

Abstract
Dengue virus causes approximately 96 million symptomatic infections annually, manifesting as dengue fever or occasionally as severe dengue1,2. There are no antiviral agents available to prevent or treat dengue. Here, we describe a highly potent dengue virus inhibitor (JNJ-A07) that exerts nanomolar to picomolar activity against a panel of 21 clinical isolates that represent the natural genetic diversity of known genotypes and serotypes. The molecule has a high barrier to resistance and prevents the formation of the viral replication complex by blocking the interaction between two viral proteins (NS3 and NS4B), thus revealing a previously undescribed mechanism of antiviral action. JNJ-A07 has a favourable pharmacokinetic profile that results in outstanding efficacy against dengue virus infection in mouse infection models. Delaying start of treatment until peak viraemia results in a rapid and significant reduction in viral load. An analogue is currently in further development.
Similar content being viewed by others

Main
Dengue is currently considered one of the top ten global health threats1. Annually, an estimated 96 million individuals develop dengue disease2, which is probably an underestimation3,4,5. The incidence has increased approximately 30-fold over the past 50 years. The virus is endemic in 128 countries in (sub-)tropical regions, with an estimated 3.9 billion people at risk of infection. A recent study6 predicts an increase to 6.1 billion people at risk by 2080. The upsurge is driven by factors such as rapid urbanization and the sustained spread of the mosquito vectors6,7,8. Dengue virus (DENV) has four serotypes (further classified into genotypes), which are increasingly co-circulating in endemic regions. A second infection with a different serotype increases the risk of severe dengue9,10. The vaccine Dengvaxia, which is approved in a number of countries for individuals aged ≥9 years, is only recommended for those with previous dengue exposure11,12,13. There are no antiviral agent s for the prevention or treatment of dengue, and the development of pan-serotype DENV inhibitors has proven challenging14,15.
JNJ-A07 is a highly potent DENV inhibitor
Following a large-scale cell-based anti-DENV-2 screen16, a hit was identified and optimized (a total of approximately 2,000 analogues were synthesized). JNJ-A07 is a representative analogue (Fig. 1a) with nanomolar to picomolar antiviral potency in various cell lines and high selectivity (Table 1, Extended Data Fig. 1). JNJ-A07 is also active in primary immature dendritic cells, which may be the initial target cells of the virus17. Potent pan-genotype and pan-serotype activities (nanomolar to picomolar potencies) were demonstrated against a panel of 21 clinical isolates covering all available genotypes within the 4 serotypes18 (Table 1). No marked antiviral activity was detected against other flaviviruses or against a selection of other RNA and DNA viruses (Extended Data Fig. 2).

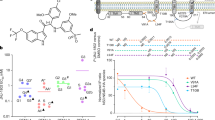
a, Molecular structure of JNJ-A07. b, Schematic of the membrane topology of DENV NS4B30,32. JNJ-A07-selected resistance mutations in orange were present in 100% of the quasispecies at end point (P43) in one of the two independently selected resistant strains (samples A and B). Resistance mutations in black were present in <100% of the quasispecies at end point; mutations in blue appeared and disappeared. TM, transmembrane. c, Mutations present in 100% of the quasispecies at end point in sample A and/or B. A and B refer to samples A and B, respectively. d, Effect of resistance mutations on replication fitness. e, Level of compound resistance imposed by NS4B resistance mutations. f, Representative western blot. The full representative western blot is depicted in Extended Data Fig. 5b. For the uncropped western blot images, see Supplementary Fig. 1. MM, molecular mass. g, Effect of JNJ-A07 on the interaction between NS3 and wild-type or mutant NS4B. For each sample, the ratio of NS3 over all NS4B species was normalized to the mean untreated wild-type ratio. h, Effect of JNJ-A07 on forming or pre-formed NS3–NS4B protein complexes. JNJ-A07-mediated treatment effect on the indicated ratios was assessed using linear mixed-effects models. A random effect for each replicate was included. Models were fitted for the three ratios separately. Sidak’s multiplicity correction was applied to the intervals to account for multiple testing. i, Model of the mode of action, whereby JNJ-A07 blocks the de novo formation of NS3–NS4B complexes (scenario 1) but does not disrupt existing ones (scenario 2). Data are the mean ± s.d. (bars in d, e) or estimated marginal means per mutation and treatment with their 95% CI (g, h) from three (h) or at least three (d, e, g) independent experiments.
JNJ-A07 targets the DENV NS4B protein
The addition of JNJ-A07 to infected cultures could be delayed without loss of antiviral potency as long as intracellular viral RNA synthesis had not been initiated to a detectable level (at 10 h after infection; Extended Data Fig. 3a, b). When the inhibitor was added after onset of viral RNA synthesis, a gradual loss of its antiviral activity was noted, which suggests that there is an interaction with the viral RNA replication machinery. A similar pattern was observed with the nucleoside analogue 7-deaza-2′-C-methyladenosine (7DMA), a broad-spectrum RNA virus inhibitor. To identify the molecular target, drug-resistant variants were selected by passaging DENV-2 in the presence of gradually increasing concentrations of JNJ-A07 (Extended Data Fig. 3c). This proved difficult in two independent efforts (A and B). As shown in the dynamics of appearance of mutations (Extended Data Fig. 3d, e), a decrease in susceptibility to the drug (32-fold) was first observed at week 15 of the selection process, and nearly 40 weeks were needed to obtain almost complete loss of antiviral activity. Multiple mutations were identified (after whole-genome sequencing) within the viral non-structural protein 4B (NS4B) at the end point, of which L94F, T108I and T216N were present in 100% of the viral population in sample A, and V91A, L94F and T108I in 100% and F47Y, P104S and T216P in <100% of the viral population in sample B (Fig. 1b, c, Extended Data Fig. 3d–f). These mutations were not present in the in-parallel-passaged untreated cultures. A close analogue of JNJ-A07 (analogue 1; Extended Data Table 1) resulted in the mutations V91A, L94F and T108I at the end point (week 29) (Extended Data Fig. 3f). Several resistance mutations occurred only at a very low frequency (that is, ≤0.5% across all 4 serotypes) in clinical isolates (Extended Data Fig. 3g), but none of these appeared together. The threonine at position 137 in NS4B (which appeared and disappeared during selection experiments in sample A) was present in 3.5% of DENV-2 clinical isolates and in 100% of the clinical isolates of the other serotypes, but it was considered a polymorphism as it did not alter the antiviral susceptibility. F47Y, S85L, V91A, L94F, P104S and T216N/P were not present in the panel of 21 clinical isolates used in this study. Isolates DENV-1/Malaysia, DENV-2/Martinique, DENV-2/Thailand, DENV-3/H87 and DENV-3/Brazil carry 108I or 108A (Table 1), which may explain the slightly decreased susceptibility of some of these viruses to JNJ-A07 compared with the other viruses from the same genotype.
To determine the replication fitness and inhibitor resistance caused by these mutations, they were inserted separately into a subgenomic DENV-2/16681 reporter replicon (Extended Data Fig. 4a). The mutations resulted in either marked attenuation of replication (F47Y, S85L, P104S and T216N) or did not affect replication (V91A, T108I, A137T and T216P) (Fig. 1d, Extended Data Fig. 4b), which did not correlate with the level of resistance imposed by these mutations (Fig. 1e). L94F conferred the highest level of resistance (950-fold), but increased the replication fitness compared with the wild type. This mutation was carried by virus strains obtained at the end point of two independent resistance-selection efforts, with the selected viruses having >50,000-fold reduced sensitivity to JNJ-A07 (Extended Data Fig. 3d, e). Although this drug-resistant virus retained full replication competence in Vero E6 cells (Extended Data Fig. 4c), it hardly replicated in C6/36 mosquito cells (Extended Data Fig. 4d–f).
JNJ-A07 blocks the NS3–NS4B interaction
As the mutations mapped to NS4B, we studied the possible effect of JNJ-A07 on the NS3–NS4B interaction. To this end, NS4B was expressed as part of the NS4A-2K-NS4B precursor19 along with the NS2B–NS3 protease–helicase complex. To facilitate NS4B-specific pull-down, a carboxy-terminal haemagglutinin affinity tag (HACt) was added to NS4B. Cells were transfected with constructs encoding the selected resistance mutants or the A137T natural polymorphism. Wild-type NS4B (NS4B(WT)) and the Q134A mutant (NS4B(Q134A), which is known to abolish the NS3–NS4B interaction20) were the positive and the negative controls, respectively, and non-HA-tagged NS4B was the technical control. Ratios of NS4B–HA and co-precipitated NS3 were measured by quantitative western blotting (Extended Data Fig. 5a, b). JNJ-A07 (at approximately 45 times the 50% effective concentration (EC50); 0.035 µM) decreased the amount of wild-type NS4B(WT) co-captured by NS3 by 95% (Fig. 1f, g), which demonstrates that it prevents the NS3–NS4B interaction. Consistently, almost complete drug-induced loss of the NS3–NS4B interaction was observed with the mutants S85L and A137T (Fig. 1g), which confer low or no drug resistance, respectively (Fig. 1e). By contrast, the NS3–NS4B interaction was barely affected by JNJ-A07 for the higher drug-resistance mutants V91A, L94F, T108I and T216N (Fig. 1f, g). Using T108I and V91A as examples of moderate and strong JNJ-A07 resistance mutations, respectively, dose–response assays were performed (Extended Data Fig. 5c–k). V91A and T108I increased the EC50 of the NS3–NS4B interaction by a factor 41 and 9, respectively, compared with the wild type (Extended Data Fig. 5e, h–k), which is in line with their effect on resistance in virus assays (Fig. 1e). Moreover, a dose-dependent decrease in the 2K-NS4B cleavage intermediate was noted (2K is a small signal sequence that translocates NS4B into the lumen of the ER), with the level of decrease following the level of resistance (Extended Data Fig. 5f, h–k). To a lesser extent, this effect was also noted for mature NS4B (Extended Data Fig. 5g–k), which indicates that JNJ-A07 slows down the cleavage kinetics of the NS4A-2K-NS4B precursor (Extended Data Fig. 5l). Next, the kinetics of JNJ-A07-induced loss of the NS3–NS4B interaction was studied (Extended Data Fig. 6a, b). Addition at 4 h after transfection significantly reduced the amounts of NS3–NS4B complexes, whereas treatment starting at 24 h had no significant effect in samples that were collected shortly thereafter (1 or 8 h) (Fig. 1h). Thus, a reduction becomes visible only at late time points (collected after 24 h), when newly formed NS3–NS4B complexes are detectable, which suggests that JNJ-A07 prevents the formation of NS3–NS4B complexes (scenario 1 in Fig. 1i) but does not disrupt them once formed (scenario 2 in Fig. 1i). Correspondingly, no reduction in co-captured NS3 was detected when a close analogue of JNJ-A07 (analogue 2; Extended Data Table 1) was added 48 h after infection (Extended Data Fig. 6c, d), and treatment of cell lysates did not disrupt already established NS3–NS4B complexes (Extended Data Fig. 6e, f).
