepLabCut model was trained, with default parameters, on 700 labelled frames from 28 videos for a total of 350,000 iterations, yielding a test error of 2.54 pixels (image size 1,440 × 430 pixels). We used this model to analyse all experimental videos. We tested 36 or 37 female flies per genotype. To analyse the behaviour with code written in Python 3.6, we first determined when the tip of the abdomen of a fly crossed the midline of a gap. Next, we determined, in a 400-ms window starting 200 ms before the midline-crossing moment, the minimum position of the abdomen tip, as a proxy of the height at which the fly crossed the gap. We then calculated the median of these crossing heights of all crossing events for single flies and compared these distributions across genotypes using a Welch test.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data shown in the main figures are available at https://doi.org/10.6084/m9.figshare.c.6145572. All other data generated in this study are available from the corresponding authors upon request.
Code availability
Custom-written software used to track the fly retina in real time is available at https://github.com/MaimonLab/EyeTrackerForm. Additional code is available upon request from the corresponding authors.
References
Burtt, E. T. & Patterson, J. A. Internal muscle in the eye of an insect. Nature 228, 183–184 (1970).
Hengstenberg, R. Das Augenmuskelsystem der Stubenfliege Musca domestica. Kybernetik 9, 56–77 (1971).
Franceschini, N., Chagneux, R., Kirschfeld, K. & Muecke, A. in Göttingen Neurobiology Report (eds. Elsner, N. & Penzlin, H.) Vol. 1, 275 (Thieme, 1991).
Hengstenberg, R. in Information Processing in the Visual Systems of Arthropods (ed. Wehner, R.) 93–96 (Springer, 1972).
Patterson, J. The eye muscle of Calliphora vomitoria L: II Transient responses to changes in the intensity of illumination. J. Exp. Biol. 58, 585–598 (1973).
Northrop, R. B. in Introduction to Dynamic Modeling of Neuro-Sensory Systems (ed. Neumann, M. R.) 298–305 (CRC, 2000).
Patterson, J. The eye muscle of Calliphora vomitoria l. Spontaneous activity and the effect of light and dark adaptation. J. Exp. Biol. 58, 565–583 (1973).
Franceschini, N. & Chagneux, R. in Göttingen Neurobiology Report (eds. Elsner N. & Wässle, H.) Vol. 2, 279 (Thieme, 1997).
Franceschini, N., Chagneux, R. & Kirschfeld, K. in Göttingen Neurobiology Report (eds. Elsner, N. & Menzel, R.) Vol. 1, 402 (Thieme, 1995).
Juusola, M. et al. Microsaccadic sampling of moving image information provides Drosophila hyperacute vision. Elife 6, e26117 (2017).
Joni, K. et al. Binocular mirror–symmetric microsaccadic sampling enables Drosophila hyperacute 3D vision. Proc. Natl. Acad. Sci. USA 119, e2109717119 (2022).
Kemppainen, J., Mansour, N., Takalo, J. & Juusola, M. High-speed imaging of light-induced photoreceptor microsaccades in compound eyes. Commun. Biol. 5, 203 (2022).
Viollet, S. Vibrating makes for better seeing: from the fly’s micro-eye movements to hyperacute visual sensors. Front. Bioeng. Biotechnol. 2, 9 (2014).
Franceschini, N. & Kirschfeld, K. Etude optique in vivo des éléments photorécepteurs dans l’œil composé de Drosophila. Kybernetik 8, 1–13 (1971).
Klapoetke, N. C. et al. Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346 (2014).
Franceschini, N. & Kirschfeld, K. Les phénomènes de pseudopupille dans l’œil composé de Drosophila. Kybernetik 9, 159–182 (1971).
Salcedo, E. et al. Blue- and green-absorbing visual pigments of Drosophila: ectopic expression and physiological characterization of the R8 photoreceptor cell-specific Rh5 and Rh6 rhodopsins. J. Neurosci. 19, 10716–10726 (1999).
Kirschfeld, K. Die Projektion der optischen Umwelt auf das Raster der Rhabdomere im Komplexauge von Musca. Exp. Brain Res. 3, 248–270 (1967).
