

Abstract
Cell death in human diseases is often a consequence of disrupted cellular homeostasis. If cell death is prevented without restoring cellular homeostasis, it may lead to a persistent dysfunctional and pathological state. Although mechanisms of cell death have been thoroughly investigated1,2,3, it remains unclear how homeostasis can be restored after inhibition of cell death. Here we identify TRADD4,5,6, an adaptor protein, as a direct regulator of both cellular homeostasis and apoptosis. TRADD modulates cellular homeostasis by inhibiting K63-linked ubiquitination of beclin 1 mediated by TRAF2, cIAP1 and cIAP2, thereby reducing autophagy. TRADD deficiency inhibits RIPK1-dependent extrinsic apoptosis and proteasomal stress-induced intrinsic apoptosis. We also show that the small molecules ICCB-19 and Apt-1 bind to a pocket on the N-terminal TRAF2-binding domain of TRADD (TRADD-N), which interacts with the C-terminal domain (TRADD-C) and TRAF2 to modulate the ubiquitination of RIPK1 and beclin 1. Inhibition of TRADD by ICCB-19 or Apt-1 blocks apoptosis and restores cellular homeostasis by activating autophagy in cells with accumulated mutant tau, α-synuclein, or huntingtin. Treatment with Apt-1 restored proteostasis and inhibited cell death in a mouse model of proteinopathy induced by mutant tau(P301S). We conclude that pharmacological targeting of TRADD may represent a promising strategy for inhibiting cell death and restoring homeostasis to treat human diseases.
Similar content being viewed by others


Main
The accumulation of misfolded proteins in human diseases promotes cell death and progressive pathology by disrupting cellular homeostasis7. As the activation of autophagy is highly effective in reducing the accumulation of misfolded proteins and restoring cellular homeostasis8,9, we sought to identify targets that could be manipulated pharmacologically to restore cellular homeostasis by inducing autophagy and inhibiting apoptosis simultaneously.
Apoptosis inhibitors that activate autophagy
No known target can simultaneously modulate autophagy and cell death; therefore, we designed and conducted a multiplexed cell-based screen to identify small molecule inhibitors of apoptosis mediated by proteasomal stress and RIPK1-dependent apoptosis (RDA) that can also activate autophagy (Extended Data Fig. 1a). This quadruplexed screen of 170,000 compounds identified two active structural analogues, ICCB-17 and ICCB-19, and an inactive close analogue, ICCB-19i (Fig. 1a). An structure–activity relationship study identified an improved derivative called apostatin-1 (Apt-1). ICCB-19 and Apt-1 inhibited Velcade (bortezomib)-induced apoptosis and RDA with a half-maximal inhibitory concentration (IC50) of about 1 μM (Extended Data Fig. 1b, c). Apt-1 showed no significant off-target activity on 97 kinases in KINOMEscan profiling (Extended Data Fig. 1d).

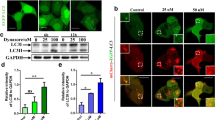
a, Chemical structures of ICCB-17, ICCB-19, ICCB-19i, and Apt-1. b, Representative images and quantification of GFP–LC3 puncta in GFP–LC3-expressing H4 cells treated as indicated. Mean ± s.e.m. of n = 5 biologically independent experiments. One-way ANOVA, post hoc Dunnett’s test. c, Scatter plot depicting changes to the interactome of beclin 1 from quantitative proteomics experiment. The targets are depicted as large red dots. See Supplementary Table 1 for details. d, Immunoprecipitation (IP)–immunoblot of MEFs. e, h, Immunoblot (IB) and quantification of LC3 II levels in MEFs of the indicated genotypes (e) or in reconstituted H4 cells (h) treated with the indicated compounds.WT, wild-type. Mean ± s.e.m. of n = 3 biologically independent experiments. f, g, K63-linked ubiquitination of beclin 1 from MEFs of the indicated genotypes treated with Apt-1. i, As in f, using reconstituted H4 cells. Compounds used at 10 μM for 6 h. For gel source data and Apt-1 synthesis, see Supplementary Figs. 1, 2.
ICCB-19 and Apt-1 effectively induced autophagy10 (Fig. 1b, Extended Data Fig. 1e–i) and the degradation of long-lived proteins11 (Extended Data Fig. 1j). The caspase inhibitor zVAD.fmk had no effect on autophagy (Extended Data Fig. 1k). ICCB-19 and Apt-1 had no effect on mTOR (Extended Data Fig. 1l). Rather, treatment of cells with ICCB-19 or Apt-1 increased levels of DsRed-FYVE dots, an indicator for phosphatidylinositol-3-phosphate (PtdIns3P), which is a critical autophagy lipid messenger generated by the ATG14L–beclin 1–VPS34–VPS15 class III phosphoinositide 3-kinase (PI3K-III) complex12 (Extended Data Fig. 1m), suggesting that this complex was activated. Treatment with ICCB-19 or Apt-1, but not ICCB-19i, increased the lipid kinase activity of VPS34 (Extended Data Fig. 1n). Treatment with Apt-1 increased the interaction of beclin 1 with TRAF2, cIAP1, and ATG14L, an important activator of the VPS34 complex13,14, but not with VPS34 (Fig. 1c, d, Extended Data Fig. 2a, b). Autophagy and degradation of long-lived proteins induced by ICCB-19 or Apt-1 were substantially reduced by genetic or pharmacological inhibition of cIAP1 and cIAP2 (here referred to as cIAP1/2) or TRAF2, and this effect was reversed by reconstitution of cIAP1 or TRAF2, respectively (Fig. 1e, Extended Data Fig. 2c–h). Thus, autophagy induced by ICCB-19 or Apt-1 involves the E3 ubiquitin ligases cIAP1 and cIAP2 and the adaptor TRAF2, which are not required for autophagy induced by inhibition of TORC1 or starvation (Extended Data Fig. 2i–l). Treatment with ICCB-19 or Apt-1, but not ICCB-19i, markedly enhanced K63-linked ubiquitination of beclin 1 (Extended Data Fig. 2m). Apt-1-induced K63-linked ubiquitination of beclin 1 was reduced by cIAP1/2 or TRAF2 deficiency and restored by reconstitution of cIAP1 or TRAF2, respectively (Fig. 1f, g, Extended Data Fig. 2n–p).
Mass spectrometry analysis identified conserved K183 and K204 residues in beclin 1 that may be modified by cIAP1 (Extended Data Fig. 3a–c). The double K183R/K204R mutant, but not the K183R or K204R single mutants, reduced K63-linked ubiquitination of beclin 1 mediated by cIAP1 (Extended Data Fig. 3d). Reconstitution of the K183R/K204R double mutant, but not of either single mutant, in beclin 1-knockdown cells blocked the induction of autophagy (Fig. 1h, Extended Data Fig. 3e–g) and reduced K63-linked ubiquitination of beclin 1 induced by Apt-1 (Fig. 1i). Thus, Apt-1 and ICCB-19 promote autophagy via K63-linked ubiquitination of beclin 1 mediated by cIAP1/2 and TRAF2.
ICCB-19 and Apt-1 indirectly inhibit RIPK1
The cleavage of caspase-3 induced by Velcade was blocked by ICCB-19 or Apt-1, but not by necrostatin-1s (Nec-1s)15 (Extended Data Fig. 4a, b). Like Nec-1s, ICCB-19 and Apt-1 protected against multiple RDA models (Extended Data Fig. 4c-m), but not against RIPK1-independent apoptosis (RIA; Extended Data Fig. 4n, o). ICCB-19 or Apt-1 also partially inhibited necroptosis (Extended Data Fig. 4p, q). However, unlike Nec-1s, ICCB-19 and Apt-1 cannot inhibit the activation of overexpressed RIPK1 (Extended Data Fig. 4r). Thus, ICCB-19 and Apt-1 are indirect inhibitors of RIPK1 kinase activity.
ICCB-19 and Apt-1 target TRADD
Stimulation with TNF promotes the formation of a transient intracellular complex (complex I) at TNFR1, which coordinates an intricate set of ubiquitination and phosphorylation events to control the activation of RIPK1, including both K63-linked ubiquitination mediated by TRAF2 or cIAP1 and M1-linked ubiquitination mediated by the LUBAC complex16,17,18,19. Treatment with ICCB-19 reduced the rapid activation of RIPK1 in complex I induced by TNF (Fig. 2a), suggesting that the target of ICCB-19 and Apt-1 may be a component of complex I. Treatment with ICCB-19 or Apt-1 increased recruitment of TRADD, HOIP, and A20, but not RIPK1, to complex I; it also increased M1-linked ubiquitination, but decreased K63-linked ubiquitination, of RIPK1 (Fig. 2b, c, Extended Data Fig. 5a–c).

a, c, Ubiquitination and activation of RIPK1 in MEFs treated with the indicated compounds. b, Complex I was isolated and analysed by mass spectrometry. Red dots, significant changes in ICCB-19-treated cells. Green or black dots, no change. See Supplementary Table 3 for details. d, f, Cell survival in MEFs (d) or Jurkat cells (f) of the indicated genotypes treated with the indicated compounds. T/5z7, TNF and 5z7 together. Mean ± s.d. of n = 3 biologically independent samples. Two-way ANOVA, post hoc Bonferroni’s test. e, g, Immunoblot and quantification of CC3 (cleaved caspase-3) in MEFs (e) or of LC3 II in Jurkat cells (g) of the indicated genotypes treated with the indicated compounds. Mean ± s.e.m. of n = 3 biologically independent experiments. h, Immunoprecipitation–immunoblot of Jurkat cells of the indicated genotypes. i, K63-linked ubiquitination of beclin 1 from control and reconstituted Tradd−/− MEFs treated with Apt-1. Compounds used at 10 μM for 6 h or as indicated. For gel source data, see Supplementary Fig. 1.
TRADD is a 34-kDa adaptor protein with an N-terminal TRAF2-binding domain and a C-terminal death domain. It is the first protein to be recruited to complex I4,5,6, and therefore may be the target of ICCB-19 and Apt-1. Tradd−/− mouse embryonic fibroblasts (MEFs) are known to be resistant to RDA20. Notably, Nec-1s, but not ICCB-19 or Apt-1, protects Tradd−/− MEFs further against RDA (Fig. 2d, e). Thus, TRADD is required for protection against RDA by ICCB-19 or Apt-1, but not Nec-1s.
In complex I, TRADD-N interacts with TRAF2 and cIAP1/2 to promote K63-linked ubiquitination of RIPK14,6,21,22. The protective effects of ICCB-19 and Apt-1, but not Nec-1s, against RDA were reduced by genetic or pharmacological inhibition of cIAP1/2 and restored by cIAP1 reconstitution (Extended Data Fig. 5d–i). Thus, cIAP1/2-mediated ubiquitination is involved in the suppression of RIPK1 activation by ICCB-19 or Apt-1. Tradd-knockout Jurkat cells were effectively protected against Velcade-induced apoptosis, and this protection could not be further enhanced by ICCB-19 or Apt-1 (Fig. 2f). TRADD, but not FADD or RIPK1, was required for this protection (Extended Data Fig. 5j, k).
In Tradd−/− MEFs, basal autophagic flux and long-lived protein degradation was increased relative to wild-type MEFs, and could not be further enhanced by ICCB-19 or Apt-1 (Extended Data Fig. 6a–c). Tradd-knockout Jurkat cells also displayed increased levels of LC3 II, which were not altered by Apt-1 (Fig. 2g). Thus, TRADD is involved in inhibiting autophagy and is required for the induction of autophagy by ICCB-19 or Apt-1. Inhibition of autophagy by Spautin-123, Atg5 knockout, or blocking lysosomal degradation prevented the activation of autophagy and protection against proteasomal stress-induced apoptosis in Tradd knockout or Apt-1-treated cells, but had no effect on RDA (Fig. 2g, Extended Data Fig. 6d–g).
Under homeostatic conditions, endogenous beclin 1 bound to cIAP1/2 and TRAF2, and binding was enhanced by TRADD deficiency (Fig. 2h). Furthermore, K63-linked ubiquitination of Beclin 1 was enhanced in Tradd−/− MEFs; this was not affected by Apt-1, but could be suppressed by adding back TRADD, which was overcome by Apt-1 treatment (Fig. 2i, Extended Data Fig. 6h). These results suggest that ICCB-19 and Apt-1 release cIAP1/2 or TRAF2 from TRADD and thereby promote the K63-linked ubiquitination of beclin 1 mediated by cIAP1/2 or TRAF2.
ICCB-19 and Apt-1 reduce inflammatory responses
Tradd−/− mice are normal and are highly resistant to multiple systemic inflammatory responses5,24,25,26. Treatment with ICCB-19 or Apt-1 minimally affected early events in the NF-κB pathway, but reduced expression of the TNF-induced inflammatory target gene products, NOS and COXII27, and of inflammatory cytokines in cells stimulated with pathogen-associated molecular patterns, including interferon γ (IFNγ), lipopolysaccharide (LPS), Pam3CSK4 (a synthetic bacterial lipopeptide), and muramyl dipeptide (MDP) (Extended Data Fig. 7a–m). Wild-type mice treated with Apt-1 showed increased survival following intravenous delivery of TNF, a mouse model of systemic inflammation (Extended Data Fig. 7n, o).
