

Abstract
Neural crest cells (NCCs) are migratory, multipotent embryonic cells that are unique to vertebrates and form an array of clade-defining adult features. The evolution of NCCs has been linked to various genomic events, including the evolution of new gene-regulatory networks1,2, the de novo evolution of genes3 and the proliferation of paralogous genes during genome-wide duplication events4. However, conclusive functional evidence linking new and/or duplicated genes to NCC evolution is lacking. Endothelin ligands (Edns) and endothelin receptors (Ednrs) are unique to vertebrates3,5,6, and regulate multiple aspects of NCC development in jawed vertebrates7,8,9,10. Here, to test whether the evolution of Edn signalling was a driver of NCC evolution, we used CRISPR–Cas9 mutagenesis11 to disrupt edn, ednr and dlx genes in the sea lamprey, Petromyzon marinus. Lampreys are jawless fishes that last shared a common ancestor with modern jawed vertebrates around 500 million years ago12. Thus, comparisons between lampreys and gnathostomes can identify deeply conserved and evolutionarily flexible features of vertebrate development. Using the frog Xenopus laevis to expand gnathostome phylogenetic representation and facilitate side-by-side analyses, we identify ancient and lineage-specific roles for Edn signalling. These findings suggest that Edn signalling was activated in NCCs before duplication of the vertebrate genome. Then, after one or more genome-wide duplications in the vertebrate stem, paralogous Edn pathways functionally diverged, resulting in NCC subpopulations with different Edn signalling requirements. We posit that this new developmental modularity facilitated the independent evolution of NCC derivatives in stem vertebrates. Consistent with this, differences in Edn pathway targets are associated with differences in the oropharyngeal skeleton and autonomic nervous system of lampreys and modern gnathostomes. In summary, our work provides functional genetic evidence linking the origin and duplication of new vertebrate genes with the stepwise evolution of a defining vertebrate novelty.
Similar content being viewed by others

Main
In model jawed vertebrates, the proper patterning and differentiation of most NCC subpopulations requires Edn signalling. Ednrs expressed by migrating and postmigratory NCCs bind Edns secreted by surrounding tissues. In zebrafish and mouse, disruption of edn1 or endothelin receptor A (ednra) results in a hypomorphic pharyngeal skeleton, skeletal element fusions and ventral-to-dorsal transformations of oropharyngeal cartilages and bones7,8,13,14,15. In edn1-mutant zebrafish, the increased dorsoventral symmetry and lack of a jaw joint causes a ‘sucker’ phenotype reminiscent of modern agnathans7. In both mouse and zebrafish, the skeletal phenotype of edn1 and ednra mutants is caused, in part, by loss of expression of dlx and hand family members in cranial NCCs8,16. In non-skeletogenic NCCs of mouse, zebrafish and Xenopus, loss of edn3 or ednrb homologues causes aberrant migration and/or loss of pigment cells10,17,18. In mammals, these defects are accompanied by deficiencies in the NCC-derived enteric nervous system (ENS)19,20.
Lampreys express homologues of edn, ednr, dlx and hand in patterns reminiscent of their gnathostome cognates21,22,23, although lamprey and gnathostome NCC derivatives differ substantially. In addition to lacking jaws, the lamprey oral skeleton consists of a specialized pumping organ made of a chondroid tissue called mucocartilage24 (Fig. 1a, Extended Data Fig. 1a, b). In the posterior pharynx, the branchial skeleton is a network of cell-rich hyaline cartilage bars and a ventral mass of mucocartilage24. In the trunk, the lamprey peripheral nervous system (PNS) lacks sympathetic chain ganglia24 and vagal NCC-derived enteric ganglia25. These differences, and the unclear phylogenetic relationships between gnathostome and lamprey edn and dlx homologues, have led to speculation that these genes acquired new roles in the NCCs of stem gnathostomes23,26.

