

Abstract
Skeletal muscle regenerates through the activation of resident stem cells. Termed satellite cells, these normally quiescent cells are induced to proliferate by wound-derived signals1. Identifying the source and nature of these cues has been hampered by an inability to visualize the complex cell interactions that occur within the wound. Here we use muscle injury models in zebrafish to systematically capture the interactions between satellite cells and the innate immune system after injury, in real time, throughout the repair process. This analysis revealed that a specific subset of macrophages ‘dwell’ within the injury, establishing a transient but obligate niche for stem cell proliferation. Single-cell profiling identified proliferative signals that are secreted by dwelling macrophages, which include the cytokine nicotinamide phosphoribosyltransferase (Nampt, which is also known as visfatin or PBEF in humans). Nampt secretion from the macrophage niche is required for muscle regeneration, acting through the C-C motif chemokine receptor type 5 (Ccr5), which is expressed on muscle stem cells. This analysis shows that in addition to their ability to modulate the immune response, specific macrophage populations also provide a transient stem-cell-activating niche, directly supplying proliferation-inducing cues that govern the repair process that is mediated by muscle stem cells. This study demonstrates that macrophage-derived niche signals for muscle stem cells, such as NAMPT, can be applied as new therapeutic modalities for skeletal muscle injury and disease.
Similar content being viewed by others


Main
Visualizing the collective cellular response to injury remains a long-term goal of the field of regenerative medicine. In pursuit of this goal, we previously developed transgenic zebrafish models of muscle injury to assay muscle stem cell (MuSC)-mediated repair2. Here, we extend this approach to document the cellular and molecular role of specific non-MuSC components of the regenerating area, with a focus on innate immune cells.
In vivo macrophage dynamics during injury
Initially, multiphoton imaging of transgenic larval zebrafish was used to document the macrophage response before, and immediately after, laser-ablation muscle injury (Fig. 1a, b and Supplementary Video 1). After injury, 34 ± 2% of injury-proximate macrophages displayed rapid and directed migration towards the wound (distance, 128.31 ± 68.03 μm; velocity, 0.149 ± 0.040 μm s−1) (Extended Data Fig. 1a and Supplementary Video 1). That not all injury-proximate macrophages responded to the lesion suggests that there is a specific macrophage subset that is primed to respond to trauma. Confocal imaging demonstrated that peak macrophage numbers were reached at 2.50 ± 0.42 hours post-injury (hpi), after which no additional macrophages entered the injury (Fig. 1c–e and Supplementary Video 2). Of the total injury-responding macrophages, 51.11 ± 1.83% remained within the wound at 24 hpi (Fig. 1d, e) and we specify these as ‘dwelling’. By contrast, macrophages that exit the wound do so before 10.48 ± 1.19 hpi, and were designated ‘transient’ (Fig. 1c, e). Transient macrophages exhibited a stellate appearance whereas dwelling macrophages had a spherical form (Extended Data Fig. 1b–d and Supplementary Video 3). This macrophage subset transition occurred irrespective of the magnitude of the injury, but scaled temporally with wound size (Extended Data Fig. 1e–h).

a, Muscle (Tg(actc1b:GFP); magenta) is patrolled by macrophages (Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry); yellow) at 4 dpf. a, After injury, macrophages migrate to the wound (arrowheads). Three different time points are shown. a, b, Frames were obtained from Supplementary Video 1 (n = 30 macrophages in n = 4 injuries). Scale bar, 100 μm. c–e, Two macrophage subsets occupy the wound at distinct times (e), an early injury-responsive transient (c) and late injury-located dwelling (d) subset (arrowheads). Scale bar, 50 μm. Frames were obtained from Supplementary Video 2 (n = 8). f–i, Dwelling macrophages are derived from transient macrophages. f, Photoconversion schematic. g, h, At 1 dpi, injury-located macrophages in Tg(mpeg1:Gal4FF/UAS:Kaede) larvae were photoconverted (g; pre-conversion, magenta (arrowheads); post-conversion, yellow) and reassessed at 2 dpi (h). Scale bar, 50 μm. i, Quantification (n = 20). Individual data points are shown. Unpaired two-tailed t-test; t38 = 0.1174, P = 0.9071. j, Retrospective tracking of dwelling macrophages using whole-larval light-sheet imaging (0.17–19.64 hpi). Top, dwelling macrophage track overlays for the duration of imaging (0.17–19.64 hpi). Bottom, dwelling (yellow mask) and transient (circled, magenta) macrophages migrate to the injury (blue) from wound-proximate origins. Frames were obtained from Supplementary Video 4 (n = 4).
To establish whether dwelling macrophages are a subset of the original pool of injury-responsive macrophages or result from an alternative migratory wave, a transgenic line—in which macrophages express the photoconvertible fluorescent protein Kaede—was used. One day post-injury (dpi), wound-localized transient macrophages were photoconverted to distinguish them from macrophages that were external to the injury (Fig. 1f, g). All dwelling macrophages within the wound site exhibited photoconverted Kaede 24 h after conversion (Fig. 1h, i), indicating that they are derived from the initial transient population. The respective contributions of tissue-resident and circulatory macrophages during repair is an area of active investigation3. Light-sheet microscopy captured the migratory events of individual macrophages at the level of the whole organism during muscle regeneration. Retrospective tracking revealed that all injury-responsive macrophages, including those that proceeded to a dwelling phenotype as well as those that did not, migrated from the vicinity of the wound and not from the circulation, and as such are tissue-resident in nature (Fig. 1j and Supplementary Video 4). This localized macrophage response is reminiscent of macrophage ‘cloaking’ behaviours, in which tissue-resident macrophages rapidly insulate muscle fibre micro-lesions4.
Macrophage–stem cell interactions in vivo
As the residency of dwelling macrophages occurred in the same temporal window as MuSC-mediated repair2, we examined whether they were required for this process. The Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry) transgenic line enables the rapid, temporally controlled, nitroreductase-mediated ablation of macrophages by timed metronidazole (Mtz) dosing (Fig. 2a and Extended Data Fig. 1i–m and Supplementary Video 5). Ablating all injury-responding macrophages and preferentially ablating dwelling macrophages both resulted in a significant regeneration deficit (Fig. 2b, c and Extended Data Fig. 1n). This deficit could not be attributed to ineffective clearance of wound debris after macrophage ablation (Extended Data Fig. 2a–f). By contrast, nitroreductase-mediated ablation of neutrophils generated no alterations to the regenerative programme (Extended Data Fig. 1o–t).

a, Mtz dosing strategy for macrophage ablation. Macrophages expressing nitroreductase are treated at the point of injury (0 dpi) (4 dpf, ablating all macrophages) or at 1.75 dpi (5 dpf, ablating only dwelling macrophages). b, c, Both ablation strategies resulted in a significant regeneration deficit. b, Relative percentage birefringence (yellow). Scale bar, 500 μm. c, Quantification (n = 24 per group). Individual data points are shown. Two-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. d, Length of macrophage–stem cell interactions after laser-ablation muscle injury for transient and dwelling macrophages (n = 5). The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Unpaired two tailed t-test; t48 = 18.45, P = <.0001. e, AiryScan microscopy revealed that dwelling macrophages maintained prolonged contacts with pax3a+ MuSCs (white arrowheads) before MuSC division. Scale bar, 20 μm. Frames were obtained from Supplementary Video 7 (n = 10). f–h, 5′-Ethynyl-2′-deoxyuridine (EdU) incorporation after ablation of dwelling macrophages reveals a requirement for this macrophage subset to maintain MuSC divisions in the wound. f, Schematic. g, All EdU+ cells in the injury zone at this time point were co-labelled with pax3a. No significant difference in proliferation is observed outside the wound in the absence of dwelling macrophages. Scale bar, 50 μm. h, Quantification (n = 13 control, 10 Mtz-treated). The black lines and grey lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test.
We next examined the spatiotemporal relationship between specific macrophage and MuSC populations during regeneration. Time-lapse imaging in a compound Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry);TgBAC(pax3a:GFP) transgenic line, which labels both macrophages and pax3a-expressing MuSCs (Extended Data Fig. 3a–d and Supplementary Video 6) revealed that pax3a+ MuSCs migrated independently of transient macrophages, displayed distinct migration kinetics and took residence at the edge of the wound by 10.25 ± 1.99 hpi (Extended Data Fig. 3a–g and Supplementary Video 6). After the transition to a dwelling state, 72.92 ± 20.83% of dwelling macrophages associated with pax3a+ cells lining the wound edge at 11.17 ± 1.13 hpi and displayed continuous interactions with these cells over the course of 5.38 ± 1.79 h (Fig. 2d, Extended Data Fig. 3d and Supplementary Video 6). These interactions are distinct in nature and duration when compared to those between transient macrophages and pax3a+ cells that are maintained only for 12.86 ± 11.95 min (Fig. 2d and Extended Data Fig. 3a–c). These observations were recapitulated using transgenic met and pax7b MuSC-labelling lines (Extended Data Fig. 3h–j and Supplementary Video 9). AiryScan confocal microscopy further revealed that macrophages enveloped MuSCs with continuous and repetitive membrane extensions (Fig. 2e, Extended Data Fig. 3k and Supplementary Videos 7, 8) and correlative-light and electron microscopy confirmed that the two cell types displayed tight membrane apposition in xyz planes (Extended Data Fig. 4a–d). Notably, these interactions are phenotypically reminiscent of dendritic cell–T cell immunological synapses within lymph nodes that lead to T cell activation5. After these protracted interactions, the associated MuSC invariably underwent cell division (Fig. 2e, Extended Data Fig. 3k and Supplementary Videos 7, 8). Dwelling macrophages interacted with a specific MuSC for 5.42 ± 1.72 h (n = 10) before division, a time frame similar to that defined for total dwelling macrophages–MuSC interactions (5.38 ± 1.79 h) (Fig. 2d and Extended Data Fig. 3d), suggesting that interactions cease upon stem cell division. No cell divisions were observed without previous prolonged interaction with a dwelling macrophage (n = 26 MuSC divisions in n = 10 larvae). Most proliferation-inducing macrophages (71%) maintained long-term associations with a single MuSC, although a small population (29%), simultaneously associated with two cells. No macrophages were observed to maintain more than two concurrent stem cell interactions before division (n = 9). After MuSC proliferation, macrophages and daughter myoblasts migrated away from each other, with macrophages moving to myoseptal locations 1.94 ± 0.94 h after MuSC division. During relocation, 44% of the macrophages interacted over 1.45 ± 0.69 h with one or both daughter myoblasts, whereas the remainder did not have such associations (n = 9) (Extended Data Fig. 3l and Supplementary Video 10). Furthermore, the ablation of dwelling macrophages severely reduced the number of proliferating pax3a+ MuSCs within the injury, lowering proliferation to the homeostatic levels that are present in uninjured regions (Fig. 2f–h). Collectively, these results reveal a surprisingly direct role for a specific macrophage subset in controlling muscle regeneration, demonstrating that a proportion of wound-attracted macrophages form a transient, pro-proliferative niche.
Macrophage diversity during regeneration
To better characterize injury-responsive macrophage populations, we carried out single-cell RNA sequencing (scRNA-seq) on injury-located macrophages isolated from discrete phases of regeneration. After muscle injury of Tg(mpeg1:mCherry) larvae, the wound region was dissected at 1, 2 and 3 dpi and macrophages were isolated (Fig. 3a). Macrophages from uninjured larvae were also included in analyses. Unsupervised clustering identified eight discrete clusters (0–7) of macrophage subtypes (Fig. 3b, c). Overlaying isolation time on UMAP (uniform manifold approximation and projection) scatter plots allowed injury times to be correlated with individual clusters (Fig. 3c). Uninjured larval macrophages formed a temporally uniform cluster (cluster 3) (Extended Data Fig. 5b and Supplementary Table 2) and unsupervised clustering revealed no further cluster complexity. A lack of predetermination in uninjured macrophages is intriguing given that only a proportion of muscle-resident, wound-proximate macrophages respond to injury, suggesting that this ability is an acquired state, at least at the level of gene expression. The majority of transient macrophages (1 dpi) also clustered together (cluster 1) (Extended Data Fig. 5b and Supplementary Table 2), suggesting a systematic activation of migratory macrophages. The remaining six clusters were composed of dwelling macrophages (2–3 dpi), highlighting their heterogeneous nature (clusters 0, 2, 4, 5, 6 and 7) (Extended Data Fig. 5b and Supplementary Table 2). Transcriptomic trajectory and pseudotime analyses revealed two ‘mature-dwelling’ macrophage subtypes represented by clusters 2 and 6. All other clusters had trajectories that suggested a transitory identity from which mature subsets arise (Fig. 3e–h and Extended Data Fig. 5c).

a, Schematic for the isolation of macrophages for scRNA-seq. b, UMAP scatter plot revealed eight spontaneous cell clusters. c, Injury time point of isolated macrophages overlaid on the UMAP plot. d, Macrophage identity validated by expression of the pan-leukocyte marker lcp1 and pan-macrophage marker cd163. Known pro-regenerative macrophage markers arg2, mmp9 and mmp13a as well as nampta are concentrated in cluster-2 macrophages. log-normalized gene read counts; scaling factor 10,000. e–h, Lineage analysis using partition-based graph abstraction (PAGA). e, Ball-and-stick representation of PAGA connectivity; pie charts represent clusters (with chart size reflective of cluster size) showing macrophage composition based on isolation time; edge thickness indicates the statistical measure of connectivity among clusters. Cells were re-embedded using a PAGA-initialized force-directed layout (force atlas 1 (FA1) and FA2 are used for graphical presentation), in which cells were grouped according to isolation time point (f), Seurat cluster identity (g) and pseudotime inferences (h). i, mmp9 expression assayed by in situ hybridization after needle-stab injury (n = 18). Scale bar, 100 μm. j, k, mmp9 expression ((TgBAC(mmp9:eGFP); magenta) is evident in a subset of dwelling macrophages (arrowheads). j, Representative images. Scale bar, 50 μm. k, Quantification (n = 7 (1 dpi), n = 11 (2 dpi)). The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Unpaired two-tailed t-test, t316 = 10.03, P < 0.0001. l–n, Ablation of mmp9+ dwelling macrophages leads to a significant muscle regeneration deficit. l, Representative images. Scale bar, 500 μm. m, Quantification (n = 24 per group). Individual data points are shown. Two-way ANOVA with Tukey’s multiple comparison test. n, EdU incorporation in mmp9+ macrophage-ablated larvae reveals a significant reduction in wound-located MuSC proliferation (n = 17 control, n = 55 Mtz-treated). Representative images are shown in Extended Data Fig. 6h. The black lines and grey lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test.
Although the simple classification of macrophage phenotypes into pro-inflammatory and anti-inflammatory subtypes does not represent in vivo diversity, specific markers can be used to broadly assign macrophage identity and cluster-2 macrophages were enriched for anti-inflammatory markers, including arginase-2 (arg2), matrix metalloproteinase-9 (mmp9) and matrix metalloproteinase-13 (mmp13) (Fig. 3d and Supplementary Table 3). As all cluster-2 cells express mmp9 (Fig. 3d and Extended Data Fig. 5c), we assessed its spatiotemporal expression after injury both by in situ hybridization (Fig. 3i and Extended Data Fig. 6a) and in a transgenic line that expresses eGFP under the control of the mmp9 promoter (TgBAC(mmp9:eGFP) (Fig. 3j, k, Extended Data Fig. 6b and Supplementary Video 11). These analyses revealed that mmp9 expression was upregulated from 2 dpi in the MuSC-associated dwelling macrophage population (Fig. 3i–k and Extended Data Fig. 6a, b). At 2 dpi, 71.09 ± 12.05% of mpeg1+ dwelling macrophages in the wound site were mmp9+ (Fig. 3j, k), a value range in line with the scRNA-seq analysis that identified 59% of 2-dpi macrophages expressing mmp9 (Extended Data Fig. 5d). The number of mmp9+ macrophages is consistent with the number of dwelling macrophages that interact with MuSCs (72.92 ± 20.83%). Furthermore, the majority of the remaining dwelling macrophages in the wound at 2 dpi expressed markers specific to cluster 6 (Pou2f3+mpeg1+, 15.10 ± 12.13% and Prox1a+mpeg1+, 23.36 ± 12.85%) (Extended Data Fig. 5e–j). This observation correlates with the trajectory and pseudotime analyses (Fig. 3e–h and Extended Data Fig. 5c), which defined clusters 2 and 6 as the two ‘mature-dwelling’ macrophage subtypes. In contrast to cluster-2 macrophages, the vast majority of cluster-6 cells were not located adjacent to pax3a+ MuSCs (2 dpi, 12 ± 10% of Pou2f3+mpeg1+ cells and 9 ± 7% of Prox1a+mpeg1+ cells were located adjacent to pax3a+ cells that were present at the wound site).
A macrophage subset required for repair
Next, we specifically ablated cluster-2 macrophages using a transgenic line expressing nitroreductase from the mmp9 promoter. Larvae were injured and treated with 5 mM Mtz at 1.75 dpi, a dose that specifically ablated 77.94% of mmp9+mpeg1+ macrophages in the injury (Extended Data Fig. 6c–e). Ablation led to significant deficits in muscle repair (Fig. 3l, m) and MuSC proliferation that were similar to those evident after ablation of mpeg1+ dwelling macrophages, as described above (Figs. 2b, c, 3g, h and Extended Data Fig. 6f–h). Subsequently, genes encoding secreted and pro-mitogenic molecules that were specifically upregulated in cluster 2 (Supplementary Table 3) were matched with receptors known to be expressed on MuSCs6,7,8, leading to the identification of a pairing between nicotinamide phosphoribosyltransferase a (Nampta) and the chemokine receptor Ccr5 (Fig. 3d and Extended Data Fig. 5d). NAMPT is a multi-functional protein with a well-characterized role in cellular metabolism and NAD regeneration9, when localized intracellularly. In addition, its secreted form is pro-mitogenic for a number of tissues and can alter the expression of myogenic regulatory factors in myoblasts10. Furthermore, CCR5—a putative cell-surface receptor for NAMPT11—is upregulated after muscle injury in mice7, and antibody inhibition of CCR5 reduces the proliferation of mouse myoblasts in vitro6.
In situ hybridization after larval muscle injury for the two nampt genes encoded in the zebrafish genome revealed that nampta (a homologue of the single mammalian NAMPT-encoding gene) was specifically upregulated within the wound at 2–3 dpi. This result was in line with the scRNA-seq analysis of cluster 2 (Fig. 3d, Extended Data Fig. 6i and Supplementary Table 3), whereas namptb (a gene only present in fish, possessing low homology to mammalian NAMPT)12 was expressed at a constant low level within the injury zone from 1 to 3 dpi (Extended Data Fig. 9i). Nampt antibody staining confirmed its localization in MuSC-associated dwelling macrophages and the extracellular space of the injury site (Fig. 4a and Extended Data Fig. 6k, l). Of the dwelling macrophages present in the injury site at 2 dpi, 72 ± 16% were Nampt+mpeg1+ (a value range in line with the scRNA-seq analysis that identified 58% of 2-dpi macrophages expressing nampta) (Extended Data Fig. 5d). This quantification is similar to both the percentage of dwelling macrophages that interact with MuSCs (72.92 ± 20.83%) and the percentage of mpeg1+ macrophages that express mmp9+ in the wound site at 2 dpi (71.09 ± 12.05%). All Nampt+mpeg1+ cells were associated with pax3a+ MuSCs (Fig. 4a). Furthermore, whole-mount immunohistochemistry on regenerating larvae revealed a reduction in Nampt levels after macrophage-specific—but not neutrophil-specific—ablation, reinforcing that the Nampt secreted within the muscle injury is macrophage-derived (Extended Data Fig. 6m, n). Next, we used the endogenous fluorescence of NADH under two-photon imaging as a surrogate, real-time readout of cellular Nampt levels within the wound milieu13. At 2 dpi, dwelling macrophages specifically expressed high levels of NADH within the wound (Extended Data Fig. 6o and Supplementary Video 12). The high levels of NADH and consequently reduced NAD+/NADH ratio (Extended Data Fig. 6p) in dwelling macrophages may result from a metabolic shift, specifically in the mmp9+ dwelling macrophage subtype. There is increasing evidence that the metabolic state of a macrophage is linked to its activation14, and an analysis of our scRNA-seq dataset revealed a potential glycolytic shift in the mmp9+ subset (Extended Data Fig. 5k, l). Alternatively, high Nampta expression may arise as a consequence of Nampta secretion, which may occur at levels high enough to deplete intracellular stores, which in turn could trigger an induction of nampta expression in dwelling macrophages.