Strong in vivo potency in mice
JNJ-A07 has a favourable pharmacokinetic profile in mice and rats, and no adverse effects were noted in rats up to doses of 300 mg per kg when given for 15 consecutive days by the oral route (Extended Data Table 2a, b). The antiviral effect was next studied in mouse infection models using the DENV-2 RL strain. First, the effect on peak viraemia (on day 3 after infection) in DENV-2-infected (106 plaque-forming units (PFU)) AG129 mice was studied. Dosing by oral gavage was initiated on the day of infection (starting about 1 h before infection) and continued twice daily until the end of the experiment (Fig. 2a). The viral RNA load in plasma dropped by 3.8 log10 copies per millilitre for the 30 mg per kg dose (P < 0.0001), 3.6 log10 for 10 mg per kg (P < 0.0001), 1.9 log10 for 3 mg per kg (P < 0.0001) and 0.8 log10 for 1 mg per kg (P < 0.05) (Fig. 2b). A dose-dependent and pronounced effect on viral RNA loads in the spleen, kidney and liver was also observed (Extended Data Fig. 7a–c). Levels of the pro-inflammatory cytokines interleukin-18 (IL-18), interferon-γ (IFNγ), tumour necrosis factor (TNF) and IL-6 were nearly normalized in the plasma of drug-treated infected mice (Extended Data Fig. 7d–g). The effect of JNJ-A07 was next assessed on virus-induced disease and mortality when dosed (oral gavage, twice daily) for just 5 consecutive days starting 1 h before infection (Fig. 2a). AG129 mice (injected on day –1 with an anti-flavivirus antibody to mimic antibody-dependent enhancement10) were challenged with 106 PFU of DENV-2. Mice were monitored for a maximum period of 25 days. In this model, the survival curve followed a biphasic pattern: early in infection, mice develop a systemic infection leading to vascular leakage, while later the virus escapes to the brain, resulting in a neurotropic infection and neurological complications. Most (19 out of 20) vehicle-treated mice had to be euthanized between day 4 and 21 after infection. At a dose of 30 mg per kg, 90% (P < 0.0001) survived the infection; at doses of 10, 3 and 1 mg per kg, the survival rate was 80% (P < 0.0001), 85% (P < 0.0001) and 75% (P < 0.0001), respectively (Fig. 2c). Viraemia on day 3 after infection was significantly reduced in all JNJ-A07 dosing groups (Extended Data Fig. 7h). Using the viraemia model, we also assessed the efficacy of the NS4B-targeting drug NITD-688 (ref. 21). Only mice treated with 100 or 30 mg per kg NITD-688 (twice daily, oral gavage) had significantly lower viral RNA levels in plasma: 4.3 log10 and 2.3 log10, respectively (Extended Data Fig. 7i).

a, Schematic of the viraemia and survival studies using AG129 mice. b, c, Effect of JNJ-A07 on viraemia on day 3 after infection (b) and on survival (c) in mice treated twice daily with 30, 10, 3 or 1 mg per kg JNJ-A07 compared with vehicle-treated mice. Treatment started 1 h before infection. In the survival study, mice received an anti-flavivirus antibody 1 day before infection. Data are from two independent studies with n = 8 (viraemia) or n = 10 (survival) mice per group. d, Schematic of the in vivo kinetics study. Each treatment group was subdivided into groups A and B (n = 8, per group) for blood collection on alternating days. e, Weight curves (mean values ± s.d.) of AG129 mice in the different treatment groups. Colours of the dots represent the different dosing groups (n = 8, per group), as specified in b; green dots represent the 0.3 mg per kg dosing group. f, g, Inhibitory effect of JNJ-A07 on viraemia in mice treated twice daily with 10 mg per kg (n = 8) or 1 mg per kg (n = 8) compared with vehicle-treated mice (n = 16). For the complete figure, see Extended Data Fig. 8. Treatment started 1 h before infection. Data (median ± s.d.) are from two independent studies. Undetermined Ct values were imputed at a Ct value of 40 (which is the LOD), corresponding to 2.6 log10 viral RNA copies per millilitre. For two-sided statistical analysis, the Kruskal–Wallis test (viraemia) or the Fisher’s exact test (survival) was used. P values were adjusted using the Holm’s multiple comparisons correction method. The mean AUC value and 95% CI was determined for f and g. In case CIs did not overlap, groups were considered to markedly differ. For b, f and g, results are shown as log10-transformed values. HEP, humane end point; LLOQ, lowest level of quantification.
The effect of JNJ-A07 was next assessed with respect to the kinetics of DENV-2 replication in AG129 mice following a non-lethal (that is, 102 PFU) viral challenge (Fig. 2d). In this model, a high peak viral RNA load (approximately 106 copies per millilitre) was achieved on day 5–6 after infection (Fig. 2f, g; for the complete figure, see Extended Data Fig. 8), which is similar to the dynamics during infection in humans22,23,24. Mice were treated with 30, 10, 3 or 1 mg per kg JNJ-A07 (orally, twice daily) for 6 consecutive days (starting about 1 h before infection). Both drug-treated and vehicle-treated mice exhibited some weight loss (<5%) (Fig. 2e) not attributable to treatment with JNJ-A07. At doses of 30, 10 and 3 mg per kg, mean viral RNA levels were mostly at the limit of detection (LOD) (Fig. 2f, Extended Data Fig. 8c–e). The viral load area under the curve (AUC) for the 30 and 10 mg per kg groups was 0% of the vehicle controls and 17% for the 3 mg per kg group. AUC confidence intervals (CIs) of the two lowest dosing groups did not differ from the controls as they overlapped with that of the vehicle group.
Finally, we explored whether the molecule is sufficiently potent to affect an ongoing, non-lethal (102 PFU) DENV-2 infection in AG129 mice (mimicking a human therapeutic setting). Administration of JNJ-A07 (30 mg per kg, twice daily for 6 consecutive days) was initiated either 1 h pre-infection or on subsequent days (Fig. 3a). Initiating treatment on the first 3 days after infection resulted in nearly complete inhibition of viral replication and markedly lower peak viraemia compared with vehicle-treated mice (Fig. 3b–d). When treatment was initiated on day 4 after infection—a time with substantial viraemia in the controls—viral loads returned to undetectable levels within 72 h (Fig. 3e). Even when treatment was first initiated on day 5 or day 6 after infection, the days on which replication reached its peak, an instant antiviral effect was observed (Fig. 3f, g). The effect on the AUC of the viraemia was determined from the day treatment was initiated until the end of the experiment. The viral load AUC of JNJ-A07-treated mice was 2% (95% CI: 0.01–1.42) of the vehicle-treated group (95% CI: 18.57–21.79) when treatment was initiated on day 0. When treatment was initiated on day 1, 2, 3, 4, 5 or 6 after infection, the viral load AUC of JNJ-A07-treated mice was 4% (95% CI: –0.56 to 3.21), 12% (95% CI: 0.95–6.05), 28% (95% CI: 4.05–12.37), 20% (95% CI: 2.81–7.87), 33% (95% CI: 5.33–10.43) and 52% (95% CI: 6.39–14.76), respectively, of that of the vehicle-treated group. Only the AUC CI of group 8 (treatment, start day 6 after infection) did not differ from group 1 (vehicle, start day 0 after infection) as both intervals overlapped.

a, Schematic of the in vivo kinetic studies in which treatment was started on various days after DENV-2 challenge (groups 3–8), while in the control groups (vehicle and JNJ-A07), treatment was started on the day of infection (groups 1 and 2, respectively). Each treatment group (n = 8, per group) was subdivided into groups A and B (n = 4, per group) for blood collection on alternating days. b–g, The inhibitory effect of JNJ-A07 on viraemia with the start of treatment at various time points after infection in AG129 mice treated twice daily with 30 mg per kg for 6 consecutive days. In the delayed-treatment groups (groups 3–8), treatment with JNJ-A07 was started on day 1 (b), day 2 (c), day 3 (d), day 4 (e), day 5 (f) or day 6 (g) after infection. As controls, two groups of mice received treatment on the day of infection: group 1 (vehicle) and group 2 (JNJ-A07). Data (median ± s.d.) are from a single experiment. Results shown as shown as log10-transformed values. Undetermined Ct values were imputed at a Ct value of 40 (which is the LOD), corresponding to 2.6 log10 viral RNA copies per millilitre. The mean AUC value and 95% CI was determined for each group. In case CIs did not overlap, groups were considered to substantially differ.
Discussion
There is an urgent need for potent and safe pan-serotype dengue antivirals for the treatment and prophylaxis of infections with DENV. Such drugs should lower viral loads during an ongoing infection, thereby reducing dengue-associated morbidity and mortality as well as transmission25,26,27. Early diagnostic testing will be key to the usability of prophylactic drugs. Prophylaxis should be beneficial during epidemics for those living in endemic regions and for those travelling to such regions. The concept behind such prophylaxis is that the drug prevents expansion of the inoculum after a mosquito bite. Prophylaxis is, for example, successfully used in the prevention of malaria28.
We reported a highly potent, pan-serotype DENV inhibitor targeting NS4B. Drug-resistant variants were only obtained in vitro following a lengthy period (up to 40 weeks) of selection, thereby demonstrating its high barrier to resistance. This is explained by the finding that a combination of three mutations in NS4B is required to reach high-level resistance. This characteristic makes it unlikely that drug-resistant variants will readily emerge in drug-treated patients. Moreover, the mutations in NS4B appeared to render the resistant variants unable to replicate in mosquito cells. This suggests that even if such mutants would develop, they may not be transmitted from human-to-human via the insect vector.
Resistance selection and reverse genetics studies pinpointed NS4B as the molecular target of JNJ-A07. NS4B is a multi-transmembrane protein residing in the endoplasmic reticulum membrane as part of the DENV replication complex. It forms a complex with NS3, which is essential for viral replication20,29. Several functions have been ascribed to NS4B30,31,32. In vitro studies revealed that NS4B dissociates NS3 from single-stranded RNA and enhances NS3 helicase activity33. No enzymatic activity has been shown to be associated with NS4B. We here demonstrated that JNJ-A07 blocks the de novo formation of the NS3–NS4B complex; conversely, established complexes appeared relatively resistant to the compound. JNJ-A07 prevented the formation of the NS3–NS4B complex, but did this inefficiently when NS4B carries mutations associated with drug resistance. In fact, a notable correlation was observed between drug resistance in a DENV replication assay on the one hand and the insensitivity of the interaction between NS3 and NS4B mutants on the other hand. This provides evidence that JNJ-A07 interferes with NS3–NS4B complex formation. L94F (the mutation conferring the highest level of resistance but resulting in increased replication fitness) has been reported as a pseudo-reversion compensating for the replication defect caused by the M142A mutation in NS4B20. As is the case for Q134A, M142A resides in the cytosolic loop of NS4B and impairs the NS3–NS4B interaction, thereby largely reducing viral replication. Our findings suggest that JNJ-A07 blocks the NS3–NS4B interaction by inducing a conformational change of the cytosolic loop. In addition, the observed accumulation of the NS4A-2K-NS4B precursor suggests that JNJ-A07 slows down the cleavage kinetics of the precursor (for example, by binding to the NS4B moiety and altering precursor folding or accessibility of the cleavage site).