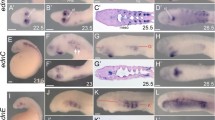
a, Illustration of the larval sea lamprey pharyngeal skeleton at stage T30 with numbered pharyngeal arch derivatives. Lat. m. pl., lateral mouth plate. b–g, Toluidine blue-stained sagittal section of the oral mucocartilage (b–d) and flat-mounted alcian blue stain of the branchial basket (e–g) at stage T30 in wild-type (WT) (b, e), Δednra (c, f) and Δednra+b (d, g) larvae. Δednra and Δednra+b exhibit reduced mucocartilage in the upper lip, lateral mouth plate (dotted lines in b–d) and first pharyngeal arch (arrows in b–d). They also display gaps in the branchial bars (arrows in f, g) and reductions in the epitrematic and hypotrematic processes (arrowheads in f, g). Δednra+b additionally lack one or more posterior branchial bars (asterisks in g). Toluidine blue staining: 3 out of 3 Δednra (c) and 4 out of 4 Δednra+b (d) exhibited reduced oral skeletons; alcian blue staining: 16 out of 16 Δednra (f) and 19 out of 19 Δednra+b (g) individuals exhibited disrupted branchial skeletons. h, Summary of expression of lamprey ednr and edn genes in the head at T25.5 (ref. 21). i–n, Expression of dlxD and hand in wild-type (i, l), Δednra (j, m) and Δednra+b (k, n) larvae. Loss of dorsoventrally intermediate dlx expression at stage T26.5 is seen in both Δednra (red arrowheads) and Δednra+b (asterisks) larvae, but is more frequent in Δednra+b individuals. By contrast, the ventral hand expression domain remains intact in Δednra and Δednra+b larvae (white arrowheads in l–n), with no measurable change in area as a proportion of total head size (Extended Data Fig. 3). Five out of 14 Δednra (j), and 7 out of 8 Δednra+b (k) individuals showed reduced dlxD expression domains; 0 out of 8 Δednra (m) and 0 out of 9 Δednra+b (p) individuals showed reduced hand expression domains. Pharyngeal arches are numbered in i. o–r, dlx3.S and hand2.L expression domains are highly reduced in X. laevis Δednra.L+S (p, r) relative to wild type (o, q). Three out of 7 Δednra.L+S (p) individuals showed reduced dlx3.S expression domains; 5 out of 8 Δednra.L+S (r) individuals showed reduced hand2.L expression domains. See Extended Data Fig. 4 for X. laevis hand2.L domain quantification. Pharyngeal arches are numbered in o. See Methods, ‘Statistics and reproducibility’ and Supplementary Tables 1–4 for detailed quantification. Anterior is towards the left in all panels. Scale bars, 100 μm.
Ednra controls head skeleton development
To better understand the functional evolution of Edn signalling, we optimized a method for efficient Cas9-mediated mutagenesis in the sea lamprey11 and used it to disrupt the function of ednr, edn and dlx genes. Recent assembly of the sea lamprey germline genome27 supports previous reports that the lamprey has one ednra, one ednrb and six edn genes21,23. Targeting two unique protein-coding sequences to control for off-target effects (Supplementary Table 1), we found that Cas9-mediated F0 mutation of ednra (Δednra) resulted in a hypomorphic pharyngeal skeleton with gaps in the branchial basket, excess and ectopic melanophores, and heart oedema (Fig. 1a–g, Extended Data Figs. 1c, 2). Whereas the Δednra phenotype resembles gnathostome ednra and edn1 mutants, including the ectopic pigment cells28, it differs from the reported effects of an Edn signalling inhibitor29, probably reflecting the specificity of CRISPR–Cas9.
In zebrafish and mouse, Ednra–Edn1 signalling acts, in part, by activating the expression of dlx paralogues in the intermediate pharynx and hand genes in the ventral pharynx8,16,30. We investigated whether lamprey ednra (Fig. 1h) regulates these genes in lamprey NCCs. Despite divergent histories of dlx duplication and loss31, lamprey Δednra larvae exhibited gaps in dlx expression in the intermediate pharynx (Fig. 1i–k, Extended Data Fig. 3a). By contrast, the ventral hand expression domain displayed no gaps, no detectable reduction in staining intensity, and no obvious reduction in size when taking into account the hypomorphic heads of mutants (Fig. 1l–n). To confirm that Ednra signalling regulates dlx and hand in gnathostomes aside from zebrafish and mouse, we used Cas9 to create Δednra.L+S and Δedn1.L+S X. laevis larvae. As in zebrafish and mouse, we observed a hypomorphic oropharyngeal skeleton, loss of the jaw joint (Extended Data Fig. 4a–c) and disruptions in dlx and hand expression that included gaps, decreased in situ hybridization signal intensity, and a reduction in the area of the hand expression domain (Fig. 1o–r, Extended Data Fig. 4d–h). These data suggest that pharyngeal expression of dlx was Edn-dependent in the last common ancestor of lamprey and gnathostomes, whereas hand regulation has diverged between X. laevis and lamprey.
Lamprey Ednr paralogues cooperate
Lamprey ednr genes are broadly coexpressed in postmigratory skeletogenic NCCs during early larval stages (Tahara32 stage 25.5 (T25.5)), a pattern not observed in any gnathostome21 (Fig. 1h). This suggests that ednra and ednrb may both function in lamprey oropharyngeal skeleton development. We thus used three separate single guide RNAs (sgRNAs) to mutagenize ednrb alone (Δednrb), and together with ednra (Δednra+b) (Supplementary Table 1). We found that, similar to gnathostome edn3 and ednrb mutants, lamprey Δednrb individuals have severe reductions in melanophores, the only discernible pigment cells in laboratory-raised lamprey larvae (Extended Data Fig. 5a). However, unlike reported gnathostome ednrb mutants, many Δednrb larvae had skeletal defects, with 27% displaying gaps in the branchial basket (Extended Data Figs. 1d, 5b, Supplementary Table 2). Furthermore, Δednra+b larvae had skeletal defects that were more frequent and severe than those in Δednra or Δednrb larvae, including the complete loss of some branchial bars (Fig. 1d, g, Extended Data Figs. 1e, 5c, Supplementary Table 2). Intermediate dlx expression was also more reduced in Δednra+b larvae than in Δednra larvae, although Δednrb individuals showed no apparent dlx reduction (Extended Data Fig. 3a, Supplementary Table 2). Similar to the single mutants, the hand expression domain of Δednra+b larvae displayed no gaps or obvious reduction in signal intensity, and image analysis confirmed that it was not significantly reduced in size relative to overall head size (Fig. 1n, Extended Data Fig. 3b, c). We also observed reduced pigmentation in Δednra+b larvae (Extended Data Fig. 5c), similar to that in Δednrb larvae, in contrast to the excess pigmentation observed in Δednra individuals. Together, these results show that ednra and ednrb cooperate to drive the differentiation of lamprey skeletogenic NCCs, whereas ednra simultaneously opposes the role of ednrb in promoting melanophore fate.
