

Abstract
Polynesia was settled in a series of extraordinary voyages across an ocean spanning one third of the Earth1, but the sequences of islands settled remain unknown and their timings disputed. Currently, several centuries separate the dates suggested by different archaeological surveys2,3,4. Here, using genome-wide data from merely 430 modern individuals from 21 key Pacific island populations and novel ancestry-specific computational analyses, we unravel the detailed genetic history of this vast, dispersed island network. Our reconstruction of the branching Polynesian migration sequence reveals a serial founder expansion, characterized by directional loss of variants, that originated in Samoa and spread first through the Cook Islands (Rarotonga), then to the Society (Tōtaiete mā) Islands (11th century), the western Austral (Tuha’a Pae) Islands and Tuāmotu Archipelago (12th century), and finally to the widely separated, but genetically connected, megalithic statue-building cultures of the Marquesas (Te Henua ‘Enana) Islands in the north, Raivavae in the south, and Easter Island (Rapa Nui), the easternmost of the Polynesian islands, settled in approximately ad 1200 via Mangareva.
Similar content being viewed by others

Main
The history of the human settlement of Polynesia has long been examined by its residents5 and has been an open question worldwide since at least the time of Captain James Cook6,7. More recently, the prevalence of certain health conditions in these island founder populations has attracted the interest of medical geneticists8. However, although essential for both medical research and historical understanding, little is known about the human genetic structure of this oceanic expanse, our planet’s last habitable region to be settled.
Background
The settlement sequence of Polynesian islands remains particularly difficult to unravel using comparative linguistic or cultural approaches owing to the rapidity of the initial expansion and the subsequent cultural exchanges between islands7,9,10,11. Meanwhile, the archaeological estimates for settlement dates remain debated, and have recently been revised forward across eastern Polynesia by up to a millennium2,3,4,12,13. Previous region-wide Polynesian genetics studies have considered only globin gene polymorphisms14 or have been restricted to near (western) Polynesia15 and the Society Islands16 and lacked an ancestry-specific approach. Meanwhile, ancient DNA studies have sequenced only four samples from one island in western Polynesia and three near-modern samples from one island in eastern Polynesia, all with low genotype density, still lower between-sample genotype overlap, and different time frames17,18. Here we use a dataset of modern samples that is two orders of magnitude larger to examine detailed intra- and inter-island population substructure across all of Polynesia (Supplementary Tables 1, 2). We leverage our sample size to perform directionality and network analyses, and leverage our high-density, overlapping genotypes from coexistent individuals to perform within-generation autosomal haplotype matching, allowing us to date and reconstruct the settlement paths of these islands for the first time. This study also allows us to demonstrate new ancestry-specific techniques for analysing genomic data from underrepresented, admixed populations.
The Polynesians are predominantly descended from Austronesian-speaking voyagers17 who trace their linguistic origins to Taiwan;9 their ancestral expansion is thought to have proceeded into Island Southeast Asia and eventually out into the Pacific19. The Austronesian-speaking settlers of the western Pacific (Fiji, Tonga and Samoa) went on to people the widely dispersed islands in the vast ocean to their east through extraordinary voyages of exploration and settlement2,20. Historians believe that family groups of 30–200 individuals sailed in double-hulled canoes across thousands of kilometres of open ocean to inhabit each new Polynesian island group21,22. The first arrivals to these isolated island groups are thought to have experienced rapid initial growth, driven by the abundant resources of unfished reefs, huge seabird colonies and flightless birds (that soon became extinct) unhabituated to humans2,7,22,23,24,25. These rapidly expanding island populations then initiated new voyages of exploration in search of—according to some theories—further untapped resources26, a model supported by early oral histories27. Geological analyses of Polynesian trade goods, particularly adzes, indicate that the remote Polynesian islands remained in trade contact with one another for several centuries26,28,29. However, these contacts were necessarily limited in frequency by the vast distances between island groups and limited in size by the capacities of the double-hulled sailing canoes21.
Under this historical model, we would expect the minor alleles on these isolated Pacific islands to be lost in a telescoping fashion following the order of the islands’ colonization—a range expansion30—owing to the compounding succession of founding bottlenecks. We confirm this hypothesis below and then use its consequence—that the genetic composition of each remote island group is dominated by the contribution of its founders (Extended Data Fig. 3), whose descendants rapidly populated it—to reconstruct the Polynesian settlement sequence. We finally evaluate this model for self-consistency to test its validity.
Dimensionality of Polynesian genetics
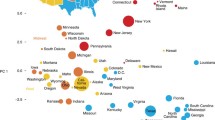
In direct contrast to continental (and nearshore island) populations, in which genetic substructure is shaped by large historical migrations, conquests and diffusions occurring freely across the two-dimensional landmass surface, thus producing two-dimensional projections of genetic variance that mirror geography31,32, we find that Polynesian population structure exhibits high dimensionality (Supplementary Fig. 1) not at all reflective of geography (Fig. 1a), with islands diverging separately in a standard principal component analysis (PCA) (Supplementary Figs. 2, 3). Indeed, the first two dimensions of major genomic variation—even in an ancestry-specific PCA of the Polynesian individuals (Fig. 1b)—do not separate islands geographically, as they do for within-continent populations33,34 (Extended Data Figs. 1, 2). Instead, each successive principal component captures the genetic drift of a particular island or island group (Fig. 1b, c, Supplementary Fig. 2), illustrating that genetic variance between these islands is dominated by their founder effects, not by diffusion clines or migration gradients. To further complicate such a standard variance-based approach (Supplementary Figs. 2–4) to genomic dimensionality reduction, the Polynesian islands differ widely in genetic diversity. Because the originating islands have much greater diversity (as discussed below), they dominate the first principal component when included in the PCA (Supplementary Fig. 3). Furthermore, many individuals, including all samples from certain islands, have some amount of non-Polynesian ancestry: European, Native American and African33. The presence of large-scale post-colonial admixture from such divergent ancestry sources completely confounds Polynesian-focused interpretations of within-island and between-island variance when these admixed samples are included in the PCA (Supplementary Fig. 4).

a–c, Ancestry-specific PCA of islanders (with non-Asian derived ancestries, such as post-colonial European ancestry, masked) shows islands (a) diverging separately along each component (b, c), owing to the independence of genetic drift from each island’s founder effect. Neither geography nor settlement sequence can be discerned. The westernmost islands are omitted, as their greater diversity would otherwise dominate the first principal component (PC) (see Supplementary Fig. 2). The per cent variance explained by each of the first four principal component dimensions is listed along each axis. Dots represent individuals, and colours represent islands. d, Ancestry specific t-SNE plot of all sampled islanders, providing superior separation of each island group. The ancestral western Pacific islands are on the left and the easternmost Polynesian island (Rapa Nui) on the right. Important patterns are now evident; for instance, Rarotonga and the Palliser group appear at the centre of the eastern Polynesian islands while the other eastern islands radiate out from them, consistent with the settlement patterns we infer below. t-SNE preserves local relationships, but not global relationships (between widely separated clusters).
To overcome these threefold obstacles to visualizing relationships between islands, we applied a novel ancestry-specific version of a nonlinear dimensionality reduction technique, t-distributed stochastic neighbour embedding (t-SNE), applying it only to the genomic segments of Polynesian ancestry in our sampled individuals and employing a matrix completion step (Fig. 1d, Supplementary Fig. 5). In a plot of this ancestry-specific t-SNE method (Fig. 1d), the islands of the ancestral west—Taiwan, Island Southeast Asia (Sumatra), Fiji, Tonga and Samoa—are grouped on the left and the more recently settled eastern islands are on the right. Islands in archipelagos, such as the Cook Islands of Mauke, Atiu and Rarotonga, form neighbouring clusters. Rarotonga and Palliser appear at the centre of the eastern Polynesian islands, with the other eastern islands radiating out from them. This pattern is consistent across alternative dimensionality-reduction methods (Methods), including our ancestry-specific formulations of uniform manifold approximation (UMAP) (Supplementary Figs. 6, 7) and self-organizing map (SOM) (Supplementary Fig. 8), as well as our genetic-drift projection method (Supplementary Fig. 9).
Tree building and path reconstruction
Because individuals from each of the islands form coherent, separate clusters in all of the non-linear, variant-based projections (t-SNE, UMAP and SOM), we can define a meaningful variant-frequency vector for each island by averaging the single nucleotide polymorphism (SNP) dosage vectors across all individuals on that island. Again, we consider only genomic segments of Polynesian origin (Supplementary Tables 3, 4), since standard non-ancestry-specific analyses are confounded by the recent introduction of highly differentiated colonial ancestry, such as European, even when the proportion of that ancestry is small (Supplementary Fig. 10). Averaging across all individuals reduces noise and produces composite Polynesian-specific frequency vectors with little to no remaining missingness from masking. Using these island-specific Polynesian-variant frequency vectors, we compute statistics for each pair of islands (Extended Data Figs. 4a–d, 7, Supplementary Figs 11–19), including the average number of pairwise differences35 (π), variant inner product36 (outgroup − F3), fixation index (Fst), and directionality index37,38 (range expansion statistic) (ψ).
The directionality index ψ (Fig. 2a) measures the aggregate increase in frequencies of retained rare variants across the genome due to founder events, following the direction of a range expansion (Fig. 2b, Supplementary Discussion, ‘On psi’). The ψ-statistic gives crucial information that is not available from any genetic distance (π, F2, MixMapper39) or inner product (F3, TreeMix40) based methods; namely, a directionality arrow delineating a parental population from its child. Most human population studies have not required such directionality, as modern human populations are generally siblings, both having genetic drift from a no-longer extant, ancient parental population. That parental population, if available from ancient samples, is clearly indicated by the arrow of time (typically carbon dating). However, among the relatively recently settled Polynesian islands, genetic drift is created not by time, but by founder effects. Thus, the undrifted (parental) populations for most of these islands are still approximately extant: they are the populations of the originating islands. When constructing a population tree, this means that our dataset contains not only the terminal (leaf) nodes, but also the internal nodes, and we know their hierarchy from the ψ statistic. This directional knowledge enables us to use a tree-building algorithm that, unlike population tree algorithms currently in use36,39,40,41 (Supplementary Figs. 20, 21), is guaranteed to find the optimal tree out of the space of all possible trees in the presence of perfect data (see Methods section ‘Migration network reconstruction’). Using this more robust directionality-based algorithm (see Supplementary Discussion, ‘On tree-building’), the settlement path of Polynesia can be reconstructed (Fig. 2a).

