

Abstract
So far, gene therapies have relied on complex constructs that cannot be finely controlled1,2. Here we report a universal switch element that enables precise control of gene replacement or gene editing after exposure to a small molecule. The small-molecule inducers are currently in human use, are orally bioavailable when given to animals or humans and can reach both peripheral tissues and the brain. Moreover, the switch system, which we denote Xon, does not require the co-expression of any regulatory proteins. Using Xon, the translation of the desired elements for controlled gene replacement or gene editing machinery occurs after a single oral dose of the inducer, and the robustness of expression can be controlled by the drug dose, protein stability and redosing. The ability of Xon to provide temporal control of protein expression can be adapted for cell-biology applications and animal studies. Additionally, owing to the oral bioavailability and safety of the drugs used, the Xon switch system provides an unprecedented opportunity to refine and tailor the application of gene therapies in humans.
Similar content being viewed by others


Main
Although viral and non-viral approaches for gene therapies have undergone substantial advancement over the past twenty years, the major focus has been on the cargo-delivery system. For example, viral capsid evolution and engineering has improved the cell and tissue targeting of adeno-associated viruses (AAVs), and the landscape of cell targeting for lentiviruses has been expanded by using different envelopes in viral production. Additionally, lipid nanoparticles have been refined for improved uptake. However, the cargo itself—and more importantly the elements that control the expression from that cargo—have not received the same attention, although engineered promoters, riboswitches or other 3′ regulatory elements that restrict expression to certain cell types have advanced1,2,3,4.
To address this gap, here we develop a method to finely control protein translation via a drug-inducible switch. The Xon system takes advantage of alternative RNA splicing5, a mechanism that provides for RNA and protein diversity through the inclusion or exclusion of different protein-coding exons, parts of exons, and different 5′ and 3′ noncoding exons. By controlling alternative splicing, we can regulate which exons are spliced in (or out). Notably, the Xon switch does not require any foreign elements for regulation, but rather takes advantage of drugs that are orally bioavailable and in human use to induce the rapid inclusion of an exon containing a start codon. As such, the Xon system can be applied to any genetic element of interest in cells or in animals.
Initially, alternative splicing for the regulated control of protein translation was achieved using drugs developed for the treatment of spinal muscular atrophy, a disorder that is caused by reduced levels of SMN1. The rationale is as follows: SMN2, a pseudogene of SMN1, is correctly spliced only around 10% of the time, resulting in an SMN2 protein that is functionally equivalent to SMN16. Two drugs, LMI070 and RG7800/RG7619, can improve the proportion of correct SMN2 splicing, and both show efficacy in animal models of spinal muscular atrophy7,8. Notably, these drugs are in later-stage clinical testing (LMI070)9 or have been approved for use in humans (RG7800/RG7619)10.
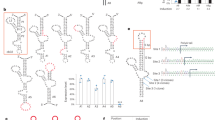
Our first derivation of a splicing switch for controlled translation used a modified SMN2 minigene, such that exon 7 (e7) exclusion and premature termination would occur in the absence of drug, but e7 inclusion and in-frame gene expression would occur in the presence of splice modifiers (Fig. 1a). For this, the original minigene was modified at the e7 acceptor splice site to reduce the approximately 10% background splicing in the native condition (indSMN2; Extended Data Fig. 1a, b). IndSMN2 was further evaluated in response to various doses of LMI070 or RG7800. Both drugs induced e7 splicing, with a complete splicing switch evident at concentrations greater than 1 μM (Extended Data Fig. 1c). For indSMN2 plus LMI070, there was an approximately 20-fold induction (Fig. 1b, Extended Data Fig. 1d). LMI070 showed greater induction activity than did RG7800 (Extended Data Fig. 1c), possibly reflecting their different mechanisms. RG7800/RG7619 binds to the SMN2 pre-mRNA sequence and promotes a conformational change that increases e7 splicing activator affinity, whereas LMI070 stabilizes U1 small nuclear RNA interactions at the e7 5′ splice site8,11.

a, Schematic depicting the SMN2-on cassette. In the absence of exon 7 (e7), a premature stop codon blocks translation of the gene of interest (GOI). Inclusion of e7 permits translation. b, Change in firefly (FF) luciferase activity in LMI070-treated (100 nM) compared with DMSO-treated cells, normalized to Renilla (Ren) luciferase activity. RLU, relative light units. Data are mean ± s.e.m. of 8 biological replicates, ****P < 0.0001 SMN2 versus indSMN2, two-way ANOVA, Bonferroni’s post-hoc test. c, SF3B3 splicing without (red; control (Ctl)) or with (blue) 25 nM LMI070 treatment. The genomic location of the LMI070 spliced-in SF3B3 exon (green bar) and intron counts are indicated. d, cDNAs for SF3B3, BECN1, GXYLT1, C12orf4 and PDXDC2 amplified from cells treated with DMSO or LMI070 (25 nM) (one representative plot from 4 RNA-seq samples). Novel splicing was confirmed by Sanger sequencing. The asterisks represent non-specific bands. e, Volcano plot illustrating differentially expressed genes between DMSO-treated and LMI070- treated cells. The red horizontal bar represents 0.05 significance on a −log10 scale. The red vertical bars indicate thresholds of −0.1 and 0.1 fold change. Genes meeting the significance and minimum fold-change thresholds are labelled (4 samples per group). f, Candidate minigenes for translation control; e1 and e2 are the flanking exons of the novel SF3B3 exon (Extended Data Tables 1, 2). g, Induction of luciferase activity in cells transfected with the SF3B3-based Xon cassette. Samples are normalized to Renilla luciferase and are relative to DMSO. Data are mean ± s.e.m. of 8 biological replicates, **P < 0.01 mCMV versus RSV or PGK; **P < 0.01 RSV versus PGK, two-way ANOVA followed by Bonferroni’s post hoc test.
Because the fully spliced-in switch required drug doses that are unlikely to translate to the clinic, we searched for exons for which splicing was responsive to therapeutically relevant conditions. HEK293 cells were treated with 25 nM LMI070 for 12 h, and RNA sequencing (RNA-seq) was performed to determine alternatively spliced exons that were present only under drug-treated conditions. Reads were aligned to the genome and splicing events that were exclusive to the LMI070-treated samples were identified. After LMI070 treatment, 45 drug-induced splicing events were above our threshold (an average of more than five novel intron-splicing events in LMI070-treated samples; Extended Data Tables 1, 2). Among them, 23 were exclusive to LMI070-treated samples and 22 were identified in all treated samples and were below our threshold criteria in control samples. To assess exclusivity, we queried the 45 events in a resource containing all exon–exon junctions from 21,504 human RNA-seq datasets (Methods). Using SF3B3 as an example, the canonical exon–exon junction in SF3B3 was observed in 12,872 datasets at an average frequency of 64 counts, whereas the LMI070-induced exon was observed in 10 and 1 dataset(s), respectively, for the 5′ and 3′ exon–exon junctions (Fig. 1c, Extended Data Tables 1, 2). The average counts per dataset for the 5′ exon–exon junction was 1.3, whereas the 3′ exon junction was observed only once. Thus, the LMI070-induced SF3B3 exon almost never occurs naturally.