Edn signalling acts through soxE and dlx
To better understand the function of Edn signalling in lamprey NCC, we analysed the expression of several NCC markers in Δednra, Δednrb and Δednra+b embryos and larvae. Expression of twistA, foxD-A and soxE2 in stage T22–23 Δednra+b embryos suggests that the specification and initial migration of cranial NCCs is largely normal in Δednr individuals (Fig. 2a, b, Supplementary Table 2). In T26.5 Δednra, Δednrb and Δednra+b larvae, expression of myc, ID, soxE1, twistA and msxA also persisted in most postmigratory NCCs, confirming largely normal cranial NCC development, although subtle migration defects cannot be ruled out (Fig. 2c, d, Extended Data Fig. 3d). By contrast, at T26.5, both Δednra and Δednrb larvae displayed clear reductions in soxE2 transcription in the forming branchial bars, and lecticanA (lecA)—a homologue of aggrecan—in the branchial bars and differentiating mucocartilage (Fig. 2e–g, i–k, Extended Data Fig. 1g–k, Supplementary Table 2). Similar reductions in soxE2 and lecticanA were observed in Δednra+b larvae, which also displayed reductions in twistA and soxE1, and localized loss of ID transcripts in oral mucocartilage precursors (Extended Data Fig. 3d). Together, these results show that mutation of either or both ednr genes results in reduced soxE expression and disruptions in skeletogenic NCC differentiation, with these effects occurring most consistently in Δednra and Δednra+b individuals (Supplementary Table 2). Reductions in postmigratory skeletogenic NCC are also seen following perturbation of edn1 or ednra in model gnathostomes13,33,34, although disrupted soxE (sox9a) expression has only been reported in zebrafish33. We thus visualized sox9.S in X. laevis Δednra larvae and found reduced expression (Fig. 2m, n). This suggests that regulation of soxE expression and NCC skeletogenesis are deeply conserved functions of Edn signalling in vertebrates.

a–d, Expression of twistA in migratory NCCs at stage T23 in wild-type (a) and Δednra+b larvae (b; 0 out of 9 with reduced expression in Δednra+b versus wild type) and expression of myc in postmigratory NCCs at stage T26.5 in wild-type (c) and Δednra+b larvae (d; 0 out of 4 with reduced expression in Δednra+b versus wild type) suggest that cranial NCC formation is largely normal in these mutants. e–l, Reduced and discontiguous expression (asterisks) of soxE2 and lecticanA in Δednra (f, j), Δednrb (g, k) and ΔdlxA (h, l) larvae at stage T26.5 versus wild-type larvae (e, i). ll, lower lip; lmp, lateral mouth pate; ul, upper lip. Reduced expression domain phenotype (asterisks) for soxE2 in n = 15 out of 21 Δednra embryos (f), n = 4 out of 8 Δednrb embryos (g) and n = 20 out of 41 ΔdlxA embryos (h). Reduced expression domain phenotype for lecticanA in n = 16 out of 16 Δednra embryos (j), n = 4 out of 8 Δednrb embryos (k) and n = 16 out of 51 ΔdlxA embryos (l). m, n, Expression of sox9.S in wild-type (k) and Δednra (l) X. laevis larvae at stages Nieuwkoop–Faber (NF) 33–34, n = 12 out of 20 Δednra.L+S individuals exhibited reduced sox9.S expression (asterisks). o, p, FGFRa expression in cardiac mesoderm is reduced in Δednra (red arrowhead in p) compared with wild type (white arrowhead in o). n = 3 out of 6 Δednra individuals exhibited reduced FGFRa expression in the heart. See Methods, ‘Statistics and reproducibility’ and Supplementary Tables 1–4 for detailed quantification. Pharyngeal arches (PAs) are numbered in e, i, m (grey numbers indicate the positions of PAs with little or no detectable expression). All panels show left lateral views. Scale bars, 100 μm.
We next investigated whether dlx genes are effectors of Edn signalling in the lamprey pharyngeal skeleton by comparing the phenotype of Δednr and Δdlx individuals. Mutation of dlxA, dlxC and dlxD alone or in combination, resulted in disruptions of soxE2 and lecticanA expression, similar to Δednr larvae (Fig. 2h, i, Extended Data Fig. 6a). At stage T30, Δdlx individuals also had hypomorphic pharyngeal skeletons with gaps in the branchial basket (Extended Data Fig. 6b), though they lacked the heart and pigment defects seen in Δednr larvae. The similar phenotypes of Δednr and Δdlx individuals suggest that, as in gnathostomes, Edn signalling works through dlx genes in lamprey skeletogenic NCCs.