JNJ-A07 had a favourable pharmacokinetic and safety profile in mice and rats and exerted strong potency in DENV-2 infection mouse models. It was highly effective in reducing viral loads (even at low doses to levels below the LOD) and virus-induced disease. Importantly, even when the start of treatment was delayed for several days after infection, a rapid and marked reduction in viral load was observed. JNJ-A07 or close analogues with comparable safety, pharmacokinetics and potency may have the potential to be effective in both prophylactic and therapeutic settings against DENV infections in humans. Recently, NITD-688 was reported as a NS4B-targeting drug, but with an unknown mechanism21. The resistance mutation profile differs from that of JNJ-A07, which is indicative of a different mode of action. JNJ-A07 was markedly more efficacious than NITD-688 in DENV-2 infection mouse models, both in a prophylactic and a therapeutic setting.
In conclusion, we demonstrated that blocking the interaction between two viral proteins (NS3 and NS4B) results in strong antiviral activity. The NS3–NS4B interaction represents a promising target for the development of pan-serotype DENV inhibitors with a high barrier to resistance. The strong potency warrants further development of this class of compounds.
Methods
Compounds
The synthesis of early chemical analogues of JNJ-A07 and its derivatives is reported elsewhere16. The synthesis of analogue 1 is described in WO/2016/050841 (compound 1A)34 and that of analogue 2 in WO/2016/050831 (compound 1A)35. The synthesis of JNJ-A07 is published in WO/2017/167951 (example 4B)36. The synthesis and chemical characterization of analogue 1, analogue 2 and JNJ-A07 are also provided in the Supplementary Methods. For in vitro experiments, compounds were dissolved in 100% dimethylsulfoxide (DMSO) as a 10 mg ml–1 or a 5 mM stock. The nucleoside analogue 7DMA (CAS number 443642-29-3) was purchased from Carbosynth. The synthesis of the DENV NS4B inhibitor NITD-688 was carried out in-house following a synthetic route as described in the literature21 and in patent WO/2019/244047 A1 (ref. 37).
Cells
Vero cells (African green monkey kidney cells; European Collection of Authenticated Cell Cultures, CL 84113001) were maintained in Eagle’s minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich), 2 mM l-glutamine and 0.02 mg ml–1 gentamicin (Thermo Fisher Scientific). Vero E6 cells (American Type Culture Collection, CRL-1586) were cultured in MEM supplemented with 7.5% heat-inactivated FBS, 2 mM L-glutamine and 100 units per millilitre penicillin–streptomycin (Sigma). In the antiviral experiments with Vero E6 cells, 2.5% heat-inactivated FBS was used. Huh-7 hepatoma-derived cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% FBS, 2 mM L-glutamine and 0.02 mg ml–1 gentamicin. In antiviral assays using Vero and Huh-7 cells, the culture medium contained 2% FBS instead of 10% FBS. Huh-7 replicon cells were cultured in the same medium as mentioned above, supplemented with 75 μg ml–1 hygromycin B (Roche). Huh-7 cells stably expressing the T7 polymerase and the DENV protease complex NS2B–NS3 (Huh7-T7/NS2B–NS3 cells) were generated by lentiviral transduction, as previously described20. Cells were cultured at 37 °C and 5% CO2 in DMEM, supplemented with 10% FBS, 2 mM l-glutamine, 100 units per millilitre penicillin, 100 μg ml–1 streptomycin, 5 μg ml–1 zeocin, 1 μg ml–1 puromycin and nonessential amino acids. Antiviral assays were performed using DMEM/2% FBS. THP-1 dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) cells (TIB-202, ATCC) were propagated in RPMI (Lonza) supplemented with 10 % heat-inactivated FBS (F7524, Sigma-Aldrich) and 0.04 % gentamicin (Gibco-Life Technologies). C6/36 mosquito cells (from Aedes albopictus; ATCC, CCL-1660) were cultivated in the absence of 5% CO2 at 28 °C in Leibovitz’s L-15 medium (Thermo Fisher Scientific), supplemented with 10% FBS, 1% nonessential amino acids (Thermo Fisher Scientific), 1% HEPES buffer (Thermo Fisher Scientific) and 1% penicillin (100 units per millilitre) and streptomycin (100 μg ml–1) solution (Thermo Fisher Scientific). Human peripheral blood mononuclear cells (PBMCs) were prepared from fresh buffy coats (obtained 24 h before preparation from the Belgian Red Cross) of healthy donors using a standard Ficoll centrifugation protocol. Monocytes were isolated from the PBMC population with Miltenyi cluster of differentiation 14 (CD14) beads (Miltenyi Biotec). Monocytes were differentiated into immature dendritic cells (imDCs) using IL-4 (R&D Systems) and granulocyte–macrophage colony-stimulating factor (GM-CSF; R&D Systems). Multiple donors were used to account for potential variation in responses due to varying representation of genetic and societal backgrounds. Cells were cultured at 37 °C and 5% CO2 unless stated otherwise. All cell lines (Vero, Huh-7, THP-1/DC-SIGN and C6/36) were regularly tested for mycoplasma contamination.
Viruses
Laboratory-adapted strain DENV-2/16681 was produced by transfection of in vitro-transcribed RNA of plasmid pFK-DVs into Huh-7 cells. This plasmid encodes the full-length DENV-2/16681. Plasmid pFK-DVs was obtained by insertion of a synthetic copy of the full-length genomic sequence of DENV-2 strain 16681 (GenBank accession NC_001474) into the low-copy plasmid vector pFK38. Moreover, the parental vector pFK was modified by insertion of the SP6 promoter upstream of the DENV 5′ nontranslated region to enable synthesis of authentic viral RNA by in vitro transcription. This plasmid was licensed to R.B.
DENV-2/16681/eGFP, carrying enhanced green fluorescent protein (eGFP) at the amino terminus of the capsid protein, was produced through the transfection of in vitro-transcribed RNA of plasmid pFK-DV-G2A in Huh-7 cells38. This plasmid encodes eGFP and the full-length DENV-2/16681. The infectious cDNA clone pFK-DVs served as the parental construct for cloning of the dengue reporter virus construct DENV-G2A. The reporter gene is followed by the 2A peptide of Thosea asigna virus to liberate the eGFP from the DENV polyprotein during/after translation. This plasmid was licensed to R.B. The resulting recombinant virus is referred to as DENV-2/16681/eGFP.
The following four DENV-1 strains were used: Djibouti (D1/H/IMTSSA/98/606), genotype 1, GenBank accession AF298808; Malaysia, produced by Infectious Subgenomic Amplicons (ISA)39, genotype 3, GenBank accession EF457905.1; Indonesia (JKT 1186 TVP 949), genotype 4, GenBank accession EU074031; and France-Toulon (CNR 25329), genotype 5, GenBank accession MF004384, obtained from the European Virus Archive (EVA).
The following six DENV-2 strains were used: Martinique (H/IMTSSA-MART/98-703), Asian America, GenBank accession AF208496; Trinidad (1751 TC 544), American, GenBank accession EU073981.1, EVA; France-Toulon (CNR 25679), Cosmopolitan, GenBank accession MF004385, EVA; Thailand (CNR 25326), Asian I, EVA; Papua New Guinea (ISA), GenBank accession FJ906959.1, Asian II; and Malaysia (ISA), GenBank accession FJ467493.1, genotype Sylvatic.
The following five DENV-3 strains were used: Malaysia (CNR 17046), genotype 1, GenBank accession MF004386, EVA; Thailand (CNR 15418), genotype 2, GenBank accession MH888332, EVA; Bolivia (strain 4025), genotype 3, GenBank accession MH888333, EVA; H87, genotype 5, GenBank accession M93130; and Brazil (ISA), genotype 5, GenBank accession JN697379.1.
The following six DENV-4 strains were used: India (strain G11337), genotype 1, GenBank accession JF262783.1; Malaysia (CNR 16861), genotype 2a, GenBank accession MH888334, EVA; Martinique (strain 017), genotype 2b, EVA; Brazil (BeH 403714), genotype 2b, GenBank accession JQ513345.1; Thailand (ISA), GenBank accession AY618988.1, genotype 3; and Malaysia, GenBank accession JF262779.1, genotype Sylvatic.
DENV-1 genotype 2 and DENV-3 genotype 4 are currently not available as full sequences in public databases such as the National Center for Biotechnology Information or Virus Pathogen Resource (ViPR; www.viprbrc.org).
The following four non-DENV flaviviruses were used: Zika virus (ZIKV; H/PF/2013, French Polynesia, GenBank accession KJ776791), Japanese encephalitis virus (JEV; CNS769-Laos 2009, Laos, GenBank KC196115), West Nile virus (WNV; R94224, CDC Human Brain 29-09-2008, Wisconsin, GenBank accession MF004388) and yellow fever virus (YFV; 88-99, Bolivia, GenBank accession MF004382).
For antiviral assays using Vero cells and C6/36 mosquito cells, time-of-drug-addition experiments, in vitro resistance-selection experiments and in vivo efficacy studies, the DENV-2 Rega Labstrain (referred to as DENV-2 RL) was used, GenBank accession MW741553. This strain was provided by V. Deubel, formerly at the Institute Pasteur, Paris. For in vivo studies, high-titre stocks were generated by propagating in C6/36 mosquito cells and subsequently concentrating either by ultracentrifugation or tangential flow filtration using tangential flow filtration capsules (Minimate TFF; Pall Life Sciences) according to the manufacturer’s protocol. Infectious virus titres (PFU per ml) were determined by performing plaque assays on baby hamster kidney cells as previously described40.
DENV-2/16681 antiviral assays
The antiviral activity of JNJ-A07 was determined against DENV-2/16681/eGFP in a phenotypic antiviral assay with eGFP readout as a measure for the amount of virus. The assay was performed on three different cell types (Vero, Huh-7 and THP-1/DC-SIGN) to exclude cell-specific activity of the compound. In brief, 2.5 × 103 Vero cells or Huh-7 cells or 7.5 × 103 THP-1/DC-SIGN cells were seeded in 384-well plates containing 9-fold serially diluted test compound. After incubating for 24 h at 37 °C, Vero and Huh-7 cells were infected with DENV-2/16681/eGFP at a multiplicity of infection (MOI.) of 1 and 5, respectively. THP-1/DC-SIGN cells were infected immediately after seeding of the cells with DENV-2/16681/eGFP at a MOI of 0.5. After 3 days of incubation at 37 °C, viral replication was quantified by measuring eGFP expression in the cells with a laser microscope. Following eGFP readout, the cytotoxic effect of JNJ-A07 was evaluated using an ATPlite cell viability luminescence assay (PerkinElmer) according to the supplier’s instructions.