a, Nampt (magenta) is expressed by macrophages (yellow; red arrowheads; Nampt+mpeg1+ macrophages) associated with MuSCs (pax3a+; cyan) within the wound (dotted line), and in the extracellular space (n = 15). Scale bar, 50 μm. The red-boxed area (left) is magnified on the right. b–g, Tissue-specific loss-of-function strategies to assess the role of Nampta in macrophages (schematic, b) and the function of Ccr5 in MuSCs (schematic, c). mpeg1+ cell-specific nampta-edited larvae and pax7b+ cell-specific ccr5-edited larvae both display regeneration deficits (representative images (d; scale bar, 500 μm) and quantification (e, n = 23 nampta gRNA injected in wild type, n = 36 uninjected and n = 47 nampta gRNA injected in mpeg1-cas9, n = 58 ccr5 gRNA injected in pax7b-cas9; individual data points are shown; two-way ANOVA with Tukey’s multiple comparison test) due to proliferation deficits in MuSCs (f (n = 12 control and n = 16 nampta gRNA-injected), g (n = 17 control and n = 27 ccr5 gRNA-injected). The black lines and grey lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test). h, hrNAMPT(1) (where (1) indicates the hrNAMPT source used; see Methods) supplementation enhanced and rescued MuSC proliferation in macrophage-ablated larvae (5 mM Mtz at 4 dpf, 0 dpi), but did not rescue proliferation in CVC-administered larvae. Treatment with mouse recombinant CCL8 (mrCCL8) enhanced proliferation; however, it did not discriminate between injured and uninjured muscle (n = 44 control, n = 29 hrNAMPT(1)-treated, n = 24 Mtz-treated, n = 19 Mtz + hrNAMPT(1)-treated, n = 23 CVC-treated, n = 12 CVC + hrNAMPT (1)-treated and n = 13 mrCCL8-treated). Representative images are shown in Extended Data Fig. 10d. The thick black lines and thin black lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. i–l, Local delivery of NAMPT promotes muscle regeneration in an adult mouse VML injury model. i, Schematic. j, VML defects treated with a fibrin hydrogel with or without NAMPT. Masson’s trichrome staining of rectus femoris muscle sections (10 days after treatment) through the middle of the defect shows that NAMPT delivery significantly increased muscle regeneration (dark red) and significantly reduced fibrosis (purple/blue, white dashed line separates fibrotic and healthy muscle, fascia in blue). Scale bar, 1 mm. k, l, Quantification of muscle regeneration (k) and fibrosis (l). The thick black lines and thin black lines within the violin plot indicate the median and quartiles, respectively. One-way ANOVA with Dunnett’s post hoc test for multiple comparisons (n = 5 per group). m, n, MuSCs show enhanced proliferation after NAMPT supplementation. The total number of PAX7+ satellite cells (m) and the number of proliferating (PAX7+Ki67+) satellite cells (n) per 10,000 cells were quantified by flow cytometry in injured tissue collected 4 days after treatment (n = 6 fibrin, n = 5 NAMPT). Gating strategy is shown in Extended Data Fig. 10e. m, n, Data are mean ± s.e.m. Two-tailed Student’s t-test. t9 = 2.803, P = 0.0206 (m); t9 = 3.254, P = 0.0099 (n).
NAMPT stimulates myoblast proliferation
Owing to the availability of molecular reagents, we next used mammalian cell culture systems to rigorously evaluate our model. We first evaluated the binding affinity of NAMPT for CCR5, as there is only a single report of this interaction11. Direct and competitive ligand-binding assays determined that human recombinant NAMPT (hrNAMPT), binds to both human recombinant CCR5 (Extended Data Fig. 7a) and mouse recombinant CCR5 (Extended Data Fig. 7b) with high affinity. Next, we used mouse cell culture models to determine whether NAMPT triggers a pro-proliferative signalling cascade. Initially we used mouse C2C12 myoblasts as they are reported either to not secrete NAMPT15,16, or to do so at very low levels17, and express CCR5 at physiologically relevant levels18. The addition of hrNAMPT protein to C2C12 cultures resulted in comparable and significant dose-dependent increases in myoblast proliferation (Extended Data Fig. 7c). To uncouple the intracellular and extracellular roles of NAMPT during proliferation, these myoblasts were treated with GMX1778, an inhibitor of the enzymatic function of NAMPT. GMX1778 had no effect on the basal level of C2C12 cell proliferation and also did not inhibit the increased proliferation elicited by NAMPT (Extended Data Fig. 7c), indicating that the pro-proliferative role of NAMPT is not reliant on its intracellular enzymatic function. This proliferative response could be recapitulated by the addition of CCR5 canonical ligands, CCL8 (also known as MCP-2) and CCL4 (also known as MIP-1β), but not the CCR2 ligand CCL2 (also known as MCP-1) (Extended Data Fig. 7c). In addition, this response was blocked in the presence of the dual CCR2/CCR5 antagonist cenicriviroc (CVC) and the CCR5-selective antagonist maraviroc (MVC), but not in the presence of the CCR2-selective antagonist PF-4136309 (Extended Data Fig. 7c).
We then used a multi-cell-type coculture system to examine the relationship between macrophages and NAMPT-mediated MuSC proliferation. We assessed the proliferation of isolated embryonic mouse PAX7+ MuSCs in three culture conditions: in single cultures of myoblasts only, and in cocultures of myoblasts with either mouse macrophages (myoblast–macrophage) or 3T3 mouse fibroblast cells (myoblast–3T3). In these experiments, we observed that the presence of macrophages promoted satellite cell proliferation (Extended Data Fig. 7g, h), a response that was not elicited when macrophages were replaced in the coculture by 3T3 cells (Extended Data Fig. 7g, h). Furthermore, addition of hrNAMPT was able to increase the proliferation of satellite cells in myoblast and myoblast–3T3 culture conditions to a level comparable to that in myoblast–macrophage cultures (Extended Data Fig. 7g, h). This effect was abolished in the presence of CVC as well as MVC, and maintained in the presence of PF-4136309 (Extended Data Fig. 7g, h). Notably, hrNAMPT did not increase satellite cell proliferation in the presence of macrophages, suggesting that there is a maximum rate of proliferation that is reached at receptor saturation. Moreover, CCL4 supplementation in cocultures mirrored the results of NAMPT addition, confirming that the proliferative effect of NAMPT is mediated by CCR5.
NAMPT induces in vivo muscle repair
Subsequently, we sought to validate these findings in vivo. We confirmed that zebrafish ccr5 was expressed in pax3a+ MuSCs at 2 dpi (Extended Data Fig. 7i–k), and that administration of CVC and MVC to larvae resulted in a significant regeneration deficit (Extended Data Fig. 7l, m). Macrophage and neutrophil migration kinetics and the macrophage transition to a dwelling state were all phenotypically wild type after CVC administration (Extended Data Fig. 7n–s). However, proliferation of pax3a+ MuSCs was severely reduced, to levels observed after ablation of dwelling macrophages (Fig. 2b, c and Extended Data Fig. 7t, u). These results were confirmed by imaging of CVC-administered, injured larvae, which displayed no discernible deficits in macrophage or MuSC migration into the injury or macrophage transition into a dwelling state at 10.20 ± 0.91 hpi. Dwelling macrophages also affiliated with MuSCs by 11.80 ± 0.57 hpi and interacted identically to control macrophages. However, in contrast to untreated larvae, dwelling macrophages remained associated with their target MuSCs until the experimental end point (18 h) and did not terminate as in control settings, in which these interactions cease and the two cell types move apart after stem cell division (Extended Data Fig. 7v and Supplementary Video 13). As such, the division of MuSCs may trigger the separation of a macrophage and its associated MuSC.
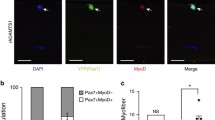
We next generated CRISPR–Cas9-mediated germline mutations in both the namtpa and ccr5 genes and assayed muscle regeneration in homozygous mutants (Extended Data Fig. 8a–c, k–m). No differences in the number of transient or dwelling macrophages (Extended Data Fig. 8d, e, n, o) and their consequent stem cell interactions (Extended Data Fig. 8f, p) were detected in either mutant when compared to wild-type siblings. However, both mutants exhibited a significant regeneration deficit (Extended Data Fig. 8g, h, q–r) and a significant decrease in proliferating MuSCs within the injury zone (Extended Data Fig. 8i, j, s, t) after muscle injury. To determine whether it is specifically macrophage-derived Nampta that is required for regeneration, we developed a macrophage-specific loss-of-function system (A.I.I. et al., manuscript submitted). Macrophage-specific expression of Cas9 was achieved by coupling Tg(mpeg1:Gal4FF) and Tg(4×UAS:NLS-cas9) transgenes. By delivering a nampta guide RNA (gRNA) using microinjection, we achieved durable macrophage-specific nampta gene editing (Fig. 4b and Extended Data Fig. 9a). Although we cannot fully exclude the possibility of ectopic low Cas9 expression in off-target tissues, immunostaining revealed a reduction in Nampt-expressing cells in nampta-gRNA-injected larvae at the wound site (Extended Data Fig. 9b). Furthermore, quantification of NAD+/NADH levels in isolated, gene-edited macrophages functionally validated this approach (Extended Data Fig. 9c). We then assayed the response of macrophages in nampta-gRNA-injected larvae to muscle injury. In injected animals, macrophages migrated normally to the wound, transitioned to a dwelling subtype and actively interacted with MuSCs within the injury (Extended Data Fig. 9d–g). However, these nampta-deficient macrophages did not induce appropriate cell proliferation and regeneration at the injury site (Fig. 4d–f and Extended Data Fig. 9h). By contrast, macrophage-specific namptb-edited larvae underwent significant muscle regeneration, albeit at a reduced rate compared with control larvae (Extended Data Fig. 9j–l). Furthermore, neutrophil-specific deletion of nampta using a Tg(mpx1:KALTA4);Tg(4×UAS:NLS-cas9) transgene combination did not result in any deficits in muscle repair (Extended Data Fig. 9m–o), confirming that macrophages are the source of wound-derived Nampta.
To confirm that Ccr5 was required on MuSCs, we used the pax7b:GAL4FF line in combination with the 4×UAS:NLS-cas9 transgenic line to mutate ccr5 in MuSCs with a dual gRNA combination (Fig. 4c). Editing ccr5 in MuSCs did not alter the number of transient and dwelling macrophages in the injury zone and did not affect associations between dwelling macrophages and stem cells (Extended Data Fig. 9p–r). However, these gene-edited larvae displayed a significant muscle repair deficit (Fig. 4d, e) and a significant MuSC proliferation deficit after muscle injury (Fig. 4g and Extended Data Fig. 9s). Both defects were similar in magnitude to those evident in germline ccr5 mutants (Extended Data Fig. 8q–t) and larvae in which Ccr5 activity was chemically inhibited (Extended Data Fig. 7l, m, t, u). Collectively, our in vitro and in vivo studies demonstrate a requirement for macrophage-derived NAMPT to stimulate MuSC proliferation in a CCR5-dependent manner (Extended Data Fig. 9t).
We next examined the effect of the addition of exogenous hrNAMPT on injured zebrafish larvae. No alternations in immune cell dynamics or lysosomal activity at the wound site were observed after hrNAMPT treatment, in contrast to previous observations in inflammatory models19 (Extended Data Fig. 10a–c). Instead, hrNAMPT supplementation led to a 30.70 ± 4.26% increase in MuSC proliferation within the wound site (Fig. 4h and Extended Data Fig. 10d) and rescued wound site proliferation in macrophage-ablated larvae, even acting to increase the proliferation above that of the control setting by 10.96 ± 3.55% (Fig. 4h and Extended Data Fig. 10d). However, hrNAMPT did not rescue the proliferation deficit when supplemented with CVC (Fig. 4h and Extended Data Fig. 10d). Notably, whereas mouse recombinant CCL8 was able to enhance the proliferative response in the wound site to a similar level to that of hrNAMPT, it also increased cell proliferation external to the injury zone by 43.03 ± 4.46% (Fig. 4h and Extended Data Fig. 10d). This highlights the specific action of NAMPT on MuSCs that are present in the wound site as being distinct from a generalized proliferative response elicited by Ccr5 activation. We additionally determined that the pro-regenerative activity of Nampt was independent of its enzymatic function in vivo, as the addition of GMX1778 to regenerating larvae did not alter MuSC proliferation (Extended Data Fig. 6q, r).
NAMPT promotes muscle regeneration in mice
We then went on to investigate whether exogenous NAMPT could facilitate regeneration in a mouse model of volumetric muscle loss (VML), an injury paradigm usually refractory to endogenous stem-cell-mediated repair processes and an area of unmet clinical need20. Notably, delivery of hrNAMPT into the muscle defect using a fibrin hydrogel—but not a fibrin-only control hydrogel—was able to fully restore muscle architecture when applied to the wound site (Fig. 4i–l). Treatment with a single dose of hrNAMPT (0.5 μg) at the point of injury led to a 3.276 ± 0.4926 mm2 increase in muscle area and a 34.76 ± 9.32% decrease in fibrotic area at the injury zone. hrNAMPT delivery resulted in an increase in both the total number and the proportion of proliferating PAX7+ satellite cells (Fig. 4m, n and Extended Data Fig. 10e, f) and also increased the number of centrally nucleated de novo muscle fibres (Extended Data Fig. 10g, h) without altering the immune cell profile of the regenerating area (Extended Data Fig. 10i, j). Angiogenesis within the wound occurred at levels similar to that described for other pro-regenerative approaches to VML injuries20,21 (Extended Data Fig. 10k, l), suggesting that angiogenesis simply scales with the level of regeneration evident in hrNAMPT-treated injuries. However, a more directed mode of action of NAMPT and CCR5 in stimulating angiogenesis cannot be formally ruled out, particularly given that both of these proteins have previously been shown to induce endothelial cell proliferation22,23,24. We also cannot rule out NAMPT regulation of other cell types present in the wound, such as fibro-adipogenic progenitors, which have also been shown to modulate muscle repair in mice. Nevertheless, these findings reveal that exogenously supplied NAMPT stimulates muscle repair in the context of an acute injury of adult mammalian muscle in a similar manner to the results that we describe for zebrafish larvae. It also reinforces the finding that it is the secreted form of NAMPT that is active in both of these settings.
Conclusion
The satellite cell is archetypal of a unipotent tissue-resident stem cell that occupies a specific anatomical niche within a differentiated tissue. Decades of research have revealed the extraordinary capacity of this system to effectively coordinate muscle repair in response to a wide variety of insults. Despite this, transplantation of isolated MuSCs has yet to provide therapeutic effects, and pro-regenerative treatments that stimulate MuSCs are lacking at this juncture. Our data suggest that even this simple stem cell system requires complex interactions with cellular components of the regenerative milieu and that macrophages are a key modulator of the regenerative process. We introduce the concept of a specific injury-located macrophage subset, acting as a transient stem cell niche that directly regulates MuSCs through the provision of mitogenic stimuli, specifically via the NAMPT–CCR5 axis. Collectively, our results suggest that providing specific macrophage-derived signals required for MuSC proliferation, such as the one we identified here, will provide an avenue to achieve better myoblast-based therapy outcomes.
Methods
Animals
Zebrafish
Existing transgenic lines used include TgBAC(pax3a:GFP)i150 (referred to as TgBAC(pax3a:GFP))25,26, Tg(mpeg1:mCherry)gl23Tg (referred to as Tg(mpeg1:mCherry))27, Tg(mpeg1:GAL4FF)gl25Tg (referred to as Tg(mpeg1:GAL4FF))27, Tg(UAS-E1b:Kaede)s1999t (referred to as Tg(UAS:Kaede))28, Tg(UAS-E1b:Eco.nfsb-mCherry)c264 (referred to as Tg(UAS:nfsb-mCherry)29, Tg(-8mpx:KALTA4))gl28Tg (referred to as Tg(mpx:KALTA4))30,31, Tg(mpx:eGFP)i114 (referred to as Tg(mpx:eGFP)32, Tg(actc1b:EBFP2)pc5Tg (referred to as Tg(actc1:BFP))33, Tg(ubi:secAnnexinV-mVenus)mq8Tg (referred to as Tg(ubi:secAnnexinV-mVenus))34, Tg(actc1b:GFP)zf10 (referred to as Tg(actc1b:GFP))35, pax7bnkgsaizGFFD164AGt (referred to as Tg(pax7b:GAL4FF))36, TgBAC(mmp9:eGFP)tyt206Tg (referred to as TgBAC(mmp9:eGFP))37, TgBAC(met:mCherry-2A-KALTA4)pc24Tg (referred to as TgBAC(met:mCherry-2A-KALTA4))2 and TgBAC(mmp9:eGFP-NTR)tyt207tg (referred to as TgBAC(mmp9:eGFP-NTR))37. All experiments were conducted in accordance with Monash University guidelines and approved by the local ethics committee. All procedures involving animals at the Hubrecht Institute were approved by the local animal experiment committees and performed in compliance with animal welfare laws, guidelines and policies, according to national and European law. Staging and husbandry were performed as previously described38. All embryos were maintained in Ringer’s solution at 28.5 °C and treated with 0.003% 1-phenyl-2-thiourea (Sigma-Aldrich) from 8 hpf.
Mice
C57BL/6J wild-type mice were sourced from Jackson Laboratories and housed in specific-pathogen-free Monash Animal Facility with ad libitum access to food and water throughout experimentation. All animal experiments were carried out with approval by Monash University Ethics Committee and conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Generation and genotyping of zebrafish mutant lines
Mutations in nampta and ccr5 were generated using the CRISPR–Cas9 system. Synthetic gRNAs targeting genes of interest were generated as crRNA:tracrRNA duplexes (Alt-R CRISPR–Cas9 system, IDT). Gene-specific crRNA sequences were selected using the Alt-R CRISPR–Cas9 custom gRNA design tool (IDT), PAM sequences are highlighted in uppercase. (nampta crRNA, 5′-acgacaagacggtcttctatGGG-3′; ccr5 crRNA 1, 5′-gtagcacccccatgcaacaaTGG-3′; ccr5 crRNA 2, 5′-attttcctgataatacatccTGG-3′). Gene-specific crRNAs were heteroduplexed to universal tracrRNA according to the manufacturer’s recommendations to generate bipartite gRNAs. Mutations were generated by injecting gRNA (nampta mutant generated using single gRNA, ccr5 mutant generated using dual gRNAs (ccr5 crRNA 1 and ccr5 crRNA 2)) and recombinant Cas9 protein (Alt-R S.pCas9 Nuclease, IDT) into the blastomere of one-cell-stage wild-type embryos. Injected embryos were grown to adulthood, outcrossed into wild-type zebrafish and screened to identify founders containing germline mutations. Identified mutants of interest; nampta c.180_182delinsTCCGTCTTGCTGACCTTTCCCCAGCAG (p.Try61Profs*4) (referred to as namptapc41) and ccr5 c.66_578delins ACCCCTATGCAACATCATTTTTACCAATGAGCAAATGGATTTAAACAAGAGAAAATCCTGCCAACTTGATTTTCCTGATAATACATAATA (p.Pro24Leufs*28) (referred to as ccr5pc42). For genotyping the nampta mutants, DNA was isolated from clipped fins (adults) or whole embryos and used in a PCR reaction with the oligonucleotides nampta_F (5′-TGCCGTGAGAAGAAGACAGA-3′) and nampta_R (5′-GCAATCAATTGCCTTACCTTTT-3′) (PCR product size, nampta 117 bp and namptapc41 141 bp). For genotyping the ccr5 mutant, a PCR was performed with the oligonucleotides ccr5_F (5′-AACGAAACTGGGCATGTAGC-3′) and ccr5_R (5′-CCGGGAATAACAAAAGCTCA-3′) (PCR product size, ccr5 618 bp and ccr5pc42 173 bp).