Conserved role for Ednra in the heart
In mouse ednra mutants, defects in cardiac NCCs and mesoderm contribute to a severe cardiac phenotype13,35. Similar to mouse, lamprey ednra transcripts mark the presumptive cardiac mesoderm and heart21, and lamprey Δednra larvae have severe heart defects (Extended Data Fig. 2a–d). We therefore examined the expression of the FGFR homologue FGFRa in Δednra larvae. In addition to being transcribed in lamprey cardiac mesoderm, functional studies suggest that FGFRa signalling is required for lamprey heart development36. We observed a strong reduction in cardiac FGFRa expression in Δednra individuals (Fig. 2o, p). This indicates that the heart oedema seen in lamprey Δednra larvae is caused in part by reduced FGFR signalling in cardiac mesoderm. Whether NCC defects are also involved in this phenotype is unclear, as cardiac NCCs have not yet been identified in lamprey.
Ednrb function in PNS has diverged
Lamprey and gnathostome ednrb genes are widely expressed in the NCCs that form the PNS21, and mammalian Edn3 and Ednrb1 mutants lack parts of their ENS37. We thus examined the expression of several PNS markers in lamprey Δednrb and Δednra+b larvae (Fig. 3a–k, Extended Data Fig. 5d–g). All PNS ganglia and nerves were easily identifiable and present in normal numbers, though select cranial ganglia were misshapen and measurably smaller in double mutants (Fig. 3a, b, g, i, Extended Data Fig. 5d, f). Recently described ENS precursors25 also appeared unaffected in mutants (Fig. 3j, k), although neurofilament-positive chromaffin-like cells in the presumptive kidney were absent38 (Fig. 3g–i, arrowheads). Because PNS defects have been reported only in mammalian Edn3 and Ednrb mutants37, we used CRISPR–Cas9 to target ednrb2 and edn3 genes in X. laevis. Targeting ednrb2 genes resulted in no obvious phenotype, probably owing to incomplete disruption of all three ednrb2 paralogues. By contrast, Δedn3.L+S individuals were frequently leucistic (Extended Data Fig. 7). Whereas all PNS components we visualized, including nascent ENS neurons, cranial nerves and dorsal root ganglia, appeared normal in Δedn3.L+S larvae (Extended Data Fig. 8a–e), subadult Δedn3.L+S frogs had a mild Hirschsprung disease-like phenotype that included missing submucosal ganglia and excess goblet cells39,40 (Extended Data Fig. 8f–q). Our results show that disrupting Ednrb signalling in lamprey and frog causes defects in distinct autonomic components of the PNS. Together with previous work in mammals, these observations imply that the development of most PNS elements was independent of Edn in the last common ancestor of lamprey and gnathostomes. They further suggest that the role of Edn signalling in development of the autonomic nervous system has diverged in tetrapods and/or lamprey. Data from other key groups—such as ray-finned fishes—should help identify ancestral and derived roles for Edn signalling in this NCC derivative.

a–d, HuC/D immunohistochemistry at stage T26.5 reveals a largely intact set of cranial ganglia (a, b) and DRGs (c, d, arrowheads) in Δednra+b larvae, although some cranial ganglia are misshapen (n = 6 out of 6 individuals). e, f, soxE2 expression in DRGs of Δednra+b larvae resembles wild type at stage T26.5. n = 10 out of 10 individuals. g–i, Neurofilament immunohistochemistry at stage T27 reveals that all major facial nerves (white arrows) are present in wild-type (g), Δednrb (h) and Δednra+b larvae (i), although presumptive chromaffin-like cells in the forming kidneys38 (black arrowheads in g) are absent in the mutants (red arrowheads in h, i); Δednrb n = 4 out of 4; Δednra+b n = 3 out of 3 individuals show this phenotype. j, k, phox2 expression at stage 26.5 reveals forming epibranchial ganglia (white arrows) and enteric neuron precursors25 (black arrows) in wild-type (j) and Δednra+b (k; n = 11 out of 11) individuals with wild-type in situ hybridization pattern. l–p, Δedn larvae phenocopy mild Δednr mutants. ΔednA larvae (m) recapitulate the hypomorphic head and heart oedema (brackets) of Δednra larvae (n), but lack the ectopic pigmentation caused by ednra disruption (arrowheads in n). ΔednE (o) exhibits reduced pigmentation, resembling Δednrb larvae (p). ΔednA, n = 22 out of 67; Δednra, n = 264 out of 325; ΔednE, n = 113 out of 154; and Δednrb, n = 177 out of 403 individuals exhibited similar phenotypes to those shown here. See Methods, ‘Statistics and reproducibility’ and Supplementary Tables 1–4 for detailed quantification. All panels show left lateral views, except c–f, which show dorsal views of the trunk (anterior on top). Scale bars, 100 μm; scale bars in a and c also apply to b and d, respectively.