The candidate exons from LMI070-treated samples share a strong 3′ AGAGUA motif that is consistent with the identified LMI070-targeted U1 RNA binding site8 (Extended Data Fig. 2a). The top five novel LMI070-induced splicing events were validated by PCR, which amplified the novel exons exclusive to cDNA samples from LMI070-treated cells (Fig. 1d). The global effect of LMI070 treatment on gene expression was also evaluated by differential expression analysis. There were 6 upregulated and 24 downregulated genes that passed the threshold for significance (P < 0.05; Benjamini–Hochberg multiple testing correction). After filtering out genes with low fold-change values (less than 0.1-fold) five upregulated and nine downregulated genes were identified (Fig. 1e).
We next used our custom pipeline to re-analyse a previous RNA-seq dataset8 obtained from human cells treated with 100 nM LMI070. We observed strong concordance with our results, detecting 70% of our candidate genes in this previous dataset. The SF3B3 and BECN1 exons were the most significantly alternatively spliced in both datasets (Extended Data Fig. 2b, c). In addition, only 7 of the 42 reference splice events8 induced by LMI070 were significantly differentially expressed in our study (Benjamini–Hochberg adjusted P value (Padj) < 0.05; Extended Data Table 3), probably because of the low LMI070 concentration used here.
We developed switch-on luciferase cassettes using the minimal intronic intervening sequences to recapitulate splicing of the drug-responsive pseudo exons in SF3B3, BECN1, C12orf4 and PDXDC2. To limit translation such that it would occur only in response to the drug, the novel exon was engineered to contain the Kozak sequence and AUG start codon (Fig. 1f). All possible downstream start codons were also removed to ensure that an AUG start would be included in response to LMI070 binding only. Increased luciferase expression was observed for each candidate cassette transfected into HEK293 cells in response to LMI070, with the SF3B3-on switch exhibiting a greater than 100-fold induction (Extended Data Fig. 3a, b), five times that of the 20-fold induction afforded by the indSMN2 minigene (Fig. 1b).
To assess background splicing from the engineered switch cassette, we performed two PCR assays. The LMI070-spliced-in exon was not detected when using primer pairs that bound the flanking exons, whereas a faint signal was observed when using primers binding within the novel exon sequence (Extended Data Fig. 3c). Thus, in the absence of LMI070, the alternative exon may be included in a small fraction of the transcripts, mirroring what was found in the Intropolis dataset. Overall, these assays showed marginal baseline luciferase activity in the absence of LMI070 for all candidate cassettes, which may reflect in-frame non-AUG start codon activity12 (Extended Data Figs. 3b, 4). The SF3B3-on switch, which showed the least background splicing, was used in all subsequent studies and is hereafter referred to as Xon.
We first compared Xon induction from the Rous sarcoma virus (RSV), the phosphoglycerate kinase (PGK) and the minimal cytomegalovirus (mCMV) promoters. All promoters drove dose-responsive inducible expression that was mirrored by splicing (Fig. 1g, Extended Data Fig. 5). The induction strength was of the order PGK ≈ RSV > mCMV, thereby providing tools for a gradient of induction.
To assess Xon in vivo, eGFP was cloned into the cassette, validated in vitro (Extended Data Fig. 6a) and the construct packaged into AAV9. AAV9-Xon-eGFP was administered intravenously to mice, and 4 weeks later the mice were given a single oral dose of either vehicle or LMI070 and eGFP expression assessed 24 h later in the liver, heart and skeletal muscle (Fig. 2a). There was notable eGFP fluorescence in liver and heart sections (Fig. 2b, Extended Data Fig. 6b), and the dose response of the eGFP signal that was observed by microscopy was confirmed by western blot (Fig. 2c, Extended Data Fig. 6c), RT–qPCR and splicing assay (Fig. 2d, Extended Data Fig. 6d–f). Notably, eGFP protein levels and novel exon splicing correlated directly to the dose of LMI070.

a, Schematic of experiments using AAV9-Xon-eGFP. Mice were injected intravenously with AAV9-Xon-eGFP, after 4 weeks they were orally dosed once with LMI070, and tissues were collected 24 h later to assess mRNA (Extended Data Fig. 6) and protein expression. vg, viral genomes. b, Representative photomicrograph showing eGFP in the liver 24 h after dosing. n = 3 mice per group. Scale bar, 100 μm. c, Representative eGFP western blot with β-catenin (β-Cat.) as the loading control. n = 4 mice per group. Form. buffer, vector formulation buffer; Veh., vehicle. d, Average (mean ± s.e.m.) cycle threshold (Ct) values for eGFP or LMI070-induced expression using TaqMan assays (Extended Data Fig. 6e). The fold change stated for the spliced product is relative to vehicle-treated, AAV9-Xon-eGFP injected mice. n = 4 mice per group. e, Schematic of the re-dosing experiment. Mice were injected intravenously and 4 weeks later were dosed with LMI070 or vehicle. LMI070-treated mice were subjected to drug washout for one week and then redosed with drug or vehicle (n = 5 mice per group). Tissues were collected 24 h after each dose. f, Representative photomicrograph of the liver showing eGFP 24 h after each dose. n = 2 mice per group. Scale bar, 100 μm. g, Representative eGFP western blot 24 h after dosing, with β-catenin as loading control. n = 3 mice per group. h, Average (mean ± s.e.m.) Ct values for eGFP and fold change relative to AAV9-Xon-eGFP-injected mice treated with vehicle (n = 3 mice per group).
We next tested the responsiveness of the Xon cassette to a second LMI070 dose. Mice were treated intravenously with AAV9-Xon-eGFP, and 4 weeks later were given one oral dose of vehicle or of LMI070. One day later, some of the mice were euthanized, and eGFP induction was assessed by histology, splicing and RT–qPCR assays. The remaining mice underwent one week of drug washout, after which they were given LMI070 or vehicle (Fig. 2e). Induction in the liver was evident after the first and the second dose by histology, western blot, splicing and RT–qPCR assays (Fig. 2f–h, Extended Data Fig. 6g, h). Induction in the skeletal muscle and the heart was also observed (Extended Data Fig. 6h, i), the relative levels of which were greatest for the latter (around 400-fold and 3,000-fold, respectively). Notably, the fold induction for each tissue was similar after each dose (Fig. 2h, Extended Data Fig. 6i). Cumulatively, these data show that eGFP expressed from an AAV-encoded Xon cassette remains undetectable in the uninduced state, and that LMI070 robustly induces expression.