ICCB-19 and Apt-1 restore proteome homeostasis
We next investigated whether ICCB-19 and Apt-1 could restore cellular homeostasis and promote the degradation of misfolded proteins. Treatment with ICCB-19 or Apt-1 reduced protein accumulation and cell death in the presence of Htt-103Q, α-synuclein (wild-type, E46K, or A53T), or tau (wild-type or P301L)28 (Extended Data Fig. 8a–f). PS19 mice, which express mutant human tau(P301S), develop progressive neuronal loss and microgliosis associated with neurofibrillary tangle (NFT)-like tau pathology29. Treatment with Apt-1 for 3 hours induced autophagy and reduced the accumulation of mutant tau in cultured brain slices from PS19 mice, and this effect was blocked by lysosomal inhibition (Extended Data Fig. 8g, h). Thus, Apt-1 can rapidly promote the degradation of accumulated mutant tau.
We tested whether Apt-1 could restore proteostasis and reduce cell death in a mutant tau fibril (pff) transmission model30. Hippocampal delivery of Apt-1 (Extended Data Fig. 8i) was able to induce autophagy and reduce the levels of tau in these mice (Fig. 3a). Treatment with Apt-1 reduced NFT-like pathogenesis induced by pffs, including a reduction of hyperphosphorylated tau+ neurons and pathological misfolded MC1+ tau31 (Fig. 3b, c). Mice injected with tau pffs showed substantial increases in activated p-RIPK1+ and apoptotic TUNEL+ cells in the CA1 hippocampus, which were inhibited by Apt-1 (Fig. 3d, e). These results suggest that RIPK1 is activated in this tauopathy model and that treatment with Apt-1 can effectively restore cellular homeostasis and block apoptosis driven by pathological tau transmission in vivo. We also tested whether Apt-1 could rescue proteostasis after NFT-like pathology had formed in PS19 mice. Treatment with Apt-1 for one week reduced pre-formed NFT-like pathology in PS19 mice that had been injected with tau pff 3 weeks earlier (Extended Data Fig. 8j, k).

a–e, Effects of Apt-1 on the pathological tangle-like tau aggregates in the hippocampus CA1 region of PS19 mice injected with tau pffs. Immunoblots of tau levels (a), immunostaining of phospho-tau (AT8) (b), p-RIPK1(S166) (d), TUNEL (e), and immunohistochemistry (IHC) for tau in pathological conformation (MC1) (c) in hippocampus CA1 region. Each dot represents the mean from an individual mouse. Mean ± s.e.m. (n = 3). Two-tailed t-test. For gel source data, see Supplementary Fig. 1.
ICCB-19 and Apt-1 interact with TRADD-N
We found that separately expressed TRADD-N (amino acids (aa) 1–197) and TRADD-C (aa 198–312) interacted, and that Apt-1 reduced this interaction (Extended Data Fig. 9a). Thus, ICCB-19 and Apt-1 might affect a previously unknown interaction of TRADD-N with TRADD-C. NanoBiT-based interaction between LgBiT-TRADD-N and TRADD-C-SmBiT was dose-dependently reduced by ICCB-19 or Apt-1, but not by ICCB-19i (Extended Data Fig. 9b–f). Direct binding of TRADD-N and TRAF221 was also reduced by Apt-1 in NanoBiT and fluorescence resonance energy transfer (FRET) systems (Fig. 4a, Extended Data Fig. 9g–k). Thus, TRADD-N and TRADD-C normally interact with each other; in cells stimulated by TNF, TRADD is recruited to TNFR1 by the binding of its death domain with the death domain of TNFR1, which frees TRADD-N to interact with TRAF2 and organize complex I recruitment. Notably, this model suggests that ICCB-19 and Apt-1 might bind to a conserved interface on TRADD-N that binds to both TRADD-C and TRAF2. Apt-1 increased the recruitment and retention of TRADD at TNFR1 and reduced binding of TRADD to TRAF2 or cIAP1, thus decreasing recruitment of TRAF2 or cIAP1 to complex I (Fig. 4b, Extended Data Fig. 9l, m).

a, The effect of Apt-1 on TRADD-N and TRAF2-C binding measured by NanoBiT assay (see Methods). Mean ± s.d. of n = 6 biologically independent samples. Two-tailed t-test. b, Immunoprecipitation–immunoblot and quantification (n = 1) of indicated proteins in MEFs treated with Apt-1, representative of three independent experiments. c, f, In vitro binding of Apt-1 to wild-type His–TRADD-N and indicated mutants as determined by thermal shift assay. d, Kinetic profile of binding of Apt-1 to TRADD-N from SPR analysis. e, Binding pose of Apt-1 in complex with TRADD-N generated by induced-fit docking. Top, shape and polarity of the ligand binding pocket surface (red, negatively charged; blue, positively charged). Bottom, details of the interaction. Apt-1 shown as cyan sticks, protein shown as pink cartoon with key residues highlighted in sticks. Dashed lines represent hydrogen bonds. g, Immunoblot and quantification of LC3 II in reconstituted Tradd−/− MEFs treated with Apt-1. S.E., short exposure; L.E., longer exposure. Mean ± s.e.m. of n = 3 biologically independent experiments. Two-way ANOVA, post hoc Bonferroni’s test. Compounds used at 10 μM for 6 h. For gel source data, see Supplementary Fig. 1.
The addition of ICCB-19 or Apt-1 to TRADD in a thermal shift assay32 increased its melting temperature (Tm) by 3.2 °C or 3.7 °C, respectively; ICCB-19i had no effect (Extended Data Fig. 10a–d). Incubation of Apt-1 with TRADD-N, but not with TRADD-C, also increased the Tm of TRADD-N by 3.7 °C (Fig. 4c, Extended Data Fig. 10e), suggesting that ICCB-19 and Apt-1 are likely to bind TRADD-N. Saturation transfer difference (STD)-NMR33 confirmed that ICCB-19 and Apt-1, but not ICCB-19i, bind with TRADD-N (Extended Data Fig. 10f–h). In addition, surface plasmon resonance (SPR) analysis determined that the binding equilibrium dissociation constant (KD) of ICCB-19 or Apt-1 with TRADD-N was 2.30 μM or 2.17 μM, respectively (Fig. 4d, Extended Data Fig. 10i, j).
We performed a 1H-15N heteronuclear single-quantum correlation NMR titration of Apt-1 with TRADD-N. The addition of Apt-1 to TRADD-N in solution perturbed the following residues: Tyr16, Ala31, His34, Gln37, Ile72, Arg119, Gly121, Ala122, Arg124, and Arg146 (Extended Data Fig. 11a). The perturbed residues localized to β-sheets 1, 3, and 4, indicating that the interface formed by these β-sheets mediates binding of Apt-1 to TRADD-N. ICCB-19, but not ICCB-19i, showed similar binding to TRADD-N (Extended Data Fig. 11b, c).
NMR-based structures for ICCB-19 and Apt-1 bound to TRADD-N generated computationally showed that Apt-1 and ICCB-19 bound with TRADD-N in similar conformations (Fig. 4e, Extended Data Fig. 11d). The binding site for ICCB-19 or Apt-1 is in the interface between TRADD-N and TRAF2-C21. The TRADD-N residues Tyr16, Phe18, Ile72, and Arg119 form a hydrophobic pocket that can bind the substituted cycloheptane of ICCB-19 or Apt-1. The amide group of Gly121 in TRADD-N forms a hydrogen bond with the carbonyl oxygen of ICCB-19 or Apt-1. The TRADD-N residues Gln142 and Asp145 form two hydrogen bonds with the heteroatoms of a 4,5-dihydro-1H-imidazole group in ICCB-19. In addition, the phenyl group of Apt-1 forms π–π stacking with Tyr16. Binding of Apt-1 with recombinant TRADD-N mutants (Y16A, F18A, I72A, R119A, and G121A) was reduced relative to binding with wild-type TRADD-N (Fig. 4f, Extended Data Fig. 11e).
Mutations to Tyr16, Phe18, or Ile72 of TRADD-N21 reduced its interaction with TRAF2-C (Extended Data Fig. 11f). TRADD-N(G121A) and TRADD-N(A122T), but not TRADD-N(R119A), showed reduced interactions with TRAF2-C. Expression of mutant TRADD (Y16A, F18A, I72A, G121A, or A122T) in Tradd-deficient cells did not suppress autophagy (Fig. 4g). The TRADD(R119A) mutant suppressed autophagy, and this effect was not overcome by Apt-1 (Fig. 4g). Consistent with a predicted H-bond between Gly121 and ICCB-19 or Apt-1, Apt-1 could not further induce autophagy in TRADD(G121A)-expressing Tradd-knockout MEFs. By contrast, Apt-1 induced autophagy in Tradd-knockout cells expressing TRADD-N(A122T) (Fig. 4g). Thus, interactions between TRADD and TRAF2 can regulate autophagy. Furthermore, ICCB-19 and Apt-1 interact with TRADD-N residues that mediate TRAF2-C binding21. Disrupting the interaction between TRADD and TRAF2 may recapitulate the induction of autophagy mediated by Apt-1 or ICCB-19.
We also characterized the interaction of TRADD-N with TRADD-C in suppressing RDA. Compared to wild-type TRADD-N, the interactions of the F18A, I72A, R119A, or G121A TRADD-N mutants with TRADD-C were all compromised (Extended Data Fig. 11g). Tradd-knockout cells complemented with mutant TRADD (Y16A, F18A, I72A, R119A, G121A, or A122T) retained partial resistance to RDA (Extended Data Fig. 12a, b). Apt-1 was unable to provide additional protection in Tradd-knockout cells expressing TRADD(G121A), and bound TRADD-N(G121A) with an estimated KD of 274 μM (compared to a KD with wild-type TRADD-N of 2 μM; Extended Data Fig. 12c–e). Apt-1 partially protected against RDA in Tradd-knockout cells complemented with the Y16A, F18A, I72A, or R119A mutant version of TRADD. RDA of Tradd-knockout cells reconstituted with double-mutant TRADD(Y16A/F18A) or triple-mutant TRADD(Y16A/I72A/R119A) could not be further protected by Apt-1 (Extended Data Fig. 12f–h). Thus, the hydrophobic pocket formed by the side chain residues of Tyr16, Phe18, Ile72, and Arg119 in TRADD may collectively stabilize the interaction of TRADD-N with ICCB-19 or Apt-1.
Conclusion
TRADD mediates extrinsic apoptosis by regulating RIPK1 activation in TNFR1 signalling5,24,25,26. Our results show that TRADD also regulates cellular homeostasis and intrinsic apoptosis mediated by proteasomal stress. ICCB-19 or Apt-1 can bind with TRADD-N, disrupting its binding to both TRADD-C and TRAF2. Binding of ICCB-19 or Apt-1 releases TRAF2 or cIAP1/2 from TRADD to promote autophagy and diminish proteasomal stress by enhancing K63-linked ubiquitination of beclin 1. In RDA, ICCB-19 or Apt-1 modulates TRADD to inhibit activation of RIPK1 in complex I and to decrease availability of TRADD to complex IIa and thereby inhibit caspase activation34 (Extended Data Fig. 12i). Targeting TRADD would protect against inflammation and cell death mediated by both RIPK1-dependent and RIPK1-independent pathways, as Tradd deficiency can protect against systemic inflammation and apoptosis in Ripk1−/− mice35. In conclusion, we identify TRADD as a novel therapeutic target that is, to our knowledge, the first of its kind to both block cell death and inflammation and concurrently activate autophagy and restore cellular homeostasis.
Methods
Animals
Wild-type (cat. no. 004781) and transgenic tauP301S (catalog no. 008169) mice were from The Jackson Laboratory. All animals were maintained in a pathogen-free environment, and the animal experiments were conducted according to the protocols approved by the Harvard Medical School Institutional Animal Care and Use Committee (IACUC). Animals were assigned randomly to experimental and control groups with the same numbers; we aimed for a number of at least three animals per group to allow basic statistical analysis. Animals were age- and sex-matched in all experiments. Where possible, the investigators were blinded during data collection and analysis of animal experiments.
A multiplexed chemical screen
The primary screen was conducted in Jurkat cells treated with the proteasomal inhibitor Velcade to induce apoptosis by proteasomal stress36. Inhibition of apoptosis by the pan-caspase inhibitor zVAD.fmk was able to partially rescue cell survival, and thus was used as a positive control. About 170,000 compounds were screened to identify hits which could inhibit apoptosis induced by proteasomal stress better than could zVAD.fmk. These positive hits were counter-screened against apoptosis induced by 5-fluorouracil (5-FU) to remove generic inhibitors of DNA damage-induced apoptosis. Hits that protected against apoptosis induced by Velcade, but not 5-FU, were further evaluated for their ability to induce autophagy using H4-GFP–LC3 cells. Finally, the hits were further tested in an RDA assay of extrinsic apoptosis in RGC5 mouse retinal ganglion cells treated with TNF and 5z-7-oxozeaenol (5z7, an inhibitor of TAK1).