a, Inferred genetic-based map of Polynesian origins for the islands sampled in our study (not to scale). The direction, line width and date for each arrow are based on inter-island statistics as described in the key and the text. For example, the widths of the arrows are inversely proportional to the value of the range expansion statistic (ψ) relative to Samoa. The order of arrow divergences indicates the order of shared drift among the child populations. Where they occur, these shared paths may indicate that one or more intermediate islands in the settlement sequence are missing from our dataset (Extended Data Fig. 5). This settlement sequence is consistent with a principal curve analysis (Extended Data Fig. 7). A sex-averaged generation time of 30 years was used, as found in several studies of pre-industrial populations (Supplementary Discussion, ‘On generation times and meiosis events’). Locations with prehistoric remains of megalithic statue building are also indicated (red asterisk). b, The range expansion statistic (ψ) shows a steady increase in retained rare variant frequencies (genetic surfing) along paths of settlement as a result of each successive founder effect. Note that each matrix element is computed on a different SNP set (rare variants found in some samples from both islands), so the matrix need not have a similar ordering across all rows or all columns—that it does is a confirmation of the range expansion process. Rapa Nui (Easter Island) is the easternmost island in our dataset with the most compounded series of founder effects. c, Example IBD segment length distributions for all pairs of individuals, one on Rapa Nui and the other on Mangareva (green), Palliser (blue), Rarotonga (red) and Samoa (black), used to fit the respective exponential decay constants (λ).
Dating
To estimate dates for the settlement events that we infer, we use a method for detecting DNA segments that have been inherited from a common ancestor (identical by descent (IBD)) for all pairs of individuals on different islands. Again, we consider only genomic segments of Polynesian ancestry. For each pair of islands A and B, we pool all of the Polynesian IBD segments shared between individuals on A paired with individuals on B, and fit an exponential curve to the resulting segment length distribution (Fig. 2c, Extended Data Fig. 4d). From the decay constant of this exponential curve, we compute the number of generations elapsed since divergence of the island pair (Extended Data Fig. 6, Supplementary Figs. 22–24). Fig. 2a shows the estimated divergence dates for all pairs of islands that are connected by a settlement path. Recent movement between islands, such as post-settlement trade contact, can introduce small numbers of longer, inter-island IBD segments, shifting the estimated divergence time towards the present, so we fit a truncated exponential. Nevertheless, these divergence dates should be seen as the terminus ante quem for the settlement of each child island (Fig. 2a, Extended Data Fig. 6, Extended Data Table 1). In the case of the most remote islands such as Rapa Nui, which are believed to have had no large-scale population exchanges with other islands, the IBD-based date should coincide closely with the actual date of settlement.
The dates that we infer from our genome-wide network analyses support the radiocarbon-based ‘short chronology’ from the comprehensive re-analysis of Wilmshurst et al.12, as corrected by Mulrooney et al.3 (Extended Data Table 1), as opposed to the previous nearly-one-thousand-year-older ‘long chronology’2,4, and as opposed to the intermediate chronology suggested by Spriggs and Anderson13 (Marquesas ad 300–600, remainder of eastern Polynesia ad 600–950). Only in the settlement of the Marquesas Island group, dated by Mulrooney to the late 1100s, and the Southern Cook Islands, dated even later by Wilmshurst to the mid-1200s, do we find different (earlier) dates. However, as Mulrooney et al. explain, the small sample size of early-dated historical sites on each island mean that new archaeological discoveries could revise Wilmshurst’s chronology (backward). Our dates, from the full island-wide ancestral history coded within modern Polynesians themselves, do not have these sampling issues affecting ancient DNA and artifacts. Indeed, modern genomes complement ancient artifacts, since issues affecting the artifacts—finding the earliest human sites on each island, determining whether objects within them are anthropogenic and determining whether those artifacts, often wood or charcoal, stem from young or old trees4,42 (inbuilt age)—do not affect the modern genomes, and vice versa.
Our date for the settlement of Rapa Nui is consistent with Wilmshurst and Mulrooney and also agrees closely with the date found by Hunt et al. (ad 1200) based on analyses of pollen in lake cores and soil erosion patterns43, as well as with recent radiocarbon dates of archaeological sites44. Furthermore, unlike the long chronology estimates (200 bc in the Marquesas), our settlement dates (ad 1140 on Fatu Hiva in the Marquesas, or 28.4 generations before 1989) agree with the genealogical oral histories of many Pacific Islanders themselves27 (ad 1005, or 29 generations before 1875, on Fatu Hiva). In the Tuamotus our dates (ad 1110, or 29.3 generations before 1989) agree even more closely with island’s oral histories45 (ad 1125, or 28 generations before 1965).
Our later divergence dates (ad 1330–1360) for some islands within archipelagos—North Marquesas (Nuku Hiva) in the Marquesas, Raivavae and Rimatara in the Australs—fall within the period of greatest inter-island trade contact in eastern Polynesia26. Either the last islands were discovered during this period of long-distance trade voyaging, as suggested by the dates of Schmid et. al. 4, or sufficient migration-to-inhabitant ratios still existed within archipelagos then to influence IBD distribution dates (Supplementary Fig. 23). Note that our reconstruction of the settlement path is independent of these date estimates, which are overlaid on it, and is more robust to later sporadic contact than IBD distributions are (see Methods sections ‘Polynesian ancestry-specific allele frequency analyses’ and ‘F4’).
Discussion
Our analyses indicate the following scenario for the settlement of eastern Polynesia. From western Polynesia, Polynesian voyagers reached Rarotonga in the Cook Islands around ad 830, having passed from Samoa along a route shared with the settlement of Fiji and Tonga. Rarotonga is the largest of the Cook Islands and has the highest elevation, with fertile volcanic soil watered by orographic rainfall26, creating distinct clouds. These clouds, together with a prominent mountain, make the island visible for long distances at sea and probably facilitated its discovery46. From this base, we find that settlers continued south around ad 1190 to Rapa Iti (a branch recently hypothesized from linguistic evidence47) and, separately, east to the smaller Cook Islands (Mauke and Atiu in our dataset).
Settlers also fanned out from Rarotonga northeast to the Society Islands (represented by Tahiti in our dataset but also containing the culturally significant island Ra‘iātea) around ad 1050, thence northeast to the Tuamotu Archipelago (represented by Mataiva in the Palliser group in our dataset) by ad 1110. At this time the widely scattered Tuamotu hub and other critical atolls in the expansion path (e.g. Nororotu in the Austral group) would have only recently emerged above falling sea levels (ad 900) and finished solidifying their topsoil and forests45,48 (Extended Data Fig. 5). Thus, our inferred dates and settlement path lend support to the idea that expansion into eastern Polynesia was mediated by the birth of those intermediary island clusters at the turn of the last millennium.
Stretching across central eastern Polynesia, the Tuamotu Archipelago was previously hypothesized to have served as a regional voyaging hub20,26,28, and our analysis indicates that it was from this hub that settlers made their way north to the Marquesas Islands (Nuku Hiva and Fatu Hiva in our dataset) and south to the Gambier Islands (Mangareva in our dataset) beginning in the mid-1100s. From Mangareva, we find that the expansion reached the easternmost inhabited Polynesian island, Rapa Nui (Easter Island), around ad 1210. This final leg had been suggested by some based on similarities between the Mangarevan and Rapanui languages49, and by similarities in their traditional stone ceremonial platforms50. This settlement sequence is also supported by our marker frequency-based genetic analyses, including ancestry-specific UMAP (Supplementary Fig. 6), drift projection (Supplementary Fig. 9), F-statistics (Supplementary Figs. 11, 12), principal curve analyses (Extended Data Fig. 7, Supplementary Fig. 17), diversity statistics (Supplementary Figs. 25–31), and ADMIXTURE clustering (Supplementary Figs. 32, 33).
Notably, we find that the population of Raivavae in the Australs arrived via the distant Tuamotus and Mangareva rather than via the other Austral islands of Tubuai and Rimatara (Fig. 1a, Supplementary Figs. 6, 7). Together with even more distant North and South Marquesas and Rapa Nui, each also with inferred settlement stemming from the Tuamotus, Raivavae had an ancient tradition of carving monumental anthropomorphic statues in stone. No other Austral island had these51; indeed, such immense sculptures are found only on those far-flung islands that we now show to have a common genetic source in the Tuamotu archipelago (Fig. 2a). It is also notable that it is only on islands that we infer were settled via the Tuamotus that pre-colonial Native American genetic contact has been identified, and its timing corresponds closely with our voyaging dates for this region33. This supports the theory that that contact occurred while the Polynesians were embarking on their easternmost, and longest, voyages of discovery.
The modern peoples of Polynesia harbour strong genetic evidence for a range expansion beginning in Samoa and propagating across eastern Polynesia through a series of telescoping founder events from the 11th and 12th centuries. Since this telescoping series of bottlenecks increased (via genetic surfing) the frequency of retained rare variants along the settlement path (see ψ-statistic, Fig. 2b), and since some of these variants are probably deleterious, future studies characterizing the individual frequencies and effects of these rare variants are desirable. We suggest that such large-scale sequencing and phenotyping studies should focus on the terminal islands in the settlement sequences that we have described, where compounded bottlenecking created the largest increase in frequencies (Fig 2b). We have shown that these particular islands also have high levels of homozygosity (Supplementary Figs. 25–27), which should increase the power to detect trait associations, and significant IBD, enabling IBD mapping, another useful approach52. Of note, two large modern Polynesian populations lie at the geographic termini of these serial bottleneck chains, Hawaii in the north and New Zealand in the south, and are thus notable candidates for such future large-scale association studies. We have introduced ancestry-specific computational methods for detailed characterization of Polynesian variant frequencies within admixed, modern samples, so potential admixture within future cohorts from such diverse populations should not be considered a barrier to designing these studies. Continued partnerships with these communities will be crucial53, since such studies will benefit both the personalized health understandings of these populations, as well as the global genetic understandings of all of us.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Sample collection and approvals
This work combines publicly available sequence data and newly generated SNP array data from samples collected over different time periods by the participating institutions (Supplementary Tables 1, 2). Written informed consent was obtained from all participants and research ethics approval and permits were obtained from the following institutions: Stanford University Institutional Review Board (IRB approval no. 20839), Oxford University Tropical Research Ethics Committee (reference no. 537-14), and the Scientific Ethics Committee of the Catholic University of Chile (reference no. 1971092). This study was also approved by the Council of Polynesian Elders for the community of Rapa Nui, along with local educational institutions, including the Lyceum Hoŋa’a o te Mana and the Lorenzo Baeza school for adults. Community engagement, including pre-participation presentations and post-participation return of results, were conducted throughout the project. Local approvals for engagement with the Rapa Nui community were obtained from the mayor (P. P. E. Paoa) of the municipality of Easter Island, and the study was registered with the National Corporation for Indigenous Development (CONADI), in accordance with the indigenous law no. 19.253. The guidelines of the UNESCO International Declaration on Human Genetic Data and the Declaration of Helsinki were followed throughout the study.