Otsuna, H. & Ito, K. Systematic analysis of the visual projection neurons of Drosophila melanogaster. I. Lobula-specific pathways. J. Comp. Neurol. 497, 928–958 (2006).
Hassan, B. A. et al. atonal regulates neurite arborization but does not act as a proneural gene in the Drosophila brain. Neuron 25, 549–561 (2000).
Land, M. F. & Nilsson, D.-E. Animal Eyes (Oxford Univ. Press, 2012).
Bahill, A. T., Clark, M. R. & Stark, L. The main sequence, a tool for studying human eye movements. Math. Biosci. 24, 191–204 (1975).
Iwashita, M., Kanai, R., Funabiki, K., Matsuda, K. & Hirano, T. Dynamic properties, interactions and adaptive modifications of vestibulo-ocular reflex and optokinetic response in mice. Neurosci. Res. 39, 299–311 (2001).
Schairer, J. O. & Bennett, M. V. L. Changes in gain of the vestibulo-ocular reflex induced by combined visual and vestibular stimulation in goldfish. Brain Res. 373, 164–176 (1986).
Cohen, B., Matsuo, V. & Raphan, T. Quantitative analysis of the velocity characteristics of optokinetic nystagmus and optokinetic after-nystagmus. J. Physiol. 270, 321–344 (1977).
Barnes, G. R. Visual-vestibular interaction in the control of head and eye movement: the role of visual feedback and predictive mechanisms. Prog. Neurobiol. 41, 435–472 (1993).
Kim, A. J., Fenk, L. M., Lyu, C., & Maimon, G. Quantitative predictions orchestrate visual signaling in Drosophila. Cell 168, 280–294 (2017).
Hateren, J. H. & Schilstra, C. Blowfly flight and optic flow. II Head movements during flight. J. Exp. Biol. 202, 1491–1500 (1999).
Cellini, B., Salem, W. & Mongeau, J.-M. Mechanisms of punctuated vision in fly flight. Curr. Biol. 31, 4009–4024 (2021).
Cellini, B. & Mongeau, J.-M. Active vision shapes and coordinates flight motor responses in flies. Proc. Natl. Acad. Sci. USA 117, 23085–23095 (2020).
Longden, K. D., Schützenberger, A., Hardcastle, B. J. & Krapp, H. G. Impact of walking speed and motion adaptation on optokinetic nystagmus-like head movements in the blowfly Calliphora. Sci. Rep. 12, 11540 (2022).
Collett, T. S. & Land, M. F. Visual control of flight behaviour in the hoverfly Syritta pipiens L. J. Comp. Physiol. 99, 1–66 (1975).
Tammero, L. F., Frye, M. A. & Dickinson, M. H. Spatial organization of visuomotor reflexes in Drosophila. J. Exp. Biol. 207, 113–122 (2004).
Hardie, R. C. & Franze, K. Photomechanical responses in Drosophila photoreceptors. Science 338, 260–264 (2012).
Tuthill, J. C., Nern, A., Holtz, S. L., Rubin, G. M. & Reiser, M. B. Contributions of the 12 neuron classes in the fly lamina to motion vision. Neuron 79, 128–140 (2013).
Strother, J. A., Nern, A. & Reiser, M. B. Direct observation of on and off pathways in the Drosophila visual system. Curr. Biol. 24, 976–983 (2014).
Sweeney, S. T., Broadie, K., Keane, J., Niemann, H. & O’Kane, C. J. Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron 14, 341–351 (1995).
Hausen, K. Motion sensitive interneurons in the optomotor system of the fly. II. The horizontal cells: receptive field organization and response characteristics. Biol. Cybern. 46, 67–79 (1982).
Schnell, B. et al. Processing of horizontal optic flow in three visual interneurons of the Drosophila brain. J. Neurophys. 103, 1646–1657 (2010).
Pick, S. & Strauss, R. Goal-driven behavioral adaptations in gap-climbing Drosophila. Curr. Biol. 15, 1473–1478 (2005).