For antiviral assays with imDCs, DENV-2/16681 was used followed by detection of DENV antigens using flow cytometry. Monocytes, isolated from PBMCs, were counted and 3 × 105 cells were seeded in wells of a 96-well plate. Next, monocytes were differentiated into imDCs by incubating them for 5 days at 37 °C in the presence of 20 ng ml–1 IL-4 and GM-CSF. The medium was then discarded and imDCs were infected with DENV-2/16681 at a MOI of 0.5 in the presence or absence of JNJ-A07. On day 2 after infection, cells were permeabilized and fixed with Cytofix/Cytoperm buffer (BD Biosciences) and stained with primary anti-prM antibody (anti-DENV complex antibody, clone D3-2H2-9-21; MAB8705, 1:400 diluted; Merck), followed by secondary goat anti-mouse Alexa Fluor-488 antibody (A-10680, 1:500 diluted; Life Technologies/Thermo Fisher Scientific). The percentage of cells expressing prM (7–11% for untreated virus control samples) was quantified using FACS on a CANTO II apparatus (BD Biosciences). Toxicity of JNJ-A07 was assessed with FACS in non-infected imDCs by measuring the viability dye eFluor 660 (Thermo Fisher Scientific) added to the cells before fixation.
Antiviral assays using DENV-2 RL strain on Vero and C6/36 cells
Virus yield reduction assays using Vero cells were performed essentially as previously described16. In brief, Vero cells were seeded at a density of 4 × 104 cells per well in 100 µl DMEM/10% FBS medium in 96-well plates. The next day, cells were infected with DENV-2 RL strain (MOI of 0.01) diluted in MEM/2% FBS assay medium (100 µl per well). Cells were incubated for 2 h, after which the viral inoculum was removed. After rinsing the cells three times with assay medium, 5-fold serial dilutions (concentration ranged from 50 to 0.0001 µg ml–1 in screening assays and from 1 to 0.000003 µg ml–1 in confirmation-of-antiviral-activity assays) of the test compounds were added to the cells. After an incubation period of 4 days, the supernatant was collected and the viral RNA load was determined by quantitative PCR with reverse transcription (RT–qPCR) as previously described41. A potential toxic effect on host cells was tested in parallel using the same protocol. For the toxicity assays, virus infection was omitted and the serial dilution of compounds was started at a higher concentration (concentration ranged from 400 to 0.001 µg ml–1 only in confirmation-of-antiviral-activity assays). After 4 days of incubation, colorimetric readout was performed using the MTS/PMS method (Promega) as previously described42.
For the antiviral assays using C6/36 mosquito cells, cells were seeded at a density of 2.5 × 105 cells per well in 100 µl culture medium (see also the section ‘Cells’) in 24-well plates. The next day, culture medium was replaced with 100 µl per well assay medium (in assay medium 10% FBS is replaced by 2% FBS) containing 2-fold serial dilutions (concentration ranged from 50 to 0.002 µg ml–1) of the test compounds. DENV-2 RL strain (MOI of 0.02; 100 µl per well), diluted in assay medium, was added to the cells. After a 7-day incubation period at 28 °C, the supernatant was collected and the viral RNA load was determined by RT–qPCR, as described for the Vero cells. A potential toxic effect on host cells was tested in parallel using the same protocol. However, virus infection was omitted and the 2-fold serial dilution of compounds ranged from 50 to 0.4 µg ml–1. On day 7 after infection, cells were fixed with 2% paraformaldehyde in PBS. Cell nuclei were stained using 4′,6-diamidino-2-fenylindool (DAPI; Thermo Fisher Scientific) and readout was performed using an ArrayScan XTI High Content Analysis Reader (Thermo Fisher Scientific). The EC50 (the compound concentration that is required to inhibit viral RNA replication by 50%) and the 50% cytotoxic concentration (the concentration that reduces the total cell number by 50%; CC50) was determined using logarithmic interpolation.
Antiviral activity against clinical isolates covering four DENV serotypes
One day before infection, 5 × 104 Vero E6 cells were seeded in 100 µl assay medium (containing 2.5% FBS) in 96-well plates. The next day, eight 2- or 3-fold serial dilutions of JNJ-A07 (for DENV: 100–0.04 nM; for other flaviviruses: 5–0.02 µM; final concentration), in triplicates or duplicates (for control), were added to the cells (25 µl per well). Four virus control wells (per virus) were supplemented with 25 µl medium and four cell control wells were supplemented with 50 µl of medium. After 15 min, 25 µl of a virus dilution was added to the wells at a MOI that was determined such that the viral growth reached its peak or the beginning of the plateau on day 4 after infection Plates were incubated at 37 °C for 4 days (DENV and ZIKV), 3 days (JEV) 2.5 days (YFV) or 2 days (WNV). After incubation, 100 µl of the supernatant was collected for viral RNA isolation.
Time-of-drug-addition assay
Vero cells were seeded at a density of 2 × 105 cells per well in a 24-well plate and the following day infected with DENV-2 RL strain (MOI of 1) in assay medium. JNJ-A07 (at a concentration of 10 times the EC50, as determined in the antiviral assay) was added at either the time point of virus infection or at 4, 10, 12, 14, 16, 18 and 22 h after infection At 24 h after infection, intracellular RNA was isolated using a RNeasy minikit (Qiagen) and DENV RNA levels were quantified by RT–qPCR. In parallel experiments, 7DMA (14 µM) was used as a reference compound. To monitor intracellular viral RNA production (that is, viral kinetics) during one replication cycle in untreated cells, confluent Vero cells in a 24-well plate (2 × 105 cells per well) were infected and incubated for 1 h. After removing the inoculum and washing the cells, assay medium was added, and the cells were collected at similar time points as indicated for the time-of-drug-addition assay. Viral RNA replication was monitored using RT–qPCR.
RNA isolation and RT–qPCR
The supernatant was transferred to 96-well S-Bloc from Qiagen (Venlo), preloaded with buffer VXL and extracted using a Cador Pathogen 96 QIAcube HT kit run on QIAcube HT automat (Qiagen) as described by the manufacturer. For DENV, purified RNA was eluted in 80 µl of AVE buffer (Qiagen); for the other flaviviruses, purified RNA was eluted in water.
DENV RNA was quantified by real-time RT–qPCR using 3.8 µl of RNA and 6.2 µl of RT–qPCR mix (GoTaq Probe one-step RT–qPCR system, Promega) and fast cycling parameters, that is, 10 min at 50 °C, 2 min at 95 °C and 40 amplification cycles (95 °C for 3 s followed by 30 s at 60 °C). Viral RNA of the other flaviviruses was quantified using 7.5 µl of RNA and 12.5 µl of RT–qPCR mix (SuperScript III Platinium one-step qRT–PCR kit with Rox from Thermo Fisher Scientific, or GoTaq Probe one-step RT–qPCR system from Promega) and standard cycling parameters, that is, 20 min at 50 °C, 3 min at 95 °C and 40 amplification cycles (95 °C for 15 s followed by 1 min at 60 °C)18.
For the DENVs, RT–qPCR reactions were loaded on a QuantStudio 12K Flex Real-Time PCR system (Applied Biosystems) and analysed using QuantStudio 12K Flex software v.1.2.3. For the other flaviviruses, RT–qPCR reactions were loaded on an ABI 7900 HT Fast Real-Time PCR system (Applied Biosystems) and analysed using SDS 1.2 Applied Biosystems software. Viral RNA was quantified using serial dilutions of a standard curve consisting of four 2-log dilutions of an appropriate T7-generated RNA standard of known quantities for each serotype or virus (100 copies to 100 × 106 copies). Inhibition values for each drug concentration were plotted using KaleidaGraph plotting software (v.4.03; Synergy Software), and the best sigmoidal curve, fitting the mean values, was used for determination of the EC50 value. The EC50 value was determined using logarithmic interpolation.
DENV-2 in vitro resistance selection
Vero cells were seeded at a density of 2 × 105 cells per well in a 24-well plate. The next day, cells were infected (MOI of 0.01; virus stock was diluted 200×) with DENV-2 RL strain and incubated with virus for 1.5–2 h at 37 °C. Virus was then removed and cells were rinsed three times using assay medium (MEM/2% FBS). Cells were further incubated in the presence of a 2-fold serial dilution of JNJ-A07 (10–0.00002 µg ml–1) for 7 days at 37 °C. After 7 days, cells were microscopically checked for cytopathogenic effects (CPEs), and the supernatant from two adjacent wells showing 30–70% CPEs was collected and pooled. The EC50 was microscopically determined as the mean concentration of the compound that was added to the two selected and pooled wells (showing 30–70% CPEs). The supernatant was used to infect freshly seeded cells using the same virus dilution (that is, 200×) as in all previous passages. The remaining supernatant was stored at –80 °C until further analysis (that is, sequencing and plaque assay). During weekly passaging of the virus, the start concentration of the compound was gradually increased. In addition, fresh compound solution was used after each tenth passage to prevent the possibility that a shift in the EC50 was the result of instability of the compound. This procedure was repeated on a weekly basis until the observed EC50 value approached the cytostatic concentration of the compound. To check for spontaneous and/or tissue-culture-adapted mutations, part of the wells served as wild-type virus controls to which no compound was added. Wild-type DENV was passaged using Vero cells in a similar way to compound-treated virus.
In vitro growth kinetics of DENV resistant to JNJ-A07
The growth kinetics of resistant viruses obtained via in vitro resistance selection with JNJ-A07 in two independent efforts (A and B) was evaluated in both Vero E6 and C6/36 cells. Vero E6 cells were seeded at a density of 4 × 105 cells per well in a 12-well plate. The next day, cells were infected with either wild-type or compound-resistant DENV-2 RL (MOI of 0.1) diluted in MEM/2% FBS assay medium. Cells were incubated for 2 h at 37 °C, after which the viral inoculum was removed and cells were washed twice with assay medium. The supernatant was collected on day 1–7 after infection, followed by the determination of the viral RNA load by RT–qPCR and plaque assay.
For evaluating the growth kinetics on C6/36 cells, the same procedure was followed but with some modifications. Cells were seeded at a density of 8 × 105 cells per well in a 12-well plate and infections were performed using a MOI of 0.01. The supernatant was collected on day 1–10 after infection for quantification of the viral RNA load by RT–qPCR. On day 11 after infection, the supernatant was collected for determination of the infectious virus titres by plaque assay.