We next assessed the efficacy of Xon for controlling erythropoietin (Epo), which is used to treat anaemia associated with chronic kidney disease13. The Xon cassette expressing mouse Epo (mEpo) was packaged in AAV8 (AAV8-Xon-Epo) and the construct was delivered intravenously to mice, which were subsequently treated with LMI070 or vehicle (Fig. 3a). mEpo plasma levels increased 25-fold to 62-fold in response to 2.5 or 10 mg kg−1 of LMI070, respectively, and returned to baseline 5 days after induction. One day after induction, Epo levels in AAV-eGFP or AAV-mEpo vehicle-treated mice were 173.6 ± 26 pg ml−1 and 446 ± 40 pg ml−1, respectively. For LMI070 treatment, mEpo levels increased in AAV8-Xon-mEpo mice to 4,333 ± 797 pg ml−1 or 10,870 ± 1,383 pg ml−1 for doses of 2.5 mg kg−1 or 10 mg kg−1, respectively (Fig. 3b, left). The elevation of mEpo induced an increase in haematocrit to 60–70% above baseline levels depending on dose, after which a slow return to basal levels was observed (Fig. 3b, right).

a, Experimental design for mEpo induction and the effect on haematocrit levels. Three weeks after AAV or buffer delivery, mice were dosed with vehicle or LMI070 at 2.5 or 10 mg kg−1 every other day for a total of 3 doses. Blood was collected for mEpo (red drop) and haematocrit (orange drop) assays as indicated. b, Left, mEpo levels 2 weeks after AAV injection, and 1, 5 and 9 days after the last dose. ****P < 0.0001, LMI070-treated versus vehicle-treated mice; ****P < 0.0001, 10 versus 2.5 mg kg−1 LMI070-treated mice. Right, haematocrit levels two weeks after AAV injection, and for 50 days after dosing. The orange shaded area shows the haematocrit range of wild-type mice. *P < 0.05 mEpo injected, LMI070-treated mice versus vehicle. c, Schematic of the re-inducibility experiment. Xon-mEpo-AAVs were delivered intravenously and mice given vehicle or LMI070 orally every other day for a total of 3 doses. After haematocrit returned to baseline levels, LMI070 was re-administered and mice were monitored as before. d, mEpo levels from 2 weeks after AAV delivery. ****P < 0.0001 AAV-Xon-mEpo-injected, LMI070-treated mice versus AAV-eGFP-injected mice. e, Haematocrit levels assessed over the time course of the experiment. *P < 0.05 mEpo-injected mice treated with LMI070 versus eGFP-injected mice. Data are mean ± s.e.m. of 9, 6 (b), 5 (d) or 4 (e) mice per group, significance was determined by two-way ANOVA followed by Bonferroni’s post hoc test.
We next tested the re-inducibility of AAV-Xon-mEpo in response to LMI070, as re-administrations are required for prolonged therapy in patients with chronic kidney disease (Fig. 3c). The re-induction of Epo generated similar results to the initial experiments, showing the practical utility of the Xon system for the regulation of therapeutic transgenes in response to repeat oral dosing (Fig. 3d, e).
To assess the applicability of our system for brain-targeted gene therapies, the Xon-eGFP cassette was packaged into AAVPHPeB14 and delivered intravenously to mice (Fig. 4a). LMI070 induced robust eGFP expression throughout the brain, and all regions evaluated showed a clear dose response according to histological assessment (Fig. 4b, Extended Data Fig. 7a), western blot (Fig. 4c), RT–qPCR and splicing assays (Fig. 4d, Extended Data Fig. 7b).

a, Schematic of the AAVPHPeB-Xon-eGFP experiment. AAVs were injected intravenously, then after 4 weeks mice were given an oral dose of LMI070, and brains were collected 24 h later to assess splicing (Extended Data Fig. 7b), transcript levels and protein expression. b, Representative compiled photomicrographs (5 mice per group) from 40-μm-thick sagittal sections. Scale bar, 3 mm. c, Representative eGFP western blots of the cortex (Cx) and the hippocampus (Hc), with β-catenin as the loading control. n = 3 mice per group. d, Average (mean ± s.e.m.) Ct values for eGFP and fold change relative to AAV-Xon-eGFP-injected mice treated with vehicle. e, Schematic depicting the assessment of AAV-Xon-PGRN for regulation of progranulin expression in the brain. Vectors were administered intravenously, then after 4 weeks mice were treated with a single oral dose of LMI070 or vehicle, and tissues were collected 24 h later. f, Progranulin protein levels in the cortex. Data are mean ± s.e.m. of 5 mice per group. ***P < 0.001 for AAV-PGRN or AAV-Xon-PGRN + 50 mg kg−1 versus mice injected with AAV-Xon-PGRN + vehicle, *P < 0.05 for AAV-Xon-PGRN + 50 mg kg−1 versus AAV-PGRN, one-way ANOVA followed by Bonferroni’s post hoc test. F.B., vector formulation buffer.
Some gene-replacement therapies have only a narrow therapeutic expression window, including the expression of progranulin (PGRN) in the treatment of PGRN-deficient frontotemporal dementia and neuronal ceroid lipofuscinosis15. For Xon-regulated delivery of PGRN, AAVPHPeB-Xon-PGRN vectors were generated and administered intravenously to mice (Fig. 4e). Mice injected with AAVPHPeB vectors that constitutively express PGRN were used as a control. Although all groups showed similar PGRN mRNA levels after AAV injection (Extended Data Fig. 7c), PGRN protein was induced to levels of 1,135 ± 183 pg ml−1 and 11,393 ± 1,153 pg ml−1, respectively, after LMI070 doses of 5 or 50 mg kg−1 (Fig. 4f). Splicing data were consistent with these results (Extended Data Fig. 7d).
It would be desirable to be able to regulate the expression of gene-editing proteins delivered by viral vectors. As such, we tested Xon for regulated Staphylococcus aureus Cas9 (SaCas9) editing in liver using Ai14 mice16. Because the Xon-SaCas9 cassette exceeds current AAV packaging limits (5.6 kb), we generated a smaller version that we denote miniXon (Extended Data Fig. 8a). Induction and splicing of miniXon was maintained and its overall amplitude was only modestly reduced relative to SF3B3-Xon; at 100 nM LMI070, induction of the miniXon was 132-fold compared with 152-fold for Xon (Extended Data Fig. 8b, c). AAV8-miniXon-SaCas9 plus single-guide RNAs targeting the loxP-STOP sequence in Ai14 mice were administered intravenously; after several weeks, LMI070 was administered and gene editing was assessed 7 days later (Extended Data Fig. 8d). The expression of tdTomato was detected by the fluorescence-activated cell sorting analysis of hepatocytes and histology on liver tissue sections (Extended Data Fig. 8e,f). Additionally, a genomic DNA PCR assay followed by Sanger sequencing confirmed gene editing (Extended Data Fig. 8g, h). Cumulatively, our results expand the utility of Xon to control Cas9 protein translation for gene-editing applications.