Tissue-specific loss-of-function zebrafish mutations
The tol2-flanked transgene Tg(4×UAS:NLS-cas9, cryaa:eGFP)gl36Tg (referred to as Tg(4×UAS:NLS-cas9, cryaa:eGFP)) and Tg(4×UAS:NLS-cas9, cmlc2:RFP)gl37Tg (referred to as Tg(4×UAS:NLS-cas9, cmlc2:RFP)) were assembled by Gateway cloning. These constructs were microinjected with tol2 mRNA into the required background. Tg(4×UAS:NLS-cas9, cryaa:eGFP) was introduced into the Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry) background, whereas Tg(4×UAS:NLS-cas9, cmlc2:RFP) was introduced into both Tg(mpx:KALTA4/UAS:nfsb-mCherry) and Tg(pax7b:GAL4FF/UAS:GFP) backgrounds. Co-segregation of the three transgenes through F0 and F1 backcrosses onto this background was achieved by selecting embryos with the appropriate fluorescence (Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry/4×UAS:NLS-cas9, cryaa:eGFP): sorted for red macrophages confirming the presence of the mpeg1:GAL4FF transgene and green lens confirming the presence of the cryaa:eGFP marker gene linked to the 4×UAS:NLS-cas9 (this line is referred to as mpeg1-cas9). Tg(mpx:KALTA4/UAS:nfsb-mCherry/4×UAS:NLS-cas9, cmlc2:RFP): sorted for red neutrophils and a red heart (this line is referred to as mpx-cas9). Tg(pax7b:GAL4FF/UAS:GFP/4×UAS:NLS-cas9, cmlc2:RFP): sorted for green muscle stem/progenitor cells and a red heart (this line is referred to as pax7b-cas9)). For gene-editing experiments, embryos incrossed from either F2 or F3 generation adults were used. For nampta and ccr5 targeting, the gRNA or dual gRNA combination used to generate mutants (as described above) were used. For namptb targeting a dual gRNA combination was used. These gRNAs were generated using namptb crRNA 1, 5′-tttctctgaccaaacacgcaAGG-3′ and namptb crRNA 2, 5′-gttgacctgtgaacgtgataGGG-3′. The nampta individual gRNA and ccr5 and namptb dual gRNA efficiencies were tested in whole-embryo gene editing and exhibited between 89 and 100% mutational efficiency. To induce tissue-specific gene editing, 3 nl of gRNA mix (3 μl of 3 μM gRNA (or if dual gRNA used 1.5 μl of each gRNA), 0.5 μl 2% phenol red, 1.5 μl 0.1 M KCl mix) was injected into the cell of one-cell-stage embryos.
Cell culture
The mouse muscle cell line C2C1239 (ECACC general cell collection, 91031101) was cultured in growth medium (DMEM (4.5 g l−1 d-glucose, no l-glutamine, no sodium pyruvate (Gibco), 20% fetal bovine serum (FBS) (Fetal Bovine Solution One Shot, Gibco), 1% GlutaMax 100× (Gibco)). Cells were maintained at 37 °C, 5% CO2. Cells at 70% confluence, passage 8, were extracted from T-75 flasks with 0.025% trypsin containing EDTA (Gibco), neutralized in growth medium, centrifuged at 180g for 5 min to pellet cells. The cells were then resuspended in 10 ml of fresh growth medium. Then, 500 μl of cells were plated on a 8-well on a coverglass II (Sarstedt) chamber slide at a density of 1 × 103 cells ml−1. Cells were left for 4 h at 37 °C to re-attach. For drug treatments, the medium was supplemented with appropriate doses and cultured for 6 h. Drug treatments were carried out in full serum medium (20% FBS).
For isolation of primary mouse myoblasts, limb skeletal muscles from embryonic day 17.5 C57BL/6J mice were minced and digested in 0.125% trypsin at 37 °C for 20 min. Fibroblasts were depleted by plating cells in 10-cm2 tissue-culture dishes (two embryos per dish) in proliferation medium (DMEM, 20% FBS) for 1 h. Medium with non-attached cells was re-plated in gelatin-coated 10-cm2 tissue-culture dishes in proliferation medium for 24 h. Myoblasts were again depleted for fibroblasts before coculturing on gelatin-coated 48-well plates in DMEM, 20% FBS, 10% L929-conditioned medium. For experiments, 100,000 myoblasts were plated with either 7,500 Mafb- and Maf-double-knockout (MAF DKO) macrophages40 (Sieweke laboratory) or 1,000 NIH 3T3 cells (Sigma-Aldrich, 93061524) per well. For drug treatments, the media were supplemented with appropriate doses of the indicated drugs and cultured for 24 h. Drug treatments were carried out in full serum medium (20% FBS).
Larval zebrafish muscle injury
Larvae (4 dpf) were anaesthetized in 0.01% tricaine (MS-222) (Sigma-Aldrich) in Ringer’s solution. Mechanical injures were targeted either to the dorsal or ventral myotomes above the cloaca when the larvae is oriented dorsal to the top, anterior to the left. Needle-stab injury was carried out as previously described2. In brief, the myotome was subjected to a single 30-gauge needle puncture that generates an extensive injury with many damaged muscle fibres. For laser-induced injury, anaesthetized larvae were mounted in a thin layer of 1% low-melt agarose in Ringer’s solution. Injuries were carried out using a UV-nitrogen laser pulsed through a coumarin 440-nm dye cell coupled to a Zeiss Axioplan microscope (MicroPoint Laser System, Andor Technology). On average, a laser injury required pulses for 5–10 s from laser beams focused through a 40× water-immersion objective. For time-lapse analysis of the immediate response to injury, muscle fibre ablations were achieved using a SIM scanner (Olympus) at 790 nm and 200 ms dwell time at 100% laser power on an Olympus FVMPE-RS upright multiphoton microscope equipped with a 25×/1.05 NA water-immersion objective. Injury-responding macrophages were tracked using the manual tracking plugin in Fiji41.
Mouse volumetric muscle loss injury
Injury
Male C57BL/6J mice aged between 10 and 12 weeks were anaesthetized and shaved on the left hindleg. A unilateral incision measuring approximately 1 cm was made exposing the underlying fascia. The left hindlimb was extended and exteriorized via the incision site by retracting the surrounding tissue. A 3 × 4-mm full thickness segment of the rectus femoris muscle was removed. Directly after, the injury site was filled with fibrin hydrogel with or without 0.2 μg or 0.5 μg of hrNAMPT source 1 (hrNAMPT(1)) (hydrogel components: 40 μl (total volume), 8 mg /ml−1 human fibrinogen (FIB3, Enzyme Research Laboratories), 4 U ml−1 bovine thrombin (T4648, Sigma-Aldrich), 5 mM CaCl2, 17 μg ml−1 of aprotinin (ab146286, Abcam)), which polymerized in the defect. Then, the soft tissue was closed with stitches.
Histology
Ten days after treatment, animals were euthanized and the wounds were collected for histological analysis. The defect site and associated proximal and distal segment of the quadriceps muscle (including the rectus femoris, vastus medialis and vastus lateralis) were excised and embedded. Histological analysis was performed on serial paraffin sections (4-μm sections collected passing the central portion of the wound). Multiple sections were stained with Masson’s trichrome (to detect collagen deposition) and the extent of fibrosis (represented by a blue/purple stain) was measured by histomorphometric analysis using ImageJ software (version 1.51h, National Institutes of Health). To maintain uniformity between samples, the length of the vastus medialis taken at multiple depths ranging from 1.0 mm to 3.0 mm serves as a reference between tissue sections to determine the depth of sectioning. For the quantification of fibrosis, the average muscle fibrotic area at each depth was scored and normalized to the area of the rectus femoris. Total muscle area was determined by calculating the average area of rectus femoris at each depth.
Immune cell profiling and PAX7+ cell quantification with flow cytometry
At 4, 6 or 8 days after treatment with either 0.5 μg of hrNAMPT(1) delivered by fibrin hydrogel or control fibrin hydrogel only, mice were euthanized via CO2 asphyxiation. The defect site and associated proximal and distal segment of the quadriceps muscles were isolated and placed in 890 μl of complete RPMI (with 10% FBS and 2 mM GlutaMax, Life Technologies). The tissue was minced with surgical scissors and 100 μl of 10 mg ml−1 collagenase II (Sigma-Aldrich) and 10 μl of 10 mg ml−1 DNase I (Biolabs), while 100 μl of dispase II (10 mg ml−1) was added into the digestion for the acquisition of PAX7+ cells. The mixture was vortexed and incubated at 37 °C for 45 min. Collagenase was then inactivated with 500 μl ice-cold PBS, 5% FBS, 5 mM EDTA and the mixture was subsequently strained through 70-μm and 40-μm filters. The cell suspension was further diluted with 1 ml of complete RPMI and centrifuged for 10 min at 300g. The supernatant was discarded and the pellet resuspended in 250 μl complete RPMI and aliquoted into wells of a 96-well U-bottom plate for antibody staining. The cell solutions were centrifuged, supernatant discarded and the pellets washed with PBS. The cell viability stain used was 100 μl of Zombie Aqua (Biolegend) Live-Dead dye diluted in PBS (1:400 dilution). Cells were incubated for 30 min at 4 °C. Cells were then blocked with FcX (anti-CD16/32 antibodies, Biolegend, 1 μg ml−1) flow cytometry buffer (PBS, 5% FBS) and kept for 20 min at 4 °C, washed with flow cytometry buffer and centrifuged. Primary surface antibody staining was performed in two separate stains with 100 μl of anti-mouse antibody cocktail (Biolegend) diluted in flow cytometry buffer: T cell stain with 2 μg ml−1 of APC anti-CD4 (clone RM4.5, 100516), Brilliant Violet 421 anti-CD8a (clone 53-6.7, 100738), and PE/Cyanine7 anti-CD3 (clone 17A2, 100220). Neutrophils and macrophages were stained with 2 μg ml−1 of PE anti-CD11b (clone M1/70, 101208), 1 μg ml−1 Brilliant Violet 421 anti-Ly6G (clone 1A8, 127628), 4 μg ml−1 Brilliant Violet 711 anti-F4/80 (clone BM8, 123147), 10 μg ml−1 APC anti-CD80 (clone 16-10A1, 104714) and 2.6 μg ml−1 PE/Cyanine7 anti-CD206 (clone C068C2, 141720). Cells were stained for 30 min on ice and washed as described above. For internal FOXP3 staining in the T cell panel, cells were fixed with 100 μl fixation/permeabilization solution (42080, Biolegend) for 35 min. Cells were then washed and resuspended in 100 μl of flow cytometry buffer with 0.5% saponin and 5 μg ml−1 FITC anti-mouse FOXP3 (clone 3G3, 35-5773-U100, Tonbo Biosciences) for 45 min and data were acquired on a Fortessa x20 (Beckman Coulter, FACSDiva (v.9.0) and FloJo (v.9.6.1) software). Satellite cell flow cytometry staining was performed with 200 μl of antibody cocktail (Biolegend) diluted in flow cytometry buffer: 5 μg ml−1 of anti-VCAM/CD106 biotin (clone 429 (MVCAM.A), 105703), 2.5 μg ml−1 of APC/Fire 750 streptavidin (405250), 2 μg ml−1 of PE/Cyanine7 anti-mouse CD45 (clone 30-F11, 103114), 2 μg ml−1 PE anti-mouse CD11b (clone M1/70, 101208), 2 μg ml−1 anti-mouse Ly6G (clone 1A8, 127607), 1 μg ml−1 PE anti-mouse CD31 (clone MEC13.3, 102507). Cells were stained for 45 min on ice and washed as described above. Cells were also stained with 200 μl flow cytometry buffer with 0.5% saponin with intracellular anti-mouse antibody cocktail: 1 μg ml−1 Brilliant Violet 421 anti-Ki67 (clone 16A8, 652411, BioLegend), 10 μg ml−1 anti-Pax7 (clone Pax7/497, NBP2-34706AF488, NovusBiologicals) for 1 h on ice. Cells were then resuspended in flow cytometry buffer (275 μl) with 25 μl of Invitrogen Count Bright Absolute Counting Beads (25,000 beads, C36950) and acquired on the Fortessa x20 (Beckman Coulter, FACSDiva (v.9.0) and FloJo (v.9.6.1) software). All events were acquired and the number of PAX7+ cell per 10,000 wound cells was calculated using the following formula: PAX7+ number in injury = 10,000 × PAX7+ cell count/((1/25,000) × beads count × (live cell percentage/100) × total cell number count after digestion). The same calculation was performed to quantify the number of proliferative PAX7+ cells, using cell count of PAX7 and Ki67 double-positive cells.
Immunofluorescence of frozen sections
Immunostaining was performed on 10-μm cryosections with antigen retrieval (10 mM sodium citrate, 0.05% Tween-20, pH 6.0). Sections were blocked with 2% BSA, 5% normal goat serum in PBS with 0.3% Triton X-100 and AffiniPure Fab Fragment Goat Anti-Mouse IgG (H+L) (Jackson Immuno Research Laboratories) to minimize unspecific binding. Antibodies: mouse anti-Pax7 (2 μg ml−1, AB_528428, DSHB) and goat anti-mouse IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 488 (1:500, A-11001, Fisher Scientific). Muscle sarcolemma were visualized by rhodamine-labelled wheat-germ agglutinin (Vector Laboratories) and nuclei were visualized by staining with DAPI (Sigma-Aldrich).
Quantification of centrally nucleated muscle fibres
Haematoxylin and eosin staining was performed on 4-μm paraffin-embedded sections. The number of nuclear centralizations within a muscle fibre were counted from five serial sections per sample by histomorphometric analysis using ImageJ software (version 1.51h, National Institutes of Health). To maintain uniformity between samples, the length of the vastus medialis taken at multiple depths ranging from 1 to 3 mm serves as a reference between tissue sections to determine the depth of sectioning. For the quantification of the average number of centrally nucleated cells, the total nuclear count at each depth was normalized to the area of the rectus femoris.
Microscopy and image analysis
Whole-larva time-lapse imaging to track injury-responding macrophages was performed using a Zeiss Light-sheet Z.1 microscope with a 5×/0.16 NA air objective and environmental controls (28.5 °C). The xy resolution was 1.14 μm, the z resolution was 5.5 μm, with a light-sheet thickness of 11.68 μm. Total imaging time per larvae was 25 h (1,000 three-dimensional stacks acquired at 1.5-min intervals), and viability of larvae was confirmed at the end of the imaging session by assessing the heart rate. For tracking, macrophage images were first filtered with a 3D median filter and then segmented with hysteresis thresholding using algorithms from the 3D ImageJ Suite42. The low and high thresholds for hysteresis were chosen visually. The datasets were then time-reversed, in order to track cells exiting the injury site. The margins of the wound were manually labelled in ImageJ. The tracking procedure was based on overlapping segmentation of cells between consecutive frames. Cases in which two cells were too close and formed one object were designated ‘merging’ as the segmentation algorithm was unable to separate them. Tracked macrophage images were reversed for the visualization of results. Subsequently, transient macrophage paths (macrophages that migrate into the injury but were not captured as dwelling) into the wound site were tracked using the manual tracking plugin in ImageJ41.
Line-scanning confocal microscopy for long-term time-lapse imaging and single z-stack acquisition was performed using a Zeiss LSM 710 upright confocal microscope equipped with a 20×/1.0 NA water-immersion objective. Kaede photoconversions were carried out using the bleaching tool with a 405-nm diode laser. Irradiation at 405 nm results in an increase in the 543 nm/488 nm fluorescence ratio of the fluorescent protein43.
Time-lapse imaging at high temporal and spatial resolution was performed on an inverted LSM 880 fast AiryScan confocal microscope equipped with a 40×/1.3 NA oil-immersion objective and piezo z-stage. The voxel size was kept constant at 0.2 × 0.2 × 1 μm and, depending on the field of view, frame rates of 3–18 frames per second were achieved. Photobleaching was assessed after imaging and determined to be minimal for imaging durations of up to 1 h.
Fixed and immunostained cell culture samples were imaged on a Leica DMi8 inverted wide-field microscope with a 10× objective.
Birefringence imaging was carried out as previously described44, using a Leica DM IRB upright microscope integrated with the Abrio LS2.2 unit (CRI Hinds Instruments) using a 5× objective. The pseudo-crystalline array of muscle sarcomeres confers to the muscle an intrinsic birefringence, making uninjured muscle appear bright against a black background when observed using polarized light, enabling non-invasive visualization of muscle integrity44. Images were analysed using Fiji41, whereby the mean grey value of the injury site birefringence is normalized to the region anterior and posterior to the myotomes injured to calculate relative birefringence of the wound site. Calculations were carried out in Microsoft Excel (v.14.4.6) and graphical visualization and statistical analysis was carried out in Prism (v.7.0c, GraphPad Software).
Microscopy images were processed in Fiji41 and Imaris 9.2 (Bitplane). Counting macrophage numbers and further three-dimensional analyses were performed by surface-rendering the cell volumes using Imaris. Sphericity analysis assessed the shape deviation of cells from a perfect sphere, which is assigned an arbitrary value of one. Sphericity values were generated as a summary statistic of the surface render. Proliferating stem cell counts were carried out on Fiji. The PAX7 and EdU acquisition channels were segmented using the threshold command. The image calculator function was used to generate a masked channel of only EdU+ PAX7 cells. The number of cells was determined using the ‘analyse particles’ command.
Chemical treatments
Cell ablation in zebrafish larvae was carried out as previously described45,46 with minor modifications. Larvae at the appropriate stage were incubated in 5–10 mM Mtz (Sigma-Aldrich) in Ringer’s solution and daily refreshed until experimental end point. Mtz addition to the Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry) transgenic line leads to efficient cell death, with macrophage ablation visible after 3 h of treatment. After 10–13 h, most of the macrophages present in the trunk had been ablated and their cell corpses cleared from the larvae (Extended Data Fig. 1k and Supplementary Video 5). Furthermore, this ablation strategy did not alter other innate immune cell responses to muscle wounding at 2 dpi (Extended Data Fig. 1l, m).
Drug treatments were carried out by incubating 4 dpf needle-stab-injured larvae in 5 and 10 μM CVC47 (Med Chem Express) and 5 and 10 μM MVC48 (Med Chem Express) in Ringer’s solution immediately after injury and refreshed daily. The effect of drug treatment on laser-ablation muscle injury was assayed by treating larvae with 5 μM CVC for 2 h before injury and with larvae being maintained in the drug until experimental end point. For the NAMPT enzymatic inhibition experiments, needle-stab-injured larvae at 5 dpf, 1.75 dpi were transferred into Ringer’s solution containing 10 μM GMX177849 (Sigma-Aldrich) and maintained as such until experimental end point. Chemokine supplementation experiments were carried out in plasticware coated with 0.1% bovine serum albumin (BSA) overnight (rinsed in Ringer’s solution before use) to minimize protein adsorption. Larvae were treated with 57 nM human recombinant visfatin source 1 (hrNAMPT(1)) (PeproTech), 57 nM mouse recombinant CCL8/MCP-2 (mrCCL8) (PeproTech) in combination with Mtz and CVC.