Whole-genome sequencing
Viral RNA was isolated from cell culture supernatant (140 µl) using a QIAamp Viral RNA Mini kit (Qiagen) per the manufacturer’s protocol with the exception that 5 µg of linear polyacrylamide (Life Technologies) was used as the carrier instead of the carrier RNA provided with the kit. All samples were subsequently treated with RNA Clean & Concentrator-5 (DNAse incuded) (Zymo Research). Viral RNA was amplified into double-stranded DNA using an Ovation RNA-Seq v.2 kit (NuGEN) per the manufacturer’s protocol. Paired-end libraries for Illumina sequencing were prepared using a Nextera XT DNA Library Preparation kit (Illumina) per the manufacturer’s protocol. Before sequencing on a MiSeq (Illumina; 150 base-paired reads), short amplification products were removed using AMPure XP beads (Beckmann Coulter). Sequence reads were binned by index read before further analysis. Poor-quality bases of each read were trimmed before alignment. Sequences were filtered for viral content by aligning the reads to genotype-specific viral genomes using the CLC genomics workbench (Qiagen). A custom script43 was used to derive the amino acid composition of each sample for all coding sequences per DENV genotype. A coverage cut-off value of 100 and a 15% read frequency cut-off were used for the reliable detection of amino acid variants.
Transient mutant replication assays to study replication fitness and compound resistance
A panel of mutant subgenomic DENV reporter replicons (sgDVs-R2A) each containing harbouring a NS4B resistance mutation was used to determine the replication fitness and compound resistance imposed by each of these mutations. First, each resistance mutation was inserted separately into the sgDVs-R2A replicon. The plasmid (denoted pFK-sgDVs-R2A) contains the non-structural genes ns1–ns5 of the DENV-2/16681 strain with cell-adaptive mutations in ns3 (A56V and H451P), ns4a (I116M) and ns5 (E892K), and the Renilla luciferase (rluc) reporter gene38. Mutations in the ns4b region were introduced by site-directed mutagenesis using a QuickChange II XL Site-Directed Mutagenesis kit according to the instructions of the manufacturer (Agilent), resulting in the respective mutant sgDVs-R2A expression plasmids. Plasmid DNA was linearized with XbaI (located at the end of the 3′ untranslated region of the viral genome) and purified using a NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel). In vitro transcription was performed with a mMESSAGE mMACHINE SP6 kit (Ambion) according to the manufacturer’s protocol. RNA was purified using acidic phenol–chloroform extraction, precipitated with isopropanol and dissolved in RNase-free water. The molecular mass and integrity were checked by agarose gel electrophoresis. In vitro-transcribed RNA of both wild-type and mutant sgDVs-R2A was transiently transfected into Huh-7 cells. To this end, 10 µg in vitro-transcribed linear RNA was electroporated into Huh-7 cells (electroporation at 975 μF and 270 V; Gene Pulser II, Bio-Rad) as previously described38,44. To determine the replication fitness, transfected cells were transferred to prewarmed complete DMEM and seeded in duplicate into 6-well plates at different densities depending on the incubation time (2 × 105 cells for 4 h and 24 h of incubation; 1 × 105 cells for 48 h and 72 h of incubations; 5 × 104 cells for 96 h of incubation). At the respective time points, cells were washed once with PBS and lysed as previously described44. Lysates were frozen immediately at –20 °C. After collection of all samples, lysates were thawed, resuspended by gentle pipetting and luciferase activity was measured for 10 s in a plate luminometer (Mithras LB940, Berthold), as previously reported44. To determine compound resistance, transfected cells (4,000 cells per well in a 384-well plate) were incubated with serial dilutions of JNJ-A07 at 37 °C. Two days after transfection, viral replication was quantified by measuring luciferase activity.
Immunoprecipitation experiments
Huh-7 cells stably expressing the T7 RNA polymerase and DENV-2 NS2B–NS3 were seeded into 10-cm-diameter cell culture dishes (2 × 106 cells per dish) 18 h before transfection. For each construct, 10 µg of plasmid DNA (plasmid encoding NS4A-2K-NS4B(-HACt) with NS4B corresponding to the wild type or containing a JNJ-A07 resistance mutation (with or without a C-terminal HA tag) was mixed with 800 µl Opti-MEM, and 30 µl of TransIT-LT1 Transfection reagent was added. The mix was equilibrated at room temperature for 20 min and added in a drop-wise manner to the cells. To study compound resistance, the indicated concentration of JNJ-A07 or an equivalent amount of DMSO without compound was added to each plate along with the transfection mix. Medium was replaced 4 h after transfection by fresh DMEM supplemented with or without the same concentration of JNJ-A07. Eighteen hours after transfection, cells were first washed with PBS and then collected. For studying the kinetics of JNJ-A07-induced block of the NS3–NS4B interaction, transfection medium without compound was replaced after 4 h. JNJ-A07 (2.8 µl of a 100 µM stock solution in DMSO) or an equivalent amount of DMSO was added either at 4 h or 24 h after transfection. Cells were collected at 1, 8 or 24 h after treatment.
Collected cells were lysed on ice for 20 min in 500 µl lysis buffer containing 150 nM NaCl, 50 mM NaF, 20 mM Tris (pH 7.5), 0.5% dodecyl β-d maltoside (DDM; w/v) and protease inhibitors (Roche). To remove cell debris, lysates were centrifuged in a pre-cooled (4 °C) benchtop centrifuge for 45 min at maximum speed (21,130g). A Bradford assay was used to determine the protein concentration of each sample and samples were adjusted to the one with the lowest concentration. For HA-specific immunoprecipitation, 30 µl of equilibrated mouse monoclonal anti-HA agarose beads (antibody concentration is 2.1 mg ml–1 settled resin, as specified by the manufacturer; A2095, Sigma-Aldrich) was added to each sample and incubated for 3 h at 4 °C. Beads were washed twice with lysis buffer and twice with PBS, and captured proteins were eluted in a first step with PBS containing 5% SDS followed by an elution step with pure PBS. Four sample volumes of acetone were added to combined eluates to perform overnight precipitation of proteins at –20 °C. Samples were centrifuged at 4 °C for 1 h at 21,130g. Pellets were air-dried, resuspended in SDS sample buffer and loaded onto a SDS–polyacrylamide gel. After electrophoresis, proteins were transferred onto an Amersham Protran 0.2-µm nitrocellulose membrane (GE Healthcare Life Sciences) for western blotting and analysed using a chemoluminescence imager (ECL ChemoCam Imager, Intas Science Imaging Instruments) as previously described19,20. NS4B- and NS3-specific bands were visualized using in-house-generated rabbit polyclonal antibodies directed against NS4B (1:1,000 dilution) or NS3 (1:2,000 dilution), respectively, as previously described19,20,30. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or β-actin served as loading controls for cell lysates (input), which were visualized using the mouse monoclonal anti-GAPDH antibody (1:1,000 dilution; sc-365062, Santa Cruz Biotechnology) or the mouse monoclonal anti-β-actin antibody (1:5,000 dilution; A5441, Sigma-Aldrich), respectively. Intensities were quantified using ImageJ2 (v.1.53j, Fiji). Statistical analysis was conducted using either R script or GraphPad Prism 7.04 software package and is specified in further detail in the legends of the respective figures. The treatment effect of JNJ-A07 on protein ratios was assessed by means of linear mixed-effects models. In addition, a random effect for replicates was included. The 95% CI values are provided, indicating the variability on the estimated effects. Sidak’s multiplicity correction was applied to the intervals to account for multiple testing.
Immunoprecipitation of NS4B–HA-containing complexes
Huh-7 cells were infected with DENV-2(NS4B-HA*), which is a replication-competent DENV carrying an internal HA tag in the NS4B protein20, or DENV-2 wild type as a control (MOI of 1). Forty-eight hours after infection, cells were treated for various time spans (1, 6 or 24 h) with either 500 nM of analogue 2 or buffer with an equivalent concentration of DMSO. Cells were collected, lysed in DDM lysis buffer and subjected to immunoprecipitation using the HA affinity tag. Captured complexes were analysed by western blotting, and intensities of NS4B- and NS3-specific bands were quantified using the ImageJ2 software package (Fiji).
In vitro drug assay
Huh-7 cells stably expressing the T7 RNA polymerase and DENV-2 NS2B–NS3 were transfected with pTM1-NS4A-2K-NS4B(-HACt) constructs using TransIT-LT1 (Mirus) according to the manufacturer’s protocol (800 μl serum-free OPTi-MEM medium, 10 μg DNA, 30 μl TransIT-LT1). After 4 h, medium was exchanged for fresh DMEM and 30 h after transfection, cells were collected, washed and resuspended in DDM lysis buffer. Lysates were treated with either 1 μM analogue 2 or an equal volume of DMSO and then incubated at various temperatures for 2 h. HA-specific complexes were analysed as described above.
Pharmacokinetics studies
All animal studies were performed with the approval of and under the guidelines of the ethics committee. The pharmacokinetics profile was evaluated in fed male CD-1 mice (n = 3 per group, 6–8 weeks old; Charles River Laboratories). Mice were intravenously injected with 2.5 mg per kg of the test compound, which was formulated as an 0.5 mg ml–1 solution in polyethylene glycol 400 (PEG400):water + NaOH (50:50), and blood samples were collected (in EDTA-containing microcentrifuge tubes) from the dorsal metatarsal vein at 0.12, 0.33, 1, 2, 4 and 7 h after dosing, or via heart puncture at 24 h after dosing. Additionally, test compound was administered by oral gavage at 1, 3, 10 and 30 mg per kg, formulated as a solution in PEG400:water + NaOH (50:50), and blood samples were collected from the dorsal metatarsal vein at 0.5, 1, 2, 4 and 7 h after dosing, or via heart puncture at 24 h after dosing. Blood samples were immediately centrifuged at 4 °C and plasma was stored at –20 °C. Compound concentrations in the plasma samples were determined using an API 4000 LC–MS/MS system mass spectrometer (Applied Biosystems). Individual plasma concentration–time profiles were subjected to a non-compartmental pharmacokinetics analysis using Phoenix WinNonlin v.6.1. (Certara).
DENV-2 infection models in mice
Breeding couples of AG129 mice (129/Sv mice deficient in both IFNα/β and IFNγ receptors) were purchased from Marshall BioResources and bred in-house. The specific pathogen-free status of the mice was regularly checked at the KU Leuven animal facility. Mice (maximum 5 mice per cage, type GM500) were housed in individually ventilated cages (Sealsafe Plus, Tecniplast) at 21 °C, 55% humidity and 12:12 h light:dark cycles. Mice were provided with food and water ad libitum as well as with cardboard play tunnels and cotton as extra bedding material. Allocation to experimental groups was performed randomly.