Triphan, T. et al. A screen for constituents of motor control and decision making in Drosophila reveals visual distance-estimation neurons. Sci. Rep. 6, 27000 (2016).
Collett, T. S. & Harkness, L. I. K. in Analysis of Visual Behaviour (eds. Ingle, D. J. et al.) 111–176 (MIT Press, 1982).
Vijayan, V. et al. A rise-to-threshold signal for a relative value deliberation. Preprint at bioRxiv https://doi.org/10.1101/2021.09.23.461548 (2021).
Rosner, R., von Hadeln, J., Tarawneh, G. & Read, J. C. A. A neuronal correlate of insect stereopsis. Nat. Commun. 10, 2845 (2019).
Land, M. F. Motion and vision: why animals move their eyes. J. Comp. Physiol. A 185, 341–352 (1999).
Gibson, J. J. The Ecological Approach to Visual Perception 1st edn (Psychology Press, 2014).
Salem, W., Cellini, B., Frye, M. A. & Mongeau, J.-M. Fly eyes are not still: a motion illusion in Drosophila flight supports parallel visual processing. J. Exp. Biol. 223, jeb212316 (2020).
Cellini, B. & Mongeau, J. Active vision shapes and coordinates flight motor responses in flies. Proc. Natl Acad. Sci. USA 117, 23085–23095 (2020).
Maimon, G., Straw, A. D. & Dickinson, M. H. Active flight increases the gain of visual motion processing in Drosophila. Nat. Neurosci. 13, 393–399 (2010).
Chiappe, M. E., Seelig, J. D., Reiser, M. B. & Jayaraman, V. Walking modulates speed sensitivity in Drosophila motion vision. Curr. Biol. 20, 1470–1475 (2010).
Pfeiffer, B. D. et al. Refinement of tools for targeted gene expression in Drosophila. Genetics 186, 735–755 (2010).
Ott, S. R. Confocal microscopy in large insect brains: zinc–formaldehyde fixation improves synapsin immunostaining and preservation of morphology in whole-mounts. J. Neurosci. Methods 172, 220–230 (2008).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Straw, A. D. & Dickinson, M. H. Motmot, an open-source toolkit for realtime video acquisition and analysis. Source Code Biol. Med. 4, 5 (2009).
Götz, K. G. Course-control, metabolism and wing interference during ultralong tethered flight in Drosophila melanogaster. J. Exp. Biol. 128, 35–46 (1987).
Green, J. et al. A neural circuit architecture for angular integration in Drosophila. Nature 546, 101–106 (2017).
Seelig, J. D. et al. Two-photon calcium imaging from head-fixed Drosophila during optomotor walking behavior. Nat. Methods 7, 535–540 (2010).
Seelig, J. D. & Jayaraman, V. Neural dynamics for landmark orientation and angular path integration. Nature 521, 186–191 (2015).
Moore, R. J. D. et al. FicTrac: a visual method for tracking spherical motion and generating fictive animal paths. J. Neurosci. Methods 225, 106–119 (2014).
Reiser, M. B. & Dickinson, M. H. A modular display system for insect behavioral neuroscience. J. Neurosci. Methods 167, 127–139 (2008).
Aronov, D. & Tank, D. W. Engagement of neural circuits underlying 2D spatial navigation in a rodent virtual reality system. Neuron 84, 442–456 (2014).
Kim, A. J., Fitzgerald, J. K. & Maimon, G. Cellular evidence for efference copy in Drosophila visuomotor processing. Nat. Neurosci. 18, 1247–1255 (2015).
Stavenga, D. G. Angular and spectral sensitivity of fly photoreceptors. II. Dependence on facet lens F-number and rhabdomere type in Drosophila. J. Comp. Physiol. A 189, 189–202 (2003).
Götz, K. G. Die optischen Übertragungseigenschaften der Komplexaugen von Drosophila. Kybernetik 2, 215–221 (1965).