Substantial improvements have been made towards the development of small, potent, ubiquitous and tissue-specific promoters. However, the lack of efficient switches with which to fine-tune protein expression limits the broader application of gene therapy for disorders in which either loss of function or overexpression is consequential. Indeed, there are many clinical scenarios in which a narrow therapeutic window exists15,17. Moreover, toxicity can emerge as a result of sustained expression at supraphysiological levels. In AAV9 therapy for spinal muscular atrophy, the initial benefits of SMN overexpression were reversed by later toxicities caused by a splicing-machinery imbalance resulting from high levels of recombinant SMN in mice18. Although our work used AAV Xon, it could also be engineered for chimeric antigen receptor (CAR) T cell therapies and applied to the setting of CAR-T cell exhaustion19 to pause CAR expression. Similarly, CRISPR-based treatments will benefit from using tunable switches, because refined control to provide a short burst of expression of CRISPR effector proteins would reduce unwanted off-target editing20, and—because the proteins would be expressed for hours rather than for months or years—immune responses to nuclease-expressing cells would be limited21,22. Additionally, the transient exposure of cells to active nucleases should reduce the genomic integration of free AAVs that is seen upon prolonged expression of editing machinery23.
Our switch system was originally designed for use with RG7800 or LMI070, but the refined system, Xon, was built around LMI070. Here we show that normally unspliced sequences that are rarely found in the transcriptome are spliced in in a dose-dependent manner. Although other splicing events induced by LMI070 represent potential drawbacks, we anticipate that most applications would entail infrequent dosing, providing substantial periods of washout that would enable the re-expression of gene products that are silenced by the inclusion of suicide exons. Moreover, the Xon platform was developed using exons that were spliced in at very low drug exposures, further minimizing unwanted splicing events.
Most strategies for the control of gene expression have focused on controlling RNA levels by including cis-regulatory elements, although an approach to control protein translation—as is achieved with Xon—has been reported24. In this system, L7Ae controls translation through binding to a specific RNA motif24; however, immune responses could compromise clinical translation. For RNA regulation, ribozyme-based switches have been engineered to self-cleave mRNA sequences in which they were embedded1. Recently, optimization of a type-III hammerhead ribozyme has been reported, with higher self-cleavage activity and tighter regulation using morpholino-blocking oligomers for in vivo application25. However, morpholinos have a slow off-rate, which renders fine control problematic. Additionally, morpholinos do not reach the brain when delivered systemically. A tetracycline-responsive ribozyme switch that combines the hammerhead ribozyme N79 from Schistosoma mansoni with a tetracycline-binding aptamer showed tetracycline induction26. However, background expression was high. Another approach uses microRNA-binding sites within the 3′ untranslated region of exogenous mRNAs to reduce transcript stability in undesired cell types by co-opting the endogenous RNA interference machinery27. However, although this controls an off state, it would have to be merged with a switch such as Xon for precision in the on state.
The Xon switch demonstrates robust control of protein expression, with the extent of induction managed by dose and by promoter strength. In this system, decreasing protein-expression levels are entirely dependent on the half-life of the protein (more than 1 week for eGFP (Fig. 2g, Extended Data Fig. 6g) and less than 5 days for mEpo (Fig. 3b, d)). Merging Xon with protein domains targeted by proteolysis-targeting chimera molecules would provide another method to manage proteins with long half-lives28. Other possible improvements include adding an endoprotease site to the N-terminal region for fused miniexon sequence cleavage. Finally, we demonstrated control of CRISPR-effector protein induction using the Xon switch, and in combination with regulated intein splicing29 or drug-dependent nuclear import30, enhanced control of CRISPR activity could be further managed.
In summary, we present a simple, highly adaptable tool for regulated protein expression. We anticipate that this system could be adapted to any biological question or application in which the fine control of expression is desired.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The in vitro and in vivo experiments were not randomized and the investigators performing molecular and biological studies were not blinded to treatment group. Individuals involved in tissue collection and histology were blinded to treatment group.
Cell culture, transfection and LMI070/RG7800 treatment
Human embryonic kidney (HEK293) cells (obtained from CHOP Research Vector Core stock) were maintained in DMEM media containing 10% fetal bovine serum (FBS), 1% l-glutamine and 1% penicillin/streptomycin at 37 °C with 5% CO2. Cells were cultured in 24-well plates and transfected at 80–90% confluence using Lipofectamine 2000 transfection reagent, according to the manufacturer’s protocol. For all experiments, 4 h after plasmid transfection, cells were treated with LMI070 (MedChemExpress, HY-19620, suspended in DMSO) or RG7800 (MedChemExpress, HY-101792A, suspended in H2O) at the indicated concentrations. Cells were tested for mycoplasma by Research Vector Core. None of the lines used here are listed in the ICLAC database of commonly misidentified cell lines.
Plasmids, primers and custom-made TaqMan gene-expression assays
Sequences encoding Xon and miniXon are available on the Davidson lab website https://www.thedavidsonlab.com and are deposited to Addgene (nos. 174659, 175660). All primer sequences and custom Taqman gene expression assays to determine SF3B3 novel exon inclusion are available upon request. Primers and custom Taqman gene expression assays were obtained from Integrated DNA Technologies (IDT).
In vitro luciferase assays
HEK293 cells were cultured in DMEM (10% FBS (v/v), 1% penicillin/streptomycin (v/v) and 1% l-glutamine (v/v) in 24-well plates. At 70–80% confluence, cells were co-transfected with the Xon. Firefly luciferase cassettes (0.3 μg per well) and a SV40p-Renilla luciferase cassette as transfection control (0.02 μg per well). Four hours after transfection cells were treated with LMI070 or RG7800 at the indicated concentrations. At 24 hours post transfection, cells were rinsed with ice-cold PBS and Renilla and firefly luciferase activities were assessed using the Dual-Luciferase Reporter Assay System (Promega) as per the manufacturer’s instructions. Luminescent readouts were obtained with a Monolight 3010 luminometer (Pharmigen). Relative light units (RLUs) were calculated as the quotient of Renilla/Firefly RLUs and results expressed relative to mock-treated control cells.
Mice and histology
Animal protocols were approved by The Children’s Hospital of Philadelphia Institutional Animal Care and Use Committee. Five to six-week-old male C57BL/6J mice used in Xon studies to control translation of eGFP, mouse erythropoietin and human progranulin were obtained from Jackson Laboratories. Five to six-week-old male and female Ai14 mice used in gene-editing studies were obtained from an in-house-bred Ai14 colony derived from mice obtained from Jackson Laboratories (stock no 007914). AAV vectors were generated at the CHOP Research Vector Core, and were administrated by retro-orbital injection at 7–8 weeks old in a volume of 150–200 μl. For the dose–response studies, several weeks after AAV administration, a single dose of LMI070 at 2.5, 5, 10 or 50 mg kg−1 (MedChemExpress, HY-19620) or vehicle solution was administrated by oral gavage. For the re-dose studies, LMI070 was re-administrated at the indicated times and concentrations. At euthanasia, mice used for non-histology work were perfused with 0.9% cold saline mixed with 2-ml RNAlater (Ambion). Brains and liver samples were collected, flash-frozen in liquid nitrogen, and stored at −80 °C until use. For histological studies, mice were perfused with 15 ml ice-cold 0.1 M PBS followed by 15 ml 4% paraformaldehyde. Sections were analysed using a DM6000B Leica microscope equipped with a L5 ET filter cube (excitation and emission wavelengths of 470 ± 20 nm and 525 ± 15 nm, respectively; dichroic 495 nm), a 10X HC PLAPO (numerical aperture (NA) 0.40), and a 40X HC PLAPO CORR (NA 0.85) lens connected to a Sola Light Engine LED light source (Lumencor). Images were collected with a Hammatsu Orca flash 4.0 monochrome camera controlled, or Leica SP8 confocal microscope equipped with a white-light laser, HyD sensors, and 40X HC PL APO CS2 (NA 0.75) lens. The excitation and emission wavelengths were set at 493 and 497–535 nm or 554 and 563–730 nm for the green and red channels. The Leica LAS X (v.3.0.3) software was used for both controlling microscopes and obtaining images.