Stereotaxic surgery and Apt-1 delivery
Intracranial injections of synthetic tau-preformed fibrils (pffs) into PS19 mice accelerate the transmission of pathological tau tangle-like aggregates throughout the brain and thus provide a model for Alzheimer’s disease pathology and tauopathy30. Eight-week-old PS19 mice (P301S tau), which express the T34 isoform of human P301S mutant tau under the control of the mouse prion promoter, were deeply anaesthetized with isoflurane and immobilized in a stereotaxic frame. The stereotaxic injections were made using predetermined coordinates with a Hamilton syringe under aseptic conditions. All injected animals were observed during and after surgery, and an analgesic was administered after surgery. T40/PS recombinant tau pffs (2 μg/μl) were injected into both sides of hippocampus of PS19 mice (ML ±1.8 mm; AP −2.2 mm; DV 1.8 mm)30. The total volume injected was 2.5 μl per injection for all mice. The mice were then either dosed immediately or left for three weeks before Apt-1 was delivered intracerebroventricularly via an ALZET micro-osmotic pump (ALZET Micro-Osmotic Model 1002). An ALZET brain infusion kit was used for delivery into lateral ventricles (ML −1.0 mm; AP −0.5 mm; DV 2.0 mm) at a rate of 0.25 μl/h. The ALZET micro-osmotic pumps were fixed to the skulls of the mice using instant adhesive and the skin incision was closed using sutures. Apt-1 (20 mM) in the ALZET micro-osmotic pumps was renewed every two days and the Apt-1 delivery was maintained for a month in the immediate dosing groups or one week in the delayed dosing group. Apt-1 treatment resulted no apparent difference in survival or behaviour of the mice. At the end of Apt-1 dosing for one month or one week, the mice were killed and perfused with PBS, and the hippocampi from half of the brains were dissected and analysed by immunoblotting after lysis in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.1% SDS). The other half of the brains were fixed in 4% paraformaldehyde and embedded in paraffin blocks from which 5-μm-thick sections were processed for immunohistochemistry (IHC) using AT8 (specific for pathological tau phosphorylated at Ser202/Thr205, 1:10,000; Invitrogen), MC1 (specific for a pathological conformation of tau, 1:2,000)31 and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labelling).
Pharmacokinetic (DMPK) study of Apt-1
Wild-type mice (2 months old) were deeply anaesthetized with isoflurane and immobilized in a stereotaxic frame using predetermined coordinates under aseptic conditions. All animals were observed during and after surgery, and an analgesic was administered after surgery. Apt-1 (20 mM, 100 μl, release rate: 0.25 μl/h) was delivered intracerebroventricularly via an ALZET micro-osmotic pump (ALZET Model 1002) and ALZET brain infusion kits into the lateral ventricles (ML −1.0 mm; AP −0.5 mm; DV 2.0 mm). The ALZET micro-osmotic pumps were fixed to the skulls of the mice with instant adhesive and the skin incision was closed using sutures. At 1 h, 6 h and 24 h after the onset of delivery, cerebrospinal fluid (CSF) was carefully withdrawn using a Hamilton syringe from the lateral ventricles (ML −1.0 mm; AP −0.5 mm; DV 2.0 mm) for DMPK analysis. The mice were killed after 24 h and the hippocampi were dissected for DMPK analysis. The number of mice was three for each time point. The concentrations of Apt-1 in collected samples were measured by HPLC as a custom service by the Scripps Research Institute Florida.
Organotypic brain slice preparation
PS19 mice (4 months old) were anaesthetized with isoflurane before decapitation. The brain was removed and immediately immersed in ice-cold cutting solution (2.5 mM KCl, 5 mM MgCl2, 11 mM d-glucose, 238 mM sucrose, 26 mM NaHCO3, 1 mM NaH2PO4, 1 mM CaCl2). The cerebellum was trimmed off and the caudal end of the brain was glued onto the cutting table of the vibratome (LEICA VT1000 S, Germany). The brain was cut in coronal slices of 350 mm with an amplitude of 1.5 mm, a frequency of 75 Hz and a velocity of 0.1 mm/s. The slices were collected and stored in ice-cold cutting solution before floating onto semi-porous membrane inserts (Millipore, Millicell-CMLow Height Culture Plate Inserts, Schwalbach, Germany). Slices were cultured at 37 °C with 5% CO2 in a culture medium consisting of 394 ml MEM, 10% normal horse serum, 5 mg/ml penicillin, 5 mg/ml streptomycin, 2.5 ml l-glutamine, 1 mM MgSO4, 11 mM d-glucose, 238 mM sucrose, 5 mM NaHCO3, 1 mM CaCl2, 26.6 mM HEPES, 0.024 ml 25% ascorbic acid and 0.5 mg insulin. The medium was changed every other day. Slices were maintained for 14 days in vitro before treatment.
Cell culture
HEK293T cells, MEFs, Jurkat cells, SH-SY5Y cells, HT-29 cells, U-937 cells, H4 cells and HeLa cells were originally obtained from the American Type Culture Collection (ATCC). HEK293T cells, MEFs and H4 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific, cat. no. 11965) with 10% (vol/vol) fetal bovine serum (FBS; Thermo Fisher Scientific, cat. no. 10082-147). Jurkat, SH-SY5Y, and U-937 cells were cultured in RPMI 1640 (Thermo Fisher Scientific, cat. no. 11875085) with 10% (vol/vol) FBS. HT-29 cells were cultured in McCoy’s 5A medium (Thermo Fisher Scientific, cat. no. 16600082) with 10% (vol/vol) FBS. Knockout MEFs were made from the indicated knockout mice and immortalized by infection of lentivirus expressing SV40T. All of the mammalian cell lines were maintained at 37 °C with 5% CO2. The cells were tested every two months using a TransDetect PCR Mycroplasma Detection Kit (Transgen Biotech, cat. no. FM311-01) to ensure that they were mycoplasma free.
Immunoblotting
Antibodies against the following proteins were used for western blot analysis: ATG14L (CST, 96752, 1:1,000), HOIP (Abcam, ab46322, 1:1,000), VPS34 (PTG, 12452-1-AP, 1:1,000), LC3B (Sigma-Aldrich L7543, 1:1,000), SQSTM1/p62 (CST 5114, 1:1,000), α-tubulin (Sigma-Aldrich T5168, 1:10,000), beclin-1 (CST 3738, 1:1,000), phospho-RIPK1 (Ser166) (CST 31122, 1:1,000), phospho-RIPK1 (Ser166) (Biolynx, BX60008-C3, 1:500 for IF), RIPK1 (D94C12, CST 3493, 1:1,000), A20/TNFAIP3 (CST 4625, 1:1,000), TRADD (Santa Cruz sc-7868, 1:500), TNF-R1 (CST 3736, 1:1,000), β-actin (Santa Cruz sc-81178, 1:5,000), caspase-3 (CST 9662, 1:1,000), cleaved caspase-3 (CST 9661, 1:1,000), tau-5 (Invitrogen AHB0042, 1:1,000), phospho-tau AT8 (Invitrogen MN1020, 1:1,000), mouse cIAP1 (homemade, 1:1,000), TRAF2 (CST 4724, 1:1,000, Santa Cruz, sc-7346, 1:1,000), phospho-AMPKα (CST 2535, 1:1,000), AMPKα (CST 2532, 1:1,000), phospho-acetyl-CoA carboxylase (CST 3661, 1:1,000), acetyl-CoA carboxylase (CST 3662, 1:1,000), phospho-S6 (CST 2211, 1:1,000), S6 (CST 2217, 1:1,000), FADD (Abcam, ab124812, 1:1,000), phospho-MLKL (CST 91689, 1:1,000), MLKL (CST 14993, 1:1,000), phospho-RIPK3 (CST 57220, 1:1,000), RIPK3 (CST 13526, 1:1,000), caspase-8 (CST 4790, 1:1,000), PARP (CST 9542, 1:1,000), GAPDH (CST 2118, 1:1,000), phospho-IκBα (CST 9246, 1:1,000), IκBα (CST 9242, 1:1,000), phospho-IKKα/β (CST 2697, 1:1,000), IKKβ (CST 2684, 1:1,000), phospho-ERK1/2 (CST 9101, 1:1,000), ERK1/2 (CST 9102, 1:1,000), phospho-SAPK/JNK (CST 9251, 1:1,000), SAPK/JNK (CST 9252, 1:1,000), phospho-p38 MAPK (CST 9211, 1:1,000), p38 MAPK (CST 9212, 1:1,000), iNOS (CST 2982, 1:1,000), COX2 (CST 4842, 1:1,000), α-synuclein (Abcam ab6162, 1:1,000), Flag (Sigma-Aldrich F3165, 1:1,000), HA (Sigma-Aldrich H6908, 1:1,000), beclin 1 (Santa Cruz, sc-48341, 1:100 for IP, CST, 3495, 1:1,000), ATG5 (Abcam, ab108327, 1:1,000). Anti-K63 Ub and anti-M1 Ub were gifts from Dr. Vishva Dixit (Genentech, 1:1,000 for IP) and anti-MC1 was a gift from Dr. Peter Davies (Feinstein Institute for Medical Research, 1:2,000 for IHC). The signals were detected using Amersham ECL Western Blotting Detection Reagent (RPN2106, GE Healthcare), SuperSignal West Pico PLUS Chemiluminescent substrate (34580, Thermo) or SuperSignal West Femto Maximum Sensitivity substrate (34095, Thermo). The membranes were reprobed after incubation in Restore Western Blot stripping buffer (21063, Thermo).
Construction and transfection of plasmids
Full-length cDNAs for mouse and human TRADD were PCR-amplified from the plasmid library and cloned into pcDNA3.1 using Phanta Max Super-Fidelity DNA Polymerase (Vazyme Biotech Co., Ltd) with appropriate tags. Human TRADD (hTRADD) mutants were generated using MutExpress II mutagenesis kit (Vazyme Biotech Co., Ltd). For protein purification, cDNA encoding truncated hTRADD (aa 1–179, wild-type or mutant) were cloned into pET-28a plasmid for expression in Escherichia coli using ClonExpress II One Step Cloning Kit (Vazyme Biotech Co., Ltd), cDNA encoding GST-tagged hTRADD (full-length or aa 180–312) was cloned into EcoRV/NotI sites in pEBG plasmid for mammalian expression, and cDNAs encoding mVenus- and Flag-tagged TRADD-N (aa 1–179) and mCerulean- and Flag-tagged TRAF2-C (aa 310–501) were cloned into pLenti plasmid for mammalian expression. For constructs used in the NanoBiT protein–protein interaction assay, TRADD-N (aa 1–179) was cloned into the pBiT1.1-N plasmid (Lg-TRADDN), and TRAF2-C (aa 310–501) and TRADD-C (aa 180–312) were cloned into the pBiT2.1-C plasmid (TRAF2C-Sm and TRADDC-Sm, respectively) according to the manufacturer’s protocol. All plasmids were verified by DNA sequencing and the details of the plasmid sequences are available upon request. Transient transfections of H4 and SH-SY5Y cells were performed using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. In brief, cells were plated at a density of 5 × 104 cells per well in a 12-well plate and transfected with a total of 1 μg DNA per well for 24 h. Medium was changed the day after transfection.
Generation of knockdown, knockout and reconstitution lines
Cells were stably infected with shRNA against mouse Traf2 (TAGTTCGGCCTTTCCAGATAA), human BECN1 3′-UTR (CTCTGTGTTAGAGATATGA) or scramble control in the pLKO.1 lentiviral background. For CRISPR–Cas9 system-mediated gene knockout, we used guide RNAs against human TRADD (sgTRADD-1 (GCGCGCAGCTCCAGTTGCAG), sgTRADD-2 (GCGCCCCCTCGCGGTAGGCG)) or ATG5 (sgATG5-1 (GCTTCAATTGCATCCTTAGA), sgATG5-2 (GTGCTTCGAGATGTGTGGTT)) in the Lenti-CRISPR v2 lentiviral background. Viral supernatant fractions were collected 48 h after transfection. Cleared supernatant fraction was filtered through a 0.45-mm filter. Polybrene (8 mg/ml) was supplemented to viral supernatant fractions. Twenty-four hours after infection, cells stably expressing shRNA or sgRNA were obtained by selection with 5 μg/ml puromycin. H4 cells expressing the BECN1 3′-UTR shRNA were infected with lentiviral particles expressing Flag–beclin 1 (wild-type or mutant). Polyclonal populations were screened until wild-type and mutant lines were generated that had near endogenous beclin 1 reconstitution levels.