Lamprey Ednrs have dedicated ligands
In vitro binding assays41 and the similarity of edn and ednr mutant phenotypes7,30,33 suggest that Edn1 is the main ligand for Ednra, whereas Edn3 is the main ligand for Ednrb. To test whether lamprey Ednra and Ednrb also have dedicated ligands, we mutated ednA, ednC and ednE, the only edn genes expressed in tissue-specific patterns during sea lamprey development21. Targeting ednC with three different sgRNAs yielded no reproducible mutant phenotype (see Methods). By contrast, lamprey ΔednA larvae displayed a combination of heart oedema and skeletal defects that resembled hypomorphic Δednra individuals, but without pigmentation defects, whereas ΔednE larvae resembled Δednrb larvae (Fig. 3l–p, Extended Data Fig. 9). The incomplete loss of melanophores in ΔednE and Δednrb larvae mimics amniote and teleost edn3 and ednrb1 mutants10,42, whereas edn3 mutant salamanders are completely leucistic43. We noted that, similar to salamanders, a high percentage of Δedn3.L+S X. laevis exhibited a complete loss of NCC-derived pigmentation (Extended Data Fig. 7, Supplementary Table 1). We conclude that all modern vertebrates have an edn that is largely dedicated to ednrb, and that NCC-derived pigment cell development in modern amphibians is particularly dependent on Edn3–Ednrb signalling.
Evolutionary history of edn and ednr genes
Despite inconclusive phylogenies6,21,23, the similarity of mutant phenotypes raise the possibility that lamprey ednA and ednE are cryptic orthologues of gnathostome edn1 and edn3, respectively. We therefore used synteny data from the recently completed sea lamprey germline genome27 to reevaluate ednr and edn phylogeny. Other than ednra, our analyses fail to support one-to-one orthology of lamprey and gnathostome ednr or edn genes (Extended Data Fig. 10), consistent with previous reports5,21 and recent genomic comparisons44. Although these analyses leave the precise history of edn and ednr duplication and loss unresolved, synteny and phylogenetic analyses of flanking genes support co-orthology of lamprey ednrb, ednE, ednA and gnathostome ednrb1 and ednrb2, edn1 and edn3, and edn2 and edn4, respectively. These relationships suggest that duplication of single primordial ednr and edn genes in the vertebrate stem yielded ednra, ednrb and two edn genes; the ancestors of the edn1–edn3–ednE and edn2–edn4–ednA clades. The conserved roles of lamprey EdnE and gnathostome Edn3 further suggest that after this initial ‘1R’ duplication, and possibly after an additional duplication event, a member of the Edn1–Edn3–EdnE paralogy group became largely specialized for Ednrb binding. Later, after the divergence of cyclostomes and gnathostomes, non-orthologous Edns (EdnA and Edn1) became independently specialized for Ednra binding in each lineage (Fig. 4f, Extended Data Fig. 10).

a, Our results suggest that the vertebrate ancestor had bona fide multipotent NCCs that activated the NCC gene-regulatory network1, but developed in the absence of Edn signalling. b, Before the first whole-genome duplication (1R) in stem vertebrates, the primordial Edn signalling system was co-opted to NCC, affecting the patterning and/or proliferation of non-neural NCC derivatives, but having little effect on the autonomic nervous system. c, Later in the vertebrate stem, duplication and specialization of the Ednra and Ednrb signalling pathways resulted in three or four NCC populations with different Edn signalling requirements, depending on when the cardiac NCC lineage arose. d, e, Changes to Ednra and Ednrb signalling targets correlate with divergence of the oropharyngeal skeleton and autonomic nervous system in the lineages leading to modern cyclostomes (d) and gnathostomes (e). f, The deduced transitional forms depicted in a–e mapped onto a phylogenetic tree of extant chordate groups. The colours of the arrows and lines reflect the steps of Edn signalling system evolution depicted in a–e. The inferred origin of edn and ednr, and their duplication during the vertebrate genome-wide duplications (1R and 2R)44, are shown with grey arrows.
Conclusions
The origin and early evolution of NCCs has been linked to the rewiring of gene-regulatory networks1, the evolution of new genes3 and genome-wide duplication events4. Although these are attractive hypotheses, functional genetic evidence conclusively linking vertebrate-specific genes and/or gene duplications to NCC evolution is sparse. This is largely owing to the difficulty of inferring ancestral gene functions using conventional genetic model organisms, which represent only a fraction of vertebrate diversity. We compared the roles of Edn signalling in lamprey, X. laevis, mammals and zebrafish to deduce when different Edn signalling functions arose (Fig. 4). All lamprey and gnathostome Edn signalling mutants have defects in the patterning, differentiation, and/or quantity of non-neural neural crest derivatives. However, early NCC development in these mutants, including lamprey and mouse ednra/ednrb double mutants45 appears largely normal, and all major NCC derivatives are discernable. This suggests that Edn signalling was probably first activated in a stem vertebrate that already had bona fide multipotent NCCs (Fig. 4a), and its recruitment affected the later patterning and/or proliferation of NCCs (Fig. 4b). The fact that all lamprey and gnathostome Ednrs function during NCC development also strongly suggests that integration of Edn signalling system into the neural crest gene-regulatory network occurred before the vertebrate genome duplications. We also find conserved specialization of the lamprey and gnathostome Ednra and Ednrb pathways in the major NCC lineages. This indicates that after the first or second vertebrate genome-wide duplication events, paralogous Edn signalling pathways acquired distinct functions in different NCC populations, as previously hypothesized6. This resulted in three, and possibly four, NCC populations with different Edn signalling requirements in stem vertebrates (Fig. 4c). We speculate that this new Edn signalling-based developmental modularity facilitated the independent evolution of these NCC populations and their derivatives. For example, after Ednra and Ednrb specialization, alterations in the Ednra-signalling pathway yielding adaptive skeletal phenotypes would be expected to have little effect on development of the PNS or pigmentation. Consistent with notion, we find differences in lamprey and gnathostome Edn signalling function that correlate to their divergent oropharyngeal skeletons and PNSs (Fig. 4d). We posit that divergence of Edn signalling targets contributed to the morphological divergence of modern jawed and jawless vertebrates. Together, our results link the stepwise recruitment, duplication and functional divergence of Edn signalling pathway components to the stepwise evolution of NCCs and their derivatives (Fig. 4f).