Cell culture drug supplementation was carried out by adding the following to the growth medium of C2C12 cells for 6 h: 1.9, 9.5 and 19 nM hrNAMPT(1) (PeproTech), 1.9 and 9.5 nM recombinant human visfatin source 2 (hrNAMPT(2), Enzo Life Sciences), 100 nM CVC (Med Chem Express), 100 nM MVC (Med Chem Express), 100 nM PF-413630950 (Med Chem Express), 9.5 nM mrCCL8, 9.5 nM mouse recombinant CCL4/MIP-1β (mrCCL4) (PeproTech), 9.5 nM mouse recombinant CCL2/MCP-1 ((PeproTech), 500 nM GMX1778 (Sigma-Aldrich) in combination with hrNAMPT(1). The following were added to the proliferation medium of primary mouse myoblast cocultures for 24 h: 9.5 nM hrNAMPT(2) (Enzo Life Sciences), 100 nM CVC (Med Chem Express), 100 nM MVC (Med Chem Express), 100 nM PF-4136309 (Med Chem Express) and 9.5 nM mrCCL4 (PeproTech).
LysoTracker assay
Larvae were incubated in 10 μM LysoTracker Deep Red (Thermo Fisher Scientific) in Ringer’s solution for 1 h in the dark and rinsed 5 times with fresh Ringer’s before imaging.
EdU labelling
Larvae
Labelling and detection was carried out as previously described51 with minor alterations. In brief, 6 dpf, 2 dpi larvae were pulsed with 50 μg ml−1 EdU (Thermo Fisher Scientific) for 1 h and chased for a further 1.5 h before fixation. EdU+ cells in a region encompassing two myotomes on either side of the injury were quantified as the number of EdU+ cells outside the injury region. EdU+ cells in the caudal haematopoietic tissue were excluded from this analysis.
Cell culture
Cells were incubated for 1 h in medium supplemented with 10 μM EdU. Following EdU pulse, C2C12 cells were immediately fixed, whereas primary mouse myoblast cocultures were rinsed in PBS, which was then replaced with medium and cultures were incubated for a further 2 h, after which cells were fixed. Samples were developed using the Click-iT EdU Alexa Fluor 647 imaging Kit (Thermo Fisher Scientific) following the manufacturer’s protocol.
Immunohistochemistry and in situ hybridization
Antibody staining of whole-mount larvae was carried out as previously described52 and on cultured myoblasts as previously described53. After in situ hybridization, antibody staining was carried out using standard procedures. Antibodies: mouse anti-Pax7 antibody (1:10, AB_528428, DSHB), chicken anti-GFP antibody (1:500, A10262, Thermo Fisher Scientific), mouse anti-mCherry antibody (1:500, ab125096, Abcam) (after in situ hybridization), rat anti-mCherry antibody (1:500, EST202, Kerafast) (immunohistochemistry), rabbit anti-PBEF1 (anti-NAMPT) antibody (1:100, AV42254, Sigma-Aldrich) (immunogen and zebrafish Nampta protein share 94% sequence homology; the antibody recapitulated the endogenous developmental expression profile of nampta12 (Extended Data Fig. 6j)), rabbit anti-POU2F3 (1:300, AV32537, Sigma-Aldrich), rabbit anti-Prox1 (1:1,000, AB5475, Sigma-Aldrich), goat anti-mouse IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 488 (1:500, A-11001, Thermo Fisher Scientific), goat anti-chicken IgY (H+L) secondary antibody, Alexa Fluor 488 (1:500, A-11039, Thermo Fisher Scientific), goat anti-mouse IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 546 (1:500, A-11003, Thermo Fisher Scientific), goat anti-rat IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 555 (1:500, A-21434, Thermo Fisher Scientific) and F(ab′)2-goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 633 (1:500, A-21072, Thermo Fisher Scientific). Nuclei were visualized with DAPI (Sigma-Aldrich). In situ hybridization and probe generation were performed as previously described54. Antisense probes used: mmp955 and namptb12. nampta (ENSDARG00000030598) PCR probe containing a T7 RNA polymerase promoter at the 3′ for the antisense probe and an SP6 RNA polymerase promoter at the 5′ for the sense probe was generated using primers 5′-GAGtatttaggtgacactatagGGTTTCATCGCAAGAGACGG-3′ and 5′-GAGtaatacgactcactatagggGCGGAAGCACCTTATAGCCT-3′. This probe recapitulates previously published12 larval zebrafish expression patterns at 1 and 3 dpf. Haematoxylin and eosin staining was performed on 10-μm cryostat cross-sections of 5- and 7-dpf larvae according to standard methods.
Enzyme-linked immunosorbent assays
hrNAMPT binding to human recombinant CCR5
Enzyme-linked immunosorbent assay (ELISA) plates (medium binding, Greiner Bio-One) were coated with 1% BSA or 20 nM of glutathione S-transferase (GST)-fused human recombinant CCR5 (hrCCR5, Abcam) in PBS overnight at 4 °C. Wells were blocked for 1 h at room temperature with 1% BSA in PBS containing 0.05% Tween-20 (PBS-T). Wells were washed three times with PBS-T and further incubated with hrNAMPT(1) (Peprotech) at increasing concentrations (0 nM to 800 nM) for 1 h in PBS-T with 0.1% BSA. Bound NAMPT molecules were detected using a biotinylated antibody for NAMPT and HRP–streptavidin (Human PBEF/Visfatin DuoSet ELISA, R&D Systems). Signals obtained on BSA-coated wells were used to remove non-specific binding for each NAMPT concentration to obtain specific binding values. Specific binding data were fitted by nonlinear regression with Prism (v.7.0c, GraphPad Software) to obtain the dissociation constant (KD) using A450 nm = Bmax × [NAMPT]/(KD + [NAMPT]).
hrNAMPT competitive binding to mouse recombinant CCR5
ELISA plates (medium binding, Greiner Bio-One) were coated with 1% BSA or 20 nM of mouse recombinant CCR5 (mrCCR5) (MyBioSource) in PBS overnight at 4 °C. Wells were then were blocked for 1 h at room temperature with 1% BSA in PBS-T. Wells were washed three times with PBS-T and further incubated with mrCCL4 (Peprotech) at increasing concentrations (0 nM to 400 nM) for 1 h in PBS-T with 0.1% BSA containing 100 nM hrNAMPT(1) (Peprotech). Bound hrNAMPT(1) molecules were detected using a biotinylated antibody for NAMPT and HRP–streptavidin (Human PBEF/Visfatin DuoSet ELISA, R&D Systems). Signals obtained on BSA-coated wells were used to remove non-specific binding for each hrNAMPT concentration to obtain specific binding values. Specific binding data were fitted by nonlinear regression with Prism v.7.0c (GraphPad Software) to obtain the half maximal inhibitory concentration (IC50) of mrCCL4 using \({A}_{450{\rm{nm}}}={A}_{450\,{\rm{nm}}}^{\mathrm{Min}}+({A}_{450\,{\rm{nm}}}^{{\rm{Max}}}-{A}_{450\,{\rm{nm}}}^{\mathrm{Min}})/(1+{10}^{(X-\log [{\text{IC}}_{50}])})\).
NAMPT in macrophage supernatant
The MAF DKO mouse macrophage cell line is known to express high levels of NAMPT56, but we sought to quantify the specific levels of NAMPT secretion under our culture conditions. MAF DKO cells and the mouse macrophage cell line Raw 264.7 (ATCC) were cultured in growth medium (DMEM, 10% FBS, 10 ng ml−1 M-CSF) for 16 h. Protein in collected supernatant was concentrated using Amicon Ultra-15 centrifugal filter with a 10-kDa nominal molecular weight limit (Merck). NAMPT in supernatant was quantified using Human PBEF/Visfatin DuoSet ELISA (R&D Systems), according to the manufacturer’s instructions. MAF DKO macrophages secreted 5.24 ± 0.67 ng ml−1 of NAMPT into the supernatant (Extended Data Fig. 7d).
Cell-surface CCR5 receptor concentration
The mouse muscle cell line C2C12 was cultured as described above. Cells were dislodged at 70–80% confluence using a cell scraper and membrane proteins were isolated using an extraction kit (Plasma Membrane Protein Extraction Kit, Abcam). CCR5 concentration in the membrane extract was then measure by ELISA (mouse CCR5 ELISA Kit, Biorbyt) and the amount of CCR5 per cell was then calculated using [CCR5](molecules/cell) = [CCR5](ng/cell)/CCR5MW × 10−9 × N0, where N0 = Avogadro constant. C2C12 cells express the CCR5 receptor at a density of 2,470 ± 441 molecules per cell (n = 6), a value in line with previously documented physiologically relevant levels of CCR518.
Mouse cytokine array
Supernatant was collected from MAF DKO cells cultured for 16 h in growth medium (DMEM, 10% FBS, 10 ng ml−1 M-CSF). The cytokine assay (Proteome profiler array, R&D systems, ARY006) was performed according to manufacturer’s instructions. In brief, nitrocellulose membranes were blocked for 1 h at room temperature on a rocking platform. During the blocking step, 0.7 ml of sample was topped up to 1.5 ml with Array Buffer and incubated with Mouse Cytokine Array Panel A Antibody Cocktail for 1 h at room temperature. Samples were then incubated on membranes overnight at 4 °C on a rocking platform. Membranes were washed before incubation with streptavidin–HRP for 30 min at room temperature. Finally, membranes were developed by adding Chemi Reagent Mix, then imaged on a Biorad Chemidoc MP system and analysed using (Image Lab software, Bio Rad). MAF DKO cells did not actively secrete the cognate ligands of CCR5 (CCL3, CCL4 and CCL5) into the supernatant (Extended Data Fig. 7e, f).
Fluorescence-activated cell sorting, scRNA-seq and analyses
Injury-responding macrophages were isolated by fluorescence-activated cell sorting (FACS) as previously described57, with the following modifications. In brief, 4 dpf Tg(mpeg1:mCherry) larvae were subject to needle-stab injury and the injured region was dissected at 1, 2 and 3 dpi and tissue dissociated into a single-cell suspension. Uninjured larval tissue was also included in the analysis. Cells were sorted using a FACS Aria II (BD Biosciences, BD FACSDiva software (v.9.0)). Live individual macrophages (based on mCherry fluorescence, DAPI exclusion and forward and side scatter properties) were sorted into pre-prepared 384-well plates containing 100–200 nl of CEL-seq primers, dNTPs and synthetic mRNA Spike-Ins contained in 5 μl of Vapour-Lock (Qiagen). Immediately after sorting, plates were centrifuged and frozen at −80 °C until sequencing.
The scRNA-seq libraries were prepared using the SORT-seq platform as previously described58. In this platform, the Cel-Seq2 protocol59 is followed with the aid of robotic liquid handlers. This protocol results in each cell being barcoded and the generation of single-cell transcriptomes of all isolated macrophages. Each time point is independently replicated and results in approximately 768 macrophages per time point (3,072 macrophages in total) to be individually sequenced. Next-generation sequencing was carried out using an Illumina NextSeq platform. Paired reads were mapped against the zebrafish reference assembly version 9 (Zv9). FASTQ files were processed as previously described58,60. Paired-end reads were aligned to the zebrafish transcriptome using bwa61 with a transcriptome dataset with improved 3′ UTR annotations to increase the mapability of transcripts62. Read 1 was used for assigning reads to correct cells and libraries (R1: 6-nt UMI + 8-nt cell barcode) while read 2 was mapped to gene models. Read counts were first corrected for UMI barcode by removing duplicate reads that had an identical combination of library, cellular and molecular barcodes that were mapped to the same gene. Gene UMI counts of all assayed plates (technical replicates of injury types) were combined into one matrix for downstream analysis. ERCC spike-in RNA controls were discarded from the expression matrices and not considered for further analysis. Downstream analysis of the combined sample gene counts were performed using Seurat (v.3.1.1)63. Gene counts were imported using the CreateSeuratObject function (min.cells = 25, min.genes = 250) and low-quality cells were discarded using the following thresholds: a minimum of 500 and maximum of 3,500 genes, a maximum of 10% of mitochondrial protein-coding gene UMIs (13 mitochondrial genes considered in total) and a minimum of 1,000 and maximum of 15,000 UMIs). The total number of cells that passed quality control, using the filters mentioned above, was 1,309. SCT transform normalization, which applies a regularized negative binomial regression and variance stabilization, was applied to the filtered dataset using the SCTransform function in Seurat, regressing the percentage of mitochondrial genes per cell (vars.to.regress = “percent.mt”). Principal component analysis was performed using the RunPCA function in Seurat. UMAP dimensional reduction was performed using the RunUMAP function (reduction.use = “pca”, dims.use = 1:50) in Seurat with the top 50 principal components. Cell-clustering analysis was performed with the FindNeighbours and FindClusters (resolution = 0.5) functions in Seurat using the same number of PCA components as for UMAP (dims.use = 1:50). Eight cell clusters were identified. To identify cluster biomarkers the FindAllMarkers (min.pct = 0.2, assay = ‘RNA’, test.use = “wilcox”) function was used to compare all clusters against each other (identified cluster-specific markers with a Bonferroni-adjusted P < 0.05 were considered to be statistically significant). Please note that an ‘RNA’ assay, with log-normalized and scaled gene-expression values, was created on the Seurat Object to perform marker identification whereas cell-specific analysis was performed on the SCT assay processed as described above. UMAP, feature plots and heat maps were created using the DimPlot, FeaturePlot and DoHeatmap functions in Seurat. To validate our sorting strategy, we examined the top-500 expressed genes common to all macrophages that passed quality control (calculated using the perFeatureQCMetrics function in scater64) (Supplementary Table 1). This list contained pan-leukocyte markers (lcp1 (lymphocyte cytosolic protein 1, L-plastin)) as well as numerous macrophage-specific genes (mpeg1, mfap4 (microfibril associated protein 4), spi1a, ptprc (protein tyrosine phosphatase receptor type C) and marco (macrophage receptor with collagenous structure))65, including those involved in antigen presentation (cd163, cd40, cd83) that have been validated as specific markers of the monocyte/macrophage lineage (Fig. 3d, Extended Data Fig. 5a and Supplementary Table 1).
Trajectory analysis was performed using PAGA66, which is part of the scanpy package (v.1.4.5.2.dev6+gfa408dc)67. PAGA is a method that explores the connectivity between identified single-cell clusters. It represents the clusters as ‘nodes’ and the potential paths connecting the clusters as ‘edges’. These edges are placed between nodes in which the cells are considered to be ‘more similar’ to each other. We chose to use PAGA as it enables trajectory construction in datasets that are more disconnected and the methodology aids in exploratory analysis. In summary, a neighbourhood graph of data points (cells) was created using the scanpy.pp.neighbours function68 (starting from the top-50 principal components, as used for UMAP dimensional reduction and number of neighbours of 35) to then run PAGA using the scanpy.tl.paga function in scanpy with the previously identified clusters as groups. Finally, PAGA cell embedding was represented using a force-directed layout initialized with PAGA coordinates using the scanpy.tl.draw_graph function in scanpy (Force Atlas 2 layout).
Pseudotime analysis was performed using diffusion pseudotime (DPT)69. The scanpy.tl.dpt function in scanpy was run using the previously defined cluster 3 (composed of cells from the uninjured time point) as root. PAGA and force-directed layout plots were generated using the sc.pl.paga sc.pl.paga_path (n_avg = 25) and scanpy.pl.draw_graph functions in scanpy.
Metascape (http://metascape.org)70 was used to carry out biological process enrichment analysis using the differentially expressed genes of each cluster or top-500 genes expressed by all macrophages in the dataset.
Electron microscopy
In brief, 4 dpf larvae were subjected to laser-ablation skeletal muscle injury. At 25 hpi, a confocal stack of the injury region was acquired on a Zeiss LSM 710 upright confocal microscope. After imaging, larvae were euthanized and fixed according standard procedures in 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 M sodium cacodylate buffer overnight at 4 °C. Larvae were next post-fixed with 1% OsO4, 1.5% K3[Fe(iii)(CN)6] and dehydration was performed with ethanol and propylene oxide. Larvae were embedded in Epon 812 and ultrathin sections of 70 nm were cut using a diamond knife (Ultra 45° Diatome) on a Leica Ultracut UCT7 and placed on 50-mesh copper grids with carbon-coated formvar support film and stained with uranyl acetate and lead citrate. Large-area scanning transmission electron microscopy (STEM) tile sets were taken on a Thermo Fisher/FEI NovaNano SEM 450 equipped with a STEM II (HAADF) detector set at 30 keV, working distance 6.8 mm. MAPS 2.1 software was used to create the tile sets and perform correlations to the confocal datasets. High-resolution electron microscopy imaging was performed on a Jeol1400Flash TEM at 80 keV.
Visualization and quantification of NAD+/NADH
To monitor the levels and source of increased NAMPT activity during the regeneration of muscle in vivo, we made use of the key physiological role of intracellular NAMPT in catalysing the rate-limiting process in the NAD salvage pathway71. Increased NAMPT activity has been documented to elevate levels of intracellular NADH72. NADH exhibits endogenous fluorescence with an excitation peak at 365 nm that is amenable to examination by two-photon excitation fluorescence microscopy73. In vivo two-photon-excited fluorescence of larval zebrafish NADH was measured on an Olympus FVMPE-RS upright multiphoton microscope using a 25×/1.05 NA water-immersion objective. A wavelength of 810 nm and a 450/70 nm bandpass filter was used. The same wavelength and a 610/70 nm bandpass filter were used to detect mCherry fluorescence. A galvo scanner was used to generate high-resolution datasets while an 8-kHz resonant scanner was used for time-lapse imaging to minimize phototoxic effects.
In vitro total NAD+ and NADH levels and their individual levels (to determine their ratio) were measured using the NAD/NADH-Glo assay (Promega) following supplier instructions. Total NAD+/NADH measurements were obtained as follows. The assay was carried out on macrophages sorted from macrophage-specific nampta-knockout larvae (mpeg1-cas9, nampta-gRNA-injected) and control (mpeg1-cas9) larvae. At 2 dpf, control larvae were soaked in either the NAMPT enzymatic inhibitor GMX1778 (10 μM) (Sigma-Aldrich) or the rate-limiting enzyme catalysis product of NAMPT, nicotinamide mononucleotide (100 μM) (Sigma-Aldrich). These were used as additional controls to identify the detection range of the assay. At 3 dpf, mpeg1+ macrophages from each group were sorted into white, flat-bottom 96-well plates (Costar) containing 50 μl PBS at a density of 2,000 cells per well. Cells were incubated in 50 μl of NAD/NADH Glo detection reagent. After 1 h incubation, luminescence was determined in a microplate reader (BMG PHERAstar; gain 3,600, 1 s integration time). Each point of measurement represents the average luminescence reaction measured in relative luminescence units.
Measurements of NAD+ and NADH individually were performed as follows. The assay was carried out on FACS-sorted mCherry+ macrophages isolated from Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry) dissected myotomal needle-stab-injured larval wound sites at 1 dpi (5 dpf) and 2 dpi (6 dpf). Cells were sorted at a density of 5,000 cells per well into white, flat-bottom 96-well plates (Costar) containing 50 μl PBS. Following the manufacturer’s instructions, two separate reactions were carried out to eliminate one of either NAD+ or NADH and the remaining metabolite was detected using 100 μl of NAD/NADH Glo detection reagent. After 2 h incubation, luminescence was determined as above and the NAD+/NADH ratios were calculated based on the levels of NAD+ and NADH.