Stavenga, D. G. in Comparative Physiology and Evolution of Vision in Invertebrates. Handbook of Sensory Physiology (ed. Autrum, H.) 357–439 (Springer, 1979).
Linneweber, G. A. et al. A neurodevelopmental origin of behavioral individuality in the Drosophila visual system. Science 367, 1112–1119 (2020).
Land, M. F. Visual acuity in insects. Annu. Rev. Entomol. 42, 147–177 (1997).
Götz, K. G. Optomotorische Untersuchung des visuellen Systems einiger Augenmutanten der Fruchtfliege Drosophila. Kybernetik 2, 77–92 (1964).
Keesey, I. W. et al. Inverse resource allocation between vision and olfaction across the genus Drosophila. Nat. Commun. 10, 1162 (2019).
Acknowledgements
We thank members of the Maimon lab for their input and constructive discussions. We thank A. Kim for help generating Supplementary Videos 5–8, A. Adachi for the immunostaining data shown in Extended Data Fig. 6, S. Thornquist for immunostaining in Fig. 5 and Extended Data Fig. 10, and A. Friedrich for immunostaining in Extended Data Fig. 10. We thank K. Fonselius for help with the pseudopupil tracking software and B. Solat for help with the behavioural experiments in Fig. 5. We thank J. Beetz, V. Bitsikas, M. Madhav, Y. Tanaka, A. Cario, P. Quinlan, C. Rusch, C. Herber, M. Ananth and L. Anneser—students in the Neural Systems and Behavior course in Woods Hole—for initial, inspiring work characterizing retinal movements at both behavioural and electrophysiological levels. We thank A. Straw and K. Panser for the GFP immunostaining data in Extended Data Fig. 2 and J. Tuthill for suggesting the use of a specific split-GAL4 line to silence early visual neurons (Extended Data Fig. 6). We thank H. Lacin for sending us the split-GAL4 lines used in Fig. 1, which also allowed us to ultimately make a more selective split-GAL4 line for retinal motor neurons. We thank W. Korff for insights into the biomechanics of the orbital ridge. We thank S. Sethi for advocating for the head-fixed gap-crossing paradigm and A. Janke for performing optomotor flight experiments that did not make it into the final paper. Fly stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) and antibodies from the Developmental Studies Hybridoma Bank at the University of Iowa (National Institute of Child Health and Human Development) were used in this study. Research reported in this publication was supported by a grant from the National Institute of Neurological Disorders and Stroke (R01NS121904) to G.M., a Leon Levy postdoctoral fellowship to L.M.F., a Kavli postdoctoral fellowship to L.M.F., and the Max Planck Society. G.M. is a Howard Hughes Medical Institute Investigator.
Author information
Authors and Affiliations
Contributions
L.M.F. first conceived the project. L.M.F. and G.M. designed the experiments. I.S. collected the anatomical data in Fig. 1. L.M.F. collected all behavioural and electrophysiological data, unless otherwise indicated, and analysed these data. A.N. collected the electrophysiological data in Extended Data Fig. 8, with the guidance of L.M.F. and G.M.; L.M.F. analysed these data. S.C.A. collected preliminary data on retinal muscle anatomy and helped to create selective split-GAL4 fly lines for retinal motor neurons together with J.L.W. J.L.W. recorded and analysed optokinetic responses of silenced and control flies in Fig. 5. G.M. and L.M.F. collected the electrophysiological data in Fig. 2b and Extended Data Fig. 2; G.M. analysed these data. G.M. collected and analysed the tethered, gap-crossing data, with extensive help from T.L.M. in fabricating the wheel and T.L.M., S.C.A. and J.L.W. in data analysis. L.D.R. collected the free-behaviour gap-crossing data; L.M.F. and L.D.R. analysed those data. L.M.F. and G.M. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Retinal muscles that attach to the front of the orbital ridge can move the entire retinal sheet coherently.