Analysis of cytotoxicity and viability
The rates of cell death were measured in triplicate or quadruplicate in a 96-well or 384-well plate using SYTOX Green Nucleic Acid Stain (Invitrogen) or ToxiLight Non-destructive Cytotoxicity BioAssay Kit (Lonza). The intensity of luminescence was determined in an EnSpire Multimode Plate Reader (PerkinElmer). Data were collected using PerkinElmer EnVision Manager Version 1.13 software. Cytotoxicity was expressed as percentages of cell death per well after deducting the background signal in non-induced cells and compared to that of the maximal cell death with 100% Lysis Reagent. The rates of cell viability were determined using CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s protocol and the results are expressed as percentages of luminescence intensity per well after deducting the background signal in blank wells and compared to that of the viability in the non-treated wells. Concentration of drugs used for inducing or inhibiting cell death: mTNF, 1 ng/ml; 5Z-7-oxozeaenol, 0.5 μM; Velcade, 50 nM; Apt-1/ICCB-19/ICCB-19i/Nec-1s, 10 μM.
Caspase-8 activity assay
Caspase-Glo 8 assay (Promega) was used to detect the activity of caspase-8 in cells and in vitro by following the manufacturer’s protocol. In brief, 2 × 105 cells (MEFs) were plated in 6-well plates and treated as indicated in 2 ml for the indicated times. After treatment, medium was removed, and 300 μl 0.5% NP-40 lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl and 0.5% NP-40) was added to each well. Cells were scraped and lysates were left on ice for 5 min. Ten microlitres of lysate per condition was transferred into a 384-well plate and 90 μl of Caspase-Glo 8 reagent supplemented with MG-132 (30 μM) was added to each well. Plates were wrapped in foil and gently mixed using a plate shaker at 300–500 rpm for 30 s. Reactions were allowed to proceed by incubation at room temperature for 1 h. Caspase-8 activity was read on a luminometer.
Complex-I/II purification
Cells were seeded in 15-cm dishes and treated as indicated with Flag–TNF (50 μg/ml). To terminate treatment, medium was removed and plates were washed with 50 ml of ice-cold PBS. Plates were frozen at −80 °C until all time points were acquired. Plates were thawed on ice and cells were lysed in 0.5% NP-40 lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl and 0.5% NP-40) supplemented with protease inhibitors and N-ethylmaleimide (2.5 mg/ml). Cell lysates were rotated at 4 °C for 30 min then clarified at 4 °C at 14,000 rpm for 30 min. Proteins were immunoprecipitated from cleared protein lysates with 20 μl anti-Flag M2 beads (Sigma) with rotation overnight at 4 °C. Four washes in 0.5% NP-40 buffer with N-ethylmaleimide were performed, and samples eluted by boiling in 50 μl 1 × SDS loading buffer. For complex-II purification, cells were seeded in 10-cm dishes and treated as indicated using medium containing TNF (10 ng/ml) and zVAD (20 μM). Cells were lysed on ice in 0.5% NP-40 lysis buffer. Cell lysates were rotated at 4 °C for 30 min then clarified at 4 °C at 14,000 rpm for 10 min. Twenty microlitres of protein G sepharose (Sigma), after pre-blocking for 1 h with lysis buffer containing 1% BSA, was incubated with FADD antibody (1.5 mg antibody per mg protein lysate) and the mixture was incubated in rotation with cleared protein lysates 4 h at 4 °C. The samples were then washed four times in lysis buffer and eluted by boiling in 50 μl 1 × SDS loading buffer. Concentration of compounds used in complex-I/II purification: 10 μM (Apt-1 or ICCB-19).
Long-lived protein degradation assay
H4 cells were cultured with l-[3,4,5-3H(N)]-leucine (0.1 μCi/ml) (PerkinElmer Life Sciences) for 24 h and chased in medium with nonradioactive leucine for 18 h. Then the medium was changed and cells were incubated for an additional 6 h along with different compounds (10 μM Apt-1, 10 μM ICCB-19, 10 μM ICCB-19i, 1 μM rapamycin). The media were recovered and treated with 10% trichloroacetic acid to separate trichloroacetic acid-soluble (amino acids) and trichloroacetic acid-insoluble (proteins) fractions. The cells were completely dissolved with 1 N NaOH. Radioactivity was measured with a liquid scintillation analyser (PerkinElmer). Long-lived protein degradation was calculated by dividing trichloroacetic acid-soluble radioactivity in the medium by total radioactivity detected in the cells and medium. The values were expressed as fold-change from the value obtained in control cells.
NanoBiT protein–protein interaction (PPI) assay
The Nano-Glo Live Cell assay kit (Promega) was used as follows: HEK293T cells were seeded at 7.5 × 103 cells per well in a white, clear-bottom 96-well plate 12 h before transfection (10 ng LgBiT-fused construct (Lg-TRADDN) and 10 ng SmBiT-fused construct (TRADDC-Sm or TRAF2C-Sm), HaloTag-Sm was used as a negative control according to the manufacturer’s protocol). After 24 h incubation, medium was removed and replaced with 100 μl Opti-MEM medium for 1 h at 37 °C. The Nano-Glo reagent was prepared according to the manufacturer’s instructions and added to each well immediately before the luminescence reading was taken. Luminescence was measured at 1-min intervals for 10 min on a plate reader and reported as relative light units (RLU). For quantitative comparison of LgBiT–SmBiT interactions, the peak values at the 2–3 min time point were used. Concentration of compounds used in NanoBiT assay: 10 μM (Apt-1, ICCB-19 or ICCB-19i).
Protein expression and purification
Recombinant wild-type and mutant His–TRADD-N (aa 1–179) protein fragments were expressed in BL21 (DE3) E. coli after induction with 0.5 mM IPTG overnight at 16 °C. 15N-labelled TRADD-N domain protein was purified from E. coli grown at 16 °C in minimal medium. Bacteria were harvested and disrupted using a high-pressure homogenizer and purified with Ni2+ affinity resin (GE Healthcare). All proteins were further purified by size exclusion chromatography on a Superdex 75 column (GE Healthcare) in a buffer containing 20 mM imidazole (pH 6.6), 200 mM NaCl, 20 mM DTT and 0.05% NaN3. All NMR samples were in the same buffer at concentrations between 0.2 and 0.4 mM with 90% H2O/10% D2O. Proteins were exchanged into assay buffer (120 mM NaCl, 20 mM NaH2PO4/Na2HPO4, pH 7.4) by dialysis for thermal shift assay.
NMR spectroscopy
The 15N-HSQC spectra of 15N-labelled TRADD-N domain protein were acquired in a buffer containing 20 mM imidazole (pH 6.6), 200 mM NaCl, 20 mM DTT and 0.05% NaN3 at 25 °C, on a 600-MHz Bruker Avance II spectrometer using a Prodigy cryoprobe. For the spectra of 0.2 mM TRADD-N with 0.5 mM Apt-1 ligand, the data were collected with eight scans for each free induction decay (FID), 512 complex points in the direct 1H dimension and 128 complex points in the indirect 15N dimension. For the spectra of 0.4 mM TRADD-N with 0.5 mM ICCB-19, or 0.5 mM ICCB-19i, the data were collected with two scans for each FID, 512 complex points in the direct 1H dimension and 128 complex points in the indirect 15N dimension. The spectra were processed and analysed using Bruker Topspin 3.6.1 software.
Saturation transfer difference-nuclear magnetic resonance (STD-NMR) spectroscopy33 was used to detect the interaction of compounds with TRADD-N. The STD spectra were acquired on a 400 MHz spectrometer (ICCB-19 and Apt-1) or 800 MHz spectrometer (ICCB-19i). The samples for STD NMR were prepared as 13 μM TRADD-N or TRADD-N(G121A) with 1 mM Apt-1, ICCB-19 or ICCB-19i in 0.5 ml PBS in D2O (10%). The on-resonance irradiation was performed at a chemical shift of −0.5 ppm, whereas the off-resonance irradiation was conducted at 37 ppm. The spectra were acquired using the following parameters: spectral window of 6.4 kHz, number of scans at 320, acquisition time of 2 s, and repetition time of 3 s. The decrease in signal intensity in the STD spectrum, resulting from the transfer of saturation from the protein to the ligand, was evaluated by subtracting the on-resonance spectrum from the off-resonance spectrum.
Thermostability shift assay
To determine stability, purified proteins were made to a final concentration of 1 μg/μl. SYPRO Orange dye was added to the protein to make a final concentration of 2×. Compounds were added to the mixture with a final concentration of 250 μM or as indicated and incubated at 4 °C for 1 h. The experiments were performed in 384-well plates specific for the real-time PCR instrument with a total volume of 20 μl/well. The assay plate was covered with a sheet of optically clear adhesive to seal each well. The assay plate was centrifuged at 800g for 2 min at 25 °C to collect solutions in the bottom of the well and remove bubbles. The assay plate was placed into the Applied Biosystems QuantStudio 6 Real-Time PCR System. The reaction was run from 25 °C, ramping up in increments of 0.05 °C/s to a final temperature of 95 °C with fluorescence detection throughout the experiment to generate a dataset. The melting temperature of the protein (Tm) was determined by performing nonlinear fitting of the data set to a Boltzmann sigmoidal curve in GraphPad Prism with the following equation: Y = bottom + (top − bottom)/(1 + exp(Tm − X/slope)), where Y is fluorescence emission in arbitrary units; X is temperature; bottom is the baseline fluorescence at low temperature; top is the maximal fluorescence at the top of the dataset; and slope describes the steepness of the curve, with larger values denoting shallower curves.
Surface plasmon resonance
The binding affinity between ICCB-19 or Apt-1 and TRADD-N was analysed at 25 °C on a BIAcore T200 machine with CM5 chips (GE Healthcare). PBS-P buffer (GE Healthcare) was used for all measurements. For SPR measurements, Flag-tagged TRADD-N protein was purified from HEK293T cells with anti-Flag affinity gel and eluted using 3 × Flag peptide. The protein was further purified by size exclusion chromatography on a Superdex 75 column (GE Healthcare) in a buffer containing 20 mM imidazole (pH 6.6), 200 mM NaCl, 20 mM DTT. The protein was dialysed into PBS and diluted to a final concentration of 40 μg/ml in NaOAc buffer (pH 4.5) before immobilization on a CM5 chip. About 5,000 response units of protein were immobilized on the chip with a running buffer composed of PBS-P. A reference was used to normalize the response unit (RU) values of protein. A series of compound concentrations ranging from 0.3125 to 10 μM was tested at 30 μl/min flow rate. The contact time was 100 s and the dissociation time was 120 s. When the data collection was finished in each cycle, the sensor surface was regenerated with PBS-P buffer. DMSO solvent correction was performed following the BIAcore T200 guide. Binding curves were displayed, and equilibrium binding constants (KD) for the interaction were determined using the steady-state affinity method incorporated in the BIAEVALUATION 4.1 software (GE Healthcare).
In vitro FRET assay
mVenus- and Flag-tagged TRADD-N (mVenus–TRADDN–Flag) and mCerulean- and Flag-tagged TRAF2C (Flag–TRAF2C–mCerulean) were expressed in HEK293T cells for 48 h. Then cells were lysed in NP-40 buffer followed by immunoprecipitation using anti-Flag affinity gel. The proteins were eluted with 5 mg/ml 3 × Flag peptide and exchanged into assay buffer (120 mM NaCl, 20 mM NaH2PO4/Na2HPO4, pH 7.4) by dialysis for FRET assay. The proteins were added into Corning black 96-well microtitre plates in triplicate at a final concentration of 1 μM. Apt-1 was incubated with the proteins for 1 h before measurement. Measurements were performed on a fluorescent plate reader (Victor3, 1420 Multilabel counter, Perkin Elmer). The following filter set was used: mCerulean filter set (excitation: 430/15 nm, emission: 460/20 nm); mVenus filter set (excitation: 485/15, emission 535/15); FRET filter set (430/15 nm, emission 450–600 nm).
Mass spectrometry and data analysis
For complex I mass spectrometry analysis, MEFs were treated with Flag–TNF in the presence or absence of ICCB-19 (10 μM) for the indicated time. Proteins that bound to TNFR1 underwent immunoprecipitation pulldown with anti-Flag-beads and were trypsin digested. The peptides were analysed on a Q Exactive HF-X Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific). Protein identification and quantification were performed by MaxQuant 1.2.1. The tandem mass spectra were searched against the UniProt mouse protein database. The precursor and fragment mass tolerances were set as 20 ppm. The false discovery rate (FDR) at peptide spectrum match level and protein level was controlled below 1%. The unique peptides plus razor peptides were included for quantification.
For mass spectrometry analysis of ubiquitination sites of beclin 1, Flag-tagged mbeclin 1 isolated from HEK293T cells expressing this construct was trypsin-digested on beads followed by immunoprecipitation. The resulting peptides were subjected to enrichment of diGly peptides using an antibody against the ubiquitin remnant motif (K-ε-GG) (PTM Biolabs Inc.). The enriched diGly peptides were analysed on the Q Exactive HF-X mass spectrometer (Thermo Scientific). The identification and quantification of diGly peptides was done by MaxQuant 1.2.1. The tandem mass spectra were searched against the UniProt mouse protein database together with a set of commonly observed contaminants. The precursor mass tolerance was set as 20 ppm, and the fragment mass tolerance was set as 0.1 Da. The cysteine carbamidomethylation was set as a static modification, and methionine oxidation and lysine with a diGly remnant were set as variable modifications. The FDR at peptide spectrum match level was controlled below 1%.