Reverse-transcription PCR
Total RNA from FACS-sorted pax3a+ cells was extracted using TRIzol reagent (Thermo Fisher Scientific). cDNA was synthesized using the iScript Advanced cDNA Synthesis Kit (Bio-Rad) following the manufacturer’s instructions. For reverse-transcription PCR (RT–PCR), 10-μl reactions were set up using GoTaq Green Master Mix (Promega). Primers used to amplify zfccr5, 5′-TTATAACCAAGAGACATGTCGGCG-3′ and 5′-ACCCAGACGACCAGACCATT-3′. The primer pair was designed to cross a 388-bp intron so that cDNA and genomic DNA (gDNA) templates would result in bands of 191 bp and 579 bp, respectively. The cycling protocol was performed as follows. Initial denaturation at 95 °C for 2 min followed by 25 cycles of denaturation at 95 °C for 30 s, annealing at 58 °C for 1 min and extension at 72 °C for 45 s followed by a final extension of 5 min at 72 °C. The cycling protocol provided an allowance for the amplification of both cDNA and gDNA templates, enabling identification of genomic DNA contaminants.
Total RNA was extracted and cDNA synthesized from 4-dpf zebrafish larval tails of different genotypes (mutant, heterozygous and homozygous siblings were identified by genotyping larval heads from heterozygous in-crosses of nampta+/pc41ccr5+/pc42). RT–PCR was carried out using the following primer pairs, all of which were designed to cross an intron; zfnampta_mutant_RT, 5′-CCGACTCCTACAAGGTCACAC-3′ and 5′-TTGACTTTTCGGGGCTTGGT-3′ (wild-type amplicon, 115 bp); zfccr5_mutant_RT, 5′-TTGAGCTGTTATAACCAAGAGACA-3′ and 5′-GAGGGAAAATTAAGCTCAGAAGG-3′ (wild-type amplicon, 657 bp); zfactc1b_housekeeping_RT, 5′-TTGACAACGGCTCCGGTATG-3′ and 5′-GCCAACCATCACTCCCTGAT-3′ (amplicon, 110 bp).
Maximum-likelihood phylogenetic tree analysis
A putative Ccr5 orthologue in zebrafish was identified by BLAST and orthology assessed by phylogenetic analysis. Amino acid sequences were aligned by MUSCLE74,75 and trimmed using GBLOCKS76,77. PHYML78 was used to generate a maximum likelihood tree using the JTT model for protein evolution (as inferred using ProTest v.3.4.2)79. Trees were visualized using iTOL (v.4.3)80.
Statistics and reproducibility
No statistical methods were used to predetermine sample size. Embryos and mice for each experimental treatment group were assigned randomly and the groups were blinded to the experimenter before analysis. All experiments were performed with a minimum of three independent biological replicates and the collated exact number of independent animals and/or samples is indicated in the figure legends. If an experiment was performed with a lower number of biological replicates it is explicitly stated. For long-term time-lapse imaging studies individual injuries were assayed in a single experimental round and each injury is an independent experimental repeat (Figs. 1a–d, 2e and Extended Data Figs. 1k, 3i, k–l). Data distributions shown as violin plots document the median (horizontal central line), interquartile range (upper quartile and lower quartile, horizontal lines above and below central median line, respectively), individual data points and frequency distribution (extending to the highest and lowest data points). Where data is presented in bar graphs each individual data point is shown. Statistical analysis was carried out using Prism (v.7.0c, GraphPad Software). Data was analysed using Student’s unpaired two-tailed t-test when comparing two conditions and ANOVA when comparing multiple conditions.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The single-cell RNA-seq dataset has been deposited at the NCBI Gene Expression Omnibus (GEO) repository under accession number GSE162979. All other data supporting the findings of this study are found within the manuscript and its Supplementary Information. Source data are provided with this paper.
Code availability
The single-cell experiment dataset was processed and analysed with programs and packages as described in the Methods. Scripts can be found at https://github.com/fjrossello/Ratnayake_et_al_2020_SC.
References
Dhawan, J. & Rando, T. A. Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 15, 666–673 (2005).
Gurevich, D. B. et al. Asymmetric division of clonal muscle stem cells coordinates muscle regeneration in vivo. Science 353, aad9969 (2016).
Dort, J., Fabre, P., Molina, T. & Dumont, N. A. Macrophages are key regulators of stem cells during skeletal muscle regeneration and diseases. Stem Cells Int. 2019, 4761427 (2019).
Uderhardt, S., Martins, A. J., Tsang, J. S., Lämmermann, T. & Germain, R. N. Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell 177, 541–555 (2019).
Miller, M. J., Safrina, O., Parker, I. & Cahalan, M. D. Imaging the single cell dynamics of CD4+ T cell activation by dendritic cells in lymph nodes. J. Exp. Med. 200, 847–856 (2004).
Yahiaoui, L., Gvozdic, D., Danialou, G., Mack, M. & Petrof, B. J. CC family chemokines directly regulate myoblast responses to skeletal muscle injury. J. Physiol. 586, 3991–4004 (2008).
Hirata, A. et al. Expression profiling of cytokines and related genes in regenerating skeletal muscle after cardiotoxin injection: a role for osteopontin. Am. J. Pathol. 163, 203–215 (2003).
Fukada, S. et al. Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells 25, 2448–2459 (2007).
Garten, A. et al. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 11, 535–546 (2015).
Krzysik-Walker, S. M. et al. Nampt/visfatin/PBEF affects expression of myogenic regulatory factors and is regulated by interleukin-6 in chicken skeletal muscle cells. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 159, 413–421 (2011).
Van den Bergh, R. et al. Monocytes contribute to differential immune pressure on R5 versus X4 HIV through the adipocytokine visfatin/NAMPT. PLoS ONE 7, e35074 (2012).
Fang, C., Guan, L., Zhong, Z., Gan, X. & He, S. Analysis of the nicotinamide phosphoribosyltransferase family provides insight into vertebrate adaptation to different oxygen levels during the water-to-land transition. FEBS J. 282, 2858–2878 (2015).
Hong, S. M. et al. Increased nicotinamide adenine dinucleotide pool promotes colon cancer progression by suppressing reactive oxygen species level. Cancer Sci. 110, 629–638 (2019).
Watanabe, R. et al. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight 3, e123047 (2018).
Henningsen, J., Rigbolt, K. T., Blagoev, B., Pedersen, B. K. & Kratchmarova, I. Dynamics of the skeletal muscle secretome during myoblast differentiation. Mol. Cell. Proteomics 9, 2482–2496 (2010).
Deshmukh, A. S., Cox, J., Jensen, L. J., Meissner, F. & Mann, M. Secretome analysis of lipid-induced insulin resistance in skeletal muscle cells by a combined experimental and bioinformatics workflow. J. Proteome Res. 14, 4885–4895 (2015).
Grube, L. et al. Mining the secretome of C2C12 muscle cells: data dependent experimental approach to analyze protein secretion using label-free quantification and peptide based analysis. J. Proteome Res. 17, 879–890 (2018).
Gilliam, B. L., Riedel, D. J. & Redfield, R. R. Clinical use of CCR5 inhibitors in HIV and beyond. J. Transl. Med. 9, S9 (2011).
Jia, S. H. et al. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J. Clin. Invest. 113, 1318–1327 (2004).
Quarta, M. et al. Bioengineered constructs combined with exercise enhance stem cell-mediated treatment of volumetric muscle loss. Nat. Commun. 8, 15613 (2017).
Corona, B. T. et al. Autologous minced muscle grafts: a tissue engineering therapy for the volumetric loss of skeletal muscle. Am. J. Physiol. Cell Physiol. 305, C761–C775 (2013).
Ming, G. F., Wu, K., Hu, K., Chen, Y. & Xiao, J. NAMPT regulates senescence, proliferation, and migration of endothelial progenitor cells through the SIRT1 AS lncRNA/miR-22/SIRT1 pathway. Biochem. Biophys. Res. Commun. 478, 1382–1388 (2016).
Sax, M. J. et al. Cancer cell CCL5 mediates bone marrow independent angiogenesis in breast cancer. Oncotarget 7, 85437–85449 (2016).
Wang, S.-W. et al. CCL5/CCR5 axis induces vascular endothelial growth factor-mediated tumor angiogenesis in human osteosarcoma microenvironment. Carcinogenesis 36, 104–114 (2015).
Seger, C. et al. Analysis of Pax7 expressing myogenic cells in zebrafish muscle development, injury, and models of disease. Dev. Dyn. 240, 2440–2451 (2011).
Relaix, F., Rocancourt, D., Mansouri, A. & Buckingham, M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 435, 948–953 (2005).
Ellett, F., Pase, L., Hayman, J. W., Andrianopoulos, A. & Lieschke, G. J. mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood 117, e49–e56 (2011).
Scott, E. K. et al. Targeting neural circuitry in zebrafish using GAL4 enhancer trapping. Nat. Methods 4, 323–326 (2007).
Pisharath, H., Rhee, J. M., Swanson, M. A., Leach, S. D. & Parsons, M. J. Targeted ablation of beta cells in the embryonic zebrafish pancreas using E. coli nitroreductase. Mech. Dev. 124, 218–229 (2007).
Okuda, K. S. et al. A zebrafish model of inflammatory lymphangiogenesis. Biol. Open 4, 1270–1280 (2015).
Evans, R. J. et al. 15-Keto-prostaglandin E2 activates host peroxisome proliferator-activated receptor gamma (PPAR-γ) to promote Cryptococcus neoformans growth during infection. PLoS Pathog. 15, e1007597 (2019).
Renshaw, S. A. et al. A transgenic zebrafish model of neutrophilic inflammation. Blood 108, 3976–3978 (2006).
Cole, N. J. et al. Development and evolution of the muscles of the pelvic fin. PLoS Biol. 9, e1001168 (2011).
Morsch, M. et al. In vivo characterization of microglial engulfment of dying neurons in the zebrafish spinal cord. Front. Cell. Neurosci. 9, 321 (2015).
Higashijima, S., Okamoto, H., Ueno, N., Hotta, Y. & Eguchi, G. High-frequency generation of transgenic zebrafish which reliably express GFP in whole muscles or the whole body by using promoters of zebrafish origin. Dev. Biol. 192, 289–299 (1997).
Pipalia, T. G. et al. Cellular dynamics of regeneration reveals role of two distinct Pax7 stem cell populations in larval zebrafish muscle repair. Dis. Model. Mech. 9, 671–684 (2016).
Ando, K., Shibata, E., Hans, S., Brand, M. & Kawakami, A. Osteoblast production by reserved progenitor cells in zebrafish bone regeneration and maintenance. Dev. Cell 43, 643–650 (2017).
Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio rerio) (Univ. Oregon Press, 2007).
Yaffe, D. & Saxel, O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270, 725–727 (1977).
Aziz, A., Soucie, E., Sarrazin, S. & Sieweke, M. H. MafB/c-Maf deficiency enables self-renewal of differentiated functional macrophages. Science 326, 867–871 (2009).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Ollion, J., Cochennec, J., Loll, F., Escudé, C. & Boudier, T. TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics 29, 1840–1841 (2013).
Ando, R., Hama, H., Yamamoto-Hino, M., Mizuno, H. & Miyawaki, A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc. Natl Acad. Sci. USA 99, 12651–12656 (2002).
Berger, J., Sztal, T. & Currie, P. D. Quantification of birefringence readily measures the level of muscle damage in zebrafish. Biochem. Biophys. Res. Commun. 423, 785–788 (2012).
Ellett, F. et al. Macrophages protect Talaromyces marneffei conidia from myeloperoxidase-dependent neutrophil fungicidal activity during infection establishment in vivo. PLoS Pathog. 14, e1007063 (2018).
Pisharath, H. & Parsons, M. J. in Zebrafish (eds Lieschke, G. et al.) 133–143 (Humana, 2009).
Lalezari, J. et al. Safety, efficacy, and pharmacokinetics of TBR-652, a CCR5/CCR2 antagonist, in HIV-1-infected, treatment-experienced, CCR5 antagonist-naive subjects. J. Acquir. Immune Defic. Syndr. 57, 118–125 (2011).
Dorr, P. et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 49, 4721–4732 (2005).
Watson, M. et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell. Biol. 29, 5872–5888 (2009).
Xue, C.-B. et al. Discovery of INCB8761/PF-4136309, a potent, selective, and orally bioavailable CCR2 antagonist. ACS Med. Chem. Lett. 2, 913–918 (2011).
Nguyen, P. D. et al. Muscle stem cells undergo extensive clonal drift during tissue growth via Meox1-mediated induction of G2 cell-cycle arrest. Cell Stem Cell 21, 107–119 (2017).
Inoue, D. & Wittbrodt, J. One for all—a highly efficient and versatile method for fluorescent immunostaining in fish embryos. PLoS ONE 6, e19713 (2011).
Figeac, N., Serralbo, O., Marcelle, C. & Zammit, P. S. ErbB3 binding protein-1 (Ebp1) controls proliferation and myogenic differentiation of muscle stem cells. Dev. Biol. 386, 135–151 (2014).
Thisse, C. & Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protocols 3, 59–69 (2008).
de Vrieze, E., Sharif, F., Metz, J. R., Flik, G. & Richardson, M. K. Matrix metalloproteinases in osteoclasts of ontogenetic and regenerating zebrafish scales. Bone 48, 704–712 (2011).
Venter, G. et al. NAMPT-mediated salvage synthesis of NAD+ controls morphofunctional changes of macrophages. PLoS ONE 9, e97378 (2014).
Ratnayake, D. & Currie, P. D. in Myogenesis (ed. Rønning S.) 245–254 (Humana, 2019).
Muraro, M. J. et al. A single-cell transcriptome atlas of the human pancreas. Cell Syst. 3, 385–394 (2016).
Hashimshony, T. et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-seq. Genome Biol. 17, 77 (2016).
Grün, D. et al. De novo prediction of stem cell identity using single-cell transcriptome data. Cell Stem Cell 19, 266–277 (2016).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Junker, J. P. et al. Genome-wide RNA tomography in the zebrafish embryo. Cell 159, 662–675 (2014).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
McCarthy, D. J., Campbell, K. R., Lun, A. T. & Wills, Q. F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017).
Rougeot, J. et al. RNAseq profiling of leukocyte populations in zebrafish larvae reveals a cxcl11 chemokine gene as a marker of macrophage polarization during mycobacterial infection. Front. Immunol. 10, 832 (2019).
Wolf, F. A. et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59 (2019).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
McInnes, L., Healy, J. & Melville, J. UMAP: uniform manifold approximation and projection for dimension reduction. Preprint at https://arxiv.org/abs/1802.03426 (2018).
Haghverdi, L., Büttner, M., Wolf, F. A., Buettner, F. & Theis, F. J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Revollo, J. R., Grimm, A. A. & Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 279, 50754–50763 (2004).
Kim, J. S., Yoon, C.-S. & Park, D. R. NAMPT regulates mitochondria biogenesis via NAD metabolism and calcium binding proteins during skeletal muscle contraction. J. Exerc. Nutrition Biochem. 18, 259–266 (2014).
Skala, M. C. et al. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc. Natl Acad. Sci. USA 104, 19494–19499 (2007).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 (2007).
Guindon, S., Lethiec, F., Duroux, P. & Gascuel, O. PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33, W557–W559 (2005).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128 (2007).
Heredia, J. E. et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153, 376–388 (2013).
Arnold, L. et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204, 1057–1069 (2007).
Revollo, J. R. et al. Nampt/PBEF/Visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab. 6, 363–375 (2007).
Zhang, L. Q. et al. Metabolic and molecular insights into an essential role of nicotinamide phosphoribosyltransferase. Cell Death Dis. 8, e2705 (2017).
Acknowledgements
We thank Monash Micro Imaging and Monash Ramaciotti Centre for Cryo-Electron Microscopy for microscopy support, Hubrecht Institute single-cell sequencing facility for sequencing service, Hubrecht Institute flow cytometry core facility and Monash FlowCore for sorting of cells using FACS and Monash Fishcore staff for technical assistance, A. Kawakami for the mmp9:eGFP and mmp9:eGFP-NTR BAC transgenic lines, F. J. Tulenko for advice on phylogenetic tree construction, H. Janovjak for advice on in vitro assays, A.-M. Tichy, E. Gerrard and A. Ruparelia for technical assistance and B. T. Kile, P. Gönczy and R. P. Currie for comments on the manuscript. P.D.N. is supported by an EMBO Long Term Fellowship (ALTF1129-2015), HFSPO Fellowship (LT001404/2017-L) and a NWO-ZonMW Veni grant (016.186.017-3). This work was supported by the National Health and Medical Research Council of Australia (APP1041885, APP1136567, APP1104190, APP1159278 to P.D.C.; APP1044754, APP1069284, APP1086020 to G.J.L.; and APP1140229, APP1176213 to M.M.M.). The Australian Regenerative Medicine Institute is supported by grants from the State Government of Victoria and the Australian Government.
Author information
Authors and Affiliations
Contributions
D.R. (live imaging, photoconversions, immunohistochemistry, in situ hybridization, scRNA-seq sample preparation, tissue-specific loss-of-function studies, zebrafish mutant studies, drug and protein supplementation studies, RT–PCR, NAD+/NADH assays) designed and performed experiments, analysed data and co-wrote the manuscript; P.D.N. (scRNA-seq), V.C.W. (live imaging), J.L.T. (mouse experimentation (NAMPT supplementation, flow cytometry, immunohistochemistry, histology), cytokine array), L.A.G. (primary myoblast coculture), A.J.W. (C2C12 cell culture), Z.J. (binding assays, ELISAs), V.O. (correlative light and electron microscopy), S.B. (histology, immunohistochemistry, zebrafish mutant studies) and C.S. (in situ hybridization) performed experiments; A.I.I. (macrophage and neutrophil lineage-specific gene-editing system) established techniques; F.J.R. (scRNA-seq analysis) and T.B. (light-sheet cell tracking) analysed data; K.L.R., C.M., G.J.L., M.M.M. and J.B. provided reagents; P.D.C., G.J.L., J.B. and M.M.M. designed and supervised experiments and P.D.C. co-wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
P.D.C., M.M.M. and D.R. through Monash University have filed provisional patent application no. 2020901237 on the clinical use of NAMPT. All other authors declare no competing interests.