(a) Schematic of the attachment of the retinal eye muscles to the orbital ridge in the Drosophila head. For an animated version see Supplementary Video 1. (b) Staining of a part of the Drosophila head including the eye and the orbital ridge in a cleared specimen using Calcofluor White. Soft tissue was removed proteolytically. It reveals the vesica piscis-shaped opening of the orbital ridge and shows strongly sclerotized parts in the frontal region, where the two muscles attach (red stars), as well as two discontinuities on the dorsal and ventral poles (arrows). One possibility is that these discontinuities decouple the front of the orbital ridge from the back, mechanically. This may allow muscles that pull the front of the retina to also move the rear by the same amount through internal cohesion within a stiff set of ommatidia, rather than through a force vector that dissipates over space from front to back. Alternatively, the inhomogeneities at the top and bottom of the orbital ridge could act as a fulcra or pivot points, leading to the rear part of the orbital ridge to move outward when the frontal part moves inward (towards the midline), which could aid coherent motion of the retina. We will test these models in biomechanical studies in the future. For an animated version of the Calcofluor White image stack see Supplementary Video 2. The same staining was also performed in a non-cleared specimen with equivalent results (data not shown). (c) We measured retinal movements using three cameras pointing at three different positions in one eye. To induce large movements, we optogenetically activated the retinal motoneurons. We expressed CsChrimson in a split-GAL4 line (w+;R44A07-AD;R13D09-DBD) and focused red light onto a spot between the fly eyes (Methods). The three plots to the right show the views of the three different cameras: we overlaid images before and after optogenetic activation and plotted the tracked centroid on top of the pseudopupil (white) to illustrate the retinal movements. (d) Optogenetic activation in this fly yielded movements roughly along the axis connecting photoreceptors 3–5, and we used this motion direction (dark arrow) for comparison across the different positions on the facet eye in (e). Schematic modified from Stavenga (1979)65 (e) Traces of the pseudopupil measured in front (blue), in the middle (orange) and in the back (green) of the fly eye. (f) Same traces as above but shifted in y (by hand) to illustrate the coherent motion of the pseudopupil across the eye.
Extended Data Fig. 2 Electrophysiological recordings from LC14 cells reveal that they have small, front facing receptive fields and that they respond strongly, in a non-direction-selective manner, to moving spots and bars.
(a) GFP expression in the VT37804-GAL4 line, characterized previously66, shows LC14 cells, which connect the lobula on one side to the contralateral lobula and medulla. Additional off-target expression in the anterior optic tubercle is also visible. (b) Example trace showing the LC14 membrane voltage (Vm) while it is modulated by a variety of visual stimuli (bars, spots and gratings). (c) Membrane potential responses from a single cell reveal strong, non-directional responses to moving spots and bars in a restricted portion of the visual field. Light gray: single trials. Black: mean response. (d) Single cell response to 100 ms spot flashes (gray region) on the LED screen. Light gray: single flashes. Black: mean response. (e) Heat map image representation of an LC14 cell’s receptive field (same cell as shown in d). The cell’s response for the heat map was determined by subtracting the mean, baseline Vm in the 100 ms before stimulus onset from the mean Vm in the 100 ms window starting 30 ms after stimulus onset. (f) Population-averaged responses to moving bars and gratings (six cells) reveal more consistent responses to bars than gratings. (g) Heat-map representations of four more LC14 cells’ receptive fields. Bottom left plot shows same data as Fig. 2b.
Extended Data Fig. 3 Optokinetic retinal tracking is interspersed with (nystagmus-like) counter-saccades and the largest counter-saccade magnitudes are observed in flight.
(a) We isolated large saccades of the right retina with a simple, threshold-crossing algorithm and plotted these alongside the concomitant retinal movements of the left retina in 10 quiescent (i.e., non-flying) flies. Data are shown in the context of full-field rightward grating motion (top) (87 saccades), rightward motion in the right visual hemisphere (middle) (56 saccades), and bilateral front-back-motion (bottom) (66 saccades). (b) Same as panel a but during tethered flight. We also show the left minus right wing beat amplitude (L–R WBA) of the flies, with rightward deflections indicating a rightward steering response and vice versa (top, middle and bottom traces include 122, 136 and 152 saccades respectively).