Housing conditions and experimental procedures were approved by the ethics committee of KU Leuven (license P169/2011 and P047/2017) following institutional guidelines approved by the Federation of European Laboratory Animal Science Associations (FELASA). AG129 mice were used to assess the activity of JNJ-A07 on viral RNA levels in plasma and several tissues (spleen, kidney and liver). To this end, female mice (7–11 weeks old, n = 8 per group) were challenged intraperitoneally (i.p.) with 106 PFU DENV-2 RL strain. Mice were treated twice daily by oral gavage for 3 consecutive days with either vehicle (PEG400:water + NaOH (50:50)) or various doses of JNJ-A07 (30, 10, 3 or 1 mg per kg dose), with the first administration 1 h before DENV challenge. On day 3 after infection, mice were euthanized, and blood, spleen, kidney and liver were collected and stored at –80 °C until further use. Viral RNA isolation from plasma and tissues was performed as previously described45.
To monitor the effect of the compound on viral RNA levels in the blood on various days after infection, an in vivo kinetics study was performed. AG129 female mice (7–11 weeks old, n = 16 per group) were inoculated i.p. with 102 PFU DENV-2 RL strain. Mice were treated twice daily via oral gavage with vehicle or JNJ-A07 using five different doses: 30, 10, 3, 1 and 0.3 mg per kg. Treatment was initiated 1 h before DENV infection and continued for 6 consecutive days. Each group was subdivided in two smaller groups (A and B; n = 8 each), from which blood was collected on alternating days: on day 1, 3 and 5 for the A groups, and on day 2, 4 and 6 for the B groups. On day 8 and day 11 after infection, mice from the A and B groups, respectively, were euthanized, and blood, spleen, kidney and liver were collected and stored at –80 °C until further use.
The protective effect of JNJ-A07 on the development of virus-induced disease was assessed in a lethal DENV challenge model (survival study). To mimic antibody-dependent-enhancement-induced dengue disease, AG129 mice (7–11 weeks old, females, n = 10 per group) were injected i.p. with 100 μl (1:50 diluted) anti-flavivirus group antigen antibody, clone D1-4G2-4-15 (‘4G2’; Millipore) 1 day before challenge with DENV-2 RL strain (106 PFU, i.p.). Mice were treated twice daily by oral gavage with either vehicle or JNJ-A07 at a dose of 30, 10, 3 or 1 mg per kg. Treatment was initiated 1 h before DENV challenge and continued for 5 consecutive days. On day 3 after infection, blood was collected for the quantification of viral RNA levels (only during one of the two studies). Mice were observed daily for body weight loss and the development of virus-induced disease. When reaching humane end points (body weight loss of ≥20%, hunched posture, ruffled fur, conjunctivitis, movement impairment, lower limb paralysis), mice were euthanized with pentobarbital. On day 25 after infection, the study was ended, and all surviving mice were euthanized with pentobarbital.
In delayed-treatment studies, AG129 female mice (7–11 weeks old, n = 8 per group) were inoculated i.p. with 102 DENV-2 RL strain. Treatment with JNJ-A07 (30 mg per kg, twice daily) was initiated on various days (day 1, 2, 3, 4, 5 or 6 after infection) and continued for 6 days. Mice treated with vehicle or JNJ-A07 on the day of infection (that is, 1 h after infection) were included as controls. Each group was subdivided in two smaller groups (A and B; n = 4 per group), from which blood was collected on alternating days: on day 1, 3, 5 and 7 after infection for the A groups, and on day 2, 4, 6 and 8 after infection for the B groups. On day 12 and day 14 after infection, mice from the A and B groups, respectively, were euthanized and blood was collected and stored at –80 °C until further use.
Cytokine measurement
Induction of pro-inflammatory cytokines was analysed in 20 μl plasma using a mouse cytokine 11-plex antibody bead kit (ProcartaPlex Mouse Th1/Th2 Cytokine Panel 11plex; EPX110-20820-901), which measures the expression of TNF, IFNγ, GM-CSF, IL-1β, IL-12p70, IL-2, IL-4, IL-5, IL-6, IL-13 and IL-18. Measurements were performed using a Luminex 100 instrument (Luminex) and were analysed using a standard curve for each molecule (ProcartaPlex). Statistical analysis was performed using a two-sided Kruskal–Wallis test, preceded by the identification of outliers using the two-sided Grubbs’ test (α = 0.05) in GraphPad Prism (GraphPad Software 9.0.0). P values were adjusted using the Dunn’s multiple comparisons correction method.
Statistical analysis for in vivo studies
Statistical power calculations considered the number of mice required to detect a significant reduction in viraemia compared with vehicle-treated controls. With groups of n = 8, a reduction of at least 0.8 log10 in viral RNA can be detected according to the independent t-test (with α = 0.05, power = 80% and a s.d. value of 0.5). In addition, statistical calculations considered the number of mice required to detect a significant improvement in survival compared with vehicle-treated controls. With groups of n = 11, a minimal survival rate of 60% for treated mice versus 0% in the untreated, infected control group can be demonstrated according to the Fisher’s exact test (with α = 0.05 and power = 80%). The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
To assess the effect of JNJ-A07 treatment on viral load in plasma, spleen, kidney and liver for each treatment group compared with the vehicle-treated mice (viraemia studies), the two-sided Kruskal–Wallis test was applied. P values from the Kruskal–Wallis test were adjusted using the Holm’s multiple comparisons correction method. To assess the effect of JNJ-A07 treatment on viral load in plasma for each treatment group compared with the vehicle-treated mice that were treated with the anti-flavivirus group antigen antibody (viraemia + 4G2 antibody), a Tobit regression model was applied. The (two-sided) P values were adjusted using the Bonferroni’s multiple comparisons correction method. For the viral kinetics studies and the delayed-treatment studies, a batch approach was applied to calculate the viral load AUC using the PΚ R package46. This package estimates a mean AUC value for settings where animals are measured at varying time points within a treatment group. Within each experiment, the mean AUC value and 95% CIs was determined for each group. The AUC was calculated using the LOD (2.6 log10 copies per millilitre) as the lowest limit. In case the CI of a compound-treated group overlapped with that of the vehicle-treated group, the groups were considered not to differ. In case the CIs did not overlap, the groups were considered to substantially differ. In the delayed-treatment studies, the viral load AUC for each of the compound-treated groups was calculated from the day treatment was initiated until the end of the study and compared with that of the vehicle-treated group. The Fisher’s exact test was used to determine whether the survival rate on day 25 for each compound treatment group differed significantly from that of the vehicle group. P values were adjusted using the Holm’s multiple comparisons correction method. P values of ≤0.05 were considered significant and P values lower than 0.0001 are depicted as P < 0.0001 in the graphs.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The genome sequence of DENV-2 RL is deposited at GenBank (accession MW741553). The synthesis and chemical characterization of all compounds described in this paper is provided as Supplementary Information (Supplementary Methods). The uncropped images of the western blots shown in Fig. 1, Extended Data Figs. 5 and 6 are presented in Supplementary Figs. 1–6. All data supporting the findings of this study are available within the Article, the source data or the Supplementary Information. Source data are provided with this paper.
Code availability
A custom script43 was used to derive the amino acid composition of each sample for all coding sequences per DENV genotype, which was not specifically developed for this research but for all similar analyses. The code for the custom script is deposited as part of the pipeline VirVarSeq but is individually accessible on the Open Source software platform SourceForge at https://sourceforge.net/projects/virtools/?source=directory. The code for this specific variant detection script is ‘codon_table.pl’. Graphs and figures were generated using Microsoft PowerPoint, GraphPad Prism (v.9.0.0) or Adobe Illustrator (v.25.4.1); the software is made available by KU Leuven through a group licence. In some figures, basic templates obtained from the Servier Medical Art library (https://smart.servier.com/) were used.
Change history
20 October 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41586-021-04123-9
References
Dengue and Severe Dengue. World Health Organization, https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue (15 April 2019).
Bhatt, S. et al. The global distribution and burden of dengue. Nature 496, 504–507 (2013).
Ayukekbong, J. A., Oyero, O. G., Nnukwu, S. E., Mesumbe, H. N. & Fobisong, C. N. Value of routine dengue diagnosis in endemic countries. World J. Virol. 6, 9–16 (2017).
Paixão, E. S., Teixeira, M. G. & Rodrigues, L. C. Zika, chikungunya and dengue: the causes and threats of new and re-emerging arboviral diseases. BMJ Glob. Health 3, e000530 (2018).
Simmons, C. P., Farrar, J. J., van Nguyen, V. & Wills, B. Dengue. N. Engl. J. Med. 366, 1423–1432 (2012).
Messina, J. P. et al. The current and future global distribution and population at risk of dengue. Nat. Microbiol. 4, 1508–1515 (2019).
Duong, V. et al. Asymptomatic humans transmit dengue virus to mosquitoes. Proc. Natl Acad. Sci. USA 112, 14688–14693 (2015).
ten Bosch, Q. A. et al. Contributions from the silent majority dominate dengue virus transmission. PLoS Pathog. 14, e1006965 (2018).
Halstead, S. B. Pathogenesis of dengue: dawn of a new era. F1000Research 4, 1353 (2015).
Katzelnick, L. C. et al. Antibody-dependent enhancement of severe dengue disease in humans. Science 358, 929–932 (2017).
Dengue vaccine: WHO position paper—July 2016. Wkly Epidemiol. Rec. 91, 349-364 (2016).
Wilder-Smith, A. et al. Deliberations of the strategic advisory group of experts on immunization on the use of CYD-TDV dengue vaccine. Lancet Infect. Dis. 19, e31–e38 (2019).
Dengue vaccine: WHO position paper—September 2018. Wkly Epidemiol. Rec. 93, 457–476 (2018).
Low, J. G., Gatsinga, R., Vasudevan, S. G. & Sampath, A. In Dengue and Zika: Control and Antiviral Treatment Strategies. Advances in Experimental Medicine and Biology (eds Hilgenfeld, R. & Vasudevan, S. G.) 319–332 (Springer, 2018).
Whitehorn, J. et al. Dengue therapeutics, chemoprophylaxis, and allied tools: state of the art and future directions. PLoS Negl. Trop. Dis. 8, e3025 (2014).
Bardiot, D. et al. Discovery of indole derivatives as novel and potent dengue virus inhibitors. J. Med. Chem. 61, 8390–8401 (2018).
Schmid, M. A. & Harris, E. Monocyte recruitment to the dermis and differentiation to dendritic cells increases the targets for dengue virus replication. PLoS Pathog. 10, e1004541 (2014).
Touret, F. et al. Phylogenetically based establishment of a dengue virus panel, representing all available genotypes, as a tool in dengue drug. Antiviral Res. 168, 109–113 (2019).