Methods
No statistical methods were used to predetermine sample size, because we could not accurately predict the general nature and thus the ‘effect size’ of the phenotypes resulting from our experimental manipulations. Because most phenotypes arising from the manipulations were visually obvious, and in order to perform in situ hybridization (ISH) and immunohistochemistry (IHC) assays truly in parallel (in the same tubes) for control, the experiments were not randomized, nor were the investigators blinded to allocation during experiments and outcome assessment.
P. marinus husbandry
P. marinus fertilizations and husbandry were carried out as described previously11. Adult spawning phase sea lampreys were housed in 200-l tanks containing reverse-osmosis-purified water with 800–1000 ppm artificial sea salt. Water in the tanks was completely replaced daily. Once ripe, the animals were stripped of gametes into Pyrex dishes, where in vitro fertilization took place in deionized water containing 400–600 ppm artificial sea salt. All animals were wild-caught from fresh water streams during their late spring–early summer spawning season, with the majority being derived from an invasive population in Lake Huron. A small fraction (1%), were trapped at the Holyoke Dam in Massachusetts. Each sgRNA was injected into clutches from at least of two different pairs of adults. Embryos and larvae were kept at 18 °C in Pyrex dishes containing deionized water and 400–600 ppm artificial sea salt. Depending on the quality of oocytes (almost all mature males produce sperm capable of fertilization) which appears determined by female broodstock health and progression of the spawning season, uninjected sea lamprey embryos display survivorship to stage T26.5 from 1–99%. Dead embryos and larvae were removed daily from each dish and the water was changed at least every other day. All P. marinus staging was as described32. All P. marinus husbandry and experiments were in accordance with CU-Boulder IACUC protocol no. 2392.
X. laevis husbandry
X. laevis fertilizations and husbandry were performed according to standard methods46. Adult females were induced to ovulate via injection of human chorionic gonadotropin, and eggs were stripped into Petri dishes. Testes were dissected from males, homogenized, and applied to the eggs for in vitro fertilization. All frog staging was according to Nieuwkoop and Faber47. All X. laevis husbandry and experiments were in accordance with CU-Boulder IACUC protocol no. 2392.
F0 mutagenesis strategy
We used CRISPR–Cas9-mediated mutagenesis to induce deletions and insertions (indels) into the protein-coding exons of injected F0 sea lamprey (P. marinus) and African clawed frog (X. laevis) embryos as previously described11,48,49,50,51,52. Although CRISPR–Cas9 is highly efficient in sea lamprey, differences in the efficiency of individual sgRNAs results in different ratios of wild-type and mutant alleles in F0 mosaic mutants. This variable mosaicism results in different sgRNAs producing phenotypically mutant individuals at different frequencies, with a range of severities. Previous work shows targeting an evolutionarily conserved, embryonically expressed gene typically results in 20–90% of injected individuals displaying a gene-specific mutant phenotype11,48,49,50,51. Work in our laboratory with 35 guides targeting 20 different developmental regulators confirms this, with an average of 46% phenotypically mutant individuals produced per gene-specific sgRNA (Supplementary Table 1, Extended Data Fig. 11).
Also as previously reported, the severity of a CRISPR–Cas9-induced phenotype correlates well with the percentage mutant alleles; with most ‘severely affected’ F0 mosaic mutants typically exhibiting 75–100% mutant (indel) alleles11,48,49,50,51. Consistent with this, the 74 severely affected phenotypic mutants selected for genotyping in this study had an average of 88% indel alleles at targeted loci (Supplementary Table 4). Importantly, every severely affected individual selected for genotyping had indel mutations at the targeted locus. Thus, as with traditional inbred mutant lines, the phenotype of CRISPR–Cas9-generated F0 mosaic mutants is a strong predictor of their genotype.