Additional information
Peer review information Nature thanks Leonard Zon and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Dwelling macrophages establish a transient niche following muscle injury that is indispensable for the regenerative process.
a, The migratory paths of injury-responding macrophages located within a 260-μm radius from the centre of the laser ablation injury (a region encompassing 2 myotomes on either side of the injury) from Supplementary Video 1 are shown, where (0, 0) is set to the centre of the wound (see still images in Fig. 1a, b). b–d, Morphological characterization of macrophage (MΦ) subsets revealed transient macrophages presenting a more stellate phenotype (b) with lower sphericity (d; 0.4163 ± 0.1260 (mean ± s.d.)), whereas dwelling macrophages appear more rounded (c) with higher sphericity (d; 0.6639 ± 0.1296 (mean ± s.d.); a 0.248 ± 0.022 higher sphericity value than to transient macrophages). Scale bar, 20 μm. d, n = 69 macrophages per group, from n = 18 injuries carried out in 6 independent experiments). The thick black lines and thin black lines within the violin plot indicate the median and quartiles, respectively. Unpaired two-tailed t-test; t136 = 11.38, P < 0.0001. e–g″, Maximum intensity projection images of the same fish transgenic for Tg(actc1:BFP) (labelling differentiated muscle fibres; magenta; e′, f′, g′), TgBAC(pax3a:GFP) (labelling pax3a+ myogenic cells; cyan; e″, f″, g″) and Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry (labelling macrophages; yellow; e‴, f‴, g‴) after a needle-stab muscle injury. Scale bar, 50 μm. h, By 2 dpi, 51.6% of the original injury-responsive macrophages remain in the wound site, and these dwelling macrophages go on to establish a transient niche that is pro-myogenic (n = 20). i–p, Establishing nitroreductase-mediated macrophage ablation parameters. The transgenic line, Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry) expresses the enzyme nitroreductase specifically within macrophages, enabling the temporally controlled genetic ablation of macrophages by addition of the pro-drug Mtz. The optimum Mtz concentration was established by treating larvae with 5 and 10 mM Mtz for 24 h (starting at 4 dpf) and visualizing (i) and quantifying (j) trunk-present macrophages. Macrophage ablation of 77% was achieved at 5 dpf by 5 mM Mtz treatment (j; n = 10). Although uninjured larvae were able to tolerate 10 mM Mtz, needle-stab injury carried out at this concentration resulted in significant lethality. Therefore, all further experiments used 5 mM Mtz for macrophage ablations. k, Titrating the Mtz dosing time to specifically ablate macrophage subsets of interest. Temporal response to 5 mM Mtz was visualized by time-lapse imaging the head and tail region of larval zebrafish immediately after Mtz addition (4 dpf). Macrophage ablation initiated at 3 h after Mtz treatment. Scale bar, 500 μm. Frames were obtained from Supplementary Video 5 (n = 8). l, m, Ablation of macrophages did not alter the neutrophil response to wounding at 2 dpi (representative images (l, scale bar, 50 μm) and quantification (m); n = 12). n, Addition of Mtz at the point of injury ablated all macrophages (yellow; shown superimposed on bright-field images of the zebrafish trunk), whereas addition of Mtz at 1.75 dpi (5 dpf) selectively ablated dwelling macrophages (n = 10). Scale bar, 500 μm. The temporal sequence of neutrophil migration in response to skeletal muscle injury was assayed (o; scale bar, 50 μm) and quantified (p; n = 14 at 5–6 hpi, n = 13 at 1 dpi and n = 12 at 2 dpi). q–t, As a control experiment for nitroreductase-mediated genetic cell ablation, neutrophils—the other key cellular component of the innate immune system—were selectively ablated using the transgenic line Tg(mpx:KALTA4/UAS:nfsb-mCherry). Ablation efficiency after 24 h of 5 mM Mtz treatment starting at 4 dpf was visualized (q) and quantified at 5 dpf (r; n = 10). After needle-stab injury, larvae were soaked in 5 mM and 10 mM Mtz (4 dpf, 0 dpi) to ablate all neutrophils and 5 mM Mtz at 5 dpf (1.75 dpi) to specifically ablate late injury-present neutrophils (Mtz treatment was maintained until experimental end point at 7 dpf, 3 dpi with daily Mtz drug refreshment) and regeneration monitored by imaging for birefringence (s; scale bar, 500 μm) and quantified (t, n = 12). Neutrophil-ablated larvae showed no regeneration defects at either Mtz dose or timing, highlighting a specific requirement for macrophages in stimulating regeneration and precluding the possibility that any regeneration defect was Mtz-related. h, j, p, r, The central lines, upper and lower lines within the violin plot indicate the median, upper and lower quartiles, respectively. One-way ANOVA with Dunnett’s post hoc test for multiple comparisons. m, The black lines and grey lines within the violin plot indicate the median and quartiles, respectively. Unpaired two-tailed t-test; t122 = 1.158, P = 0.2591. t, Individual data points are shown. Two-way ANOVA with Tukey’s multiple comparison test.
Extended Data Fig. 2 Macrophage ablation does not disrupt tissue debris clearance after muscle injury.
a–f, Clearing of wound site debris is mediated by a number of cell types81, but largely attributed to phagocytic macrophages82. Absent or ineffective clearance might be one explanation for the lack of regeneration observed when macrophage populations are ablated early in the regenerative process. At 3 dpf, macrophages within the Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry) transgenic line were ablated in larval zebrafish by the addition of 5 mM Mtz. Macrophage-ablated larvae were then subject to a needle-stab skeletal muscle injury at 4 dpf and the wound site assayed for clearance of damaged muscle fibres by a number of criteria. a, Fluorescently conjugated phalloidin staining to detect the actin cytoskeleton of remnant necrotic skeletal muscle fibres in the injury site. No difference is evident between control and macrophage-ablated larvae. n = 3 independent injuries per time point presented, n = 12 injuries per time point imaged. Scale bar, 50 μm. b, c, Annexin-V marking the apoptotic and phagocytic signalling process was visualized in vivo using the transgenic line Tg(ubi:secAnnexinV-mVenus) (magenta, n = 3 independent injuries per time point presented; b; scale bar, 50 μm) and quantified (c). d, e, Acidic membranous organelles and the autophagocytic process were marked by LysoTracker fluorescent vital dye (magenta), imaged (d; scale bar, 50 μm) and quantified (e). The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test (c) or unpaired two-tailed t-test; t12 = 0.3311, P = 0.7463 (e). No difference is evident between control and macrophage-ablated larvae. f, Haematoxylin and eosin-stained cross sections through needle-stab-injured myotomes demonstrates cleared wound site (black arrowheads) at 3 dpi in macrophage-ablated larvae, demonstrating the removal of dead and dying fibres evident in the wound site at 1 dpi (n = 3 larvae per group).
Extended Data Fig. 3 Dwelling macrophage–MuSC associations precede MuSC proliferation.
a–d, Macrophage and MuSC dynamics after injury were assayed using the Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry);TgBAC(pax3a:GFP) (yellow, macrophages; cyan, muscle stem/progenitor cells) compound transgenic line. The pax3a:GFP reporter is particularly useful in this context as it is expressed in both the MuSC compartment and the dividing progenitor population. After laser-ablation muscle injury (white dotted line) transient macrophages migrate into the wound site (a–c). Simultaneously, activated pax3a+ myogenic stem cells from the injured and adjacent myotomes travel to line the edge of the injury site (a–c, white arrowheads). Once macrophages transition to a dwelling subtype, they initiate interactions with wound-edge-lining pax3a+ cells (d, orthogonal views). Scale bar, 50 μm. Frames were obtained from Supplementary Video 6 (n = 5). e–g, MuSCs migrate into the injury site independent of macrophage-derived signals. As macrophages and MuSCs migrate and populate the injury site simultaneously, the dependence of one on the other was assessed. e, f, Both control larvae and macrophages-ablated larvae (5 mM Mtz added at the point of injury on 4 dpf to ablate all macrophages) displayed pax3a+ stem cells in the injury site (white dotted line) at 2 dpi (white arrowheads) after needle-stab skeletal muscle injury (e; scale bar, 50 μm) and MuSCs lining the edge of the injury site (yellow asterisks) after laser-ablation muscle injury at 14 hpi (f; scale bar, 50 μm). g, Quantification of laser-ablation muscle-injury-responsive macrophages. The thick black lines and thin black lines within the violin plot indicate the median and quartiles, respectively. Not significant in unpaired two-tailed t-test; t12 = 0.1715, P = 0.8667. In the control setting (e, f, top), these cells were associated with macrophages. e, At 3 dpi after needle-stab injury, control larvae displayed regenerated cyan-fluorescence-persistent muscle fibres (red arrowheads) that arose from pax3a+ MuSCs present in the wound, a hallmark of a healing muscle injury. By contrast, although the wound site was still occupied by pax3a+ stem cells at 3 dpi in the macrophage-ablated larvae, there were no nascent pax3a+ stem cell-derived muscle fibres present, highlighting that the presence of macrophages in the wound site is not prerequisite for pax3a+ myogenic cell migration; however, it is required for those migrated cells to enter the myogenic replenishment program (n = 12). h–j, The pax3a+ myogenic cells that dwelling macrophages interact with also express the MuSC markers met (h, i) and pax7b (j). h, After needle-stab skeletal muscle injury (white dotted line) in larvae transgenic for TgBAC(pax3a:GFP) (cyan) and Tg(met:mCherry-2A-KALTA4/UAS:nfsb-mCherry) (magenta), wound-present MuSCs are pax3a+met+. These cells undergo both symmetric expansion (white open arrowheads) and asymmetric divisions (closed yellow arrowheads), giving rise to two pax3a+met+ daughter cells or a pax3a+met+ and a pax3a+met− daughter cell, respectively. The broad myogenic marker pax3a+ visualizes each of these events (n = 20, scale bar, 50 μm). i, Time-lapse imaging was used to visualize MuSC (TgBAC(pax3a:GFP)/Tg(met:mCherry-2A-KALTA4/UAS:nfsb-mCherry), cyan/yellow double-positive cells) and dwelling macrophage (Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry), yellow) interactions after a laser-ablation skeletal muscle injury (white dotted line). Dwelling macrophages (white closed arrowhead) interact with pax3a+met+ MuSCs (magenta open arrowhead), after which the MuSC undergoes symmetric division to generate two pax3a+met+ progenitors. Scale bar, 30 μm. Frames were obtained from Supplementary Video 9 (n = 6). j, Dwelling macrophages (Tg(mpeg1:GFP); yellow) that are present at the wound site (white dotted line) interact with MuSCs that also express the marker pax7b (cyan; Tg(pax7b:GAL4FF/UAS:nfsb-mCherry)). In this muscle stem/progenitor cell marker gene trap line, GAL4FF is integrated into the pax7b gene (referred to as Tg(pax7b:GAL4FF)) and used to drive Tg(UAS:nfsb-mCherry). Scale bars, 50 μm (n = 9 per time point). k, Dwelling macrophages interact with wound-responsive MuSCs. Examples of three independent laser-ablation muscle injury sites showing dwelling macrophage–stem cell associations followed by stem cell divisions (arrowheads). Scale bars, 15 μm (injury 1), 30 μm (injuries 2 and 3). Frames were obtained from Supplementary Video 7. l, After MuSC division, the associated dwelling macrophage, and generated daughter cells, migrate away from each other. Dwelling macrophages interact with MuSCs located within the injury zone (white dotted line). After MuSC division (closed white, magenta, yellow and grey arrowheads highlight four independent stem cells), movements of the daughter cells (open white, magenta, yellow and grey arrowheads highlighting the daughter cells that arose from the stem cells indicated by closed arrowheads of the same colour) within the wound site are highlighted such that their relation to the dwelling macrophages can be visualized. Cell movements are tracked until dwelling macrophages localize to the vertical myosepta. Scale bar, 40 μm. Frames were obtained from Supplementary Video 10. Representative of n = 9 stem cell divisions assayed in n = 5 long-term time lapses.
Extended Data Fig. 4 Correlative light and electron microscopy analyses of the transient macrophage–stem cell niche reveals that macrophages and stem cells maintain direct heterocellular surface appositions in xyz planes.
a, Confocal microscopy of the wound site at 25 h after laser-ablation muscle injury in the compound transgenic zebrafish line, Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry);TgBAC(pax3a:GFP), labelling macrophages (yellow) and pax3a+ muscle stem/progenitor cells (cyan). Scale bar, 50 μm (n = 1). b, Large STEM tile set of the same trunk region of the larva illustrated in a generated after Epon-embedding and sectioning. This dataset was used to correlate and identify the highlighted macrophage–stem cell interaction of interest (white asterisk in a, b). Dotted square marks the area that was further examined by transmission electron microscopy (TEM) in c, d. c, Region of interest encompassing a macrophage and stem cell that maintain a close interaction examined through z-depth. The cells of interest and interaction area are segmented (macrophage cytoplasm, yellow; macrophage nucleus, orange; stem cell cytoplasm, cyan; stem cell nucleus, blue; cell–cell interaction surface, magenta). d, High-resolution images of two planes through the z-depth (plane shown correlated with red dotted lines to the segmentation images) further demonstrate the close association between the two cells. Scale bar, 5 μm (n = 1).
Extended Data Fig. 5 Prox1a+Pou2f3+ macrophages (cluster 6) identified by scRNA-seq are a dwelling macrophage subset independent from Mmp9+ macrophages (cluster 2).
a, After appropriate filtering, 1,309 macrophages were analysed by scRNA-seq. These cells express antigen processing and presentation genes, confirming their macrophage character. Feature plots highlighting antigen-presenting genes expressed by the majority of cells (cd83, cd81a and cd40) as well as genes differentially expressed by individual subsets. The percentage of cells expressing a gene of interest is also recorded (maroon, information extracted from Supplementary Tables 1, 3). b, Graphical representation of UMAP scatter plot clusters. Uninjured macrophages cluster together (cluster 3). Macrophages isolated from a ‘transient’ time point (1 dpi) also predominantly cluster together (cluster 1). The remaining six clusters (0, 2, 4, 5, 6, 7) are mainly composed of macrophages isolated from ‘dwelling’ time points (2–3 dpi). c, Lineage analysis using PAGA visualizes gene expression changes leading to the definition of an mmp9+ dwelling macrophage subset along path 3-0-4-2 of the PAGA cluster and a prox1a+pou2f3+ dwelling macrophage subset along path 3-5-1-6 of the PAGA cluster. The numerical scale (right) expresses normalized gene expression from Seurat, whereas clusters (bottom) indicate subsets along the PAGA path with length being proportional to the cell numbers in each cluster. d, Violin plots show the expression pattern of mmp9 and nampta based on cluster identity and isolation time point. The percentage of cells expressing the markers at 2 dpi are documented. e–j, Violin plots (percentage of cells in cluster 6 expressing a specific marker recorded as percentage (e, h)) and antibody staining for a subset of the cluster-6 markers (Pou2f3 (f) and Prox1a (i)) after needle-stab muscle injury (white dotted line), showing a specific, late-injury-dwelling macrophage population (Supplementary Table 3 (worksheet 7)). Antibody staining against mCherry was used to identify all mpeg1+ macrophages in the Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry) (yellow) line. After injury, at 1 dpi, almost no Pou2f3+mpeg1+ (f; scale bar, 50 μm) or Prox1a+mpeg1+ (i; scale bar, 50 μm) macrophages are present in the wound site; their numbers start to increase slightly at 2 dpi with the highest percentage being present at 3 dpi, identifying this subset as a late-injury-dwelling macrophage subset. g, j, Quantification (n = 10 at 2 and 3 dpi in g and n = 7, n = 11 and n = 7 at 1, 2 and 3 dpi, respectively, in j). The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Unpaired two-tailed t-test; t17 = 4.574, P = 0.0003 (g) or one-way ANOVA with Dunnett’s post hoc test for multiple comparisons (j). k, l, Cluster-2 macrophages display a metabolic shift towards glycolysis at the gene expression level. KEGG pathway analysis of cluster-2 differentially expressed genes (Supplementary Table 3) identified genes associated with glycolysis (term: dre00010) and oxidative phosphorylation (term: dre00190) pathways. Genes associated with glycolysis were upregulated (k), whereas the majority of genes associated with oxidative phosphorylation were downregulated (l), highlighting a metabolic shift towards glycolysis in cluster-2 mmp9+ dwelling macrophages.
Extended Data Fig. 6 The intracellular enzymatic function of NAMPT is not needed for its pro-proliferative activity during skeletal muscle regeneration.
a–c, mmp9 labels a subset of wound-dwelling macrophages. a, These mmp9-expressing macrophages (magenta) associate with wound-present pax3a+ stem cells (cyan; wound site, white dotted line; n = 15). b, After laser-ablation skeletal muscle injury (white dotted line), a subset of macrophages (Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry); yellow) starts expressing mmp9 (TgBAC(mmp9:eGFP); magenta) upon dwelling (yellow/magenta double-labelled cells). After injury, mpeg1+ macrophages respond to wounding. At the same time, mmp9-expressing neutrophils are found to be present in the injury (magenta only; green open arrowheads); these neutrophils have a cellular phenotype and wound site dynamics that are distinct from macrophages. The mmp9-expressing neutrophils co-express the neutrophil-specific marker mpx (data not shown; n = 16) and exit the wound site by 5.21 ± 1.27 hpi (n = 6). Once macrophages start to dwell, a subset of mpeg1-expressing macrophages also start to express mmp9 (white closed arrowheads). Frames were obtained from Supplementary Video 10 (n = 6). c, Titrating the Mtz dose enables specific ablation of mmp9-expressing immune cells in the TgBAC(mmp9:eGFP-NTR) (magenta) transgenic line. After treatment with 5 mM Mtz for 6 h (5 dpf, 1.75 dpi to 6 dpf, 2 dpi) uninjured larvae show ablation of mmp9-expressing immune cells (phenotypically distinct, stellate morphology, mmp9 high-expressing cells), whereas skin cells—which express mmp9 at significantly lower levels (phenotypically distinct, hexagonal morphology)—are resistant to ablation. After needle-stab skeletal muscle injury (4 dpf, 0 dpi), larvae treated with Mtz at 5 dpf, 1.75 dpi demonstrate an absence of mmp9 high-expressing cells, specifically in the wound site (dotted white line) (n = 12). d, e, The dose and duration of Mtz treatment used specifically ablates 77.94% of mmp9+mpeg1+ macrophages (mmp9−mpeg1+ macrophages, 6.65 ± 1.50 in Mtz-treated versus 5.47 ± 2.75 in control; mmp9+mpeg1+ macrophages, 2.18 ± 1.19 in Mtz-treated versus 9.87 ± 2.72 in control). d, Representative images, with white arrowheads identifying mmp9+mpeg1+ (magenta and yellow, respectively) macrophages within the needle-stab skeletal muscle injury zone (white dotted line). e, Quantification. The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. f–h, Ablation of mmp9+ macrophages leads to a significant reduction in muscle stem/progenitor cells present in the wound site. After Mtz treatment there is a significant reduction in Pax7+ MuSCs located in the wound site (white dotted line) (f). g, Quantification. The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Unpaired two-tailed t-test; t41 = 6.196, P < 0.0001. This reduction is not due to a stem-cell migration deficit, as Mtz treatment is carried out after MuSC migration (MuSCs lining the wound edge in the Mtz-treated larvae is shown in f), but rather due to a MuSC proliferation deficit within the wound site (yellow dotted line) as observed by assaying EdU incorporation (h; 3 independent examples of larval injuries for both control and Mtz-treated larvae are shown; images are representative of the quantification presented in Fig. 3r). i, nampta mRNA expression is specifically upregulated in the injury site from 2 dpi onwards (black arrowhead, n = 18) in larval zebrafish. j–n, Assessing Nampt expression in larval zebrafish. Nampt antibody staining carried out in a developmental series (1, 3 and 4 dpf) confirmed a match between the distribution pattern of the protein and the previously published RNA transcript data12 and demonstrated ubiquitous expression during early development, with enrichment at the somite boundaries (white closed arrowheads) at 1–4 dpf and further enrichment in the intestine (white open arrowhead) from 3 dpf onwards (n = 10) (j). k, l, After needle-stab skeletal muscle injury, Nampt protein expression in the wound site is upregulated starting at 6 dpf, 2 dpi. l, Quantification (n = 9 control, n = 10 Mtz-treated). The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Unpaired two-tailed t-test; t18 = 5.430, P < 0.0001. m, n, Nampt upregulation in the wound site (white dotted line) is of macrophage origin, as selectively ablating macrophages (5 mM Mtz added at the point of injury (4 dpf, 0 dpi) to Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry) larvae to ablate all macrophage) leads to a significant reduction in Nampt expression in the wound site. This is further confirmed by Nampt levels being unperturbed after selective ablation of neutrophils (Tg(mpx:KALTA4/UAS:nfsb-mCherry) larvae used in neutrophil ablation). n, Quantification. The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. o, p, Increased NAMPT activity leads to elevated levels of intracellular NADH. o, NADH autofluorescence (magenta) displays a localized upregulation in dwelling macrophages (yellow), indicating that these cells are the primary source of wound-present Nampt (n = 15). Scale bar, 20 μm. p, This was quantitatively confirmed by means of a bioluminescence-based assay, which demonstrated wound-located dwelling macrophages have a 0.6781 ± 0.1650 lower NAD+/NADH ratio compared with transient macrophages. The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Unpaired two-tailed t-test; t8 = 4.110, P = 0.0034. q, r, Inhibiting the enzymatic function of Nampt does not affect MuSC proliferation. After needle-stab skeletal muscle injury (white dotted line) TgBAC(pax3a:GFP) (cyan) larvae were treated with the small-molecule, competitive inhibitor GMX1778 from 5 dpf, 1.75 dpi untill experimental end point (6 dpf, 2.5–2.75 dpi). GMX1778 selectively inhibits the rate-limiting enzymatic function of Nampt in NAD+ biosynthesis and at the administered concentration results in a severe reduction in larval NAD+/NADH levels (Extended Data Fig. 9c). q, Inhibiting the enzymatic function of Nampt had no effect on MuSC proliferation in the injury zone, highlighting that the functionality of Nampt during stem cell proliferation is distinct to its intracellular role in energy metabolism. r, Quantification (n = 17 control, n = 13 GMX1778-treated). The central lines, upper and lower lines within the violin plot indicate the median, upper and lower quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. a–d, f, h, i–k, m, q, Scale bars, 50 μm.