RNA extraction, RT–PCR and splicing assays
Total RNA was extracted using TRIzol (Life Technologies) according to the manufacturer’s protocol, with the exception of 1 μl Glycoblue (Life Technologies) in addition to the aqueous phase in the isopropanol precipitation step and a single wash with cold 70% ethanol. To determine splicing of the SMN2-on and Xon switches, 2 μg of total RNA from HEK293 cells or tissue samples was treated with DNaseI Free kit (Thermo Fisher) followed by cDNA generation using the High capacity cDNA kit (Thermo Fisher). Splicing was determined by PCR using the Phusion HighFidelity polymerase (Thermo Fisher) and PCR products separated on a 2.5% agarose gel pre-stained with EtBr and spliced-in and spliced-out band densitometry performed using with the ChemiDoc Imaging System (BioRad) and Image Lab analysis software. Splicing induction from mouse tissues was determined using two custom TaqMan assays designed to determine total or LMI070-spliced in mRNA transcripts. The percentage of induction was determined by dividing the average Ct novel exon and average Ct total, relative to control mice injected with AAV virus plus vehicle.
Genomic DNA extraction and genome-editing analysis
Genomic DNA from liver samples was extracted using a Monarch genomic DNA purification kit (NEB) according to the manufacturer’s instructions. Deletion of the loxP-STOP-loxP region of the transgenic ROSA26 locus in the Ai14 mice was confirmed by PCR using primers binding outside the intervening segment cleaved by the sgRNA/SaCas9 complex pair (forward: 5′-GCT GGT TAT TGT GCT GTC TCA TC-3′ and reverse: 5′-CAT GAA CTC TTT GAT GAC CTC CTC-3′). PCR products were cloned into TOPO plasmids using the TOPO TA Cloning Kit and subsequently transformed into DH5α competent cells. Individual colonies were analysed using Sanger sequencing to determine editing of the loxP-STOP cassette.
Western blots
Tissues were homogenized in RIPA buffer (final concentration: 50 mM Tris, 150 mM NaCl, 1% Triton-X100, 0.1% SDS, 0.5% sodium deoxycholate, with Complete protease inhibitors (Roche)) and samples were incubated for 1 h rotating at 4 °C then clarified by centrifugation at 10,000g for 10 min. Total protein concentration was determined by DC protein assay (BioRad) and 30 μg was loaded onto 4–12% NuPAGE Bis-tris gels in MES buffer (Novex Life Technologies) to determine eGFP and β-catenin levels. After electrophoresis, proteins were transferred to 0.2 μm PVDF (Bio-Rad). Membranes were blocked with 5% milk in PBS-T and then blotted with rabbit anti-β-catenin (Ab2365, dilution 1:4,000; Abcam), rabbit anti-GFP (A11122, dilution 1:5,000; Invitrogen) followed by horseradish-peroxidase-coupled antibodies (goat anti-rabbit: 111-035-144, dilution 1:50,000; Jackson ImmunoResearch). Blots were developed with ECL Plus reagents (Amersham Pharmacia) and imaged on the ChemiDoc Imaging System (BioRad). eGFP and β-catenin protein levels were determined using the same PVDF membrane (whole blot). Restore western blot stripping buffer (21063, Thermo Fisher Scientific) was used to remove primary eGFP and secondary antibodies before β-catenin detection.
Haematocrit and plasma epo assays
At each indicated time point, 70 μl of blood was collected from the eye into heparin-treated microhaematocrit capillary tubes (Fisher Scientific, 22-362-566) and centrifuged using a micro-haematocrit centrifuge (International Equipment Company model MB, 341118722). Haematocrit counts were then measured using a micro-haematocrit card reader (LW scientific, ZCP-EZRD-HEM7). Plasma samples in the capillary tubes were collected and stored at −80 °C. Two- or fourfold-diluted (control and vehicle treated mice, respectively) or tenfold-diluted plasma samples (LMI070 treated mice) were used for measuring plasma Epo protein concentration using a Mouse Erythropoietin Quantikine ELISA Kit (R&D), and ELISA samples measured with a SpectraMax M5 plate reader (Molecular Devices). The normal haematocrit range was obtained from ref. 31.
Quantification of human progranulin by time-resolved FRET immunoassay
Brain samples were homogenized with Precellys in 400 μl of STEN buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.6, 2 mM EDTA, 1% NP-40) with Complete Protease Inhibitor (Thermo, A32955) and PhosSTOP phosphatase inhibitor (Thermo, A32957). Brain extracts were then clarified by centrifugation for 20 min at 15,000 rpm at 4 °C. Supernatant was collected, and total protein concentration was quantified by BioRAD DC protein assay kit (Bio-Rad, 5000116). Samples were aliquoted and frozen at −80 °C for future use as needed. Human progranulin antibodies (R&D Systems AF2420 and R&D Systems MAG2420) were labelled with donor Tb-fluorophore and acceptor d2-fluorophore, respectively (Cisbio). After optimization of antibody titres and incubation conditions, quantification of human progranulin levels was performed in a 384-well low-volume ProxiPlate (Perkin Elmer, 6008238). Then, 16 μl of diluted sample was loaded in duplicate followed by the addition of 2 μl antibody mix. A standard curve was prepared using Recombinant Human Progranulin protein standard (R&D Systems, 2420-PG-050). All sample and standard dilutions were performed in 1× PBS with 0.1% bovine serum albumin. For brain tissue, the standard was spiked with STEN buffer homogenized mouse brain extract to account for the matrix effects. Plates were then incubated at room temperature for 1 h. Time-resolved fluorescence resonance energy transfer readout was performed with an EnVision Reader (PerkinElmer). After the excitation of the donor fluorophore Tb at 320 nm and a time delay of 100 μs, the resulting Tb and d2 emission signals were read at 615 nm and 665 nm, respectively.