Genotyping
Sampled populations and genotyping platforms are detailed in Supplementary Tables 1, 2. A total of 26 populations were genotyped at the University of California, San Francisco (UCSF) using Affymetrix Axiom LAT-1 arrays. Genotype calling was performed following default parameters using Affymetrix’s Genotyping Console software. The average call rate was 98.5% for all newly genotyped samples. Before filtering and merging, the total number of SNPs called was 813,036. The resulting SNP density after merging with different reference panels varied across working datasets for downstream analyses, as detailed throughout the methods below.
Data curation
Quality control filters were applied across all sampled individuals using the Plink 1.9 package54, removing individuals with >1% of genotyped sites missing (mind .01), removing genotyped sites missing in > 1% of individuals (geno .01), and removing sites (18 SNPs) with extreme deviations from Hardy–Weinberg equilibrium (P-value less than 10 × 10−110). The independence of drift between these separated, small island populations leads us to expect some deviation from Hardy-Weinberg in Polynesia, so we do not apply a typical threshold here. All samples were analysed on the GRCh37 (hg19) genome build55. REAP56 was used to determine kinship coefficients using the ADMIXTURE clustering results discussed below; individuals with a kinship coefficient of >0.2 (first-degree relatives) were iteratively removed. Total numbers of individuals from each population after all filters were applied are given in Supplementary Table 1. After merging reference sequence data with sample genotyped data, strand inconsistencies were flipped when unambiguous, while ambiguous SNPs were removed, leaving 689,899 SNP sites. The recombination map from the 1000 Genomes project was used to assign genetic positions57 in centimorgans (cM).
Admixture analyses
Principal component analysis
EIGENSOFT 7.2.158 was used for all PCA. Linkage disequilibrium pruning (LD-pruning) was used across sliding 50-SNP windows with 10-SNP steps to remove variants with >0.5 squared correlation (-indep-pairwise 50 10 .5), leaving 461,571 SNPs for PCA. Plots were made with ggplot2 3.1.059 using R 3.5.260.
Global ancestry clustering analysis
Unsupervised ancestry clustering was performed using ADMIXTURE 1.3.061 on the LD-pruned dataset described above for PCA combining samples from all Pacific island populations together with continental references from Africa (Yoruba), Europe (Britain and Spain), East Asia (Japan and China) and the Americas (Aymara, Mapuche, Huilliche and Pehuenche) for a total of 686 samples. The numbers of samples from each population are given in Supplementary Tables 1, 2. An elbow62 was found in the cross-validation error plot at K = 7 clusters, with larger numbers of clusters delivering little improvement (Supplementary Fig. 32).
Local ancestry analysis
Semi-supervised local ancestry inference was performed for all filtered Pacific island samples (430 samples, 689,899 SNP sites) using RFMix v1.5.463 with two expectation-maximization (EM) iterations and references from the five ancestry clusters, namely African (60 West African Yoruba individuals), European (30 Spanish and 30 British individuals), Native American (60 Native American individuals from Puno, Peru), Ni-Vanuatuan (all 19 individuals from Vanuatu) and Remote Oceanian (60 individuals with <1% ancestry from outside the Pacific islands as identified by ADMIXTURE). The existence of these five ancestries within the Pacific island samples had been indicated by the K = 7 unsupervised global ADMIXTURE clustering run discussed above (Supplementary Fig. 32). The recommended RFMix settings (two EM iterations and a 0.2-cM window size) were used, and unphased samples were first phased by SHAPEITv2.837 with default settings64. A few Pacific island individuals, particularly in the Marquesas, were found to also have >5% East Asian ancestry in the ADMIXTURE results. This is likely owing to the post-colonial movement to those islands of Hakka immigrants from China for work in the 19th century65. Those individuals were removed, so that this sixth ancestry did not need to be separately resolved by local ancestry analysis.
Masking
As discussed above, modern Pacific Islanders are often admixed, possessing European and occasionally Native American and African ancestries (Supplementary Fig. 32). European ancestries entered Polynesia during the colonial period with the first European explorer (Magellan) arriving in the 16th century and significant immigration commencing in the early 19th century65. Native American ancestry, particularly from emigration of admixed Hispanic individuals from Chile, which annexed Rapa Nui (Easter Island), and African ancestry also entered33. Because ancestries fully (or partially) introduced via colonial settlement did not necessarily follow the same island settlement process (or founder sizes and dates) as the original Polynesian settlement, such ancestries need to be distinguished, necessitating an ancestry-specific approach66 (Supplementary Figs. 10, 20). For this reason we removed European chromosomal segments, as well as African and Native American, from the Pacific island samples. This step is called masking67,68, since variants located in certain ancestry segments (identified above by RFMix via haplotype sequence pattern matching) are masked (removed) from the analysis. We refer to the remaining (unmasked) chromosomal segments as Polynesian ancestry chromosomal segments (Supplementary Table 3), and we refer to analyses that use only these segments as Polynesian ancestry-specific analyses. (Such analyses may still include as references non-Polynesian populations, such as from Europe or Taiwan. These reference populations will of course have non-Polynesian ancestry and are not masked.) A description of which analyses were performed masked and which unmasked, and when references were used, is given in Supplementary Table 4.
Polynesian ancestry-specific allele frequency analyses
Treemix analysis
Treemix40 was run on the combined set of Pacific island and reference populations (Supplementary Tables 1, 2) using raw marker counts for each population. It was also run on the Pacific island populations using only the counts of markers found in Polynesian-ancestry chromosomal segments for each population, as described above.
Creation of Polynesian ancestry-specific genotype frequency vectors and matrix
For each of an individual’s two haplotypes, variants located in non-Polynesian ancestry segments were masked, as described above. The two haplotypes for each individual were then averaged to create a genotype frequency vector having, for each site, 0 when no alternate allele was present, 0.5 when one alternate allele and one reference allele were present, and 1 when no reference allele was present. Some sites, where an individual had no Polynesian variant on either haplotype, remained missing. These missing values were accounted for in the following manner. The genotype frequency vector for each individual from the dataset was placed into the row of an N individuals × p genotyped markers matrix and the nuclear norm regularized matrix completion algorithm of Mazumder et. al was applied to create a reduced rank approximation to the original, incomplete 689,899-dimensional masked genotype matrix33,69. Unlike earlier methods70,71,72, this method permits the use of all samples rather than only a panel of reference samples for the completion step; thus, far more data is used allowing for more accurate completion. In addition, instead of using haplotypes (haploid genomes) as the unit of analysis, this method uses genotype frequency vectors (frequency vectors for the diploid genome). Since there is no linkage present in the genome across chromosome boundaries (owing to independent assortment of chromosomes), population phasing cannot resolve parental haplotypes across these boundaries. Thus, a genome-wide haplotype vector constructed by assembling all chromosomes sequentially into a single row vector will switch phase arbitrarily across chromosome boundaries and so is already a mixture of an individual’s two true parental haplotypes. Further, by explicitly averaging an individual’s two haplotype vectors to form a single genotype frequency vector for that individual, we are able to fill in much of the masked data that is missing from either of the two haplotypes.
Ancestry-specific drift projection
Each Pacific island individual’s Polynesian ancestry-specific genotype frequency vector, described above, was projected onto the axis (drift axis), defined as the axis between the centroid of the indigenous Taiwan (Atayal and Paiwan) genotype frequency vectors and the centroid of the Rapa Nui (Easter Island) genotype frequency vectors. Each Pacific island individual’s genotype frequency vector was also projected onto the first principal component of the subspace orthogonal to this axis to provide a second coordinate for two-dimensional visualization. The first principal component of this orthogonal subspace is computed by finding the residual of each data point after subtracting off its component parallel to the drift axis and then determining the direction of greatest variation for these residuals. The per cent variance explained by each dimension was computed directly by finding the variance of the projections on that dimension.
Ancestry-specific t-SNE
The number of significant (P > 0.05) dimensions for the genotype frequency matrix, described above, was determined (n = 14) using a Tracy–Widom distribution58 and verified via a scree plot73. To ensure that all population structure was captured, the genotype frequency matrix was projected onto its first twenty principal component axes. A t-SNE was generated by applying the Barnes–Hut t-SNE implementation to this projected matrix using: theta = 0, perplexity = 15, exaggeration factor = 10, max iter = 10,000, and lying iter = 1,000 parameters74,75. Both a two-dimensional and three-dimensional embedding were created. Projections onto fewer dimensions yielded similar results, with some clusters beginning to disappear in the range 12–15 dimensions, as predicted by the Tracy–Widom analysis.
Ancestry-specific UMAP
The left singular vectors of the completed genotype frequency matrix were used as input for computing a two-dimensional UMAP with a Manhattan distance metric and 80 nearest neighbours76,77.
Ancestry-specific SOM
A two-dimensional SOM of the genotype frequency matrix was produced on a 100 × 100 rectangular grid using a Gaussian neighbourhood78. The package Somoclu, a massively parallel implementation of SOM, was used for optimization with parameters: 10 epochs, stdcoef 0.5, and linear cooling79.
Principal coordinate analysis and principal curves
Principal coordinate analysis (PCoA) and principal curves were constructed from the relevant distance matrices (either π or F3, described below) using R 3.5.2 together with the package buds80.
Population statistics
All population statistics described below (ψ, π, F3, F4, Fst and heterozygosity) were computed on population variant frequency vectors created by computing, for each site, \({\mathop{p}\limits^{ \sim }}_{i}=\frac{{\mathop{a}\limits^{ \sim }}_{i}}{{\mathop{n}\limits^{ \sim }}_{i}}\), where ãi is the minor allele count at the site aggregated across all individuals’ haplotypes (two haploid genomes per individual) having that site located in a Polynesian chromosomal segment for population i, and ñi is the total count of Polynesian minor and major alleles for i. A tilde is used to denote counts from Polynesian-specific chromosomal haplotypes. Any sites not located in a Polynesian segment for any of the individuals within a population (or located in only one haplotype within the entire population) were removed from the dataset for all populations, so as to have no populations with one or fewer total allele observations at any site. This filtering resulted in the loss of 60,377 SNPs (8.75% of the total 689,899 SNPs), leaving 629,522 SNPs across all populations for computation of population allele frequency statistics.