Based on these observations, we devised a strategy for creating, selecting, and analysing CRISPR–Cas9-generated sea lamprey and X. laevis mutants. First, two or more unique sgRNAs were designed against protein-coding exons of the gene of interest. When possible, we selected unique, but evolutionarily conserved regions to increase the chances that in-frame deletions will disrupt functionally critical domains and yield loss-of-function alleles. Second, individual sgRNAs were co-injected with Cas9 protein or mRNA into zygotes or, in the case of X. laevis, zygotes and two-cell stage embryos. Third, F0 injected embryos were monitored daily and scored for morphological defects. Fourth, morphological defects associated with two or more sgRNAs targeting the same gene were designated as the putative ‘mutant phenotype’ for that gene. For example, the unique pigmentation defect seen when targeting ednrb exons was deemed the putative ‘ednrb mutant phenotype’ only after two different sgRNAs targeting the ednrb locus produced the same defect. Fifth, mutagenesis of the targeted loci was confirmed by genotyping several representative severely affected phenotypic mutants (see below for genotyping method). Sixth, once mutant genotype and mutant phenotype were linked by showing all selected mutants had mutant alleles, severely affected phenotypic mutants were picked for analyses via in situ hybridization, alcian blue staining, immunohistochemistry and toluidine blue staining (see below for protocols). For dlx sgRNAs, which resulted in unusually high mortality before larval stages, probably owing to the early function of dlx genes in neurectoderm patterning, severe phenotypic mutants were lightly fixed and genotyped after in situ hybridization analysis as recently described49,50. This additional step was performed to re-confirm the link between mutant phenotype and mutant genotype in the relatively small number of surviving dlx mosaic mutants.
P. marinus sgRNA and Cas9 injections
We mutagenized the P. marinus dlxA, dlxC, dlxD, ednA, ednC, ednE, ednra and ednrb loci by injecting zygotes with at least two unique sgRNAs per gene (Supplementary Table 1). To create ednra+b double mutants, zygotes were injected with four different combinations of ednra and ednrb guides. dlxA+C+D triple mutants were created using a single sgRNA 100% complementary to dlxA and dlxD, with one mismatch to dlxC (Supplementary Table 1). As previously described, sgRNA target sites were chosen using all available transcriptome sequence data to avoid protein-coding off-targets11. In brief, candidate sgRNA sequences demonstrating off-target matches with >80% overall identity in the target site, and >90% identity in the 3′ half of the target site (closest to the protospacer adjacent motif (PAM) site) to any off-target sequence (with an NGG PAM site) were not used. Lamprey zygotes were injected as previously described with approximately 5 nl of a solution containing 400 pg sgRNA, and either 800 pg Cas9 protein (a 2:1 ratio of protein:sgRNA by mass) or 1 ng Cas9 mRNA, 5 mg ml−1 lysinated rhodamine dextran (LRD) and nuclease free water. For ednra + ednrb combined experiments, 200 pg of each of two sgRNAs were used with 800 pg of Cas9 protein. Approximately 200–500 zygotes were injected per experiment, and each sgRNA–Cas9 combination was injected into zygotes from at least two different pairs of wild-caught sea lampreys.
As in other vertebrates53, microinjection of lamprey embryos causes increased mortality before gastrulation and developmental delay compared to uninjected sibling controls11. Owing to differences in the quality of female broodstock (we see no difference in sperm quality among mature males), person injecting and progression of the spawning season, this microinjection-induced mortality can range from 10–90%. However, after gastrulation, clutches of microinjected embryos have a survivorship to early larval stages (T26–T30) similar to uninjected siblings, typically around 90%. This was true for all sgRNAs tested in this study, except for the dlx sgRNAs, which had substantially increased mortality to larval stages compared to uninjected siblings, resulting in 30–40% survival to T26.5. We suspect this is due to the early roles of dlx genes in neurectoderm patterning.
X. laevis sgRNA and Cas9 injections
In the tetraploid frog X. laevis, both the ‘long’ (L) and ‘short’ (S) homeologues (following the gene nomenclature convention of Session et al.54) of X. laevis edn1, edn3 and ednra were simultaneously targeted52 (Supplementary Table 1). Zygotes or two-cell embryos were injected with a 5 nl droplet containing 800 pg of a single sgRNA targeting both edn3.L and edn3.S, or 400 pg of each of two sgRNAs targeting edn1.L and edn1.S, or ednra.L and ednra.S, alongside either 1 ng of Cas9 mRNA, or 1.6 ng of Cas9 protein. X. laevis injection mixes were supplemented with 5 mg ml−1 LRD and/or 300 pg eGFP mRNA (per 5 nl injection droplet). Approximately 50–200 zygotes were injected per experiment.
P. marinus CRISPR–Cas9 controls
To demonstrate that the phenotypes associated with each sgRNA injected were due to disruption of the targeted genes, rather than to off-targets, each P. marinus gene was targeted with at least two unique sgRNAs. All sgRNAs targeting the same gene produced the same mutant phenotype, though usually with different efficiencies (Supplementary Table 1).