Extended Data Fig. 7 NAMPT binds to the CCR5 receptor present on MuSCs and induces proliferation.
a, b, NAMPT selectively binds to CCR5. a, ELISA plates were coated with human recombinant CCR5 (hrCCR5) or BSA and further incubated with hrNAMPT(1) at increasing concentrations (0 nM to 800 nM). NAMPT molecules bound to CCR5 were detected using a biotinylated antibody. NAMPT binds to CCR5 with a KD of 172 ± 18 nM (n = 2 independent experiments performed in triplicate). The graph shows a representative binding curve from which non-specific binding to BSA was deducted. Data are mean ± s.e.m. b, NAMPT is a highly evolutionary conserved protein, with human NAMPT sharing 96% and 88.24% identity with mouse NAMPT and zebrafish Nampta proteins, respectively. As we use hrNAMPT in mouse cell line assays, we also assayed the affinity of hrNAMPT for mrCCR5 by means of a competitive ligand-binding assay with the cognate ligand, CCL4. ELISA plates were coated with mrCCR5 or BSA and incubated with mrCCL4 at increasing concentrations (0 nM to 400 nM) along with 100 nM hrNAMPT(1). hrNAMPT(1) molecules bound to CCR5 were detected using a biotinylated antibody. mrCCL4 shows an IC50 of 34.4 ± 2.2 nM (n = 2 independent experiments performed in triplicate) revealing both hrNAMPT’s strong affinity for mrCCR5 and its binding to the receptor via the same sites used by CCL4. The graph shows a combined binding curve from which non-specific binding to BSA was deducted. Data are mean ± s.e.m. c, Exogenous NAMPT supplementation enhances myoblast proliferation. In vitro assay assessing the effects of exogenously introduced factors on C2C12 myoblast proliferation. Proliferation is identified by EdU incorporation. NAMPT administration (two commercially available NAMPT sources tested, hrNAMPT(1) and hrNAMPT(2)) leads to a dose-dependent increase in myoblast proliferation. This effect is specifically mediated by the CCR5 receptor. Co-administration of NAMPT with the CCR2/CCR5 dual inhibitor CVC and CCR5-specific inhibitor MVC abolishes the pro-proliferative response of NAMPT, whereas co-administration with the CCR2 inhibitor PF-4136309 (PF4) does not hinder the stimulatory effect of NAMPT on myoblast proliferation. In agreement with this finding, the endogenous ligands of CCR5, mrCCL8 and mrCCL4, functioned to enhance C2C12 proliferation, whereas the CCR2-specific ligand mouse recombinant CCL2 did not to increase proliferative rates beyond that of the control. The pro-proliferative function of NAMPT is separate from its intracellular role in energy metabolism, as co-administering NAMPT with the NAMPT enzymatic inhibitor (GMX1778) did not affect its effect on myoblast proliferation. The central lines, upper and lower lines within the violin plot indicate the median, upper and lower quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. n = 20 control, n = 10 1.9 nM hrNAMPT(1), n = 10 9.5 nM hrNAMPT(1), n = 10 19 nM hrNAMPT(1), n = 9 1.9 nM hrNAMPT(2), n = 10 9.5 nM hrNAMPT(2), n = 10 100 nM CVC, n = 10 100 nM MVC, n = 10 100 nM PF4, n = 10 100 nM CVC + 9.5 nM hrNAMPT(1), n = 10 100 nM MVC + 9.5 nM hrNAMPT(1), n = 9 100 nM PF4 + 9.5 nM hrNAMPT(1), n = 10 9.5 nM mrCCL8, n = 10 9.5 nM mrCCL4, n = 10 9.5 nM mrCCL2, n = 10 500 nM GMX1778, n = 10 500 nM GMX1778 + 9.5 nM hrNAMPT(1). d, Cultured MAF DKO macrophages actively secrete NAMPT. ELISA-quantified NAMPT concentration in the supernatants of 16-h M-CSF-stimulated MAF DKO and Raw 264.7 macrophages (n = 1 experiment in triplicate, stimulated with 10 ng ml−1 M-CSF). The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Unpaired two-tailed t-test; t4 = 0.3302, P = 0.7578. e, f, Cultured MAF DKO macrophages do not actively secrete the cognate ligands of CCR5, CCL3, CCL4 and CCL5. e, Representative array spots detecting CCR5 ligands expressed by MAF DKO macrophages after 16 h stimulation with M-CSF (10 ng ml−1). f, Quantification. Data are mean ± s.e.m. (n = 1 array per group performed in duplicate). g, h, PAX7+ satellite cells in mouse primary myoblast monocultures display enhanced proliferation upon CCR5 receptor signalling, mediated by either exogenous NAMPT (g, h) or CCL4 (h) supplementation. Coculturing myoblasts with macrophages stimulated satellite cell proliferation. This pro-proliferative response is abolished by administrating CVC (g) and MVC (h), whereas PF-4136309 had no negative effect (h), confirming that the pro-proliferative function of macrophages on stem cells is mediated by CCR5 signalling. In addition, coculturing myoblasts with 3T3 cells that do not naturally secrete NAMPT83 does not stimulate satellite cell proliferation, suggesting that NAMPT is the macrophage-derived pro-proliferative cue that drives stem cell proliferation. g, n = 18 control, n = 18 9.5 nM hrNAMPT(2), n = 18 100 nM CVC, n = 18 100 nM CVC + 9.5 nM hrNAMPT(2) for myoblasts, myoblast + macrophages and myoblasts + 3T3 cells); h, n = 35 control, n = 36 9.5 nM hrNAMPT(1), n = 36 100 nM MVC, n = 36 100 nM PF4, n = 55 9.5 nM mrCCL4 for myoblasts, n = 42 control, n = 36 9.5 nM hrNAMPT(1), n = 36 100 nM MVC, n = 31 100 nM PF4, n = 46 9.5 nM mrCCL4 for myoblast + macrophages and n = 36 control, n = 36 9.5 nM hrNAMPT(1), n = 36 100 nM MVC, n = 36 100 nM PF4, n = 51 9.5 nM mrCCL4 for myoblast + 3T3 cells. The black lines and grey lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. i–m, Inhibiting Ccr5 receptor activation does not affect injury-responsive immune cell dynamics in larval zebrafish. i, j, The predicted orthologous Ccr5 protein in zebrafish was identified by determining the appropriate best hit for the protein most similar to the human CCR5 amino acid sequence in zebrafish using BLAST. A maximum-likelihood phylogenetic tree constructed using protein sequences of CCR5 positioned the putative zebrafish Ccr5-like (Ccr5l) sequence as a homologue of mammalian CCR5 with strong bootstrap support. Bootstrap values with 500 replicates are documented below the branches. CCR7 was used as an outgroup. The accession numbers for genes included in our analysis are provided in the table (j). k, pax3a+ MuSCs express the ccr5 receptor at 2 dpi as detected by RT–PCR (n = 3). l, m, Larvae soaked in CVC or MVC displayed a marked regeneration deficit revealed by birefringence imaging (l; scale bar, 500 μm) and quantification (m; n = 20 control, n = 22 5 μM CVC, n = 14 10 μM CVC, n = 16 5 μM MVC, n = 12 10 μM MVC). Individual data points are shown. Two-way ANOVA with Tukey’s multiple comparison test. n–s, Ccr5 inhibition by CVC does not affect macrophage (Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry); yellow) migration into the injury site (skeletal muscle labelled using Tg(actc1b:BFP); magenta) or the successful transition into a dwelling macrophage subtype (n; scale bar, 50 μm). o, Quantification (n = 10 per group at each time point). The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. CVC-treated macrophages appear morphologically indistinguishable from controls, with transient macrophages possessing lower sphericity values then their dwelling counterpart (p; scale bar, 20 μm). q, Quantification (n = 45 transient control, n = 42 transient 5 μM CVC, n = 50 dwelling control, n = 39 dwelling 5 μM CVC). Data are mean ± s.d. Two-way ANOVA with Tukey’s multiple comparison test. r, CVC treatment does not alter the response of neutrophils (Tg(mpx:eGFP); magenta) to needle-stab muscle injury (white dotted line). Scale bar, 50 μm. s, Quantification (n = 11 control, n = 14 5 μM CVC). The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. Unpaired two-tailed t-test; t23 = 0.5838, P = 0.5650. t–u, pax3a+ myogenic stem cell (TgBAC(pax3a:GFP); cyan) proliferation is inhibited by CVC addition as demonstrated by decreased EdU incorporation (magenta) of these cells in the wound site (white dotted line) after injury (t; scale bar, 50 μm) and quantification (u, n = 19 per group). The black lines and grey lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. Two-way ANOVA with Tukey’s multiple comparison test. v, CVC treatment (pre-treatment for 2 h and maintained after laser ablation skeletal muscle injury (white dotted line) until experimental end point) does not interfere with the initiation and maintenance of dwelling macrophage (white arrows)–MuSC (white arrowheads) associations in the wound site. Scale bar, 50 μm. Frames were obtained from Supplementary Video 13.
Extended Data Fig. 8 Zebrafish germline nampta and ccr5 mutants present severe skeletal muscle regeneration deficits in response to acute muscle injury.
a–j, CRISPR–Cas9 was used to target exon 2 of nampta, which resulted in a germline deletion–insertion mutation that produced an altered amino acid sequence and a subsequent premature stop codon (asterisks) (nampta p.Try61Profs*4, referred to as namptapc41). a, Sanger trace of the genotyping PCR amplicon demonstrating the effect of Cas9–gRNA-induced mutation at a DNA and amino acid sequence (AA) level in the target nampta locus (n = 3). b, Schematic representation of the nampta gene highlighting its exons, mutation site, transcript and primer-binding sites for RT–PCR. c, RT–PCR for nampt cDNA demonstrates a reduction in the level of mutant transcript, encoding a truncated protein, highlighting that it is targeted for degradation by nonsense-mediated decay. actc1b transcript levels act as a loading control. d–f, In contrast to mice, where unconditional Nampt gene knockout results in embryonic lethality84, larval zebrafish are able to survive nampta germline knockout. This is probably due to the fact that Namptb can functionally compensate and fulfil, at least in part, the enzymatic role of Nampta in the NAD+ salvage pathway12. Macrophage and stem cell dynamics are unaffected in the nampta mutant. d, Both nampta heterozygous and homozygous mutants have wound site (white dotted line) macrophage (yellow) dynamics that are comparable to wild-type siblings, with macrophages transition to a dwelling state at 2 dpi. e, Quantification (n = 12 1dpi, n = 19 2 dpi nampta, n = 23 1 dpi, n = 43 2 dpi nampta+/pc41, n = 12 for both 1 and 2 dpi namptapc41). f, Furthermore, dwelling macrophages in the heterozygous and homozygous nampta mutants go on to interact with Pax7+ MuSCs that are present in the wound site. Representative of n = 20 observations. Homozygous mutants present a severe regeneration deficit after needle-stab muscle injury as observed by birefringence imaging (g; scale bar, 500 μm) and quantified (h; n = 12 nampta, n = 13 nampta+/pc41, n = 11 namptapc41). Individual data points are shown. Two-way ANOVA with Tukey’s multiple comparison test. i, This repair deficit could be correlated to a significant proliferation deficit (EdU, white) observed within the injury site after needle-stab skeletal muscle injury (myosin heavy chain (MyHC) to visualize skeletal muscle, magenta). Proliferation within the injury zone in mutant larvae decreased to homeostatic levels observed external to the wound site, highlighting that the mutants failed to elicit the additional proliferative response needed to sustain repair. Furthermore, these observations recapitulate what is seen after ablation of dwelling and mmp9-expressing macrophages. j, Quantification (n = 23 nampta, n = 19 nampta+/pc41, n = 18 namptapc41). k–t, CRISPR–Cas9 was also used to target exon 2 of ccr5 (using two gRNAs) resulting in a deletion–insertion mutation that induced a frame shift and subsequent premature stop codon (ccr5 p.Pro24Leufs*28, referred to as ccr5pc42) (k). l, Schematic of the two exons of ccr5, along with the site of mutation (513-bp deletion and 90-bp insertion) and mRNA transcript. The primer-binding sites for RT–PCR are also documented. m, RT–PCR analysis of ccr5 cDNA demonstrates a 234-bp product, corresponding to the mutant transcript (red arrowhead). No wild-type transcript corresponding to a 657-bp product (red arrow) is present in the mutant. Both PCR fragments have been sequence-verified. The mutant transcript lacks the majority of the chemokine domain encoded by exon 2 of ccr5, and as such the mutant protein, if translated, would be non-functional as it lacks the ligand-binding site. actc1b transcript levels act as a loading control (n = 3). n–p, As for the nampta mutant described above, the ccr5 mutant presented with macrophage dynamics (representative images (n) and quantification (o; n = 8 for both 1 dpi and 2 dpi ccr5, n = 20 1 dpi, n = 23 2 dpi ccr5+/pc42, n = 8 for both 1 and 2 dpi ccr5pc42)) and macrophage–stem cell interactions (p, representative of n = 20 observations) comparable to that of their wild-type siblings. q–t, Furthermore, this mutant mirrored the phenotypic defects described above for the nampta mutant and presented with a significant skeletal muscle-repair deficit upon injury (q (scale bar, 500 μm); quantification (r; n = 11 ccr5, n = 26 ccr5+/pc42, n = 9 ccr5pc42; individual data points shown; two-way ANOVA with Tukey’s multiple comparison test)), due to a wound-site MuSC-proliferation defect (s, muscle labelled by phalloidin; quantification (t; n = 12 ccr5, n = 25 ccr5+/pc42, n = 8 ccr5pc42)). e, j, o, t, The central lines, upper and lower lines within the violin plot indicate the median, upper and lower quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. h, r, Data are mean ± s.d. Two-way ANOVA with Tukey’s multiple comparison test. d, f, i, n, p, s, Scale bars, 50 μm.
Extended Data Fig. 9 Immune-cell-specific gene editing of nampta and namptb and MuSC-specific gene editing of ccr5 in larval zebrafish.
a–c, Validating the macrophage-specific nampta gene editing strategy. a, Macrophages were isolated by FACS at 3 dpf from nampta-gRNA-injected mpeg1-cas9 larvae. DNA isolated from these cells was used to generate a PCR amplicon of the region encompassing the gRNA target site. Sanger sequencing of the amplicon confirmed the presence of sequence disruptions, starting from a few base pairs upstream of the PAM site. b, Nampt protein expression in the wound site was assessed in the nampta-gRNA-injected mpeg1-cas9 larvae after needle-stab muscle injury (white dotted line). The gene-edited larvae presented observably reduced Nampt expression (magenta) within the injury zone (n = 12). Scale bar, 50 μm. c, FACS-isolated macrophages from gene-edited larvae were assayed for Nampt functionality by measuring NAD+/NADH levels using a luminescence-based assay. Macrophages isolated from control, uninjected larvae were used to measure the baseline NAD+/NADH levels for larval zebrafish macrophages. Macrophages from larvae treated with the Nampt enzymatic inhibitor GMX1778 were used to identify the NAD+/NADH levels present in macrophages in the absence of Nampt function. Furthermore, macrophages from larvae treated with nicotinamide mononucleotide (NMN), the main product of the rate-limiting enzymatic reaction of Nampt were used to establish the maximum threshold of the sensitivity of the assays. Macrophages isolated from nampta-gRNA-injected mpeg1-cas9 larvae presented with a reduction in NAD+/NADH levels, reflective of a loss-of-function of Nampt activity within macrophages present in these larvae. This assay would also detect the residual enzymatic activity of Namptb, which would not be affected by this gene-specific targeting approach. The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. One-way ANOVA with Dunnett’s post hoc test for multiple comparisons. d–g, Macrophage (yellow) dynamics in the nampta-gene-edited larvae were assayed after wounding and demonstrated no observable deviation from the control larvae (d, n = 10). nampta-gene-edited larval macrophages, located within the injury zone (white dotted line), transitioned to a dwelling state at 2 dpi (e (scale bar, 50 μm); f, quantification (n = 16 at 1 and n = 2 dpi control, n = 11 at 1 dpi and n = 14 at 2 dpi nampta gRNA-injected)). These dwelling macrophages go on to interact with pax3a+ MuSCs present in the wound (g; scale bar, 50 μm; representative of n = 20 observations). h, Macrophage-specific nampta-knockout larvae were unable to elicit the required proliferative response in the injury zone. Scale bar, 50 μm. Representative of the quantification shown in Fig. 4f. i–l, In zebrafish, Nampta and not Namptb governs the regenerative role of Nampt in muscle regeneration. i, Lateral view of namptb expression by in situ hybridization in the wound site demonstrates constitutive expression from 1 to 3 dpi in the injury site (scale bar, 100 μm). mpeg1-cas9 larvae injected with two gRNAs targeting namptb (schematic, j) demonstrated modest skeletal muscle regenerative abilities by birefringence imaging (k; scale bar, 500 μm) after needle-stab muscle injury. l, Quantification (n = 36 control, n = 34 namptb gRNA-injected). Individual data points are shown. Two-way ANOVA with Tukey’s multiple comparison test. m–o, Macrophages, not neutrophils, are the primary and functional source of Nampta in muscle regeneration. nampta was specifically knocked down in neutrophils (using the mpx-cas9 line; schematic, m), the other key innate immune cell type present in the regenerating area. Using this approach, no regeneration deficit was observed after needle-stab muscle injury, as observed by birefringence imaging (n; scale bar, 500 μm). o, Quantification (n = 14 control, n = 40 nampta gRNA-injected). Individual data points are shown. Two-way ANOVA with Tukey’s multiple comparison test. p–s, MuSC-specific ccr5 gene editing does not affect injury-responsive (white dotted line demarcates wound site boundaries) macrophage dynamics and the transition to a dwelling phenotype at 2 dpi (p (scale bar, 50 μm); q, quantification (n = 11 at 1 dpi and n = 13 at 2 dpi control, n = 9 at 1 dpi and n = 16 at 2 dpi ccr5 gRNA-injected)). Furthermore, these ccr5-gene-edited pax7b+ MuSCs (cyan) display phenotypically wild-type interactions with dwelling macrophages (yellow) located in the injury zone (r; scale bar, 50 μm; representative of n = 20 observations). However, pax7b+ cell-specific ccr5-knockout larvae are not able to maintain the required proliferative response in wound-resident MuSCs (s; scale bar, 50 μm; representative for the quantification in Fig. 4g). f, q, The continuous lines and dotted lines within the violin plot indicate the median and quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. l, o, Data are mean ± s.d. Two-way ANOVA with Tukey’s multiple comparison test. t, Summary schematic of the role of injury-responding macrophages in modulating MuSC proliferation. Following acute muscle trauma (1), macrophages and MuSCs migrate into the injury zone (2). Around half of these injury-responsive macrophages dwell in the wound site for the duration of repair (3). A subset of the dwelling mmp9+mpeg1+ macrophages start to actively interact with pax3a+ MuSCs that are present in the wound (4). These macrophages secrete Nampta, which in turn binds to Ccr5 expressed on MuSCs (4′) and activates a signalling cascade that results in MuSC proliferation (5).