The effect of Apt-1 on the binding partners of beclin 1 was characterized by mass spectrometry. Proteins obtained by immunoprecipitation against Flag-tagged beclin 1 in cells with or without Apt-1 treatment were trypsin digested. The resulting peptides in three replicates were analysed on a Thermo Scientific Orbitrap Fusion Tribrid mass spectrometer. The protein identification and quantification were done by MaxQuant 1.2.137. The tandem mass spectra were searched against the UniProt human protein database and a set of commonly observed contaminants. The precursor mass tolerance was set as 20 ppm, and the fragment mass tolerance was set as 0.5 Da. The cysteine carbamidomethylation was set as a static modification, and the methionine oxidation was set as a variable modification. The FDR at the peptide spectrum match level and protein level was controlled to be <1%. The unique peptides plus razor peptides were included for quantification. The summed peptide intensities were used for protein quantification.
Molecular modelling and docking methods
The 3D atom coordinates of TRADD and TRAF2 interaction complex were obtained from the Protein Data Bank (PDB; https://www.rcsb.org) with PDB ID 1F3V. The TRADD part of this 3D structure served as the protein receptor in the following induced-fit docking procedure performed with the molecular simulation software suite Schrödinger (version 2018-1, Schrödinger, LLC, New York, NY, 2018). The receptor was first prepared with the Protein Preparation Wizard. The structure was preprocessed following default settings except that no waters were deleted at this step, then hydrogen bond assignment and restrained minimization were performed in the refinement step, followed by removing water molecules with fewer than three H-bonds to non-waters. The 3D structures of the small molecules were next prepared using LigPrep with no ionization but stereoisomers were generated. The prepared structures of the TRADD receptor and small molecules were then submitted for induced-fit docking to predict the binding modes. At the beginning of this step, to define the binding site, we inspected the interface of the TRADD–TRAF2 interaction and set the docking pocket as the cavity around the centre of residues Ile72, Ala122 and Arg146. Considering the surface residue flexibility, we specified refinement of the residues within 9 Å of the ligand during the induced-fit docking process.
Fluorescence microscopy
Cells were seeded at 2.5 × 104 cells per well on poly-l-lysine coated glass cover slips and transfected as described. Cells were fixed in 4% paraformaldehyde, followed by permeabilization with 0.1% Triton X-100. Nuclei were stained using DAPI (Sigma). Cells expressing GFP- or RFP-fusion proteins were imaged with an Olympus Fluoview FV1000 confocal microscope (Olympus) using a 40× objective. Images were taken using Olympus FV10-ASW 3.0 software. For quantification of GFP-LC3 and DsRed-FYVE puncta, the average spot intensity in 1,000 cells from each indicated sample was determined. Images were processed using ImageJ and Photoshop CC. Concentrations of compounds used to induce or block autophagy: 10 μM (Apt-1, ICCB-19, ICCB-19i or spautin-1), for 6 h or as indicated.
In vivo delivery of TNF
Wild-type mice (n = 10, male, 8 weeks of age) were injected intravenously via the tail vein with mTNF (9.5 μg/mouse) 30 min after an intraperitoneal injection of Apt-1 (20 mg/kg). Control mice (n = 9) received an equal amount of vehicle 30 min before mTNF challenge. Kaplan–Meier survival curves were determined.
VPS34 lipid kinase assay
HEK293T cells were transfected with Flag–beclin 1 for 18 h and then treated with ICCB-19, ICCB-19i, or Apt-1 (10 μM) for another 6 h. Flag–beclin 1 was immunoprecipitated with anti-Flag to isolate the beclin 1–VPS34 complex. Immunoprecipitated beads were added with sonicated phosphatidylinositol (1 μl of 5 mg) and ATP (1 μl of 10 mM) in 30 μl reaction buffer (40 mM Tris (pH 7.5), 20 mM MgCl2, 1 mg/ml BSA) for 30 min at room temperature. Wortmannin (10 μM) was used as a control and added to the reaction to inhibit VPS34. The conversion of ATP into ADP was measured using an ADP-Glo Kinase Assay Kit (Promega) according to the manufacturer’s instructions.
KINOMEscan profiling
KINOMEscan profiling was used to assess the interaction of Apt-1 with a panel of 97 kinases as a custom service (DiscoverX/Eurofins, San Diego, CA USA). In brief, DNA-tagged recombinant kinases were produced in E. coli. The assay plates with kinases were incubated at room temperature with shaking for 1 h and the affinity beads were washed with wash buffer (1 × PBS, 0.05% Tween 20). The beads were then re-suspended in elution buffer. The kinase concentration in the eluates was measured by qPCR. Apt-1 was screened at 10 μM, and the results for primary screen binding interactions are reported as percentage of control, where lower numbers indicate stronger hits in the matrix.
Quantitative reverse-transcription PCR
Total RNA from skin tissue was extracted with Trizol reagent (Life Technologies) and RNeasy Columns (Qiagen) and cDNA was prepared using a Superscript III cDNA-synthesis kit (Life Technologies). Quantitative reverse-transcription PCR of Tnf was performed using a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems). Data were collected using QuantStudio 12K Flex software version 1.3 (Applied Biosystems). Gapdh was used as a reference gene. Data were analysed according to the ΔCT method.
Quantification and statistical analysis
All cell death data are presented as mean ± s.d. of one representative experiment. Each experiment was repeated at least three times. Mouse data are presented as mean ± s.e.m. of the indicated n values. All immunoblots were repeated at least three times independently with similar results. Quantifications of immunoblots were performed using ImageJ 1.52a, and the densitometry data were adjusted to loading control and normalized to control treatment. Error bars for immunoblot analysis represent the s.e.m. between densitometry data from three unique experiments. Curve fitting and statistical analyses were performed using GraphPad Prism 8.0 or Microsoft Excel 2016 software, using either unpaired two-tailed Student’s t-test for comparison between two groups, or one-way ANOVA with post hoc Dunnett’s test for comparisons among multiple groups with a single control, or two-way ANOVA with post hoc Bonferroni’s test for comparisons among different groups. Statistical comparisons for series of data collected at different time points were conducted by two-way ANOVA. The significance of in vivo survival data was determined using the two-sided log-rank (Mantel–Cox) test. Differences were considered statistically significant if P < 0.05. *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The 3D atom coordinates of the TRADD–TRAF2 interaction complex were obtained from PDB (https://www.rcsb.org) with PDB ID 1F3V. All source data associated with the paper are provided as part of the paper. Source data for all figure plots are provided with the paper. The full gel blots and the proteomics datasets are provided in the Supplementary Information. Source data are provided with this paper.
References
Wallach, D., Kang, T. B., Dillon, C. P. & Green, D. R. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 352, aaf2154 (2016).
Shan, B., Pan, H., Najafov, A. & Yuan, J. Necroptosis in development and diseases. Genes Dev. 32, 327–340 (2018).
Ashkenazi, A. & Dixit, V. M. Death receptors: signaling and modulation. Science 281, 1305–1308 (1998).
Hsu, H., Xiong, J. & Goeddel, D. V. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell 81, 495–504 (1995).
Pobezinskaya, Y. L. et al. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat. Immunol. 9, 1047–1054 (2008).
Hsu, H., Shu, H. B., Pan, M. G. & Goeddel, D. V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84, 299–308 (1996).
Sweeney, P. et al. Protein misfolding in neurodegenerative diseases: implications and strategies. Transl. Neurodegener. 6, 6 (2017).
Morishita, H. & Mizushima, N. Diverse cellular roles of autophagy. Annu. Rev. Cell Dev. Biol. 35, 453–475 (2019).
Menzies, F. M. et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034 (2017).
Yoshii, S. R. & Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 18, 1865 (2017).
Zhao, J., Zhai, B., Gygi, S. P. & Goldberg, A. L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl Acad. Sci. USA 112, 15790–15797 (2015).
Noda, T. & Yoshimori, T. Molecular basis of canonical and bactericidal autophagy. Int. Immunol. 21, 1199–1204 (2009).
Zhong, Y. et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1–phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 11, 468–476 (2009).
Matsunaga, K. et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11, 385–396 (2009).
Degterev, A. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008).
Wertz, I. E. et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 528, 370–375 (2015).
Haas, T. L. et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36, 831–844 (2009).
Ofengeim, D. & Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 14, 727–736 (2013).
Vucic, D., Dixit, V. M. & Wertz, I. E. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 12, 439–452 (2011).
Guo, X. et al. TAK1 regulates caspase 8 activation and necroptotic signaling via multiple cell death checkpoints. Cell Death Dis. 7, e2381 (2016).
Park, Y. C. et al. A novel mechanism of TRAF signaling revealed by structural and functional analyses of the TRADD-TRAF2 interaction. Cell 101, 777–787 (2000).
Shu, H. B., Takeuchi, M. & Goeddel, D. V. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc. Natl Acad. Sci. USA 93, 13973–13978 (1996).
Liu, J. et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147, 223–234 (2011).
Chen, N. J. et al. Beyond tumor necrosis factor receptor: TRADD signaling in toll-like receptors. Proc. Natl Acad. Sci. USA 105, 12429–12434 (2008).
Michallet, M. C. et al. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity 28, 651–661 (2008).
Ermolaeva, M. A. et al. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat. Immunol. 9, 1037–1046 (2008).
Callejas, N. A., Casado, M., Boscá, L. & Martín-Sanz, P. Requirement of nuclear factor kappaB for the constitutive expression of nitric oxide synthase-2 and cyclooxygenase-2 in rat trophoblasts. J. Cell Sci. 112, 3147–3155 (1999).
Apostol, B. L. et al. Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum. Mol. Genet. 15, 273–285 (2006).
Yoshiyama, Y. et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351 (2007).
Iba, M. et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 33, 1024–1037 (2013).
Jicha, G. A., Bowser, R., Kazam, I. G. & Davies, P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J. Neurosci. Res. 48, 128–132 (1997).
Grøftehauge, M. K., Hajizadeh, N. R., Swann, M. J. & Pohl, E. Protein-ligand interactions investigated by thermal shift assays (TSA) and dual polarization interferometry (DPI). Acta Crystallogr. D 71, 36–44 (2015).
Cho, H. J. et al. Probing the effect of an inhibitor of an ATPase domain of Hsc70 on clathrin-mediated endocytosis. Mol. Biosyst. 11, 2763–2769 (2015).
Micheau, O. & Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003).
Anderton, H. et al. RIPK1 prevents TRADD-driven, but TNFR1 independent, apoptosis during development. Cell Death Differ. 26, 877–889 (2019).
Voortman, J., Checińska, A. & Giaccone, G. The proteasomal and apoptotic phenotype determine bortezomib sensitivity of non-small cell lung cancer cells. Mol. Cancer 6, 73 (2007).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Acknowledgements
This work was supported in part by grants from UCB, FBRI-Fidelity, and the Quadrangle Fund for Advancing and Seeding Translational Research at Harvard Medical School (Blavatnik-Accelerator of Harvard and HMS/Q-FASTR) (to J.Y.). The work of D.X., L.Y., and B.S. during revision was supported in part by grants from the National Key R&D Program of China (2016YFA0501900), the China National Natural Science Foundation (31530041, 91849204 and 21837004), the Chinese Academy of Sciences (XDB39030000 and XDB39030600) and Shanghai Science and Technology Development Funds (20QA1411500). We thank L. Thompson for HTT(Q103)-PC12 cells; Z. Liu for Tradd−/− MEFs; V. Dixit for M1 and K63 ubiquitin antibodies; P. Davies for the MC1 antibody; G. Cuny for advice on chemistry; Z.-F. Wang and G. Heffron for advice on SPR and NMR; J. Smith for help with compound screening; J. Waters for help with microscopy; the EQNMR facility at HMS for their assistance; and C. Wang and C. Cao for the use of BIAcore.
Author information
Authors and Affiliations
Contributions
This project was conceived, designed, and directed by J.Y. D.X. and H. Zhao designed and conducted most of the experiments. D.X. and H.Z. made equal contributions as joint first authors and are listed alphabetically in the author list. M.J., H. Zhu, J.G. and S.A.D. conducted the multiplexed small molecule screen and contributed to early study of the compounds. Z.J.S. and G.W. assisted with NMR analysis of the TRADD–Apt-1 complex. J.X. conducted computational analysis of the TRADD–Apt-1 complex. Z.H. and V.M.Y.L. directed the tau transgenic mouse model study. Y.Q. and L.T. synthesized ICCB-19 and ICCB-19i for revision. B.S., P.A., L.M., M.G.N., A.N., L.Y., J.L., X.H., H.W., M.Z. and V.J.M. conducted some of the experiments. The manuscript was written by J.Y., D.X. and H. Zhao.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. Harvard University has filed a patent for the ICCB-19 and Apt-1 series of small molecules.