Psi (ψ)
The range expansion statistic (ψ) of Peter et al. 37 (see Supplementary Discussion, ‘On directionality’ and ‘On psi’) was computed first by polarizing all markers (identifying the minor allele) using the indigenous Taiwanese samples (Atayal and Paiwan) as an outgroup. To investigate the effect of using a different outgroup in a separate analysis a repolarization was performed using the western islands (Tonga, Samoa, Fiji) as an outgroup. The latter calculation reduced the standard errors for the range expansion statistic on islands settled subsequent to western Polynesia, that is, the eastern Polynesian islands; nevertheless, the general ordering of islands in the range expansion was the same for both calculations (see comparison Supplementary Fig. 14). Because allele frequencies drifted during the Pacific island settlement process, some minor alleles in Taiwan would have become major alleles by the time the settlers reached western Polynesia (see Supplementary Discussion, ‘On psi’), so the intermediate repolarization using Tonga, Samoa, and Fiji as an outgroup increased the resolution of the range expansion statistic (reduced standard errors) for downstream islands. The larger number of samples from Tonga, Samoa, and Fiji (51), as opposed to Taiwan (22), also contributed, as it allowed us to set a more permissive bound for confirming that an allele observed minor in the outgroup samples was also minor in the outgroup population. This in turn increased the number of markers present in the latter analysis. (A 0.1 or lower minor allele frequency was required in the merged Tonga, Samoa, Fiji outgroup samples, yielding 228,262 SNPs, as opposed to the more stringent requirement of minor alleles being fixed in the Taiwan outgroup samples, following the procedure of Zhan et. al. 38, which yielded only 137,383 SNPs.) ψ was calculated using the formula of Peter et. al,
where the sum is taken only over SNPs shared polymorphic in both the population A sample and the population B sample81. When using Taiwan as an outgroup, we masked the small Ni-Vanuatuan segments seen within Polynesia, since these segments trace their predominant ancestral origin back to a Papuan outgroup in New Guinea, rather than to the Austronesian outgroup, Taiwan. (Admixture between populations stemming from these two sources occurred on Vanuatu and other Melanesian islands in the thousand years before the settlement of Polynesia and was carried into Polynesia during its settlement17,82.) However, both masking methods (both the Taiwan and the Tonga, Samoa, Fiji outgroup polarizations) gave the same ordering of islands settled.
Pi (π)
This quantity is the average number of pairwise differences per pair of haplotypes (haploid genomes) selected at random, one from each population, normalized by the number of sites35,83,84. Also known as the nucleotide diversity85, it can be computed by first taking the ratio of the total number of mismatch combinations at a site to the total number of combinations, that is, where a1 is the number of alleles of one type in population 1 at a biallelic marker, b1 is the number of the other type, n1 = a1 + b1 is the total number of haplotypes in population 1, and thus p1 = a1/n1 is the allele frequency in population 1, then at this site
This is an unbiased estimator that can be averaged over all sites to find the average number of pairwise differences per haplotype pair per site35,85. Using this frequency-based formulation, this estimator can be generalized to Polynesian-specific allele frequencies for each island \({\mathop{{\rm{p}}}\limits^{ \sim }}_{i}\).
F 3
The F3 shared drift statistic of Patterson et al.86 was computed using the formula
where \({\mathop{{\rm{p}}}\limits^{ \sim }}_{A}\) = \({\mathop{{\rm{a}}}\limits^{ \sim }}_{A}\)/\({\mathop{{\rm{n}}}\limits^{ \sim }}_{A}\) is the sample allele frequency in the ancestry of interest in population A (\({\mathop{{\rm{n}}}\limits^{ \sim }}_{A}\) total observations and \({\mathop{{\rm{a}}}\limits^{ \sim }}_{A}\)observations of the allele a) and
and similarly for B and C. For multiple sites these values are computed for each site and then averaged across all sites36.
F 4
To detect departures from the reconstructed settlement tree (inter-island admixture), the F4 statistic was computed for each site using the formula of Patterson et al.36
and was then averaged across all sites. The F4 statistic is expected to be zero unless groups A and B do not form a separate clade from C and D within the actual population tree. Thus, when computing statistics of the form F4(parental_island, child_island; Samoa, X), where X varies across all islands that are not descended from parental_island in our model, a zero value of F4 is expected if the data completely support our settlement model. This is because all non-descendant islands (X) must lie in a common clade with outgroup Samoa; that is, external to the parental_island, child_island subclade. We look for significant evidence (P < 0.001) of departure from this model for each parental_island, child_island pair in our settlement sequence, and across all possible non-descendant islands X, while accounting for the multiple tests (n = 52) with a Bonferroni correction. We find deviations from our settlement tree only for 3 of its branches: Mangareva–Raivavae (migration from Tahiti), Mangareva–Palliser (migration from Tahiti), and North Marquesas–Palliser (migration from Tahiti and also from the Cooks). The Tahitian migrations go only to French Polynesian islands and likely reflect modern (see Supplementary Fig. 23) introgression to those islands from Tahiti, the modern capital, source of teachers, ministers, and civil servants, and centre of employment, transportation, and residential education for French Polynesia. The migration from the Cooks directly to North Marquesas (bypassing the Palliser group) is intriguing, especially in light of our late dated Palliser–North Marquesas connection (ad 1330). It could be that North Marquesas (Nuku Hiva) was settled earlier more directly from the Cooks, whereas South Marquesas (Fatu Hiva) was, we have found, settled early (ad 1140) from Palliser. Later within-island-group migration between these neighbouring islands may have led North Marquesas to exhibit these two origin signals, one from Palliser and one from the Cooks. If so, North and South Marquesas would be an unusual case, where two neighbouring islands were settled from different parental islands, then, because they were not separated by large oceanic distances, were able to exchange enough subsequent migrants to leave a notable genetic trace within their post-growth population base.
F st
The Hudson estimator for Fst is
for a given SNP. For multiple sites, the numerator and the denominator (unbiased estimators of the variance between populations and the variance in the ancestral population respectively) are averaged across all SNPs separately before taking the ratio to create a consistent estimator87.
Heterozygosity
The unbiased estimator for heterozygosity, first given by Nei and Roychoudhury88, for a specific site is
where \({\mathop{p}\limits^{ \sim }}_{\ell }\) is the frequency of the \({\ell }\)th allele at the site, and N is the total number of alleles at that site (two for each of our SNPs). This estimator was aggregated across each SNP locus k using
for all r of our SNP loci35,88.
Standard errors
Standard errors for all allele frequency-based statistics were computed using the block bootstrap using 100 replicates and a block size of 1,000 markers89. This gives better variance estimates than the jackknife for these pairwise allele frequency comparisons35. The markers are bootstrapped together as long contiguous blocks to preserve the effects of linkage on the variance of the estimates36.
Migration network reconstruction
The various population measures of distance and directionality (ψ, π, F3) between all pairs of islands define together tensors that annotate the complete graph of island connectivity. It remains to prune this graph judiciously to arrive at the tree representing the branching settlement process of the serially founded Pacific islands; that is, a tree describing which islands were settled from which other islands (Supplementary Discussion, ‘On differences between range expansion trees and typical population trees’ and ‘On tree building’).
In brief, we use the range expansion statistic ψ (Fig. 2b) to determine the upstream islands along the range expansion; that is, the set of potential parent islands for each island. Beginning with the island with the largest ψ (measured against Samoa), we work backward in order of decreasing ψ (Fig. 2b, Extended Data Fig. 4a), joining each still orphaned island (j) to its closest related potential parent island (i) as defined by ψ. To measure genetic distance (closeness), we use the average number of pairwise differences πij (Extended Data Fig. 4b, Supplementary Fig. 17), since πij has been shown to have higher correlation with the divergence time between two populations (i and j) than the outgroup-F3 statistic84 (Supplementary Discussion, ‘On different drift distance metrics’), although the same settlement sequence is also returned when using the latter metric instead (Supplementary Fig. 12).
Begin with the island with the most potential parents (at the end of the range expansion) (Fig. 2b) or, in other words, the largest ψ, Rapa Nui. Consistent with its terminal position in the range expansion, Rapa Nui also has the lowest heterozygosity (Supplementary Fig. 31) and the highest intra-island IBD (Supplementary Figs. 25, 26). Starting with this terminal island, we consider all potential parent islands as indicated by the ψ directionality index, and connect Rapa Nui to the most closely related potential parent as indicated by the smallest average number of pairwise differences (π). We then proceed to the island with the second most potential parents according the ψ-statistic (here Raivavae (Fig. 2b)) and repeat. For Samoa, Fiji, Tonga and upstream islands, we use the ψ directionality index polarized using the Taiwan outgroup. For islands downstream of Samoa, as indicated by the Taiwan-polarization ψ, we use a ψ-statistic repolarized using the more proximal Samoa, Fiji and Tonga outgroup, since it has smaller standard errors (see ψ discussion above).
This recursive algorithm for building the branching settlement path of the Pacific islands is a form of the Chu–Liu–Edmonds algorithm, which is guaranteed to produce the minimum spanning tree of a directed acyclic graph90,91 (Supplementary Discussion, ‘On tree-building’). That our graph is acyclic can be shown (proof derived in Supplementary Discussion, ‘On the acyclicity of psi’) from the formal definition of ψ, which defines our edge directionality. The lack of significant internal cross-migration edges was determined by our F4 analysis above.
In the case of parental islands with multiple child islands, we can now use an inner product measure, the F3 statistic (Extended Data Fig. 4c), which measures shared genetic drift, to determine whether any of those child islands share additional drift with each other beyond what they share with their common parent (Extended Data Fig. 7). Such additional shared drift is indicated in Fig. 2a by branching arrows; that is, arrows from a parent island that share an initial path before later branching to each child island. The order of arrow divergence indicates the ordering of shared drift among the child populations. These shared paths may suggest that intermediate islands in the settlement sequence are missing from our dataset (Extended Data Fig. 5), since the founding bottleneck of an intermediate island could account for the additional shared drift. To further verify our settlement sequence, and to look for signs of post-settlement inter-island admixture, we compute F4 statistics of the form F4 (parental island, child island; Samoa, X) with X ranging over all Polynesian islands not stemming from parental island in the settlement tree (described above). These F4 statistics indicate whether there is statistically significant evidence for deviations from our settlement model; that is, later migrations across the ocean of sufficient size to significantly alter the genetic base of the post-growth island populations. Only three branches in our settlement sequence show any significant deviations, and each of these indicate a migration from Tahiti to an outlying French Polynesian island, consistent with Tahiti’s recent role as the capital of French Polynesia.
Principal curve analysis
To independently verify our settlement sequence map, we compute unsupervised principal curves78,80 between the islands using genetic distances defined by both the outgroup-F3 and π metrics (Extended Data Fig. 7 and Supplementary Fig. 17, respectively).
IBD analyses
In highly related populations, such as populations that have passed through a population size bottleneck in the recent past, individuals will share many ancestors, and thus many identical-by-descent (IBD) genetic fragments92. In such cases, for example serially founded small island populations, IBD-based analyses become a powerful tool for reconstructing migrations.