Extended Data Fig. 10 NAMPT supplementation after acute skeletal muscle injury enhances proliferation, specifically in the injury zone.
a–c, NAMPT supplementation does not alter the immune cell response to injury. Needle-stab muscle-injured larvae (white dotted line) were immediately supplemented with hrNAMPT(1) (4 dpf, 0 dpi) and their immune cell dynamics were assayed at 2 dpi. The number of macrophages (Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry); yellow) and neutrophils (Tg(mpx:eGFP); cyan) were comparable to untreated controls (a; scale bar, 50 μm). b, Quantification (n = 11 for neutrophils and n = 23 for macrophages in control, n = 6 for neutrophils and n = 23 for macrophages in 57 nM NAMPT(1)). The central lines, upper and lower lines within the violin plot indicate the median, upper and lower quartiles, respectively. Two-way ANOVA with Tukey’s multiple comparison test. a, Furthermore, the autophagocytic process within the injury zone was compared by LysoTracker (magenta) and found to be similar in control and hrNAMPT(1)-supplemented larvae a. c, Quantification (n = 6 control, n = 23 57 nM hrNAMPT(1)). The central lines, upper and lower lines within the violin plot indicate the median, upper and lower quartiles, respectively. Unpaired two-tailed t-test; t27 = 1.143, P = 0.2629. d, The effect on wound site (white dotted line; skeletal muscle visualized by phalloidin staining, magenta) proliferation (EdU, white) was assayed following NAMPT and combination drug supplementation in the Tg(mpeg1:GAL4FF/UAS:nfsb-mCherry) line. Four individual examples are provided for each group and these images are representative of the quantification presented in Fig. 4h. NAMPT supplementation increases wound site proliferation in control settings and rescues the proliferative deficit that occurs after macrophage ablation. However, as NAMPT acts on the Ccr5 receptor to elicit its proliferative response, inhibiting this receptor by CVC treatment resulted in a proliferation deficit that was resistant to NAMPT-mediated rescue. Furthermore, although the CCR5 ligand, CCL8, functioned to increase proliferation in the wound site, it also increased proliferation external to the injury zone, highlighting that it lacks the wound specificity that NAMPT is able to exert. Scale bar, 50 μm. e, Proliferating satellite cells in mouse muscle injuries supplemented with NAMPT. The gating strategy to isolate the proliferating satellite cell population is shown. Cells that are CD45−, CD11b−, Ly6G−, CD31−, VCAM-1+ and PAX7+ are considered to be satellite cells. Proliferating satellite cells are additionally Ki67+. Quantification at 4 days after treatment is shown in Fig. 4m, n. f, Representative muscle regeneration cryosections stained for PAX7 (satellite cells, yellow), wheat-germ agglutinin (WGA, magenta) and nuclei (DAPI, blue) for tissues collected 6 days after treatment. Scale bar, 50 μm. g, h, Representative tissue sections stained with haematoxylin and eosin (g; scale bar, 100 μm) used to quantify centrally nucleated muscle fibres in the injury zone at 6 days after treatment (n = 6 mice per group) (h). The central lines, upper and lower lines within the violin plot indicate the median, upper and lower quartiles, respectively. Unpaired two-tailed t-test; t10 = 2.770, P = 0.0198. i, j, Immune cell profile after mouse muscle injury supplemented with NAMPT. i, Gating strategy to analyse wound-associated immune cell subsets in muscle injury. The gating strategy is shown for the neutrophil, macrophage and T cell subset panels. j, Percentages were calculated over total live cells in the collected tissue (n = 5 mice for day 6, n = 6 mice for day 8). Data are mean ± s.e.m. Two-way ANOVA with Bonferroni post hoc test for pair-wise comparisons. k, l, Angiogenesis following mouse VML injury treated with NAMPT. k, Muscle injuries were treated with NAMPT (0.5 μg) delivered in fibrin or fibrin-only control and tissues were collected at 6 days after treatment. Representative regenerated muscle cryosections were stained for CD31 (endothelial cells; yellow) and laminin (magenta). Scale bar, 50 μm. l, Quantification of the CD31+ area (n = 6 mice per group). Data are mean ± s.e.m. Unpaired two-tailed t-test; t8 = 3.275, P = 0.00113.
Supplementary information
Supplementary Table 1
Top 500 most expressed genes in all sequenced macrophages. Excel worksheet listing the most expressed genes common to all macrophages analysed. Biological process enrichment analyses on these top 500 genes describe the overall nature of the isolated and sequenced cells.
Supplementary Table 2
UMAP cluster composition. Excel worksheet documenting the composition of the 7 clusters identified by UMAP scatter plot. The macrophage frequency, isolation time point and percentage make up in relation to total number of analysed macrophages is presented.
Supplementary Table 3
Differentially expressed genes and biological enrichment analysis for macrophage subsets identified by UMAP scatter plot. Excel workbook of the differentially expressed genes for each individual cluster (8 worksheets). Biological processes enriched in each individual cluster are presented in the form of a bar graph of the top non-redundant enrichment terms and their relationships in the form of network visualisation.
Video 1
: Following injury, macrophages in the vicinity of the wound site mount an immediate wound migratory response. The video begins by showing a 4 dpf zebrafish larva’s unperturbed skeletal muscle (Tg(actc1b:GFP), magenta) with patrolling muscle resident macrophages (Tg(mpeg1:GAL4FF/UAS:NfsB-mCherry), yellow). Three frames into the video, the larva is subject to an acute laser ablation injury targeting the ventral myotome. Following injury, a subset of macrophages near the wound site actively migrate into the injury site (see stills in Fig. 1a-b’’). Migrating macrophages project large cytoplasmic protrusions on route to the wound site. Macrophages migrate along the mid line as well as over both the dorsal and ventral myotomes. Each migrating macrophage path is shown in individually tracked colours (see stills Extended Data Fig. 1a). Maximum intensity projections of confocal z-stacks imaged at each time point. Larval view: lateral, anterior to left. Video length: 1.02 h, 4 frames/min. Video representative of n=4 observations.
Video 2
: A subset of injury-responding macrophages ‘dwell’ in the wound site for the duration of the regenerative process. The video shows two clips presented side-by-side, extracted from a single continuous 24 h confocal time-lapse movie of macrophages (Tg(mpeg1:GAL4FF/UAS:NfsB-mCherry), yellow) responding to skeletal muscle injury (Tg(actc1b:GFP), magenta). The left clip illustrates the early macrophage response to injury from 2 to 9 hours following injury. Macrophages migrate into the wound site extending long cytoplasmic protrusions, resulting in an initial increase in the wound-located macrophages. These transient macrophages remain in the wound site up to 9.80 h (see stills in Fig. 1c). The right clip shows the late injury-present dwelling macrophages, which are composed of 50% of the original macrophages that migrated into the wound site (see stills in Fig. 1d). Dwelling macrophages remain in the wound site until injury resolution is complete (see macrophage injury residing time quantification in Fig. 1e). Maximum intensity projections of confocal z-stacks imaged at each time point. Larval view: lateral, anterior to left, laser ablation targeted to ventral myotome. Video length: left clip 7 h, right clip 6 h, 6 frames/h. Video representative of n=8 observations.
Video 3
: Transient and dwelling macrophages are phenotypically distinct. This video illustrates the morphological differences between early wound-located transient macrophages and late wound remaining, dwelling, macrophages. The video is presented in a tabular format with the left column highlighting a transient macrophage and the right a dwelling macrophage. The bottom row shows the fluorescent pre-processed image of the macrophage (Tg(mpeg1:GAL4FF/UAS:NfsB-mCherry), yellow) while the top row shows the processed surface rendered image. The surface rendering is pseudocoloured with a sphericity scale where a perfect sphere would have a sphericity value of 1. Transient macrophages have lower sphericity values while dwelling macrophages appear more rounded having significantly higher sphericity values (see graph in Extended Data Fig 1d).
Video 4
: Macrophages that go onto dwell are initially located in close proximity to the wound site at the point of injury. Light-sheet microscopy time-lapse imaging video of a whole 4 dpf Tg(mpeg1:GAL4FF/UAS:NfsB-mCherry)/Tg(actc1b:GFP) (grey) zebrafish larvae, documenting the macrophage response to laser ablation injury from 0.17 h post laser ablation injury until 19.04 h post injury. Retrospective tracking identifies macrophages that go on to dwell within the wound site. The video was processed such that dwelling macrophages are highlighted in yellow and their migration into the injury site (blue) can be observed over the imaged time course. The migratory paths into the injury zone of macrophages that do not adopt a dwelling phenotype (transient macrophages) are also visualised (multi-colour paths). Maximum intensity projection of confocal z-stacks imaged at each time point. Lateral view: anterior to the top, dorsal to the right. Video length: 19 h, 0.67 frames/min. Video representative of n=4 observations.
Video 5
: Timeline of nitroreductase-mediated macrophage ablation in larval zebrafish. Video consists of two clips presented one after the other, the first clip shows the head region and the second the tail region of a 4 dpf compound Tg(mpeg1:GAL4FF/UAS:NfsB-mCherry);Tg(actc1b:GFP) (yellow macrophages, magenta skeletal muscle) larval zebrafish. 5 mM metronidazole (Mtz) is added to the Ringer’s solution in which the anesthetised larvae are maintained for the duration of imaging. Following Mtz addition, larvae are continuously imaged to visualise the temporal response of macrophage ablation (see stills in Extended Data Fig. 1k). Maximum intensity projections of confocal z-stacks imaged at each time point. Lateral view: anterior to the top, dorsal to the right. Each clip length: 13.5 h, 0.67 frames/h. Video representative of n=8 observations.
Video 6
: Once macrophages transition to a dwelling state, they actively start interacting with pax3a+ muscle stem cells present in the wound site. Video consists of two clips extracted from two time-lapse movies taken in immediate succession of the same larvae revealing macrophages (Tg(mpeg1:GAL4FF/UAS:NfsB-mCherry), yellow) and pax3a+ muscle stem/progenitor cells (TgBAC(pax3a:GFP), cyan) responding to muscle injury (muscle injury shown in differential contrast). The first clip is taken immediately following injury and reveals that both macrophages and pax3a+ cells migrate into the injury site independently of each other. pax3a+ cells originate from the undamaged regions of the ablated myotome as well as from adjacent myotomes and migrate to line the edge of the wound site (pax3a+ cells that go onto line the wound edge are indicated by magenta arrowheads) (see stills in Extended Data Fig. 3a-c). At the beginning of the second clip (10.25 hpi) macrophages have transitioned to a dwelling state. Dwelling macrophages initiate intense and continuous interactions with pax3a+ cells lining the wound edge (see stills in Extended Data Fig. 3d). A surface rendered image of these interactions is shown to further highlight the specific nature of these associations. Maximum intensity projections of confocal z-stacks imaged at each time point. Larval view: lateral, anterior to left, laser ablation to ventral myotome. Video length: 1st clip 4 h, (1.54 frames/min), 2nd clip 3.75 h (0.5 frames/min). Video representative of n=5 observations.
Video 7
: Following dwelling macrophage interactions, associated myogenic cells undergo cell division. Video documents an example of an injury-present dwelling macrophage (Tg(mpeg1:GAL4FF/UAS:NfsB-mCherry), yellow) interacting with a pax3a+ cell (TgBAC(pax3a:GFP), cyan) which consequently undergoes cell division (see stills in Fig. 2e). The video shows, in high resolution, the spatial control macrophages exert on specific myogenic cells present in the wound site. Maximum intensity projections of confocal z-stacks imaged at each time point. Video commences at 20 hpi. Larval view: lateral, anterior to left, laser ablation to ventral myotome. Video length: 0.65 h, 14.75 frames/min. Video representative of n=10 time-lapse videos documenting n=26 cell divisions.
Video 8
: Further documentation of dwelling macrophage associated pax3a+ cells undergoing cell division. Video showing dwelling macrophage-myogenic cell contacts leading to the associated myogenic cell undergoing cell division in 3 independent larval laser ablation injuries. Maximum intensity projections of confocal z-stacks imaged at each time point. White arrowheads point to cells undergoing division. Larval view: lateral, anterior to left. Clip 1 (see stills in Extended Data Fig. 3k, injury 1), video commences at 18.5 hpi, video length: 0.4 h, 15 frames/min. Clip 2 (see stills in Extended Data Fig. 3k, injury 2), video commences at 17 hpi, video length: 0.32 h, 7.25 frames/min. Clip 3 (see stills in Extended Data Fig. 3k, Injury 3), video commences at 26 hpi, video length: 5.91 h, 1.4 frames/min. Video’s representative of n=10 time-lapse videos documenting n=26 cell divisions.
Video 9
: Dwelling macrophages interact with pax3a+/met+ muscle stem cells in the wound site. Video documents dwelling macrophage (Tg(mpeg1:GAL4FF/UAS:NfsB-mcherry), yellow)-muscle stem cell (TgBAC(pax3a:GFP);Tg(met:mCherry-2A-KALTA4/UAS:NfsB-mcherry), cyan and yellow double labelled cells) associations within the injury site (white line demarcates boundaries of the injury, video starts 9.33 h post-laser ablation skeletal muscle injury in a 4 dpf larval zebrafish). The video is presented such that two smaller single fluorescence channel windows (cyan channel: pax3a, yellow channel: met and mpeg, stem cells and macrophages, respectively, are distinguished based on morphology and co-labelling with pax3a for muscle stem cells) are placed above the main video of the merged channels. White arrowheads highlight two pax3a+/met+ muscle stem cells that dwelling macrophages are interacting with. During the video, both of these muscle stem cells undergo symmetric cell divisions (highlighted in single fluorescent channels by asterisks). See stills in Extended Data Fig. 3i. Maximum intensity projection of confocal z-stacks imaged at each time point. Lateral view: anterior to the top, dorsal to the right. Video length: 6.34 h, 6 frames/h. Video representative of n=6 observations.
Video 10
: Following muscle stem cell division the pre division-associated dwelling macrophage migrates away from the daughter cells. Video documents the muscle stem cells (cyan, TgBAC(pax3a:GFP)) and dwelling macrophages (yellow, Tg(mpeg1:GAL4FF/UAS:NfsB-mCherry)) response to laser ablation skeletal muscle injury from 11 hpi to 16.5 hpi in a 4 dpf zebrafish larvae. The video is presented such that two smaller single fluorescent channel windows are placed bellow the main video of the merged channel. The video captures two dwelling macrophages in the midst of each simultaneously interacting with two wound-present muscle stem cells. These interactions lead to 4 successive muscle stem cell divisions (white arrows). Following division, the dwelling macrophage on the left maintains short-lived interactions with the stem cell division-generated daughter cells prior to migrating away and taking up a myoseptal position (magenta arrowhead). Following associated-stem cell divisions, the dwelling macrophage on the right maintains cursory interactions with one pair of daughter cells, and consequently actively migrates away from the other pair of daughter cells and relocates to the vertical myosepta. See stills in Extended Data Fig. 3l. Maximum intensity projection of confocal z-stacks imaged at each time point. Lateral view: anterior to the top, dorsal to the right. Video length: 5.53 h, 15 frames/h. Video representative of n=5 observations.
Video 11
: A subset of macrophages that dwell express mmp9. Video captures the response to laser ablation skeletal muscle injury (white line demarcates boundaries of the injury) starting at 1 hpi in a 4 dpf larval zebrafish. A compound transgenic larvae is used to visualise yellow macrophages (Tg(mpeg1:GAL4FF/UAS:NfsB-mCherry) and magenta mmp9-expressing cells (TgBAC(mmp9:EGFP). At the start of the video, mmp9+ neutrophils are seen in the wound site; these cells are morphologically distinct from macrophages and have exited the wound site prior to 4.5 hpi when mmp9 expression is initiated in a subset of mpeg+ macrophages. The video also presents a single magenta channel window of the wound site superimposed towards the top of the main video. White arrowheads identify macrophages that will go onto express mmp9. When a macrophage starts expressing mmp9, the arrowhead changes colour to magenta. This moment is also highlighted in the single channel window by a magenta asterisks. See stills in Extended Data Fig. 6b. Maximum intensity projections of confocal z-stacks imaged at each time point. Lateral view: anterior to the top, dorsal to the right. Video length: 11.83 h, 3 frames/h. Video representative of n=6 observations.
Video 12
: Dwelling macrophages express high levels of NADH. Multiphoton microscopy was used to capture the intrinsic two-photon autofluorescence of NADH (magenta) in a compound transgenic line labelling both macrophages (yellow, Tg(mpeg1:GAL4FF/UAS:NfsB-mCherry)) and muscle stem/progenitor cells (cyan, TgBAC(pax3a:GFP)) following laser ablation skeletal muscle injury. The video captures stem/progenitor cell-associated dwelling macrophages expressing high levels of NADH. Maximum intensity projection of z-stacks imaged at each time point. Lateral view: anterior to the top, dorsal to the right. Video length: 24.5 min, 2 frames/min. Video representative of n=15 observations.
Video 13
: Inhibition of Ccr5 signalling does not affect dwelling macrophage-muscle stem cell interactions following laser ablation skeletal muscle injury. Video consists of two representative clips extracted from two independent time-lapse movies of Tg(mpeg1:GAL4FF/UAS:NfsB-mCherry)/TgBAC(pax3a:GFP) (macrophages in yellow and stem cells in cyan) compound transgenic larvae pre-treated with the Ccr5 inhibitor cenicriviroc (5 µM, pre-treated for 2 h prior to injury and maintained in the drug till experimental end point) and subject to laser ablation skeletal muscle injury (white line demarcates boundaries of the injury). The first clip is taken immediately following muscle injury and documents both macrophages and muscle stem cells migrating into the injury zone. Over time, a subset of the injury-responsive macrophages are seen to take on a more rounded dwelling macrophage phenotype and initiate interactions with wound site-located muscle stem cells (white arrow). At the beginning of the second clip (10 hpi) dwelling macrophages are actively interacting with wound-present muscle stem cells (white arrows). These interactions persist until the end of the time-lapse video (23 hpi). However, none of these interactions are seen to terminate with the associated muscle stem cell undergoing proliferation. Maximum intensity projections of confocal z-stacks imaged at each time point. Larval view: lateral, anterior to left, laser ablation to dorsal myotome. Video length: 1st clip 14 h, (2 frames/h), 2nd clip 13 h (3 frames/h). Video representative of n=11 observations.
Source data
Rights and permissions
About this article

Cite this article
Ratnayake, D., Nguyen, P.D., Rossello, F.J. et al. Macrophages provide a transient muscle stem cell niche via NAMPT secretion. Nature 591, 281–287 (2021). https://doi.org/10.1038/s41586-021-03199-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03199-7
- Springer Nature Limited
This article is cited by
-
Zebrafish nampt-a mutants are viable despite perturbed primitive hematopoiesis
Hereditas (2024)
-
Macrophages in tissue repair and regeneration: insights from zebrafish
Cell Regeneration (2024)
-
Skeletal muscle regeneration after extensive cryoinjury of caudal myomeres in adult zebrafish
npj Regenerative Medicine (2024)
-
fhl2b mediates extraocular muscle protection in zebrafish models of muscular dystrophies and its ectopic expression ameliorates affected body muscles
Nature Communications (2024)
-
From inflammation to bone formation: the intricate role of neutrophils in skeletal muscle injury and traumatic heterotopic ossification
Experimental & Molecular Medicine (2024)