Additional information
Peer review information Nature thanks Kalle Gehring, Ingrid Wertz and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Isolation of ICCB-19 and Apt-1 for modulating both apoptosis and autophagy.
a, A multiplex chemical screening scheme for compounds that can modulate cellular homeostasis by activating autophagy and also block apoptosis. Primary screen: Jurkat cells were treated with Velcade (50 nM) and individual compounds (10 μM) in the library for 25h and cell viability was measured. 710 compounds which could protect against Velcade-induced apoptosis were selected. Secondary counterscreen: HCT116 cells were treated with 5-fluorouracil (5-FU) (100 μM) and individual compounds selected from the Primary screen (10 μM) for 24 h and cell viability was measured. The hits which protected against apoptosis induced by 5-FU were eliminated from further studies. Tertiary screen: H4-GFP-LC3 cells were treated with individual compounds (10 μM) for 24 h and GFP-LC3 dots were quantified. Quaternary screen: RGC-5 cells were treated with mTNF (0.5 ng/ml), TAK1 inhibitor (5Z)-7-Oxozeanol (0.5 μM) and individual compounds (10 μM) for 8 h and cell viability was measured. b, IC50s of ICCB-19 and Apt-1 protecting Velcade-induced apoptosis (50 nM) in Jurkat cells treated with indicated compounds for 24 h and cell viability was measured. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. c, IC50s of ICCB-19 and Apt-1 protecting RDA in MEFs were treated with mTNF (1 ng/mL) and 5Z-7-Oxozeaenol (0.5 μM) in the presence of indicated compounds at different concentrations for 8 h and cell survival was measured. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. d, KINOMEscan profiling of Apt-1 (10 μM) against a panel of 97 kinases. Binding interactions reported as % Ctrl, where lower numbers indicate stronger hits. Negative control = DMSO (100% Ctrl); positive control = control compound (0% Ctrl); 0 ≤ %Ctrl <0.1 Very Strong; 0.1 ≤ %Ctrl <1 Strong; 1 ≤ %Ctrl <10 Medium; 10 ≤ %Ctrl <35 Weak; %Ctrl ≥ 35 No effects. No significant binding of Apt-1 to this panel of 97 kinases was detected. e, H4 cells were treated with indicated concentrations of Apt-1. Autophagy was determined by LC3 II levels using immunoblotting. s.e. = shorter exposure, LE = longer exposure. Mean ± s.e.m. of n = 3 biologically independent experiments. f-i, SH-SY5Y (f), HeLa (g), HT-29 (h), and Jurkat (i) cells were treated with Apt-1 (10 μM), NH4Cl (20 mM) as indicated for 6 h. Autophagy was measured by LC3 II induction and p62 reduction by immunoblotting. The levels of LC3 II in cells treated with both Apt-1 and NH4Cl, the latter of which inhibits lysosome, were higher than that treated with either Apt-1 or NH4Cl alone. Thus, ICCB-19/Apt-1, but not ICCB-19i, induce autophagic flux. Mean ± s.e.m. of n = 3 independent experiments. Two-tailed t-test. **P = 0.0037 (f), 0.0024 (g), 0.0027 (h), 0.0036 (i). j, Effects of ICCB-19/Apt-1 on long-lived protein degradation. The rates of long-lived protein turnover in H4 cells treated with indicated compounds (10 μM, 6 h); rapamycin as positive control. Values expressed as fold changes relative to normal control cells. Mean ± s.e.m. of n = 4 biologically independent samples. One-way ANOVA, post hoc Dunnett’s test. k, MEFs and Jurkat cells treated with zVAD.fmk (20 μM) for 6 h.Levels of LC3 II determined by immunoblotting. l, MEFs were treated with vehicle (0 h), ICCB-19 (10 μM), or Apt-1 (10 μM) for indicated times. Cell lysates analysed by immunoblotting using indicated antibodies. m, H4-DsRed-FYVE cells were treated with indicated compounds for 6 h and imaged; representative cells shown. Average DsRed-FYVE puncta per 1000 cells from each sample was determined using ImageJ. Mean ± s.e.m. of the puncta per cell from n = 5 biologically independent experiments. One-way ANOVA, post hoc Dunnett’s test. **P = 0.0034; ***P = 0.0004; n.s. not significant (P = 0.6502). n, Beclin 1/Vps34 kinase complex isolated from Flag-Beclin 1 transfected HEK293T cells treated with ICCB-19, Apt-1, or ICCB-19i (10 μM) for 6 h. PI3P kinase activity was measured by in vitro lipid kinase assay using ADP-Glo Kinase Assay Kit. Wortmannin (10 μM) was used as a control to inhibit Vps34 kinase activity. Mean ± s.e.m. of n = 4 biologically independent samples. One-way ANOVA, post hoc Dunnett’s tests. ***P = 0.0003; 1 × 10−15 (left to right). For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 2 cIAP1/2 and TRAF2 are required for induction of autophagy by ICCB-19/Apt-1.
a, HEK29T cells were transfected with Flag-Beclin 1 for 12 h, then treated with Apt-1 (10 μM) for another 12 h. Cell lysates were immunoprecipitated using anti-Flag beads. cIAP1 and TRAF2 levels were determined by immunoblotting. b, MEFs were treated with indicated concentrations of Apt-1 for 12 h. Cell lysates were immunoprecipitated using anti-Beclin 1 antibody. cIAP1 and TRAF2 levels were determined by immunoblotting. c, d, Long-lived protein turnover rates in MEFs with indicated genotypes treated with indicated compounds. Expressed as fold changes relative to normal control cells. Mean ± s.d. of n = 4 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA, post hoc Bonferroni’s tests. ***P = 3 × 10−14, 6 × 10−10, 1 × 10−7, 2 × 10−8 (left to right, c, d). e, MEFs were pre-treated with SM-164 (1 μM) for 1 h, then treated with Apt-1 for 6 h. LC3 II levels were determined by immunoblotting. Mean ± s.e.m. of n = 3 biologically independent experiments. Two-tailed t-test. ***P = 0.0003. f, shRNA-mediated TRAF2 stable knockdown MEFs were treated with Apt-1 (10 μM) for 6 h. LC3 II levels were determined by immunoblotting. Mean ± s.e.m. of n = 3 biologically independent experiments. Two-tailed t-test. **P = 0.0012. g, h, cIap1−/− and Traf2−/− MEFs reconstituted with HA-mcIAP1 and HA-mTRAF2, respectively, were treated with Apt-1 (10 μM) for 6 h. LC3 II levels were determined by immunoblotting. Mean ± s.e.m. of n = 3 biologically independent experiments. Two-tailed t-test**P = 0.0057 (g), ***P = 0.0007 (h). i, j, MEFs with indicated genotypes were treated with rapamycin (1 μM) for indicated time. LC3 II levels were determined by immunoblotting. The quantification of each experiment was shown on the right (n = 1), representative of 3 independent experiments. k, l, MEFs with indicated genotypes were incubated in HBSS for indicated time. LC3II levels were determined by immunoblotting. The quantification of each experiment was shown on the right (n = 1), representative of 3 independent experiments. m, MEFs were treated with indicated compounds for 6 h, then cell lysates were tandem-immunoprecipitated with anti-Beclin 1 antibody and denatured in 3 M urea. Anti-K63-linkage specific polyubiquitin antibody was used to conduct secondary immunoprecipitation. Samples were then immunoblotted with anti-Beclin 1 antibody to measure the K63-linkage specific ubiquitination of Beclin 1. n, MEFs were pretreated with SM-164 (1 μM) for 1 h, then treated with Apt-1 (10 μM) for 6 h, then K63-linkage specific ubiquitination of Beclin 1 was analysed as in (m). o, p, Reconstituted MEFs were treated with Apt-1 (10 μM) for 6 h, then K63-linkage specific ubiquitination of Beclin 1 was analysed as in (m). For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 3 cIAP1 mediates K63-linked ubiquitination of Beclin 1 at K183 and K204.
a, Schematic representation of mass spectrometry assay to determine K63-linked ubiquitination sites of Beclin 1 by cIAP1. b, The quantitative mass spec analysis of K63-linked ubiquitination of each lysine site. c, Sequence alignment of key ubiquitination sites (K) within Beclin 1 orthologues from different species. See supplementary Table 2 for details. d, HEK293T cells were transfected with indicated plasmids for 24 h. Cells were lysed in 6 M urea and lysates were subjected to pull-down with Ni2+ beads and analysed by immunoblotting with anti-Beclin 1 antibody to detect ubiquitylated Beclin 1. e, Validation of Beclin 1 expression in Beclin 1-silenced H4 cells. f, Control and Beclin 1-silenced H4 cells were treated with Apt-1 (10 μM) for 6 h. LC3 II levels were determined by immunoblotting. g, Beclin 1-silenced H4 cells reconstituted with WT and mutants Beclin 1 were treated with Apt-1 (10 μM) for 6 h. LC3 II levels were determined by immunoblotting. Mean ± s.e.m. are quantified from n = 3 biologically independent experiments in graphs. Two-tailed t-test. **P = 0.0022 (d), 0.0049 (f), 0.0024 (g); *P = 0.0309, 0.0195 (left to right, d), 0.0126 (g); n.s. not significant, (P = 0.6959) (f). For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 4 ICCB-19/Apt-1 block Velcade-induced apoptosis, RIPK1-dependent apoptosis and necroptosis.
a, b, Jurkat (a) or SH-SY5Y (b) cells were stimulated by Velcade (50 nM) in the presence of Apt-1 (10 μM), Nec-1 s (10 μM), or zVAD (20 μM) for 12 h and 24 h. The levels of cleaved caspase-3 were determined by immunoblotting. c, Tak1−/− MEFs were treated with 1 ng/ml mTNF in the presence of indicated compounds for 3 h. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. One-way ANOVA, post hoc Dunnett’s tests. ***P = 1 × 10−15 (left to right); n.s. not significant, (P = 0.7989). d, Tak1−/− MEFs were treated as in (a), the cell lysates were analysed by immunoblotting using indicated antibodies. e, MEFs were treated with mTNF (1 ng/ml) and 5Z-7-Oxozeaenol (0.5 μM) in the presence of indicated compounds for 1 h and 2 h and the cell lysates were analysed by immunoblotting using indicated antibodies. f, g, ICCB-19/Apt-1 inhibit RDA, including complex IIa formation (f) and caspase-8 activation (g). MEFs treated as in (e) were lysed with IP buffer and FADD was immunoprecipitated by anti-FADD antibody. Total lysates and IP samples were analysed by immunoblotting to determine the recruitment of RIPK1 to FADD in complex IIa (f). MEFs were treated with mTNF (1 ng/ml) and 5Z-7-Oxozeaenol (0.5 μM) in the presence of ICCB-19 (10 μM), Apt-1 (10 μM), Nec-1 s (10 μM), or zVAD.fmk (20 μM) for 4 h and the activity of caspase-8 was determined using Caspase-Glo 8 Assay Systems (g). Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. One-way ANOVA, post hoc Dunnett’s tests. ***P = 1 × 10−15 (g). h-k, RDA was induced in Tbk1−/− MEFs (h, i), Nemo−/− MEFs (j, k) by the treatment with mTNF (10 ng/ml) together with ICCB-19 (10 μM) and Nec-1 s (10 μM) at indicated times and cell death was determined by SYTOX Green (h, j) and caspase-3 cleavage (CC3) immunoblotting (i, k). Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments (h, j).). Two-way ANOVA. ***P = 5 × 10−7 (h), 4 × 10−7 (j). l, RDA was induced in WT MEFs by the treatment with mTNF (10 ng/ml) and IKK inhibitor TPCA-1 (5 μM) in the presence of ICCB-19 (10 μM) or Nec-1 s (10 μM) for indicated times and cell death was determined by SYTOX Green. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA. ***P = 1 × 10−5. m, Recombinant active caspase-8 was incubated with vehicle, ICCB-19 (10 μM), Apt-1 (10 μM), or zVAD.fmk (20 μM) for 1h and the activity of caspase-8 was determined using Caspase-Glo 8 Assay Systems. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. One-way ANOVA, post hoc Dunnett’s tests. n.s. not significant, (P = 0.9931, 0.9215 (left to right)). n, WT MEFs were treated with mTNF (1 ng/ml) and cycloheximide (CHX, 1 μg/mL) to induce RIA in the presence or absence of ICCB-19 (10 μM) or Nec-1 s (10 μM) for indicated time and cell survival was determined by CellTiter-Glo assay. Mean ± s.d. of n = 8 biologically independent samples, representative of 3 independent experiments. One-way ANOVA, post hoc Dunnett’s tests. n.s. not significant, (P = 0.9962). o, p65/p50 DKO MEFs were treated with mTNF (1 ng/ml) together with ICCB-19 (10 μM) for indicated times and cell survival was determined by CellTiter-Glo assay. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA. n.s. not significant, (P = 0.1895). p, MEFs were treated as indicated and the cell survival was measured by CellTiter-Glo assay. The concentrations of reagents used: mTNF: 1 ng/mL; (5Z)-7-oxozeaenol: 0.5 μM; zVAD: 20 μM; ICCB-19: 10 μM; Apt-1: 10 μM; Nec-1 s: 10 μM. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. q, Necroptosis of MEFs was induced by the treatment with TNF/5z7/zVAD in the presence of indicated compounds for indicated hours and the activation of RIPK1(p-S166), RIPK3(p-T231/S232), and MLKL(p-S345) was determined by immunoblotting. r, HEK293T cells were transfected with Flag-RIPK1 expression construct for 12 h in the presence of Nec-1 s (10 μM), ICCB-19 (10 μM), or Apt-1 (10 μM). The activation of RIPK1 was determined by immunoblotting using p-RIPK1(S166) antibody. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 5 The protection of RDA by ICCB-19/Apt-1 requires TRADD.