Hepatocyte isolation and flow cytometry
To isolate hepatocytes from Ai14 mice, whole livers were perfused (perfusion solution: cold 0.5 mM EDTA in Ca2+-, Mg2+-free HBSS), excised and disrupted with forceps. They were then minced into very small pieces using a razor blade and enzymatically digested for 2 h at 37 °C with shaking (digestion solution: 3 mM CaCl2, 200 units per ml collagenase IV, and 1.5% BSA in HBSS). The final hepatocyte suspension was passed through a 70-μm nylon mesh cell strainer and rinsed to finish straining cells through the filter (washing solution: 1.5% BSA in HBSS (Ca2+, Mg2+-, no phenol red). Cells were pelleted by centrifugation and washed, before fixation at room temperature for 15 min (fixing solution: 4% PFA in HBSS). Fixed cells were pelleted, resuspended in FACS buffer (0.5% BSA, EDTA (2 mM), in PBS), and counted using Trypan Blue (1:10 dilution) to determine the total count of live cells. For flow cytometry, cells were aliquoted and stained with primary (ASGR1, Proteintech, 11739-1-AP) and secondary (Alexa647, Thermo Fisher, A32733) antibodies, all diluted at 1:100 in 0.1% PBS/BSA for 30 min at room temperature. Flow cytometry data was acquired using a BD FACSAria Fusion (BD Biosciences). ASGR1/A647 fluorescence was detected using a 640-nm laser and a 670/30 filter. tdTomato fluorescence was detected using a 561-nm laser and a 582/15 filter. Data were analysed using FlowJo 10 software (FlowJo LLC).
RNA-seq
RNA-seq libraries were constructed using the Illumina TruSeq Stranded Total RNA with Ribozero gold protocol at the Iowa institute of Human Genomics. Data from four LMI070-treated (25 nM) and four DMSO-treated HEK293 cell groups were obtained after sequencing across two lanes run on an Illumina Hi-Seq 4000 150PE. The resulting fastq files were aligned to the GRCh38 human genome obtained from Ensembl using the STAR aligner32. Splice junction output by STAR were quantified using a custom R script designed to identify splicing events unique to LMI070 treatment. Top ranking LMI070-exclusive splice events were manually assessed for their applicability to function as a splicing switch. A primary requirement of this evaluation was that two splice events (donor and acceptor) were identified as exclusive or enriched in LMI070-treated cells creating inclusion of a pseudoexon of reasonable size. Candidate splice events were visually evaluated using the Sashimi plot function available in IGV33,34,35. Previously published RNA-seq results from ref. 8 (NVS-SM1 100nM, and DMSO) were obtained as fastq files from NCBI Gene Expression Omnibus. Splicing analysis was performed on these datasets using our analytical pipeline (as described above). NVS-SM1, also known as LMI070, induced splicing events from ref. 8 were compared against induced events obtained from our study. To assess the exclusivity of LMI070-induced splice sites to LMI070 treatment we evaluated the frequency with which candidate splice junctions were previously identified in diverse human RNA-seq datasets deposited in the Sequence Read Archive (SRA). This analysis was performed using Intropolis, a database of exon–exon junctions from 21,504 human RNA-seq samples in the SRA archive. The Intropolis database is indexed by GRCh37 genomic position so we first converted our GRCh38 positions to GRCh37 using the LiftOver tool from the UCSC genome browser36. We then queried LMI070-induced splice sites against the Intropolis database using a custom Python script. The results for each LMI070 candidate splice event are summarized in Extended Data Tables 1, 2. Differential gene expression analysis was performed using DESeq2 to compare samples from the LMI070 and DMSO conditions37. To visualize the abundance of meaningfully differentially expressed genes we generated a volcano plot with a Benjamini–Hochberg-adjusted P value threshold of 0.05 and a 0.1 log fold-change threshold.
Statistical analysis
Statistical analyses were performed using GraphPad Prism v.7 software. Outlier samples were detected using the Grubb’s test (a = 0.05). Normal distribution of the samples was determined using the D’Agostino and Pearson normality test. Data were analysed using one-way ANOVA or two-way ANOVA followed by a Bonferroni’s post hoc test. Statistical significance was considered when P < 0.05. All results are shown as mean ± s.e.m.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
RNA-seq datasets are archived in the NCBI Gene Expression Omnibus under accession number GSE176527. Sequences encoding Xon and miniXon along with primer sequences and details on the custom TaqMan gene expression assays to determine SF3B3 novel exon inclusion are available on the Davidson lab website (https://www.thedavidsonlab.com). Plasmids encoding Xon and miniXon are available from Addgene (nos. 174659, 175660).
Code availability
The R code to characterize novel LMI070-induced splicing events from RNA-seq data is available at https://github.com/DavidsonLabCHOP/Xon.
Change history
01 February 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41586-021-04366-6
References
Yen, L. et al. Exogenous control of mammalian gene expression through modulation of RNA self-cleavage. Nature 431, 471–476 (2004).
Dow, L. E. et al. Inducible in vivo genome editing with CRISPR–Cas9. Nat. Biotechnol. 33, 390–394 (2015).
Brown, B. D., Venneri, M. A., Zingale, A., Sergi Sergi, L. & Naldini, L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat. Med. 12, 585–591 (2006).
Domenger, C. & Grimm, D. Next-generation AAV vectors—do not judge a virus (only) by its cover. Hum. Mol. Genet. 28 (R1), R3–R14 (2019).
Berget, S. M., Moore, C. & Sharp, P. A. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc. Natl Acad. Sci. USA 74, 3171–3175 (1977).
Cartegni, L. & Krainer, A. R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 30, 377–384 (2002).
Naryshkin, N. A. et al. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 345, 688–693 (2014).
Palacino, J. et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 11, 511–517 (2015).
An Open Label Study Of LMI070 (Branaplam) in Type 1 Spinal Muscular Atrophy (SMA); https://clinicaltrials.gov/ct2/show/NCT02268552
FDA Approves Oral Treatment for Spinal Muscular Atrophy (7 August 2020); https://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy
Wang, J., Schultz, P. G. & Johnson, K. A. Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc. Natl Acad. Sci. USA 115, E4604–E4612 (2018).
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. & Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009).
Eschbach, J. W., Kelly, M. R., Haley, N. R., Abels, R. I. & Adamson, J. W. Treatment of the anemia of progressive renal failure with recombinant human erythropoietin. N. Engl. J. Med. 321, 158–163 (1989).
Chan, K. Y. et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 20, 1172–1179 (2017).
Amado, D. A. et al. AAV-mediated progranulin delivery to a mouse model of progranulin deficiency causes T cell-mediated toxicity. Mol. Ther. 27, 465–478 (2019).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010).
Collins, A. L. et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 13, 2679–2689 (2004).
Van Alstyne, M. et al. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat. Neurosci. 24, 930–940 (2021).
Weber, E. W. et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 372, eaba1786 (2021).
Fu, Y. et al. High-frequency off-target mutagenesis induced by CRISPR–Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826 (2013).
Charlesworth, C. T. et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 25, 249–254 (2019).
Wagner, D. L. et al. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 25, 242–248 (2019).