Extended Data Fig. 4 Retinal saccade magnitudes and peak velocities are tightly correlated, akin to the main sequence in human saccades.
(a) Saccade peak velocity plotted against saccade amplitude of the saccades of the right retina from Extended Data Fig. 3a. Data are shown in the context of full-field rightward grating motion (top) (87 saccades), rightward motion in the right visual hemisphere (middle) (56 saccades), and bilateral front-back-motion (bottom) (66 saccades). (b) Same as panel a but for the saccades of the right retina from Extended Data Fig. 3b, which occurred during tethered flight (top, middle and bottom traces include 122, 136 and 152 saccades respectively). The mean saccade amplitudes for flight and quiescence were 2.6° and 1.1° for full-field rightward motion, 1.6° and 0.7° for rightward motion in the right hemisphere and 2.7° vs 0.6° deg for bilateral front-to-back motion. All saccade-magnitude differences between flight and quiescence were highly significant (two-sided Welch-test: p = 9·10−14, 7·10−16, 5·10−55 for top, middle, and bottom panels respectively).
Extended Data Fig. 5 The gain of optokinetic tracking is below unity and was significantly modulated by flight for bilateral front-to-back motion.
(a) The initial pseudopupil velocity for full-field 15°/s leftward grating motion when analyzing the left eye (orange) and for full-field 15°/s rightward grating motion when analyzing the right eye (blue). Data for optokinetic responses made during quiescence and tethered flight are shown separately. Each point represents data from one fly. (b) Initial pseudopupil velocities for unilateral motion stimuli: 15°/s grating motion in the left side for the left eye and right side for the right eye. (c) Initial pseudopupil velocities for bilateral front-back-motion. The initial pseudopupil velocities during flight and quiescence were not significantly different for rotational stimuli (a, b) but they were significantly bigger in flight for bilateral front-to-back motion, which simulates forward translation (two-sided Wilcoxon signed-rank test, left eye: p = 0.005, right eye: p = 0.007).
Extended Data Fig. 6 Silencing neurons in the early fly visual system abolishes optokinetic responses to visual motion but preserves spontaneous retinal movements in flying flies.
(a) Immuno-stain of the split-GAL4 line R34G07-AD;R9B08-DBD, characterized previously36, labelling L1–L4 visual neurons (green) and neuropil (anti-brp, magenta). (b) Example traces showing retinal movement in a control fly, expressing inactive tetanus toxin in the L1–L4 cells, and an experimental fly, expressing active tetanus toxin in the same neurons, in flight and non-flight. All behavioral responses were made in the context of a full-field grating rotating at 15°/s (1 Hz temporal frequency). (c) Single fly averaged (thin lines) and population averaged (thick line) retinal responses for 8 control flies and 10 experimental flies during non-flight/quiescence. Right panel: Optomotor index (Methods ) quantifying the response strength to visual motion in the expected optokinetic direction. (d) Same as panel c, but in flying flies. Black: Left minus right wingbeat amplitude (L–R WBA).
Extended Data Fig. 7 In both D. melanogaster and D. suzukii, the sign and magnitude of retinal optokinetic responses and walking optomotor responses are consistent with non-hyperacute sampling of the visual world by the fly retina.