Germline
GERMLINE 1.5.3 was run on the phased Pacific islander samples to find all IBD shared segments of 5 cM or greater using the -min_m flag. Fragments shorter than this length are prone to false positives owing to insufficient SNPs93,94,95. Up to four homozygous marker mismatches were permitted per IBD slice (-err_hom), and one heterozygous marker mismatch was permitted per IBD slice (-err_het). For a demonstration that our results are robust to IBD breaks due to phasing errors, see Extended Data Fig. 6.
Polynesian ancestry-specific filtering
To deconvolve Polynesian ancestral history from later (colonial and post-colonial) ancestry histories (for instance, European) we used an ancestry-specific approach to IBD66. Inter-island IBD segments lying wholly within post-colonial ancestries, or spanning post-colonial and pre-colonial ancestries, are necessarily the result of post-colonial inter-island contact events and were discarded. IBD segments lying wholly within chromosomal regions of known pre-colonial ancestry sources, that is Polynesian ancestry, were identified and analysed together.
Runs of homozygosity
Polynesian runs of homozygosity (ROH) were computed by summing together only Polynesian-specific IBD segments found shared between an individual’s two haploid genomes, then normalizing by the effective fraction of homozygous Polynesian ancestry segments found in that individual. These are the only segments of the diploid genome that could have shared a Polynesian ancestry ROH. Population Polynesian-specific ROH values were computed by averaging these values for all individuals within each island population. Standard errors were calculated by using the jackknife over individuals in a population96.
Ancestry-specific sum of IBD segment lengths
When analysing IBD segments, it has been typical to sum the total length (Wab) of segments shared between a pair of individuals (a and b), one from each of a pair of populations (A and B), and then sum over all such pairs to arrive at a total sum of IBD sharing between each pair of populations97. This sum can be normalized, dividing by the total number of possible cross-population pairs of individuals, one from each of the populations (nAnB), to give the average total IBD length shared (WAB) per cross-population individual pair94,97,98
This normalization can also be performed over the total number of cross-population haplotype (haploid genome) pairs (\(2{n}_{A}\cdot 2{n}_{B}\)), rather than all individual pairs66 (nAnB).
When considering only IBD segments found in those portions of both individuals’ genomes that belong to a particular ancestry, the normalization must be modified to reflect the reduced fraction of the pairs’ genomes that were considered. Thus, we replace the number of cross-population pair comparisons by an effective number of pair comparisons. If \({f}_{a}\) is the fraction of the genome of a particular ancestry in individual a, and similarly for \({f}_{b}\), then the expected fraction of pairwise overlap between the two individuals is \({f}_{a}{f}_{b}\), rather than 1 as it is for non-admixed individuals. The denominator of the normalization above is now modified by the factor \(\bar{{f}_{A}}\bar{{f}_{B}}\), where \(\bar{{f}_{A}}\) is the average fraction of the ancestry of interest in population A
Within a single non-admixed population, the normalized intra-population IBD length sharing per haplotype pair is,
The ancestry-specific normalization factor for intra-population IBD in an admixed population can be derived by considering the sum of all possible same-ancestry haplotype pair comparisons within the population of interest
These ancestry-specific normalization factors make clear that, although the normalized total length of IBD sharing between two populations gives a measure of the relatedness of the populations, it is quite sensitive to an accurate estimation of the average fraction in each population of the ancestry of interest.
A heat map showing the normalized Polynesian-specific IBD sum values for each pair of Pacific islands in our dataset is displayed in Supplementary Fig. 24. Trends of increasing IBD sharing along the course of the inferred settlement chain (see the map in Extended Data Fig. 5) are evident, but there is significant noise.
IBD segment length distributions
A better approach is to compute the distribution of lengths of IBD segments shared between pairs of individuals, one from each of the two populations being compared. Although the total count (integral) of this distribution will be influenced by the fraction in each population of the ancestry of interest, the shape (decay rate) of the distribution will not be. Such robustness to the estimate of each population’s ancestry fraction, which can vary by a few per cent between different ancestry inference methods, is of great benefit. In addition, the shape of the IBD length distribution (decay rate) changes steadily each generation. It does not depend, as genetic drift does, on the fluctuations, which are generally unknown, of the historical population sizes.
Assuming no interference, recombination can be modelled as a Poisson process occurring along the genome at a rate of one recombination break per generation per unit of genomic length (measured in Morgans)99. Thus, the length of a recombination segment, that is the distance between recombination events, is the waiting time of a Poisson process of rate T, where T is measured in generations. Hence, the distribution of the length of fragments (x) from a particular ancestor T generations ago will be exponential with λ = T decay rate100
If we are considering recombination segments shared between two present day individuals stemming from the same common ancestor, that is IBD segments, we must adjust the rate for the number of recombination events per unit length that have occurred down both sides of the pedigree from this common ancestor, which gives a λ of 2T total95,98,101. Each of these 2T opportunities for recombination to occur along the genome is called a meiosis event. For our empirical calculations (and all plots), we use cM, rather than M, so the λ rate constant is divided by 100, yielding T/50.
The total distribution of tract lengths shared between all individuals can be viewed as independent samples from the same exponential distribution. Ralph and Coop have shown that the decay rate parameter λ of this distribution is a weighted average of the distribution of times to the most recent common ancestor across all genomic sites102. This distribution of times can be a complicated function of the demography, when the latter is not simple, leading to an ill-conditioned inverse problem102. However, for our problem—dating the founding of an isolated island group—the demography is amenable. Consider a parent island whose Polynesian explorers crossed thousands of kilometres of Pacific waves to discover and colonize a child island during the Polynesian settlement process. A pair of present-day Pacific Islanders, one from the child island and one from the parent island, cannot share a common ancestor at any site (in their Polynesian ancestry segments) more recently than the founding date of the child island. Moreover, because of the small size of the founding populations arriving on double-hulled sailing canoes2,7,21,22, all individuals on the child island will share ancestors with one another dating to at least the time of this founding bottleneck before which time they coalesce with the ancestors of modern individuals on the parent island. Thus, the decay rate parameter λ will measure the time (T/50 with T in generations) to the split of the parent and child island populations.
Example IBD length distributions for all pairs of individuals in our dataset—with one individual from Rapa Nui (Easter Island) and one from Mangareva, Palliser, Rarotonga, or Samoa—are plotted in Supplementary Fig. 22. Note that altering the normalization factor based on the estimated fractions of Polynesian ancestry amounts to a rescaling of the y-axis in Supplementary Fig. 22a or a translation of the y-axis in Supplementary Fig. 22b. This alters the amplitude, but not the decay rate shape parameter λ, of the exponential in Supplementary Fig. 22a, or, equivalently, the intercept, but not the slope of the lines in Supplementary Fig. 22b, thus demonstrating graphically that λ is robust to noise (errors) in ancestry normalization. However, the sum of IBD lengths, which is the integral of the curves in Supplementary Fig. 22a, is clearly not robust to such errors in the normalization (rescaling the y-axis).
Our empirically observed IBD length distributions are left truncated at 5 cM, since fragments shorter than this length are prone to false positives due to insufficient SNPs93,94,95. The distributions are also right truncated at 15 cM, because outlier segments longer than this are expected to stem from recent contact, dating to less than 10 generations ago (18th century or later) as computed from the expected generation time (g) based on a single fragment length \({\ell }\) in Morgans
where 1M is one Morgan94.
Such occasional post-colonial, inter-island Polynesian contact is not the focus of our Polynesian settlement analysis, so we filter out these few outlier inter-island IBD segments. Not removing these outliers does not change our island settlement dates significantly, but, by distorting the tail of the exponential decay distributions, does increase our standard error (see Supplementary Discussion, ‘On quantification of error in IBD dating’).
To estimate each pairwise λ, we use the maximum likelihood estimator for a left and right truncated exponential distribution. Since the exponential distribution is memoryless, the left truncation is trivially handled by translation of the distribution. That is, the distribution of the length in excess of 5 cM for each fragment is also exponential with the same decay constant λ. For what follows we assume that the IBD lengths have been thus recentred by subtracting 5 cM. The right truncation is less elegant to handle, yielding an equation for the maximum likelihood estimator (\(\hat{\lambda }\)) of λ given by103
where \({ {\mathcal L} }_{n}\) (the sample likelihood for the n IBD segments) is the product of their individual likelihoods \( {\mathcal L} \), \(\bar{x}\) is the (recentred) mean IBD length, x0 is the (recentred) right truncation point and n is sample size (number of IBD segments). This transcendental equation must be solved numerically. The standard error (SE) can be obtained directly from the observed Fisher Information I(\(\hat{\lambda }\))
since
where the observed Fisher Information is found by
Using this method, the estimated λ values for the exponential distributions of Polynesian-specific IBD segment lengths for pairs of individuals, one from Rapa Nui (Easter Island) and one from each of the other remote Pacific islands in our dataset, are shown in Extended Data Fig. 4d. The pattern confirms the results of our drift statistics; Mangareva is the island most recently connected to Rapa Nui, while Samoa, the root of the expansion into remote Polynesia, is the most archaic connection.
A few caveats remain. The model of a Poisson process of recombination events along a continuous genome holds for small IBD segment lengths, that is, T > 5, but for more recent relatedness, leading to very long IBD segments, one must consider the finite size of the chromosomes themselves when computing the IBD length distribution104. In addition, the model of IBD segment independence holds only for (N >> T), where N is the population size and T is the number of generations100. Fortunately, our dataset has T values of 25−30 generations and N values in the thousands21, so we do not fall into these problematic regimes. However, because of the founding bottlenecks for each island, there is some intra-island IBD shared between pairs of individuals on the island (Supplementary Figs. 25, 26), so some recombination events will be non-productive. That is, when a haplotype stemming from one islander recombines with a haplotype from another with a recombination event occurring in the midst of an IBD segment on one haplotype that is shared with a third individual, the recombination break will occasionally not break up the IBD sequence, since the two different recombining haplotype segments might themselves be identical (IBD) at the recombination point and thus both in IBD with the third individual. This will happen with a frequency equal to the percentage of the genome shared IBD on average between pairs of individuals on the same island. Therefore, we can correct for the frequency of these non-productive recombination events at each meiosis event. The correction factor ρ (the proportion of the genome shared on average intra-island) is specific to each island (dependent on the intra-island IBD on each island accrued through preceding founding bottlenecks) and so must be applied separately to the two branches of the pedigree, one from the common ancestral population on the parent island down to the present population on the parent island (A) and from the common ancestral population down to the child island (B). The number of effective meioses, which is equal to λ, can be expressed after correction as
where g is the number of generations to a common ancestor of populations A and B, ρA is the average fraction of the genome in IBD between pairs of individuals on A, equivalent to the probability of non-productive recombination on A, and similarly for ρB.