Hanlon, K. S. et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 10, 4439 (2019).
Saito, H. et al. Synthetic translational regulation by an L7Ae-kink-turn RNP switch. Nat. Chem. Biol. 6, 71–78 (2010).
Zhong, G. et al. A reversible RNA on-switch that controls gene expression of AAV-delivered therapeutics in vivo. Nat. Biotechnol. 38, 169–175 (2020).
Strobel, B. et al. A small-molecule-responsive riboswitch enables conditional induction of viral vector-mediated gene expression in mice. ACS Synth. Biol. 9, 1292–1305 (2020).
Brown, B. D. et al. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 25, 1457–1467 (2007).
Winter, G. E. et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015).
Buskirk, A. R., Ong, Y. C., Gartner, Z. J. & Liu, D. R. Directed evolution of ligand dependence: small-molecule-activated protein splicing. Proc. Natl Acad. Sci. USA 101, 10505–10510 (2004).
Roper, J. et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol. 35, 569–576 (2017).
O’Connell, K. E. et al. Practical murine hematopathology: a comparative review and implications for research. Comp. Med. 65, 96–113 (2015).
Dobin, A. & Gingeras, T. R. Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinformatics 51, 11.14.1–11.14.19 (2015).
Thorvaldsdóttir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Katz, Y. et al. Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics 31, 2400–2402 (2015).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Nellore, A. et al. Human splicing diversity and the extent of unannotated splice junctions across human RNA-seq samples on the Sequence Read Archive. Genome Biol. 17, 266 (2016).
Kearse, M. G. & Wilusz, J. E. Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev. 31, 1717–1731 (2017).
Acknowledgements
We thank E. Lysenko, Z. Hilfinger and J. Weber of the Davidson laboratory, as well as N. Hoque of the Novartis Institute for Biomedical Research for technical assistance. This work was supported in part by the Hereditary Disease Foundation (A.M.M. and P.T.R.), T32 HG009495 (National Institutes of Health) (P.T.R.), NBIR (R.S., D.L., A.M.M. and B.L.D.) and the Children’s Hospital of Philadelphia Research Institute.
Author information
Authors and Affiliations
Contributions
A.M.M. designed the research, performed experiments, analysed data and wrote the manuscript. A.A.H., E.L. and D.L. performed experiments. P.T.R. performed all bioinformatic analyses and wrote the manuscript. L.T. performed all histology analysis and contributed to statistical analyses. A.M. contributed to all in vivo studies. R.S. and D.L. assisted with progranulin studies. B.L.D. designed and supervised the research, analysed data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
B.L.D. serves on the advisory boards and/or has sponsored research from Homology Medicines, Intellia Therapeutics, Spirovant Sciences, Resilience Bio, Patch Bio, Saliogen, Moment Bio, Triplet Biosciences, Panorama Medicines, Roche and NBIR. A.M.M. has sponsored research from NBIR.
Additional information
Peer review information Nature thanks Alessandra Biffi, Mikhail Shapiro and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 In vitro assessment of SMN2-on cassettes.
a, Cartoon depicting SMN2 exon 7 in its native sequence or with splice-site modifications introduced to reduce background levels of exon 7 inclusion (3′ acceptor splice site, indSMN2). b, Representative RT–PCR reaction showing exon 7 inclusion with the SMN2-on cassettes in the absence of LMI070. The quantification of the exon 7 spliced-in or -out transcripts is depicted as the mean ± s.e.m. of 6 biological replicates. c, Exon 7 splicing of the SMN2 and indSMN2 cassette in response to LMI070 or RG7800. Representative RT–PCR reaction showing exon 7 inclusion as a function of LMI070 or RG7800 dose. The quantification of the exon 7 spliced-in or spliced-out transcripts are the relative transcript levels presented as the mean ± s.e.m. of 8 biological replicates. d, Luciferase activity of the SMN2 and indSMN2 cassettes in response to LMI070. The graph shows relative expression of firefly luciferase expressed from the SMN2-on or indSMN2-on cassettes in cells treated with DMSO or LMI070 (100 nM). The activity of the transfection control Renilla luciferase cassette is represented as a line above the bar graph. Data are the mean ± s.e.m. of 8 biological replicates.
Extended Data Fig. 2 Comparison between induced splice junctions in a previous study and in this work.
See ref. 8. a, Sequence logo of U1 RNA site targeted by LMI070 from 45 spliced-in exons identified by RNA-seq. b, To identify splice junctions with the highest induction across both studies we plotted the mean splice junction counts from our study against the mean splice junction counts from the dataset in ref. 8 reprocessed using our pipeline. The datasets correlate with Pearson’s r = 0.7, indicating that induced junctions performed similarly by rank across both datasets. Additionally, the top ranked splice junctions associated with SF3B3 and BECN1 were consistent across both datasets. c, Sashimi plots depicting novel LMI070-spliced in exons for the SF3B3 and BECN1 genes identified in our study and in ref. 8 by RNA-seq. Genomic location, position of the LMI070 spliced in exon, and intronic counts observed are indicated.
Extended Data Fig. 3 Candidate minigene cassette responsiveness to LMI070 and splicing response to LMI070 of the SF3B3-Xon cassette.
a, Luciferase induction of the minigene cassettes for SF3B3, BECN1, C12ORF4 and PDXDC2. The fold-change luciferase activity in LMI070-treated samples (depicted as +) is relative to DMSO-treated (depicted as −) cells, with data normalized to Renilla luciferase expression. Data are the mean ± s.e.m. of 8 biological replicates. ****P < 0.0001 for SF3B3 versus other candidate exons, ***P < 0.001 C12orf4 versus PDXDC2, one-way ANOVA with Bonferroni’s post hoc test. b, Luciferase activity of the minigene cassettes for SF3B3, BECN1, C12ORF4 and PDXDC2. Data show expression of firefly luciferase from the minigenes in response to DMSO (−) or LMI070 (+) treatment relative to Renilla luciferase activity. Data are mean ± s.e.m. of 8 biological replicates. c, Splicing analysis of the SF3B3-Xon cassette. Representative RT–PCR splicing assay (6 biological replicates) showing inclusion of the LMI070-induced SF3B3 exon in response to DMSO or LMI070 treatment. Inclusion of the LMI070 spliced-in exon was detected using primers binding the exons flanking the LMI070-induced exon (left), or using primers binding within the novel exon sequence (right).
Extended Data Fig. 4 Depiction of the frequency that non-AUG start codons are used determined by ribosome profiling.
a, Translation frequency from AUG and non-AUG start codons determined from ribosome profiling39. b, Depiction of non-AUG start codons in frame luciferase transcripts expressed under control of the SF3B3, BECN1, C12orf4 or PDXDC2 minigenes.