Płaszczyca, A. et al. A novel interaction between dengue virus nonstructural protein 1 and the NS4A-2K–4B precursor is required for viral RNA replication but not for formation of the membranous replication organelle. PLoS Pathog. 15, e10077362019 (2019).
Chatel-Chaix, L. et al. A combined genetic–proteomic approach identifies residues within dengue virus NS4B critical for interaction with NS3 and viral replication. J. Virol. 89, 7170-7186 (2015).
Moquin, S. A. et al. NITD-688, a pan-serotype inhibitor of the dengue virus NS4B protein, shows favorable pharmacokinetics and efficacy in preclinical animal models. Sci. Transl. Med. 13, eabb2181 (2021).
Clapham, H. E., Tricou, V., Van Vinh Chau, N., Simmons, C. P. & Ferguson, N. M. Within-host viral dynamics of dengue serotype 1 infection. J. R. Soc. Interface 11, 20140094 (2014).
Sim, S. et al. Tracking dengue virus intra-host genetic diversity during human-to-mosquito transmission. PLoS Negl. Trop. Dis. 9, e0004052 (2015).
Simmons, C. P. et al. Recent advances in dengue pathogenesis and clinical management. Vaccine 33, 7061–7068 (2015).
Nguyen, N. M. et al. Host and viral features of human dengue cases shape the population of infected and infectious Aedes aegypti mosquitoes. Proc. Natl Acad. Sci. USA 110, 9072–9077 (2013).
Simmons, C. P. et al. Therapeutics for dengue: recommendations for design and conduct of early-phase clinical trials. PLoS Negl. Trop. Dis. 6, e1752 (2012).
Low, J. G., Ooi, E. E. & Vasudevan, S. G. Current status of dengue therapeutics research and development. J. Infect. Dis. 215, S96–S102 (2017).
Unlocking the Potential of Preventive Therapies for Malaria. World Health Organization, https://www.who.int/malaria/media/preventive-therapies/en/ (7 April 2017).
Zou, J. et al. Characterization of dengue virus NS4A and NS4B protein interaction. J. Virol. 89, 3455–3470 (2015).
Miller, S., Sparacio, S. & Bartenschlager, R. Subcellular localization and membrane topology of the dengue virus type 2 non-structural protein 4B. J. Biol. Chem. 281, 8854–8863 (2006).
Chatel-Chaix, L. et al. Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe 20, 342–356 (2016).
Zmurko, J., Neyts, J. & Dallmeier, K. Flaviviral NS4B, chameleon and jack-in-the-box roles in viral replication and pathogenesis, and a molecular target for antiviral intervention. Rev. Med. Virol. 25, 205–223 (2015).
Umareddy, I., Chao, A., Sampath, A., Gu, F. & Vasudevan, S. G. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J. Gen. Virol. 87, 2605–2614 (2006).
Janssen Pharmaceuticals & KU Leuven. Preparation of mono- or di-substituted derivatives as dengue viral replication inhibitors. Patent WO/2016/050841 (2016).
Janssen Pharmaceuticals & KU Leuven. Preparation of mono- or di-substituted derivatives as dengue viral replication inhibitors. Patent WO/2016/050831 (2016).
Janssen Pharmaceuticals & KU Leuven. Preparation of substituted indoline derivatives as dengue viral replication inhibitors. Patent WO/2017/167951 (2017).
Novartis. N-substituted tetrahydrothienopyridine derivatives as antiviral agents and their preparation. Patent WO/2019/244047 (2019).
Fischl, W. & Bartenschlager, R. High-throughput screening using dengue virus reporter genomes. Methods Mol. Biol. 1030, 205–219 (2013).
Aubry, F. et al. Single-stranded positive-sense RNA viruses generated in days using infectious subgenomic amplicons. J. Gen. Virol. 95, 2462–2467 (2014).
Kum, D. B. et al. A yellow fever–Zika chimeric virus vaccine candidate protects against Zika infection and congenital malformations in mice. NPJ Vaccines 3, 56 (2018).
De Burghgraeve, T. et al. 3′,5′Di-O-trityluridine inhibits in vitro flavivirus replication. Antiviral Res. 98, 242–247 (2013).
Kaptein, S. J. F. et al. A derivate of the antibiotic doxorubicin is a selective inhibitor of dengue and yellow fever virus replication in vitro. Antimicrob. Agents Chemother. 54, 5269–5280 (2010).
Verbist, B. M. et al. VirVarSeq: a low-frequency virus variant detection pipeline for Illumina sequencing using adaptive base-calling accuracy filtering. Bioinformatics 31, 94–101 (2015).
Münster, M. et al. A reverse genetics system for Zika virus based on a simple molecular cloning strategy. Viruses 10, E368 (2018).
Zmurko, J. et al. The viral polymerase inhibitor 7-deaza-2′-C-methyladenosine is a potent inhibitor of in vitro Zika virus replication and delays disease progression in a robust mouse infection model. PLoS Negl. Trop. Dis. 10, e0004695 (2016).
Jaki, T. & Wolfsegger, M. J. Estimation of pharmacokinetic parameters with the R package PK. Pharm. Stat. 10, 284–288 (2011).
Acknowledgements
This work was supported by a Seeding Drug Discovery Strategic Award from the Wellcome Trust (grant 089328/Z/09 and grant 106327/Z/14) and received funding from the Flanders Agency Innovation & Entrepreneurship (VLAIO O&O grants IWT 150863 and HBC.2019.2906). Part of this research work was performed using the ‘Caps-It’ research infrastructure (project ZW13-02) that was financially supported by the Hercules Foundation (FWO) and Rega Foundation, KU Leuven. We thank M. Flament, R. Pholien, C. De Keyzer, C. Vanderheydt, E. Maas, S. Claes and the staff at the Rega animal facility at KU Leuven; E. Peeters, S. De Bruyn, N. Verheyen, C. Van Hove, E. Coesemans, B. De Boeck, A. Beckers, P. Gysemberg, T. Loomans and K. Allaerts at Janssen Pharmaceutica; and J. Fortin, F. Doublet and P. Muller at Janssen-Cilag for technical assistance.
Author information
Authors and Affiliations
Contributions
S.J.F.K. and J.N. planned, coordinated and executed the experimental virology work at KU Leuven. O.G. and M.V.L. planned, coordinated and executed the experimental virology work at Janssen Pharmaceutica. P.G. executed the experimental virology work at Janssen Pharmaceutica. D.K., L.C.-C., M.M. and R.B. planned, coordinated and executed the experimental virology work at Heidelberg University. D.B. planned, coordinated and executed the medicinal chemistry work at Cistim. K.D. did experimental work at KU Leuven and advised on the design of experiments. K.T. performed whole-genome sequence analysis at Janssen Pharmaceutica. M.C. performed the statistical analyses at Janssen Pharmaceutica. G.Q., F.T. and X.d.L. planned, coordinated and executed the experimental work at UVE. B.K., J.-F.B., T.H.M.J. and P.R. planned, coordinated and executed the medicinal chemistry work at Janssen Pharmaceutica. B.S. planned and coordinated the pharmacokinetics and pharmacodynamics work at Janssen Pharmaceutica. A.M. and P.C. coordinated and guided the experimental medicinal chemistry work at CD3. P.C. and J.N. designed and initiated the project. K.S. and M.V.L. initiated the project at Janssen Pharmaceutica. J.N., P.C., A.M., S.J.F.K., O.G., R.B., M.V.L. and K.S. secured funding from external organizations. S.J.F.K., O.G., M.V.L. and J.N. wrote the manuscript with contributions from K.S., D.K. and R.B., and comments from all authors.
Corresponding authors
Ethics declarations
Competing interests
S.J.F.K., O.G., A.M., B.K., J.-F.B., D.B., B.S., T.H.M.J., K.D., P.R., K.S., P.C., M.V.L. and J.N. have filed a patent application claiming the discovery of this class of antiviral molecules as DENV replication inhibitors (WO/2017/167951). The other authors declare no competing interests.
Additional information
Peer review information Nature thanks Brett Lindenbach, R. Guy and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Dose-response curves of the antiviral activity of JNJ-A07 against DENV-2 on various cell types.
a-f, The antiviral effect (% Inhibition viral RNA replication, % Inhibition eGFP expression, or % Inhibition of infected cells) is depicted by white dots. The effect of JNJ-A07 on cell growth is depicted by grey dots. Assays were performed on Vero cells (a and c), Huh-7 hepatoma cells (b), C6/36 mosquito cells (d), human monocytic leukemia THP-1 cells expressing the DC-SIGN receptor (e), and immature dendritic cells (imDCs) (f). Cells were infected with either the DENV-2/16681/eGFP strain (a-b, e), DENV-2/16681 (f) or the DENV-2 RL strain (c-d). Data represent mean values ± s.d. from two (for Vero and C6/36 cells using DENV-2 RL, and for imDCs using DENV-2/16681), three (for THP-1/DC-SIGN cells using DENV-2/16681), and at least five (for Vero and Huh-7 cells using DENV-2/16681) independent experiments.
Extended Data Fig. 2 JNJ-A07 is highly specific for DENV.
a, Antiviral activity of JNJ-A07 against various of RNA and DNA viruses. CHIKV, chikungunya virus; HadV, human adenovirus; HBV, hepatitis B virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; hRV, human rhinovirus; IVA, influenza virus A; IVB, influenza virus B; RSV, respiratory syncytial virus; rVSV, recombinant vescular stomatitis virus; VACV, vaccinia virus; JEV, Japanese encephalitis virus; WNV, West Nile virus; YFV, yellow fever virus; ZIKV, Zika virus. ND, not determined. b, NS4B sequence alignment of related flaviviruses. The NS4B protein sequence of DENV-2/16681 was aligned with corresponding sequences from JEV strain JEV CNS769 Laos 2009 (GenBank KC196115), tick-borne encephalitis virus strain Oshima 5.10 polyprotein gene (GenBank MF374487), WNV isolate R94224 CDC polyprotein gene (GenBank MF004388), YFV isolate Bolivia 88 1999 polyprotein gene (GenBank MF004382) and ZIKV strain HPF 2013 (GenBank KJ776791) using Clustal Omega Version 2.1. Post-processing was conducted with Jalview 2.11.1.3. The DENV NS4B topology model was added manually based on Miller et al.30. Black arrowheads are pointing at locations associated with compound-resistance.