The correction factor ρ can be found by dividing the average total sum of IBD segments (S) between pairs of individuals on an island by the length of the genome93,105 (35.3 M). Since we empirically observe only the sum of IBD segments longer than 5 cM, we must extrapolate the total sum of all IBD fragments by integrating our fitted exponential distribution of IBD segment lengths (for example, Supplementary Fig. 22a). Thus, the total sum of IBD for N total IBD segment matches in the population (generally unknown) is given by
and the truncated sum of IBD is given by
By inspection we can see that
The extrapolated sums of IBD in the Polynesian component on average between pairs of individuals on each island as a per cent of the genome are plotted in Supplementary Fig. 26, showing that these correction factors represent an adjustment of only a few per cent.
We can now construct the symmetric matrix of Polynesian-specific pairwise island λ values (shown in Supplementary Fig. 23), and convert it, using our ρ adjustment factors for each island, to a generation count to common ancestor for each island pair.
For a detailed discussion of the uncertainty in our estimates of these dates see Supplementary Discussion, ‘On quantification of error in IBD dating’ and Extended Data Fig. 6.
Some island pairs, particularly distantly related islands or island pairs each with small numbers of samples, have large standard errors. Removing all entries corresponding to λ values that have standard errors above 0.07 (representing errors larger than 15% of the average lambda value), creates a matrix of more precise generation values, but with some missing entries. Because this is a distance matrix, entries must be consistent with the triangle inequality, so we can impute these missing entries using triangulation from the precisely known entries.
In fact, we can use something stronger than the standard triangle inequality
for distances d(i,j) between two islands i and j and an third island k. We can instead use the ultrametric inequality
This holds in our case, because all samples were taken from contemporary populations, and so all are leaf nodes of the ancestry tree dating to the same period (the present). Thus, so long as we use a distance metric d(i,j) that is uniform in time for each population, for instance the ρ-adjusted generation (or year) matrix, the distances back from each pair of populations to their common ancestor will be identical, yielding an ultrametric tree. This works because the per generation recombination rate is constant over time, so the distance from an ancestral population to any of its sampled contemporary descendants is the same when measured by segment length distributions. (Note that this matrix, measuring the total distance along the tree from one island to another, is twice the matrix measuring how many generations have passed since an island pair split, since the former sums down both tree branches descending from the split.) To complete the proof of the ultrametric inequality we notice that for any two contemporary populations i and j and a third population k, k must either coalesce in ancestry with i first, with j first, or after i and j have themselves first coalesced. In the last case, d(i,j) is clearly less than both d(i,k) and d(j,k), so the inequality above holds. In the first case, where k coalesces first with i, i and k have a shared common ancestor (m) before coalescing with the branch to j, so writing d(i,j) = d(i,m) + d(m,j), and noting that we said the distance to a common ancestor must be identical for terminal nodes d(i,m) = d(k,m) when using years, we have d(i,j) = d(i,m) + d(m,j) = d(k,m) + d(m,j) = d(k,j). Hence, d(i,j) is equal to d(j,k) in case one (and similarly it is equal to d(i,k) in case two), making the bound of the ultrametric inequality valid (tight) for both cases.
Using the ultrametric inequality, we can impute unknown distances d(i,j) simply by searching across all intermediate populations k and finding the minimum106
From this completed distance matrix of generations, we can apply dates to each of the migrations using the average human generation time (see Supplementary Discussion, ‘On generation times and meiosis events’).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The samples for this project were collected by the University of Oxford, Stanford University and the University of Chile as part of different studies. SNP data for all newly genotyped individuals are available through a data access agreement to respect the privacy of the participants for the transfer of genetic data from the European Genome-Phenome Archive under accession number EGAS00001005362.
Code availability
All new techniques described in Methods are available from https://github.com/AI-sandbox and all existing software packages and versions used are noted in Methods.
References
Low, S. Hawaiki Rising: Hōkūle‘a, Nainoa Thompson, and the Hawaiian Renaissance (Univ. of Hawaii Press, 2019).
Kirch, P. V. On the Road of the Winds (Univ. of California Press, 2017).
Mulrooney, M. A., Bickler, S. H., Allen, M. S. & Ladefoged, T. N. High-precision dating of colonization and settlement in East Polynesia. Proc. Natl Acad. Sci. USA 108, E192–E194 (2011).
Schmid, M. M. E. et al. How 14C dates on wood charcoal increase precision when dating colonization: the examples of Iceland and Polynesia. Quat. Geochronol. 48, 64–71 (2018).
Kahō‘āli‘i Keauokalani, K. Kepelino’s traditions of Hawaii. Bernice P. Bishop Museum Bulletin 206 (1932).
Cook, J. The Journals of Captain James Cook on his Voyages of Discovery (Cambridge Univ. Press, 1955).
Kirch, P. V. & Green, R. C. Hawaiki, Ancestral Polynesia (Cambridge Univ. Press, 2001).
Minster, R. L. et al. A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat. Genet. 48, 1049–1054 (2016).
Gray, R. D., Drummond, A. J. & Greenhill, S. J. Language phylogenies reveal expansion pulses and pauses in Pacific settlement. Science 323, 479–483 (2009).
Walworth, M. Eastern Polynesian: the linguistic evidence revisited. Ocean. Linguist. 53, 256–272 (2014).
Martinsson-Wallin, H., Wallin, P. & Anderson, A. Chronogeographic variation in initial East Polynesian construction of monumental ceremonial sites. J. Island Coastal Archaeol. 8, 405–421 (2013).
Wilmshurst, J. M., Hunt, T. L., Lipo, C. P. & Anderson, A. J. High-precision radiocarbon dating shows recent and rapid initial human colonization of East Polynesia. Proc. Natl Acad. Sci. USA 108, 1815–1820 (2011).
Spriggs, M. & Anderson, A. Late colonization of east Polynesia. Antiquity 67, 200–217 (1993).
Hill, A. V. S. et al. Polynesian origins and affinities: globin gene variants in eastern Polynesia. Am. J. Hum. Genet. 40, 453–463 (1987).
Wollstein, A. et al. Demographic history of Oceania inferred from genome-wide data. Curr. Biol. 20, 1983–1992 (2010).
Hudjashov, G. et al. Investigating the origins of eastern Polynesians using genome-wide data from the Leeward Society Isles. Sci. Rep. 8, 1823 (2018).
Skoglund, P. et al. Genomic insights into the peopling of the Southwest Pacific. Nature 538, 510–513 (2016).
Posth, C. et al. Language continuity despite population replacement in Remote Oceania. Nat. Ecol. Evol. 2, 731–740 (2018).
McColl, H. et al. The prehistoric peopling of Southeast Asia. Science 361, 88–92 (2018).
Emory, K. P. The Tuamotuan creation charts by Paiore. J. Polynesian Soc. 48, 1–29 (1939).
Hunt, T. & Lipo, C. The Statues that Walked (Free Press, 2011).
Whyte, A. L. H., Marshall, S. J. & Chambers, G. K. Human evolution in Polynesia. Hum. Biol. 77, 157–177 (2005).
Duncan, R. P., Boyer, A. G. & Blackburn, T. M. Magnitude and variation of prehistoric bird extinctions in the Pacific. Proc. Natl Acad. Sci. USA 110, 6436–6441 (2013).
Steadman, D. W. Extinction and Biogeography of Tropical Pacific Birds (Univ. of Chicago Press, 2006).
Kirch, P. V. et al. Human ecodynamics in the Mangareva Islands: a stratified sequence from Nenega-Iti Rock Shelter (site AGA-3, Agakauitai Island). Archaeol. Oceania 50, 23–42 (2015).
Rolett, B. V. Voyaging and interaction in ancient East Polynesia. Asian Perspect. 41, 182–194 (2002).
Handy, E. S. C. The Native Culture in the Marquesas (The Bishop Museum, 1923).
Weisler, M. I. et al. Cook Island artifact geochemistry demonstrates spatial and temporal extent of pre-European interarchipelago voyaging in East Polynesia. Proc. Natl Acad. Sci. USA 113, 8150–8155 (2016).
Collerson, K. D. & Weisler, M. I. Stone adze compositions and the extent of ancient Polynesian voyaging and trade. Science 317, 1907–1911 (2007).
Slatkin, M. & Excoffier, L. Serial founder effects during range expansion: a spatial analog of genetic drift. Genetics 191, 171–181 (2012).
Stephens, M. & Novembre, J. Interpreting principal component analyses of spatial population genetic variation. Nat. Genet. 40, 646–649 (2008).
Wang, C. et al. Comparing spatial maps of human population-genetic variation using procrustes analysis. Stat. Appl. Genet. Mol. Biol. 9, 13 (2010).
Ioannidis, A. G. et al. Native American gene flow into Polynesia predating Easter Island settlement. Nature 583, 572–577 (2020).
Novembre, J. et al. Genes mirror geography within Europe. Nature 456, 98–101 (2008).
Nei, M. & Kumar, S. Molecular Evolution and Phylogenetics (Oxford Univ. Press, 2000).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Peter, B. M. & Slatkin, M. Detecting range expansions from genetic data. Evolution 67, 3274–3289 (2013).
Zhan, S. et al. The genetics of monarch butterfly migration and warning colouration. Nature 514, 317–321 (2014).
Lipson, M. et al. Efficient moment-based inference of admixture parameters and sources of gene flow. Mol. Biol. Evol. 30, 1788–1802 (2013).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012).
Leppälä, K., Nielsen, S. V. & Mailund, T. admixturegraph: an R package for admixture graph manipulation and fitting. Bioinformatics 33, 1738–1740 (2017).
Anderson, A. J., Conte, E., Smith, I. & Szabo, K. New excavations at Fa’ahia (Huahine, Society Islands) and chronologies of central East Polynesian colonization. J. Pac. Arch. 10, 1–14 (2019).
Hunt, T. L. & Lipo, C. P. Evidence for a shorter chronology on Rapa Nui (Easter Island). J. Island Coast. Archaeol. (2008).
Mulrooney, M. A. An island-wide assessment of the chronology of settlement and land use on Rapa Nui (Easter Island) based on radiocarbon data. J. Archaeol. Sci. 40, 4377–4399 (2013).
Pirazzoli, P. A. & Montaggioni, L. F. Late Holocene sea-level changes in the northwest Tuamotu islands, French Polynesia. Quat. Res. 25, 350–368 (1986).
Di Piazza, A., Di Piazza, P. & Pearthree, E. Sailing virtual canoes across Oceania: revisiting island accessibility. J. Archaeol. Sci. 34, 1219–1225 (2007).
Walworth, M. The Language of Rapa Iti (Univ. Hawaii, 2015).
Dickinson, W. Pacific atoll living: how long already and until when. Geol. Soc. Am. Today 19, 4–10 (2009).
Fischer, S. R. Mangarevan doublets: preliminary evidence for proto-southeastern Polynesian. Ocean. Linguist. 40, 112–124 (2001).