Extended Data Fig. 5 Analysis of SF3B3-Xon expressed from different promoters.
a, Firefly luciferase of the Xon cassettes in response to varying doses of LMI070 relative to Renilla luciferase (grey line). The data are the mean ± s.e.m. of 8 biological replicates. b, Representative RT−PCR for assessment of LMI070-induced pseudo exon expression. Exon inclusion in the Xon cassette was detected using primers flanking the pseudo exon. Data are the mean ± s.e.m. of 8 biological replicates. c, Representative gels from RT−PCR analysis for assessment of the LMI070-induced pseudo exons expressed from the noted promoters in response to varying doses of LMI070. Pseudo exon inclusion was detected using primers binding within the LMI070-induced pseudo exon and the downstream exon. Splicing was quantified and transcript levels presented as the mean ± s.e.m. of 8 biological replicates.
Extended Data Fig. 6 Assessment of the SF3B3-Xon cassette to control translation of eGFP in vitro and in vivo.
a, eGFP expression in HEK293 cells transfected with the SF3B3 minigene cassette (SF3B3-Xon-eGFP) and treated 24 h later with DMSO (left) or LMI070 (right). Bright-field panels are shown below (4 technical replicates). b, Representative photomicrograph of heart tissue sections showing eGFP in heart 24 h after treatment with LMI070 at 50 mg kg−1 (n = 2 mice). Scale bar, 200 μm. Inset, eGFP expression in cardiomyocytes at a higher magnification. Scale bar, 50 μm. c, Extended exposure of the western blot from Fig. 2c (4 mice per group). d, PCR assay demonstrates splicing activity in liver, heart and skeletal muscle in response to LMI070 (4 mice per group). e, Cartoon depiction of the Xon assays designed to quantify the LMI070-induced transcripts and eGFP expression levels from the Xon cassette after AAV9-Xon-eGFP gene transfer. f, Data show average Ct values for eGFP or LMI070-induced expression in heart and skeletal muscle. Fold change of the spliced-in expression cassette is shown relative to basal levels in mice injected with AAV9-Xon-eGFP and treated with vehicle (4 mice per group). g, Extended exposure of the western blot from Fig. 2h (3 mice per group). h, PCR assay demonstrates splicing activity in liver, heart and skeletal muscle after each LMI070 dose (3 mice per group). i, Data show average Ct values for eGFP or LMI070-induced expression in heart and skeletal muscle after each dose. Fold change of the spliced expression cassette is shown relative to basal levels in mice injected with AAV9-Xon-eGFP and treated with vehicle (3 mice per group).
Extended Data Fig. 7 In vivo activity of Xon in brain.
a, Representative photomicrographs (5 mice per group) showing eGFP expression from mice treated intravenously 4 weeks earlier with AAVPHPeB-Xon-eGFP, and 24 h after treatment with vehicle or LMI070 at 5 or 50 mg kg−1. Thalamus (Th), hippocampus (Hc), cerebellum (Cb) and facial motor nucleus (VII), cortex (Cx), striatum (Str), substantia nigra (SN), and medial vestibular nucleus (MV) are shown. Scale bar, 100 μm; inset scale bar, 25 μm. In the hippocampus, * and ** denote the polymorphic and CA3 areas, respectively. In the cerebellum, * denotes the deep cerebellar nuclei. b, Splicing assays for exon inclusion in the cortex and the hippocampus of mice injected with AAVPHPeB-Xon-eGFP (3 mice per group). c, RT–qPCR of human progranulin expression. Data are mean ± s.e.m. of 5 mice per group, ****P < 0.0001 vehicle versus AAV-treated groups, one-way ANOVA followed by Bonferroni’s post-hoc test. d, Splicing assays for exon inclusion in cortex samples of mice injected with AAVPHPeB-Xon-PGRN (3 mice per group).
Extended Data Fig. 8 Generation of the miniXon cassette and assessment of miniXon control of SaCas9 for in vivo gene editing in liver.
a, Cartoon depicting the AAV genome size with the SF3B3-Xon and SF3B3-miniXon cassettes. b, Luciferase induction in HEK293 cells transfected with SF3B3-miniXon-luciferase or SF3B3-Xon-luciferase in response to varying doses of LMI070. All samples are normalized to Renilla luciferase activity and are relative to DMSO treated cells. Data are the mean ± s.e.m. of 8 biological replicates (***P < 0.001 versus SF3B3.Xon, two-way ANOVA followed by Bonferroni’s post hoc test). c, Splicing inclusion assays of the LMI070-induced exon at 100 nM LMI070. Pseudo exon inclusion in the Xon cassette was detected using primers flanking the pseudoexon (left) or by priming within the novel exon sequence (right; 4 technical replicates). d, Experimental design. Mice were injected with AAV8-miniXon-SaCas9 plus AAV8-sgAi14-eGFP (1 × 1012 viral genomes, 1:1 ratio) and 2 weeks later dosed with vehicle or LMI070 at 50 mg kg−1 to induce SaCas9 expression and editing of the loxP-STOP cassette (guides: sgA14_1: 5′-CTCTAGAGTCGCAGATCCTC-3′, sgAi14_2: 5′-ACGAAGTTATATTAAGGGTT-3′). One week later, mice were euthanized, and livers processed to assess gene editing by genomic DNA PCR, histology and FACS of isolated hepatocytes. e, Representative FACS analysis of hepatocytes obtained from Ai14 mice after LMI070 or vehicle treatment. The gating/sorting strategy (above), and the percentage of tdTomato expressing cells for each condition (below) is shown (4 biological replicates). f, Representative photomicrographs of liver sections obtained from AAV injected Ai14 mice 1-week after LMI070 treatment. tdTomato expression (red) is evident in LMI070 treated mice (5 mice per group). Scale bars, 100 μm. g, SaCas9 mediated editing of the loxP-STOP cassette in Ai14 mice as detected by PCR assay of liver genomic DNA (3 of 5 mice with guides plus LMI, 2 of 4 mice with guides plus vehicle, 1 of 2 untreated mice are shown). A PCR product of 355 bp size corresponding to the edited Ai14 ROSA Locus was observed in the LMI070-treated mice. h, Sanger sequencing of the 355 bp PCR product confirmed targeted deletion of the loxP-STOP cassette and DNA repair of the Ai14 reporter locus.
Supplementary information
Rights and permissions
About this article

Cite this article
Monteys, A.M., Hundley, A.A., Ranum, P.T. et al. Regulated control of gene therapies by drug-induced splicing. Nature 596, 291–295 (2021). https://doi.org/10.1038/s41586-021-03770-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03770-2
- Springer Nature Limited
This article is cited by
-
Control of mammalian gene expression by modulation of polyA signal cleavage at 5′ UTR
Nature Biotechnology (2024)
-
Control of neuronal excitation–inhibition balance by BMP–SMAD1 signalling
Nature (2024)
-
Adeno-associated virus as a delivery vector for gene therapy of human diseases
Signal Transduction and Targeted Therapy (2024)
-
Synthetic macromolecular switches for precision control of therapeutic cell functions
Nature Reviews Bioengineering (2024)
-
Specificity, synergy, and mechanisms of splice-modifying drugs
Nature Communications (2024)