(a) Experimental setup: pin-tethered flies walked on floating ball. We recorded the ball’s rotations as a readout of the flies’ turning velocity (black) alongside the displacements of the deep pseudopupil (left: orange, right: blue). Visual stimuli were presented on a conical screen using a projector and consisted of full-field rotating gratings at varying spatial wavelengths (always 4 Hz temporal frequency) for 5 s. (b) The inter-ommatidial angle Δϕ of the compound eye limits the spatial wavelength λ of a grating that can be properly resolved (modified from Land 199767). Below the cut-off wavelength of λL = 2·Δϕ, direction-selective motion responses of the visual system are predicted to invert due to spatial aliasing67,68. (c) Top and middle: displacements of the left and right pseudopupils during the stimulus period as a function of the grating wavelength, λ (1 to 40°) for left- and rightward motion. Bottom: concomitant average walking velocity during the stimulus period. Thin lines are the trial-averages from 11 single flies. Thick lines are population averages. Note the sign inversion in the range of λ = 5–10° for both the retinal optokinetic reflex and the walking optomotor response. (d) Average responses for both retinas for front-to-back (thicker line) and back-to-front motion for Drosophila melanogaster (top, data as in b) in comparison to Drosophila suzukii (bottom). D. suzukii showed inverted responses at smaller wavelengths than D. melanogaster, i.e. had a higher spatial acuity, consistent with the fact that D. suzukii have approximately twice the number of ommatidia as D. melanogaster69. The sign-inversion of behavioral responses around the critical wavelength inferred from the optics of the eye argues that any retinal movements flies are making in the context of moving gratings are not allowing them to perceive the motion direction of fine gratings better than would be expected from the first-order optics of the eye. The lack of evidence for hyperacuity in this context does not exclude it existing in other circumstances.
Extended Data Fig. 8 Visual neurons activate during spontaneous saccades with direction-selective responses.
(a) Left: sample Vm responses of an HS cell on the left side of the brain to rightward followed by leftward grating motion (1 Hz temporal frequency). (b) Vm of the same cell, alongside the x-movements of the left retina (orange), in the context a stationary vertical grating. Arrows indicate moments of spontaneous saccades. (c) Trial-averaged Vm of single flies (gray, N = 12 HS or VS cells) and population averaged Vm (black) for left-eye retinal saccades (orange). Data from a dark arena (left), a uniformly lit arena (middle), and a stationary grating (right) are shown. Top plots shown downward retinal movements for VS cells and leftward retinal movements for HS cells, which should produce visual motion in the preferred direction. Bottom plots show the opposite, null-direction retinal movements. The direction-selective responses to gratings argue that HS and VS cells respond to the visual motion induced on the retina by < 1° eye movements. The weak response to eye movements in darkness, or with a uniformly lit screen, is opposite in sign to that observed with a grating, which may represent an efference copy of the predicted motion signal arriving to HS/VS cells with each eye movement. This efference copy is potentially superseded by the actual, grating motion input with a high contrast grating, in the rightmost column.
Extended Data Fig. 9 Evidence that flies are genuinely in the dark during the lights-off epoch of the gap crossing experiments.
(a) Flies walking on the gap-crossing wheel (Fig. 5) were presented with a grating printed on paper that was physically moved back and forth in front of the right eye with a motorized manipulator. A small slit in the printed grating allowed us to slide an InfiniStix lens through it, abutting the fly’s right eye, to track the deep pseudopupil. (b) Example traces showing the horizontal shift of the right retina in two flies (blue) together with the grating position (black). (c) We observed a clear optokinetic response with the lights on, but not during darkness, demonstrating that there was genuinely no light available for flies to see with the lights off, even after being dark adapted for 30 min. Light blue: single flies. Dark blue: population mean. Five repetitions of the grating’s movement were presented and averaged for each fly, in each 5-min. epoch shown. (d) We quantified the number of gap crossings from the data presented in Fig. 5 and observed a ~30%, statistically significant, drop in the rate of gap crossing during the dark period (two-tailed paired t-test, p = 0.013 when comparing lights on #1 with darkness and p = 0.0056 when comparing lights on #2 with darkness).
Extended Data Fig. 10 Anatomical characterization of two the split GAL4 lines used for silencing retinal motor neurons.