Flenley, J. & Bahn, P. The Enigmas of Easter Island (Oxford Univ. Press, 2003).
Buck Te Rangi Hīroa, P. H. Vikings of the Sunrise (J. B. Lippincott, 1938).
Belbin, G. M. et al. Toward a fine-scale population health monitoring system. Cell 184, 2068–2083.e11 (2021).
Claw, K. G. et al. A framework for enhancing ethical genomic research with Indigenous communities. Nat. Commun. 9, 2957 (2018).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Tyner, C. et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 45, D626–D634 (2017).
Thornton, T. et al. Estimating kinship in admixed populations. Am. J. Hum. Genet. 91, 122–138 (2012).
The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Wickham, H. ggplot2 (Springer, 2016).
R Core Team. R: a Language and Environment for Statistical Computing https://www.R-project.org/ (2017).
Alexander, D. H. & Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinf. 12, 246 (2011).
Holmes, S. & Huber, W. Modern Statistics for Modern Biology (Cambridge Univ. Press, 2019).
Maples, B. K., Gravel, S., Kenny, E. E. & Bustamante, C. D. RFMix: A discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 93, 278–288 (2013).
O’Connell, J. et al. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 10, e1004234 (2014).
D’Arcy, P. The Chinese Pacifics: a brief historical review. J. Pacific Hist. 49, 396–420 (2014).
Browning, S. R. et al. Ancestry-specific recent effective population size in the Americas. PLoS Genet. 14, e1007385 (2018).
Schroeder, H. et al. Origins and genetic legacies of the Caribbean Taino. Proc. Natl Acad. Sci. USA 115, 2341–2346 (2018).
Moreno-Estrada, A. et al. The genetics of Mexico recapitulates Native American substructure and affects biomedical traits. Science 344, 1280–1285 (2014).
Mazumder, R., Hastie, T. & Tibshirani, R. Spectral regularization algorithms for learning large incomplete matrices. J. Mach. Learn. Res. 11, 2287–2322 (2010).
Reich, D. et al. Reconstructing Native American population history. Nature 488, 370–374 (2012).
Skoglund, P. et al. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science 336, 466–469 (2012).
Moreno-Estrada, A. et al. Reconstructing the population genetic history of the Caribbean. PLoS Genet. 9, e1003925 (2013).
Nguyen, L. H. & Holmes, S. Ten quick tips for effective dimensionality reduction. PLoS Comp. Biol. 15, e1006907 (2019).
Maaten, L. V. D. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Van Der Maaten, L. Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res. 15, 3221–3245 (2014).
McInnes, L., Healy, J. & Melville, J. UMAP: Uniform manifold approximation and projection for dimension reduction. Preprint at https://arxiv.org/abs/1802.03426 (2018).
Diaz-Papkovich, A., Anderson-Trocmé, L., Ben-Eghan, C. & Gravel, S. UMAP reveals cryptic population structure and phenotype heterogeneity in large genomic cohorts. PLoS Genet. 15, e1008432 (2019).
Hastie, T., Tibshirani, R. & Friedman, J. The Elements of Statistical Learning (Springer, 2009).
Wittek, P., Gao, S. C., Lim, I. S. & Zhao, L. Somoclu: an efficient parallel library for self-organizing maps. J. Stat. Softw. 78, https://doi.org/10.18637/jss.v078.i09 (2017).
Nguyen, L. H. & Holmes, S. Bayesian unidimensional scaling for visualizing uncertainty in high dimensional datasets with latent ordering of observations. BMC Bioinf. 18, 65–79 (2017).
Peter, B. M. & Slatkin, M. The effective founder effect in a spatially expanding population. Evolution 69, 721–734 (2015).
Pugach, I. et al. The complex admixture history and recent southern origins of Siberian populations. Mol. Biol. Evol. 33, 1777–1795 (2016).
Takahata, N. & Nei, M. Gene genealogy and variance of interpopulational nucleotide differences. Genetics 110, 325–344 (1985).
Peter, B. M. Admixture, population structure, and F-statistics. Genetics 202, 1485–1501 (2016).
Nei, M. Molecular Evolutionary Genetics (Columbia Univ. Press, 1987).
Patterson, N. et al. Reconstructing Indian population history. Nature 461, 489–494 (2009).
Bhatia, G., Patterson, N., Sankararaman, S. & Price, A. L. Estimating and interpreting FST: the impact of rare variants. Genome Res. 23, 1514–1521 (2013).
Nei, M. & Roychoudhury, A. K. Sampling variances of heterozygosity and genetic distance. Genetics 76, 379–390 (1974).
Davison, A. C. & Hinkley, D. V. Bootstrap Methods and Their Application (Cambridge Univ. Press, 1997).
Chu, Y. J. & Lui, T. H. On the shortest arborescence of a directed graph. Science Sinica 14, 1396–1400 (1965).
Edmonds, J. Optimum branchings. J. Res. Natl. Bur. Stand. 71B, 233–240 (1967).
Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M. & Wilson, J. F. Runs of homozygosity: windows into population history and trait architecture. Nat. Rev. Genet. 19, 220–234 (2018).
Huff, C. D. et al. Maximum-likelihood estimation of recent shared ancestry (ERSA). Genome Res. 21, 768–774 (2011).
Baharian, S. et al. The Great Migration and African-American genomic diversity. PLoS Genet. 12, e1006059 (2016).
Gusev, A. et al. Whole population, genome-wide mapping of hidden relatedness. Genome Res. 19, 318–326 (2009).
Efron, B. Bootstrap methods: another look at the jackknife. Ann. Stat. 7, 1–26 (1979).
Botigué, L. R. et al. Gene flow from North Africa contributes to differential human genetic diversity in southern Europe. Proc. Natl Acad. Sci. USA 110, 11791–11796 (2013).
Atzmon, G. et al. Abraham’s children in the genome era: major Jewish diaspora populations comprise distinct genetic clusters with shared Middle Eastern ancestry. Am. J. Hum. Genet. 86, 850–859 (2010).
Jobling, M., Hurles, M. & Tyler-Smith, C. Human Evolutionary Genetics (Garland Science, 2013).
Liang, M. & Nielsen, R. The lengths of admixture tracts. Genetics 197, 953–967 (2014).
Palamara, P. F., Lencz, T., Darvasi, A. & Pe'er, I. Length distributions of identity by descent reveal fine-scale demographic history. Am. J. Hum. Genet. 91, 809–822 (2012).
Ralph, P. & Coop, G. The geography of recent genetic ancestry across Europe. PLoS Biol. 11, e1001555 (2013).
Deemer, W. L. Jr & Votaw, D. F. Jr Estimation of parameters of truncated or censored exponential distributions. Ann. Math. Stat. 26, 498–504 (1955).
Hill, W. G. & White, I. M. S. Identification of pedigree relationship from genome sharing. G3 Genes Genom. Genet. 3, 1553–1571 (2013).
McVean, G. A. T. et al. The fine-scale structure of recombination rate variation in the human genome. Science 304, 581–584 (2004).
Makarenkov, V. & Lapointe, F.-J. A weighted least-squares approach for inferring phylogenies from incomplete distance matrices. Bioinformatics 20, 2113–2121 (2004).
Fehren-Schmitz, L. et al. Genetic ancestry of Rapanui before and after European contact. Curr. Biol. 27, 3209–3215 (2017).
Crowe, A. Pathway of the Birds (Univ. Hawai'i Press, 2018).
Marck, J. C. Topics in Polynesian Language and Culture History (The Australian National Univ., 2000).
Niespolo, E. M., Sharp, W. D. & Kirch, P. V. 230Th dating of coral abraders from stratified deposits at Tangatatau Rockshelter, Mangaia, Cook Islands: implications for building precise chronologies in Polynesia. J. Archaeol. Sci. 101, 21–33 (2019).
Kirch, P. V. Tangatatau Rockshelter: The Evolution of an Eastern Polynesian Socio-ecosystem (Cotsen Institute of Archaeology Press, 2017).
Sear, D. A. et al. Human settlement of East Polynesia earlier, incremental, and coincident with prolonged South Pacific drought. Proc. Natl Acad. Sci. USA 117, 8813–8819 (2020).
Kahn, J. G. & Sinoto, Y. Refining the Society Islands cultural sequence: colonisation phase and developmental phase coastal occupation on Mo'orea Island. J. Polynesian Soc. 126, 33 (2017).
Conte, E. & Molle, G. Reinvestigating a key site for Polynesian prehistory: new results from the Hane dune site, Ua Huka (Marquesas). Archaeol. Oceania 49, 121–136 (2014).
Allen, M. S. Marquesan colonisation chronologies and postcolonisation interaction: implications for Hawaiian origins and the ‘Marquesan homeland’ hypothesis. J. Pac. Arch. 5, 1–17 (2014).
Prebble, M. & Wilmshurst, J. M. Detecting the initial impact of humans and introduced species on island environments in Remote Oceania using palaeoecology. Biol Invasions 11, 1529–1556 (2009).
Anderson, A., Kennett, D. J., Culleton, B. J. & Southon, J. in Taking the High Ground (eds. Anderson, A. & Kennett, D. J.) 288 (ANU Press, 2012).
Kirch, P. V., Conte, E., Sharp, W. & Nickelsen, C. The Onemea Site (Taravai Island, Mangareva) and the human colonization of Southeastern Polynesia. Archaeol. Oceania 45, 66–79 (2010).
Allen, M. S. & Steadman, D. W. Excavations at the Ureia site, Aitutaki, Cook Islands: preliminary results. Archaeol. Oceania 25, 24–37 (1990).
Matisoo-Smith, E. et al. Patterns of prehistoric human mobility in Polynesia indicated by mtDNA from the Pacific rat. Proc. Natl Acad. Sci. USA 95, 15145–15150 (1998).
Acknowledgements
We thank the participants and volunteers who donated DNA samples for this study and the local community leaders and authorities who provided approval and logistics support in the various sampling sites, including T. Teariki (Cook Islands), S. Foliaki (Tonga), N. Tuuau (Samoa), H.-W. Peng (Taiwan), J. Roux (French Polynesia), E. Paoa (Rapa Nui), as well as J. Martinson, D. Weatherall and J. Clegg for their pioneering fieldwork in the region that ultimately led to this work. We thank H. Kane and S. Kahanamoku for critiques and suggestions on the text, as well as M. Spriggs for his comments on the manuscript concerning archaeological dates in Polynesia. We thank the Core Staff at the UCSF Institute for Human Genetics for contributing genotyping capacity, and the Stanford Center for Computational, Evolutionary and Human Genomics (CEHG) for supporting the initial stages of this project. We thank J. Cervantes for IT support and M. Ortega and G. Mireles for technical support at LANGEBIO’s Genomics Core Facility at CINVESTAV, Mexico. This work was supported by the George Rosenkranz Prize for Health Care Research in Developing Countries, Mexico’s CONACYT Basic Research Program (grant number CB-2015-01-251380), and the International Center for Genetic Engineering and Biotechnology (ICGEB, Italy) Grant CRP/MEX15-04_EC (each awarded to A.M.-E.); the Chilean funding programs FONDEF, FONDECYT and CONICYT (grants D10I1007, 1130303 and USA2013-0015, respectively); the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC) and a Wellcome Trust Fellowship with reference 106289/Z/14/Z (A.J.M.); together with National Library of Medicine (NLM) training grant T15LM007033 and an American Society of Engineering Education NDSEG Fellowship (each awarded to A.G.I.). Views expressed are those of the author(s) and not necessarily those of the NIHR, the NHS, or the Department of Health.