a, b, Mass spectrometry analysis shown in Fig. 2b using ICCB-19 (a) or Apt-1 (b) were confirmed by immunoprecipitation-immunoblotting using indicated antibodies, quantified on the right (n = 1), representative of 3 independent experiments. c, MEFs were treated with Flag-mTNF (50 ng/ml) in the presence of Apt-1 (10 μM) for indicated time. The complex I was isolated by anti-Flag beads and denatured in 6 M urea. The complex I was further analysed by immunoprecipitation using anti-M1 (6 M urea) or K63 (3 M urea) ubiquitin antibody under denatured condition. The levels of RIPK1 ubiquitination were analysed by immunoblotting. d, WT and Traf2−/− MEFs were stimulated by mTNF (1 ng/ml) and 5Z-7-Oxozeaenol (0.5 μM) in the presence of indicated compounds for 8 h. Cell survival was determined by CellTiter-Glo assay. Mean ± s.d. of n = 4 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA, post hoc Bonferroni’s tests. ***P = 0.0001. e, WT and cIap1/2−/− MEFs were stimulated by mTNF (10 ng/ml) in the presence of vehicle, ICCB-19 (10 μM) or Nec-1 s (10 μM) for indicated time. Cell death was determined by CellTiter-Glo assay. Mean ± s.d. of n = 4 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA. f, MEFs were pretreated with SM-164 (50 nM) for 1 h, then stimulated by mTNF (10 ng/ml) in the presence of vehicle, ICCB-19 (10 μM) or Nec-1 s (10 μM) for indicated time. Cell death was determined by SYTOX Green assay. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA. ***P = 4 × 10−5; n.s. not significant, (P = 0.1772). g, cIap1/2−/− MEFs were stimulated with Flag-TNF (50 ng/ml) for indicated minutes in the presence of vehicle or ICCB-19 (10 μM) and the complex I was pulled down using anti-Flag beads. The levels of activated RIPK1 and total RIPK1 were determined by immunoblotting. h, i, cIap1/2−/− MEFs (h) and cIAP1-reconstituted cIAP1/2 DKO MEFs (i) were stimulated with Flag-TNF (50 ng/ml) for indicated minutes in the presence of vehicle or Apt-1 (10 μM) and the complex I was pulled down using anti-Flag beads. TRADD recruitment to complex I was determined by immunoblotting, quantified on the right (n = 1), representative of 3 independent experiments. j, k, Fadd-deficient and Ripk1-deficient Jurkat cells were treated with Velcade (50nM) in the presence of ICCB-19 (10 μM), Nec-1 s (10 μM), NAC (100 μM), or zVAD.fmk (20 μM). The activation of caspase-8, PARP cleavage (j), and activation of caspase-3 (k) were determined by immunoblotting. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 6 Autophagy is required for ICCB-19/Apt-1 to inhibit Velcade-induced apoptosis.
a, Tradd+/+ and Tradd−/− MEFs were treated with vehicle or ICCB-19 (10 μM) for 6 h. Autophagy levels were determined by immunoblotting using anti-LC3 antibody. Mean ± s.e.m. of n = 3 biologically independent experiments (right). Two-tailed t-test. n.s. not significant, (P = 0.9172). b, Tradd+/+ and Tradd−/− MEFs were treated with Apt-1 (10 μM), NH4Cl (10 mM) as indicated for 6 h. Autophagy levels were determined by immunoblotting using anti-LC3 antibody. Mean ± s.e.m. of n = 3 biologically independent experiments. Two-tailed t-test. n.s. not significant (P = 0.4064, 0.8913 (left to right)). c, Long-lived protein turnover rates in Tradd+/+ and Tradd−/− MEFs. Expressed as as fold changes relative to Tradd+/+ cells. Mean ± s.e.m. of n = 5 biologically independent samples, representative of 3 independent experiments. Two-tailed t-test. ***P = 0.0001. d, WT and Tradd-KO Jurkat cells were treated with Apt-1 (10 μM) and Spautin-1 (10 μM) followed by Velcade (50 nM) for 24 h. Cell survival was determined by CellTiter-Glo assay. Mean ± s.d. of n = 4 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA, post hoc Bonferroni’s tests. ***P = 2 × 10−5, 3 × 10−15 (left to right). e, Jurkat cells were treated with ICCB-19 (10 μM), Apt-1 (10 μM), Chloroquine (50 μM), E64d (5 μg/ml) followed by Velcade (50 nM) for 24 h. The cell survival was determined by CellTiter-Glo assay. Mean ± s.d. of n = 4 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA, post hoc Bonferroni’s tests. ***P = 4 × 10−14, 1 × 10−15 (left to right). f, Atg5-WT and Atg5-KO Jurkat cells were pretreated with Apt-1 (10 μM) or zVAD (20 μM) for 1 h, then stimulated by Velcade (50 μM) for 24 h. Cell survival was determined by CellTiter-Glo assay. Mean ± s.d. of n = 4 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA, post hoc Bonferroni’s tests. ***P = 1 × 10−15. Validation of Atg5 knockout was determined by immunoblotting and shown on the right. g, Atg5+/+ and Atg5−/− MEFs were stimulated by TNF (1 ng/ml) and 5z7 (0.5 μM) for 8 h in the presence or absence of Apt-1 (10 μM). Cell survival was determined by CellTiter-Glo assay. Mean ± s.d. of n = 4 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA, post hoc Bonferroni’s tests. n.s. not significant, (P = 0.2568, 0.0822 (left to right)). h, HEK293T cells were transfected with indicated expression plasmids for 24 h. The whole-cell lysate lysed in 6 M urea was subjected to pull-down with Ni2+ beads and analysed by immunoblotting with anti-Beclin 1 antibody to detect ubiquitylated Beclin 1. The ubiquitination of Beclin 1 by cIAP1 was reduced upon overexpression of TRADD, which was restored by Apt-1. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 7 ICCB-19/Apt-1 inhibit inflammatory responses.
a, MEFs were stimulated by mTNF (10 ng/ml) in the presence of vehicle or ICCB-19 (10 μM) for indicated time. NF-κB and MAPKs activity were determined by immunoblotting using indicated abs. b, MEFs were stimulated by mTNF (10 ng/ml) in the presence of vehicle or ICCB-19 (10 μM) for indicated time. The protein levels of iNOS and Cox2 were determined by immunoblotting. c-g, BV2 cells (a microglial-like cell line) (c, d, e) and BMDMs (bone marrow-derived macrophages) (f, g) were treated with IFNγ (c, 1 unit/μl), Pam3CSK4 (ligand for TLR2) (e, 10 ng/ml), LPS (ligand for TLR4) (f, 10 ng/ml), or MDP (ligand for NOD2/RIPK2 pathway) (d, g, 10 μg/ml) for indicated time. The mRNA levels of TNF were determined using quantitative PCR. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments (c-g). h, BV2 cells were treated with IFNγ (1 unit/μl) together with Apt-1 (10 μM) or Nec-1 s (10 μM) for 24 h. TNF production was determined by ELISA. i, BV2 cells were pretreated with Apt-1 (10 μM) or Nec-1 s (10 μM) for 1 h and then MDP (10 μg/ml) was added to cells together with transfection reagent for 7 h. TNF production was determined by ELISA. j, BV2 cells were pretreated with Apt-1 (10 μM) or Nec-1 s (10 μM) and then treated with Pam3CSK4 (10 ng/ml) for 8 h. TNF production was determined by ELISA. k, BMDMs were pretreated with Apt-1 (10 μM) or Nec-1 s (10 μM) for 1h and then treated with LPS (10 ng/ml) for 7 h. TNF production was determined by ELISA. l, BMDMs were first primed with IFNγ (10 ng/ml) for 2 h and then removed. The cells were then treated with Apt-1 (10 μM) or Nec-1 s (10 μM) for 1 h and MDP (10 μg/ml) was added to cells directly and treated for 8 h. TNF production was determined by ELISA. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments (h-l). m, BMDMs were treated with LPS (10 ng/ml) in the presence of vehicle control or Apt-1 (10 μM) or Nec-1 s (10 μM) for indicated time. NF-κB and MAPKs activity were determined by immunoblotting using indicated abs. n, Body temperature was measured in male mice (n = 10, 8 weeks) injected with mTNF (9.5 μg, i.v.) after pretreatment with Apt-1 (20 mg/kg) 30 min before. Control mice (n = 9) received an equal amount of vehicle before mTNF challenge. Mean ± s.e.m. Two-way ANOVA. ***P = 0.0007. o, Kaplan–Meier Survival Curve was measured on mice treated as in (n). Two-sided log-rank (Mantel-Cox) test. ***P = 1 × 10−5. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 8 Restoring proteome homeostasis by ICCB-19/Apt-1.
a, b, Parallel wells of PC12/Htt-Q103 cells were cultured with Vehicle, ICCB-19 (10 μM), ICCB-19i (10 μM), Apt-1 (10 μM), Nec-1 s (10 μM), and zVAD (20 μM) as indicated before the addition of Ponasterone A (5 μM) for 48 h. Nuclei were labelled with DAPI. The amount of Htt-Q103-EGFP aggregates per mm2 was quantified using ImageJ. Cell viability was measured by CellTiter-Glo assay. Survival rate is compared with vehicle-treated cells (b). Mean ± s.d. of n = 3 (a), n = 4 (b) biologically independent samples, representative of 3 independent experiments. One-way ANOVA, post hoc Dunnett’s tests. ***P = 1 × 10−5 (left to right, a, b); n.s. not significant, (P = 0.8556, 0.9195, 0.8613, 0.5687, 0.5304, 0.9998, 0.6029, 0.2821 (left to right, a, b)). c, d, SH-SY5Y cells were transfected with expression vectors for RFP-α-Synuclein WT, E46K, or A53T for 24h and then treated with vehicle or Apt-1 (10 μM) for 24 h. RFP-α-Synuclein was quantified by Fluorescence/Cell (RLU) by ImageJ. The cells treated as in (c) were lysed and analysed by immunoblotting for the levels of α-Synuclein (d). Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA, post hoc Bonferroni’s tests. ***P = 8 × 10−5; *P = 0.0262, 0.0367 (left to right, c). e, f, Quantification of GFP-Tau fluorescence (e) or immunoblots of tau levels (f) in H4 cells treated with Apt-1. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. Two-way ANOVA, post hoc Bonferroni’s tests. ***P = 8 × 10−5, 8 × 10−4 (e, left to right). g, Representative images and quantification of cultured brain slices from PS19 mice (4 months old) stained with phospho-tau (red) and cell nuclei (DAPI/blue). Mean ± s.e.m. of n = 5 biologically independent samples. Two-tailed t-test. h, Immunoblots of tau levels in cultured PS19 mouse (4 months old) brain slices treated with indicated compounds. i, Pharmacokinetics of Apt-1 over 24 h dosing period in cerebrospinal fluid (CSF) and hippocampus. Apt-1 was delivered using intracerebroventricular Alzet micro-osmotic pump (20 mM Apt-1, 100 μl, release rate: 0.25 μl/h). CSF was collected at 1 h, 6 h, and 24 h. Hippocampi were collected at 24 h. The concentrations of Apt-1 were measured by HPLC. The concentration of Apt-1 in hippocampus at 24 h was 6.27 μM. Mean ± s.e.m. (n = 3 mice in each group). j, k, Synthetic preformed fibrils (pffs) [5μg full length tau (2N4R) with P301S mutation (T40/PS) per injection] or vehicle were injected into the hippocampi of PS19 mice (8 weeks old). Three weeks after the pffs injection, Apt-1 was delivered intracerebroventrically by Alzet micro-osmotic pumps (20 mM Apt-1, release rate 0.25μl/h) for one week before sacrificing. The hippocampi were isolated from the mice for immunoblotting using TAU-5 (Thermo Fisher) (j), or immunostaining for phospho-Tau (AT8) (k). Dots represent the mean from individual mice. Mean ± s.e.m. (n = 3 mice in each group). Two-tailed t-test. ***P = 0.0007. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 9 ICCB-19/Apt-1 reduce the interaction of TRADD and TRAF2.