We visualized expression in these two split-Gal4 lines by driving CsChrimson-tdTomato in R44A07-AD;R13D09-DBD (a) and R14B04-AD;R13D09-DBD (b). Maximum z-projections of the brain are shown over roughly the posterior and anterior halves to better visualize the branching. VNC maximum projections are shown over the full stack. The green cells on the right and left side of the SEZ (arrows) are retinal motor neurons, based on their dendritic arborization, location of cell body and their axons leaving the brain just below the antennal lobe. We could optogenetically induce retinal movements via expression of CsChrimson expression in both split lines. Expression levels were low and stochastic, particularly in line R44A07-AD;R13D09-DBD. We could clearly detect motoneurons in on both sides of the brain in 4/14 imaged brains and one motoneuron in 7/14 brains. For the lines shown in (b) we could detect motoneurons bilaterally in 16/16 brains. The stacks shown here and in in Fig. 5f are particularly clear examples chosen to best illustrate the location branching pattern if the neurons. (c) We checked for Kir2.1/Kir2.1-mutant expression in our experimental flies and could detect the motoneurons in both split GAL4 lines, again with low expression. We stained three brains for R44A07-AD;R13D09-DBD and detected the motoneurons bilaterally in 2/3 (one shown) and unilaterally in one brain (not shown). For line R14B04-AD;R13D09-DBD, we detected motoneurons bilaterally in 3/3 imaged brains (only one is shown).
Supplementary information
Supplementary Information
Supplementary Discussion and References.
Supplementary Video 1
Schematic of the attachment of the retinal eye muscles to the orbital ridge in the Drosophila head. Animated schematic of the retinal muscles in the fly head.
Supplementary Video 2
3D rendering of the orbital ridge. Cuticular stain of a part of the Drosophila head, including the eye and the orbital ridge, in a cleared specimen using Calcofluor white.
Supplementary Video 3
3D rendering of the retinal muscles. 3D rendering of a portion of the fly head, near one eye, showing immunohistochemical labelling of both retinal muscles using phalloidin (red) and chitin using Calcofluor white (blue).
Supplementary Video 4
Simultaneous tracking of photoreceptor tips using a water immersion objective and the deep pseudopupil using an air lens. Simultaneous imaging and tracking of the deep pseudopupil seen through an air objective (top left) and photoreceptor tips seen through water immersion (top right).
Supplementary Video 5
Retinal movements in response to panoramic visual motion in a pin-tethered, non-flying fly (4× speed). Top panel: visual stimulus. Top inner panels: right and left side of the fly head. Top outer panels: Zoom-in on the two deep pseudopupils. The tracked centroids of the right and left deep pseudopupils are marked with blue and orange dots, respectively. Lower traces show the horizontal movements of the left (orange) and right (blue) deep pseudopupil over time.
Supplementary Video 6
Retinal movements and a wing-steering signal in response to panoramic visual motion in a pin-tethered, flying fly (4× speed). Same as Supplementary Video 5, but for a flying fly. The lower trace shows the L–R WBA signal.
Supplementary Video 7
Retinal movements in response to unilateral visual motion in a pin-tethered, non-flying fly (4× speed). Same as Supplementary Video 5, but for visual motion presented to only one eye at a time.
Supplementary Video 8
Spontaneous retinal movements in a pin-tethered, flying fly in the context of varying, stationary visual scenes (2× speed). Same as Supplementary Video 6, but in the context of stationary visual stimuli (dark screen, uniformly lit screen, and vertical grating).
Supplementary Video 9
First example video showing retinal movements during tethered gap crossing (0.25× speed). Top left: tracked centroids of the right (blue) and left (orange) deep pseudopupil. Grey regions reflect a 2D histogram of pseudopupil positions for each eye (100 × 100 bins), showing all bins with more than 25 counts as grey. Below: video of the tethered fly walking on a 3D-printed wheel with two gaps (one with a horizontal grating painted on the wall and one with a vertical grating). Traces on the right show the x movement of the right (blue) and left (orange) pseudopupil and the angular position of the wheel (black) over time.
Supplementary Video 10
Second example video showing retinal movements during tethered gap crossing (0.25× speed). Same as Supplementary Video 9, but for a second example fly.
Supplementary Video 11
Third example video showing retinal movements during tethered gap crossing (0.25× speed). Same as Supplementary Video 9, but for a third example fly.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fenk, L.M., Avritzer, S.C., Weisman, J.L. et al. Muscles that move the retina augment compound eye vision in Drosophila. Nature 612, 116–122 (2022). https://doi.org/10.1038/s41586-022-05317-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-05317-5
- Springer Nature Limited