Extended Data Fig. 3 Time-of-drug-addition and in vitro resistance selection.
a, Experimental setup of the time-of-drug-addition assay (TOA). b, TOA and in vitro kinetics of DENV-2 replication. In vitro DENV RNA replication in the absence of compound is depicted by the red curve. Onset of intracellular viral replication is at 10 h post-infection (p.i.), as shown in the inset. The inhibitory effect of JNJ-A07 on DENV replication when added at different time points p.i. is depicted by the blue curves (0.0001 µM, light blue; 0.001 µM, dark blue). The broad-spectrum RNA virus inhibitor 7-deaza-2’-C-methyladenosine (7DMA) served as positive control (black curve with white circles). Data (mean ± s.d.) from at least three independent experiments. c, Experimental approach of in vitro resistance selection. d-e, The dynamics of appearance of mutations was studied using whole genome sequencing. JNJ-A07 selected for mutations in NS4B, which were not present in the wild-type (WT) viruses that were passaged along without any drug pressure, of two independently selected resistant strains. Results for the A and B sample are shown in (d-e), respectively. Each coloured line shows the dynamics of appearance of a certain mutation during passaging of the virus in presence of JNJ-A07; the mutation is depicted in the same colour. Whole genome sequencing was performed on DENV variants harvested at every 5th passage (P) and at the end of the experiment (i.e., passage 43). One passage represents a one-week time span. The dotted line represents the cut off (15%) for the detection of variants compared with WT in the virus population. The increasing EC50 values, as determined by microscopic evaluation of virus-induced CPE, are depicted below the graphs. f, Mutations in DENV NS4B identified at end point after in vitro resistance selection using JNJ-A07 and Analogue 1. g, Natural occurrence of the NS4B mutations in clinical isolates.
Extended Data Fig. 4 Replication properties of resistant subgenomic replicons and DENV strains.
a, Schematic representation of the subgenomic DENV-2/16681 reporter replicon sgDVs-R2A38. b, Effect of resistance mutations in NS4B on replication fitness. Resistance mutations identified in Extended Data Fig. 3 were introduced into sgDVs-R2A. A replication-deficient replicon containing an inactivating mutation in the NS5 RNA-dependent RNA polymerase domain (GND) served as negative control. Huh-7 cells were transfected with in vitro transcribed RNA of WT or mutant sgDVs-R2A and lysed at the time points given at the top right and Renilla luciferase activity was measured as marker of replication. Relative light units (RLU) were normalized to the 4 h value, reflecting transfection efficiency. Plotted are the mean ± s.d. from at least three independent experiments, each carried out with independent RNA preparations. c, In vitro growth kinetics of resistant DENV (blue bars) compared to WT DENV (grey bars) on Vero E6 cells for the A sample. The virus titer in the supernatant was determined by plaque assay. d, In vitro growth kinetics of resistant DENV (blue bars) compared to WT DENV (grey bars) on C6/36 cells for the A sample. Viral RNA load in the supernatant was determined by RT-qPCR. e, Infectious virus on day 11 p.i. in the supernatant of C6/36 cells infected with resistant DENV (blue bars) or WT DENV (grey bars) for the A sample, as determined by plaque assay. f, In vitro growth kinetics of resistant DENV (dark blue bars) compared to WT DENV (light grey bars) on C6/36 cells for the B sample. Viral RNA load in the supernatant collected on day 1–10 p.i. was determined by RT-qPCR. Data are from a single experiment (d-f) or mean values ± s.d. from three independent experiments (c). LOD, limit of detection; LLOQ, lowest limit of quantification.
Extended Data Fig. 5 Impact of JNJ-A07 on the interaction between NS3 and various NS4B species.
a, Experimental design to study the impact of JNJ-A07 on the interaction between NS3 and WT or mutant NS4B. b, Captured protein complexes were analyzed by Western blot. A representative Western blot is shown. c-d, Western blot analysis to determine the NS3-NS4B interaction strength. The uncropped images of b-d are presented in Supplementary Fig. 1-3. Numbers on the left are molecular weights (kDa). GAPDH served as loading control for cell lysates (input). e-g, Signal intensities (from three independent blots) of NS3, NS4A-2K-NS4B, 2K-NS4B and NS4B were normalized to WT NS4A-2K-NS4B-HACt in DMSO-treated control cells. Protein ratios (mean ± s.e.m.) were calculated for each sample. Repeated measures one-way ANOVA with subsequent Dunnett’s multiple comparisons test was used for statistical analysis. ns, not significant. h-j, Protein intensities (mean ± s.e.m.; three independent experiments) for WT (h) and compound-resistant NS4B-mutants V91A (i) and T108I (j), normalized to an untreated WT control. For statistical analysis, JNJ-A07-treated samples were compared with the corresponding untreated control (left of the dashed line) using ordinary one-way ANOVA with subsequent Dunnett’s multiple comparisons test. k, EC50 values (mean ± s.e.m.) for protein ratios in (e-g) obtained by fitting four-parameter dose-response curves to the results from each individual experiment. Fold change in EC50 is the ratio between the mean EC50 for WT and the respective mutant constructs. l, JNJ-A07 potentially slows down the processing dynamics of the NS4A-2K-NS4B precursor (illustrated by the hourglass icon), which is first cleaved by the NS2B-NS3 protease at the NS4A-2K cleavage site. 2K-NS4B is subsequently processed by the host signal peptidase complex into mature NS4B and 2K.
Extended Data Fig. 6 JNJ-A07 does not disrupt existing NS3–NS4B complexes.
a, Experimental setup to study the kinetics of JNJ-A07-induced block of the NS3–NS4B interaction. b, Impact of JNJ-A07 on forming or pre-formed NS3–NS4B complexes. Huh-7 T7 NS2B-NS3 cells treated with 0.035 µM JNJ-A07 or equal amounts of DMSO were harvested at 1, 8, or 24 h after drug addition. Lysates were subjected to HA-specific pull-down and analyzed by Western blot (enrichment factor 5). A representative Western blot is shown. Numbers on the left represent molecular weights (kDa). c, Experimental setup of the in cellulo assay, in which Huh-7 cells were infected with DENV-2(NS4B-HA*)20 at an MOI of 1. At 48 h p.i., cells were treated for given periods with 500 nM of Analogue 2 or DMSO. d, NS3–NS4B complexes were enriched by NS4B-HA* pull-down and total lysates (input) and immune complexes (pull-down) were analysed by Western blot. NS3/NS4B ratios (depicted below the picture) were normalized to non-drug treated samples (n = 1). e, Experimental setup of the in vitro drug assay to investigate the effect of the drug on established NS3–NS4B complexes. Cell lysates were treated with 1 µM of Analogue 2 or equal amounts of DMSO added to the lysis buffer and incubated for 2 h at different temperatures in order to test complex stability. Subsequently, NS4B-HACt pull-down was performed. f, Western blot analysis (n = 1) analogous to the one shown in (d). The lower protein amount observed with the sample incubated at 37 °C was most likely due to proteolytic degradation in spite of adding protease inhibitors. For uncropped images of the representative blots in (b, d, f), see Supplementary Fig. 4-6. GAPDH (b, f) or β-actin (d) served as loading control for cell lysates (input).
Extended Data Fig. 7 In vivo efficacy of JNJ-A07 on viral RNA and cytokine levels.
a-c, Inhibitory effect of JNJ-A07 on viral RNA levels in spleen (a), kidney (b) and liver (c) on day 3 p.i. from AG129 mice treated twice-daily with 30 (white dots), 10 (light blue dots), 3 (dark blue dots) or 1 (grey dots) mg/kg JNJ-A07, as compared to vehicle-treated mice (red dots). Data are from two independent studies with n = 8 per group in each study. d-g, IL-18 (d), IFNγ (e), TNF (f), and IL-6 (g) levels in plasma on day 3 p.i. (from one of the viremia studies in Fig. 2b). h, Inhibitory effect of JNJ-A07 on viral RNA levels in plasma on day 3 p.i. in the survival study (also see Fig. 2c). Dosing groups were similar to those in (a-c). Treatment started 1 h before infection. Mice were injected with the Anti-Flavivirus antibody one day before infection. Data are from one study with n = 10 per group. i, Inhibitory effect of NITD-688 on viral RNA levels on day 3 p.i. in AG129 mice treated twice-daily with 100 (yellow dots), 30 (white dots), 10 (light blue dots) or 3 (dark blue dots) mg/kg NITD-688, as compared to vehicle-treated mice (red dots). Treatment started 1 h before infection. Data are from one study with n = 8 per group. Individual data and median values are presented. Undetermined Ct values were imputed at a Ct value of 40 (which is the LOD), corresponding to 2.6 log10 viral RNA copies/mL. Statistical analysis was performed using the two-sided Kruskal-Wallis test (a-c, i) or a Tobit regression model (h). P values were adjusted using the Holm’s (a-c), Dunn’s (d-g) or Bonferroni’s (h-i) multiple comparisons correction method. ns, not significant, as compared to vehicle-treated mice. LLOQ, lowest level of quantification.
Extended Data Fig. 8 Efficacy of JNJ-A07 in the in vivo kinetics study.
a, Schematic outline of the in vivo kinetics study. Each treatment group was sub-divided in group A and B (n = 8, per group) for blood collection on alternating days. b, Weight curves (mean values ± s.d.) of AG129 mice for the different treatment groups during the in vivo kinetics study (two independent studies). Colours of the dots represent the different treatment groups, as specified in (c-g). c-g, Inhibitory effect of JNJ-A07 on viremia on various days p.i. in mice treated twice-daily with 30 mg/kg (white dots, n = 8), 10 mg/kg (light blue dots, n = 8), 3 mg/kg (dark blue dots, n = 16), 1 mg/kg (grey dots, n = 8), or 0.3 mg/kg (green dots, n = 8) JNJ-A07, as compared to vehicle-treated mice (red dots, n = 16). Treatment was initiated 1 h before intraperitoneal infection. Data (median ± s.d.) are from two independent studies. Undetermined Ct values were imputed at a Ct value of 40 (which is the limit of detection), corresponding to 2.6 log10 viral RNA copies/mL. The mean AUC value and 95% CI was determined for each group. In case the CIs did not overlap, groups were considered to differ markedly. LLOQ, lowest level of quantification.
Supplementary information
Supplementary Information
This file contains Supplementary Methods and Supplementary Figs. 1–6.
Source data
Rights and permissions
About this article

Cite this article
Kaptein, S.J.F., Goethals, O., Kiemel, D. et al. A pan-serotype dengue virus inhibitor targeting the NS3–NS4B interaction. Nature 598, 504–509 (2021). https://doi.org/10.1038/s41586-021-03990-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03990-6
- Springer Nature Limited
This article is cited by
-
The endoplasmic reticulum (ER): a crucial cellular hub in flavivirus infection and potential target site for antiviral interventions
npj Viruses (2024)
-
mRNA-encoded Cas13 can be used to treat dengue infections in mice
Nature Microbiology (2024)
-
Challenges in combating arboviral infections
Nature Communications (2024)
-
Multiplexed multicolor antiviral assay amenable for high-throughput research
Nature Communications (2024)
-
A robust mouse model of HPIV-3 infection and efficacy of GS-441524 against virus-induced lung pathology
Nature Communications (2024)