a, Expression constructs encoding Flag-TRADD-N (1-179) and HA-TRADD-C (180-312) were transfected into HEK293T cells for 18 h. Then the cells were treated with Apt-1 (10 μM) or vehicle for another 6 h. The binding between Flag-TRADD-N (1-179) and HA-TRADD-C (180-312) was analysed by Co-IP assay as indicated. b-d, The effect of Apt-1 and ICCB-19 on the binding between TRADD-N and TRADD-C was determined by NanoBiT assay. Constructs were made encoding LgBiT and SmBiT fused to the N and C termini of TRADD-N and TRADD-C, respectively. HEK293T cells were transfected with these two plasmids for 24 h and then treated with indicated compounds for 6 h. The luminescence indicating the interaction of TRADD-N and TRADD-C was measured using Nano-Glo Live Cell Reagent. e, Test of the Nano-Bit system assay using a known binding pair: PRKAR2A and PRKACA. f, Apt-1 (10 μM) does not affect the Nano-Bit system assay as determined by using the known binding pair: PRKAR2A and PRKACA. g, Apt-1 reduces the binding between TRADD-N and TRAF2-C in a dose-dependent manner as determined by NanoBiT assay. h, Schematic representation of a cell-free Förster resonance energy transfer (FRET)-based assay to detect TRADDN and TRAF2C interaction. i, Purification of indicated proteins for FRET assay expressed in HEK293T cells. Proteins were pulled down by anti-Flag affinity gel and eluted by 3 × Flag peptide. CBB staining of the proteins are shown on the right. j, FRET-based assay to measure the direct interaction of TRADD-N and TRAF2-C was developed in which the donor Flag-TRAF2C-mCenulean (TRAF2C-mC) was excited at 430 nm, and the emission was measured from 450 to 600 nm. When FRET occurs, the acceptor mVenus-TRADDN-Flag (mV-TRADDN) emission will increase and the donor emission will decrease. Apt-1 was added to the system with indicated concentration and incubated for 1h, then subjected to FRET assay. k, Effect of TRAF2 on the binding between TRADD-N and TRADD-C was determined by NanoBiT assay. HEK293T cells were transfected with the plasmids as indicated for 24h and then treated with Apt-1 (10μM) for 4h, then luminescence was measured. l, U937 cells were stimulated with TNF (10 ng/ml) for indicated minutes in the presence of vehicle or Apt-1 (10 μM) and the complex I was pulled down using anti-TNFR1. Levels of cIAP1, TRAF2, and TRADD recruitment were determined by immunoblotting. Quantifications of cIAP1, TRAF2, and TRADD are shown below each blot. m, Due to the lack of a good anti-TRADD antibody for immunoprecipitation, Tradd−/− MEFs were reconstituted with Flag-mTRADD. Cells were treated with indicated concentrations of Apt-1 for 12 h, then co-IP was performed using anti-Flag antibody followed by immunoblotting using indicated antibodies. Mean ± s.d. of n = 3 (e, f), n = 4 (c, g), n = 5 (b), n = 6 (d, k) biologically independent samples, representative of 3 independent experiments. One-way ANOVA, post hoc Dunnett’s test. ***P = 1 × 10−15 (b, left to right), ***P = 1 × 10−15, n.s. not significant, (P = 0.3357) (d). ***P = 1 × 10−15 (k, left to right). For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 10 ICCB-19/Apt-1 bind to TRADD-N.
A fluorescence-based thermal shift assay was developed to quantify ICCB-19/Apt-1 binding to TRADD by measuring changes in thermal denaturation temperature (Tm). a, CBB staining of GST-tag and GST-TRADD purified from HEK293T cells. b, in vitro binding of GST-TRADD (50 μM) with ICCB-19 (250 μM) and Apt-1 (250 μM) was determined by thermal shift assay. Thermal unfolding of GST-TRADD is monitored using SYPRO Orange. Data were collected in the presence of ICCB-19 and Apt-1, leading to a rightward shift in the unfolding transition. The apparent melting temperature (Tm) is the peak in the derivative of the unfolding curve (dF/dT), which is used as an indicator of thermal stability. c, GST-tag (50 μM) does not bind to the compounds (250 μM) as determined by thermal shift assay. d, ICCB-19i does not bind to GST-TRADD as determined by thermal shift assay. e, GST-TRADD-C (50 μM) alone does not bind to either ICCB-19 (250 μM) or Apt-1 (250 μM) as determined by thermal shift assay. f-h, TRADD-N/ICCB-19 (f), TRADD-N/Apt-1 (g), and TRADD-N/ICCB-19i (h) samples used for STD NMR experiments were prepared as 1 mM ICCB-19 (f), 1 mM Apt-1 (g), 1 mM ICCB-19i (h), and 13 μM TRADD-N in 0.5 mL of PBS in D2O (10%). The on-resonance irradiation of TRADD-N was performed at a chemical shift of -0.5 ppm, whereas the off-resonance irradiation was conducted at 37 ppm. Spectra were acquired using the following parameters: spectral window of 6.4 kHz, number of scans at 320, acquisition time of 2 s, and repetition time of 3 s. The decrease in signal intensity in STD spectrum, resulting from the transfer of saturation from the protein to the ligand, is evaluated by subtracting the on-resonance spectrum from the off-resonance spectrum. This subtraction yields a positive signal from a bound ligand. The asterisks indicate the signals of the compounds. The STD data suggest that both ICCB-19/Apt-1, but not ICCB-19i, bind with TRADD-N. i, CBB staining of 6 × His- and Flag-tagged TRADD for SPR purified from HEK293T cells. The proteins were pulled down by anti-Flag affinity gel and eluted by 3 × Flag peptide. The proteins were further purified by size exclusion chromatograph on a Superdex 75 column (GE Healthcare) in a buffer containing 20 mM imidazole (pH 6.6), 200 mM NaCl, 20 mM DTT. j, BIAcore SPR analysis of ICCB-19 binding to TRADD-N. The kinetic profile of ICCB-19 binding to TRADD-N is shown. A series of concentrations of ICCB-19 (ranging from 0.3125 to 10 μM) was used to measure the binding kinetics, with TRADD-N immobilized on the CM5 chip.
Extended Data Fig. 11 The binding mechanism of ICCB-19/Apt-1 on TRADD.
a-c, Superposition of 2D 1H-15N HSQC spectra of 15N-labelled His-TRADD-N (250 μM) in the presence (red) and absence (blue) of Apt-1 (500 μM) (a), ICCB-19 (500 μM) (b) or ICCB-19i (500 μM) (c). The close-up view of the region exhibited large perturbations was shown right. d, Binding pose of ICCB-19 in complex with TRADD-N was generated by induced-fit docking. The left panel demonstrated the shape and polarity of the ligand binding pocket surface, with red regions indicating negatively charged and blue positively charged. The right panel showed details of the interactions between the compound and TRADD-N. The compound was shown as cyan sticks, and the protein was shown as pink cartoon with key residues highlighted in sticks. Hydrogen bonds were shown as red dashed lines. e, Coomassie blue staining of WT and each mutant protein for thermal shift assay. f, g, HEK293T cells were seeded at 7.5 × 103 cells per well in a white, clear-bottom 96-well plate 24 h before transfection. Cells were then transfected with the indicated plasmids for 24h. Medium was removed and replaced with Opti-MEM medium (100 μl) for 1 h at 37 °C. The Nano-Glo reagent was prepared and added to each well immediately before the luminescence reading was taken. Luminescence was measured immediately on a plate reader and reported as relative light units (RLU). Mean ± s.d. of n = 6 biologically independent samples, representative of 3 independent experiments.
Extended Data Fig. 12 A mutagenesis study of ICCB-19/Apt-1 binding with TRADD to inhibit RDA and activate autophagy.
a, Tradd−/− MEFs were reconstituted with Flag-tagged WT or mutant TRADD as indicated. Expression levels of TRADD were determined by immunoblotting. b, Tradd−/− MEFs transfected with Flag-tagged WT or indicated TRADD mutants were stimulated by TNF/5z7 for 9 h in the presence or absence of Apt-1 (10 μM). Cell survival was determined by CellTiter-Glo assay. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. Two-tailed t-test. c, d, TRADD-N(G121A)/Apt-1 (c), and TRADD-N(G121A)/ICCB-19 (d) samples for STD-NMR analyses were prepared as that of WT TRADD in (10f) with 1 mM Apt-1 (c), 1 mM ICCB-19 (d), and 13 μM TRADD-N(G121A) in 0.5 mL of PBS in D2O (10%). e, BIAcore SPR analysis of Apt-1 binding to TRADD-N(G121A). The kinetic profile of Apt-1 binding to TRADD-N(G121A) is shown. A series of concentrations of Apt-1 (ranging from 0.15625 to 5 μM) was used to measure the binding kinetics, with TRADD-N(G121A) immobilized on the CM5 chip. f, Tradd−/− MEFs were reconstituted with Flag-tagged WT or indicated TRADD mutants. The expression levels of TRADD were determined by immunoblotting. g, h, Tradd−/− MEFs reconstituted with Flag-mutant TRADD (Y16A/F18A or Y16A/I72A/R119A) were stimulated by TNF/5z7 for indicated time in the presence or absence of Apt-1 (10 μM). The cell survival was determined by CellTiter-Glo assay. Mean ± s.d. of n = 3 biologically independent samples, representative of 3 independent experiments. i, A model for mechanism by which Apt-1 targets TRADD to inhibit RDA and activate autophagy. In TNF-stimulated cells: Apt-1 binds to TRADD-N to reduce its binding with TRADD-C which stabilizes the binding of TRADD mediated by its DD in TRADD-C with the DD in TNFR1. The binding of Apt-1 with TRADD in complex I modulates the K63/M1-linked ubiquitination of RIPK1 by reducing the binding of TRADD with TRAF2/cIAP1/2 which increases the recruitment of A20 and HOIP to inhibit the activation of RIPK1 kinase. Increased retention of TRADD in complex I also decreases cytosolic availability of TRADD for the formation of complex IIa in which TRADD is known to be a key component. Treatment with Apt-1 also reduces the activation of NF-κB in TNF-stimulated cells. In autophagy pathway: TRADD normally binds to TRAF2 and cIAP1/2 homeostatically. Apt-1 can release TRAF2 and cIAP1/2 from their binding with TRADD. Released TRAF2/cIAP1/2 in turn mediates K63-linked ubiquitination of Beclin 1 to promote the formation of Vps34 complex, production of PtdIns3P, and activation of autophagy. For gel source data, see Supplementary Fig. 1.
Supplementary information
Supplementary Figure
Supplementary Figure 1. Full scan of all gel blots. Full blot images of all cropped bands presented in data figures, unless shown elsewhere already. Note that all the loading controls were on the same gel as target protein.
Supplementary Figure
Supplementary Figure 2. Synthesis and chemical structure characterization of Apt-1. Synthesis of Apt-1 and chemical structure characterization of Apt-1, including 1H NMR spectra and LRMS spectra of Apt-1.
Supplementary Table
Supplementary Table 1. Proteomics analysis of Beclin 1-VPS34 complex. Related to Fig. 1c. An excel file of the proteomics data showing the quantification of the abundance of each protein interacted with Beclin 1 in the presence or absence of Apt-1.
Supplementary Table
Supplementary Table 2. Proteomics analysis to identify the ubiquitination sites of Beclin 1 mediated by cIAP1. Related to Extended Data Fig. 3b. The quantitative mass spec analysis of K63 ubiquitination of each lysine site of Beclin 1 in the presence or absence of cIAP1.
Supplementary Table
Supplementary Table 3. Proteomics analysis of TNF-RSC complex (complex I). Related to Fig. 2b. An excel file of the proteomics data showing the quantification of the abundance of each protein in complex I in the presence or absence of ICCB-19.
Source data
Rights and permissions
About this article

Cite this article
Xu, D., Zhao, H., Jin, M. et al. Modulating TRADD to restore cellular homeostasis and inhibit apoptosis. Nature 587, 133–138 (2020). https://doi.org/10.1038/s41586-020-2757-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2757-z
- Springer Nature Limited
This article is cited by
-
A guide to cell death pathways
Nature Reviews Molecular Cell Biology (2024)
-
NAP1L1 regulates BIRC2 ubiquitination modification via E3 ubiquitin ligase UBR4 and hence determines hepatocellular carcinoma progression
Cell Death Discovery (2024)
-
Pharmacological targeting of adaptor proteins in chronic inflammation
Inflammation Research (2024)
-
Deciphering a cell death-associated signature for predicting prognosis and response to immunotherapy in lung squamous cell carcinoma
Respiratory Research (2023)
-
TRAF2 promotes M2-polarized tumor-associated macrophage infiltration, angiogenesis and cancer progression by inhibiting autophagy in clear cell renal cell carcinoma
Journal of Experimental & Clinical Cancer Research (2023)