Author information
Authors and Affiliations
Contributions
A.M.-E. and A.G.I. conceived the study. A.G.I., A.J.M., C.D.B. and A.M.-E. provided overall project supervision and management. A.M.-E., A.G.I., E.H., K.S., A.J.M. and C.R.G. contributed to study design. A.S., C.E., S.H., E.G.B., C.D.B. and A.M.-E. carried out genotyping experiments and quality control. A.G.I., J.B.-P., C. B.-J., J.E.R.-R., C.D.Q.-C., J.R.H. and A.M.-E. analysed the data. A.G.I. developed the analytical methods. A.G.I., J.B.-P. and A.M.-E. interpreted the results. A.M.-E., K.S., E.H., A.V.S.H., J.F.M.-P., K.A., T.P., K.R., M.C.A.-A., A.S., A.S.M.S., C.E., S.H., E.G.B., R.A.V., M.M., A.J.M. and C.D.B. contributed to acquisition of the data. A.G.I. wrote the manuscript, and A.M.-E., E.H., K.F. and S.H.-C. provided feedback on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
C.D.B. is a member of the scientific advisory boards for Liberty Biosecurity, Personalis, 23andMe Roots into the Future, Ancestry.com, IdentifyGenomics, Genomelink and Etalon, and is a founder of CDB Consulting. C.R.G. owns stock in 23andMe and is member of the scientific advisory board for Encompass Bioscience.
Additional information
Peer review information Nature thanks Patrick Kirch, Benjamin Peter and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Comparison of genetic and geographic coordinates for European vs. Polynesian samples.
a, A principal component analysis of samples from Europe (15 from each nation) is shown to closely fit the geography of Europe. (See Extended Data Fig. 2 for a quantitative comparison.) b, c, A principal component analysis (b) of samples from Polynesia (with non-Polynesian ancestry masked) is shown not to match the vast geography of the Pacific (c), and instead splits out island groups one at a time, reflecting the founder effects that dominate the variance of these populations.
Extended Data Fig. 2 Permutation test for fit between genetic and geographic coordinates.
100,000 random permutations of the population labels were created for the European populations’ genetic data (blue, left) versus the Polynesian populations (orange, right). For the European populations, out of 100,000 random permutations of the population labels on the genetic PCA, none better fits the geography of Europe (after fitting using a Procrustes analysis32), than the correct labels, showing that the genetic coordinates of Europeans fit the geographic coordinates of Europe better than chance every time. However, for the Polynesian data 5% of the random permutations of the labels on the genetic PCA fit the geographic coordinates of the Pacific islands better (after fitting using Procrustes), showing that the genetic data in Polynesia does not fit Polynesia’s geography better than random chance. In the box and whiskers plots the mean and upper and lower quartiles of the rms error of the fits of the random permutations of population labels are indicated by horizontal lines. The fits of the actual population labels are indicated by asterisks.
Extended Data Fig. 3 Continuity between ancient and modern Polynesian island populations.
F3 statistics were computed between ancient Rapanui samples and the Polynesian component from modern samples from each island in our dataset (top)107. Indigenous Austronesian language speakers from Taiwan (the Atayal) were used as an outgroup. The ancient Rapanui were found to be the most similar genetically to the modern Rapanui, indicating genetic continuity. A similar comparison was performed between the only other ancient samples from an island in our study, Tonga (bottom)18. Again, the modern Tongans appear most similar genetically; however, all islands downstream from Tonga in our inferred settlement path also share the same amount of genetic drift with the ancient Tongan samples (to within one standard error), as they should, since they are all descendants of these ancient Tongan sample according to our settlement reconstruction.
Extended Data Fig. 4 Statistics used for settlement path inference.
All statistics are based on the Polynesian-specific aggregate SNP frequency vectors computed for each island from all sampled individuals. The number (n) of individuals used are given for each island in Supplementary Table 1. a, Directionality index (ψ), used to define sets of potential parent islands, plotted for each island relative to Samoa (equivalent to the top row of the matrix in Fig. 2b). b, Average number of pairwise differences (π), measuring genetic distance and used to select the closest of potential parents, plotted for each island relative to Rapa Nui. c, F3 statistic, used to find additional shared genetic drift, plotted for each island relative to Rapa Nui, with Taiwan as an outgroup. Standard errors in a–c were determined by a block bootstrap analysis. d, Exponential decay constant (λ) for the Polynesian-specific IBD fragment length distributions between all pairs of individuals from Rapa Nui and each plotted island. The λ values can be used to calculate the number of generations elapsed since each pair of island populations were joined. Error bars show 95% confidence intervals of the maximum likelihood estimates determined analytically from the Fisher Information.
Extended Data Fig. 5 Settlement map with candidate intermediate islands added.
A reproduction of the map of Fig. 2a showing intermediate islands that are in the settlement path but not in our dataset that are possible candidates for explaining the additional shared drift observed in the corresponding colored settlement branches, that is, genetic drift shared between the child islands but not shared with the parent island. The additional shared drift of the Austral islands (Rimatara and Tubuai) with the Society islands (Tahiti) and Tuamotus (Palliser) beyond what they each share with their parental island (Rarotonga in the Cooks) could indicate that there exists a shared intermediate island in their settlement path that we do not have in our dataset, for instance Mangaia108. Geological analyses of ancient tools found on Mangaia (green) have shown that it served as a connection between the Cook islands and remote eastern Polynesia28, now uninhabited Nororotu (Maria Atoll) is also believed to have played a role as an intermediary island108. Traditional histories give Raiatea (pink) and its surrounding islands a role in the settling of remote eastern Polynesia108. Finally, linguistic studies have found connections between Marquesic languages (Marquesas and Mangareva) and the central Tuamotus (orange)109. North Marquesas, South Marquesas, and Mangareva share drift with one another beyond what they share with Palliser, the westernmost island group in the Tuamotus, which could indicate that these three populations shared a common settlement path eastward through some of the Tuamotu Archipelago before diverging. Another possible explanation for additional shared drift is the settlement of each child island from a common subpopulation within the parental island, such as from the same clan or village.
Extended Data Fig. 6 Effect of phasing errors on IBD dates.
IBD segments on the island of Rapa Nui were identified between all male X chromosomes. The log of the number of IBD segments (y axis) of a given genetic length (x axis) is plotted (orange; bottom left). The expected exponential decay of IBD segment lengths (linear semilog plot) is seen. The slope of this line (−0.161) is the exponential (decay) constant lambda. Since the X chromosome is perfectly phased in men, because it is haploid, the identification of these IBD segments is unaffected by errors introduced through phasing algorithms. To quantify the effect of such errors, synthetic-female individuals were constructed by combining two male X chromosomes to make a diploid pair and to erase the phase information by recording only the genotype. The unphased diploid genotypes so constructed were phased and IBD segments were again identified and plotted (green; bottom right). The difference between the exponential decay constant (−0.166) of these statistically phased genotypes and the previous one is seen to be minor (top panel), amounting to three per cent (3.01%), which corresponds to a difference of around 25 years for dates approximately eight hundred years ago (as in Polynesia). Uncertainty in the slope of the lines (equivalent to the uncertainty in the estimate exponential decay constant) is shaded.
Extended Data Fig. 7 Polynesian ancestry-specific shared drift ordination plot with principal curve.
A principal coordinate analysis (PCoA) projection of the pairwise shared drift distances (the Polynesian ancestry-specific outgroup-F3) between each Pacific island population using Taiwan as an outgroup (Supplementary Fig. 12). This PCoA projection uses only the pairwise distance matrix and is fully unsupervised; that is, it does not presuppose that Rapa Nui is a terminal island along some settlement path. Nevertheless, it shows the same ordering as in Supplementary Fig. 9, confirming that Rapa Nui is indeed the terminal island in our dataset along the longest drift path, and confirming the drift ordering along that path. For further confirmation, a principal curve was also fit to the full dimensional space (Supplementary Fig. 12) and then projected into the two-dimensional PCoA space for visualization. The orthogonal projections of each island onto the principal curve are shown as thinner grey lines. This fully unsupervised principal curve confirms the visually apparent path from Island Southeast Asia (Sumatra, far right) through Samoa, Fiji, Tonga and ending in Raivavae, Mangareva, and Rapa Nui (far left) in that order (cf. migration map in Fig. 2a). This projection of the high dimensional principal curve does not double back on itself, showing that the apparent ordering in this projection is consistent with the original high dimensional ordering. Note that this principal curve is able to fit only one settlement path (the principal one, that is, the longest drift path), which ends in Rapa Nui. Other settlement paths that branch away from this principal (longest) path appear simply as clusters projected onto the principal curve, since islands on those paths share no further drift with the principal path. That is, islands settled along secondary branching paths appear as clusters lying very close to one another along the principal curve. For example, Rapa Iti, which branches off from Rarotonga separately from the main settlement path (Fig. 2a), appears here as coincident with Rarotonga along the principal curve. The eigenvalue for PC1 over the sum of eigenvalues is .997 and for PC 2 is .002 (all eigenvalues are non-negative).
Supplementary information
Supplementary Information
This file contains supplementary text, Figs. 1–33, Tables 1–4, discussion and references.
Rights and permissions
About this article

Cite this article
Ioannidis, A.G., Blanco-Portillo, J., Sandoval, K. et al. Paths and timings of the peopling of Polynesia inferred from genomic networks. Nature 597, 522–526 (2021). https://doi.org/10.1038/s41586-021-03902-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03902-8
- Springer Nature Limited
This article is cited by
-
Ancient Rapanui genomes reveal resilience and pre-European contact with the Americas
Nature (2024)
-
Ecological dynamics of moa extinctions reveal convergent refugia that today harbour flightless birds
Nature Ecology & Evolution (2024)
-
Deep history of cultural and linguistic evolution among Central African hunter-gatherers
Nature Human Behaviour (2024)
-
Ancient DNA reveals rapid natural selection during the Black Death
Nature (2022)
-
Principal component analysis
Nature Reviews Methods Primers (2022)