To further validate sgRNA specificity in P. marinus, and to ensure that the CRISPR–Cas9 method does not artefactually cause any of the described defects, we used two negative control strategies. In addition to the negative control sgRNA described in our methods paper11, we tested an intron-spanning sgRNA partially complementary to two separate exons of the P. marinus ednrb gene (see Supplementary Table 1 for sequence). Neither sgRNA produced a phenotype (Supplementary Table 1), though both resulted in a slight developmental delay, as previously reported11. In addition to these ‘untargeted’ sgRNA negative controls, we also injected more than 20 other sgRNAs complementary to the exons other P. marinus developmental genes (Extended Data Fig. 11). These sgRNAs were designed to disrupt developmental regulators expressed in the developing head at the same time as ednr, edn and dlx. None of these negative control sgRNAs yielded the ednr or edn mutant phenotypes, though three sgRNAs (a2cg1, p19g1 and w11g3) produced phenotypes grossly similar to dlx mutants (Extended Data Fig. 11).
Severe heart oedema (approximate heart cavity volume greater than 3× normal by visual inspection) is part of both the ednra and ednA mutant phenotypes, and occurs at a high frequency in embryos injected with sgRNAs targeting fgf8/17/1811 (Extended Data Fig. 11). This raised the possibility that heart oedema could be a non-specific side-effect of sgRNA–Cas9 injection. To test this, we counted the number of negative control larvae, aside from those injected with fgf8/17/18 sgRNA, displaying heart oedema (Extended Data Fig. 11). Of 21 pools of larvae injected with 21 different negative control sgRNAs, 9 pools displayed no detectable heart oedema, while 11 displayed heart oedema of various severities at a frequency of 7.7% or lower. One sgRNA yielded severe heart oedema at a frequency of 27%. These data show that severe heart oedema is not a general side effect of the CRISPR–Cas9 method in lamprey.
X. laevis CRISPR–Cas9 controls
An edn3 morphant phenotype was previously reported in X. laevis18. An sgRNA designed to simultaneously target the edn3.L and edn3.S homeologues yielded a severe version of the X. laevis edn3 morphant phenotype that mimicked salamander edn3 mutants43, confirming its specificity. For edn1 and ednra, we designed separate sgRNAs against the L and S homeologues and performed negative controls by individually injecting each sgRNA separately as reported previously52. This strategy relies on redundancy of the X. laevis homeologues to show that neither sgRNA alone causes any spurious morphological defects. The fact that defects are only obtained by simultaneous disruption of homeologues, serves as a control showing that the phenotype is specifically due to a loss of edn1 and ednra function52.
Scoring of mutant phenotypes
Successfully injected embryos were identified by fluorescence of the LRD lineage tracer at 4–6 days post fertilization and dead and LRD-negative larvae were discarded. Successfully injected embryos and larvae were then monitored for morphological abnormalities as they developed. Suites of morphological defects associated with injection of a particular sgRNA, and also seen when injecting one or more other sgRNAs targeting the same gene, were designated as the ‘mutant phenotype’ for that gene. Of embryos and larvae displaying the ‘mutant phenotype’, we deduced, based on previous work, that most severe had more than 75% mutant alleles and were likely near null-mutants11,48,49,50,51. This assumption was supported by genotyping representative severe mutants for all targeted genes (see ‘Genotyping’).
For each gene, we focused on ‘severely affected’ mutants for detailed morphological and histological analyses. The severe mutant phenotype of all genes was apparent at pharyngula stages onward (stage T26.5 for lamprey, stage 41 for X. laevis) and defined as follows. For X. laevis Δedn1 and Δednra the severe mutant phenotype was defined as a reduction in head size (all structures anterior to the heart) to approximately 70% of WT size or smaller. For P. marinus ΔednA the severe mutant phenotype was defined as a reduction in head size to approximately 70% of its WT size or smaller, together with heart oedema. For Δednra, the severe mutant phenotype was defined as a reduction in head size to approximately 70% of its WT size or smaller, together with heart oedema, and ectopic pigmentation around the heart. For ΔdlxA, ΔdlxC, and ΔdlxD, severe mutants were defined as having a head reduced to approximately 70% of WT size or smaller. For Δ Δedn3.L+S, ΔednE, and Δednrb severe mutants were defined as having a 50% reduction in the number of melanophores or greater (in the case of X. laevis injected unilaterally at the 2 cell stage, this applies only to the injected side). For the Δednra+b, the severe mutant phenotype was defined as an approximately 70% reduction in head size, heart oedema, and approximately 50% reduced pigmentation. All larvae demonstrating a ‘severe mutant phenotype’ were counted and are presented as fraction of the total number of LRD-positive embryos and larvae that survived to fixation at a stage were phenotype could be scored (Supplementary Tables 1 and 2).
As in other vertebrates53, sea lamprey embryos injected with negative control sgRNAs, DNA constructs, or any other synthetic oligonucleotide, display a slight developmental delay. In sea lamprey we find that a delay of ~5% is typical, that is, 10-day-old injected embryos and larvae typically appear 9.5 days old compared to unmanipulated siblings. Thus, developmental events such as somite segregation, yolk absorption, gill openings and melanin deposition32 were used, rather than days post-fertilization, to stage-match mutant and negative control embryos.
Statistics and reproducibility
See Supplementary Tables 1–4 for quantification, statistics, and experiment information for all assays in this work, including: larval phenotype frequencies observed for each sgRNA and the number of times each sgRNA was injected for this work (Supplementary Table 1); ISH, IHC and histological assay total numbers observed and assigned as affected, and the number of times each experiment was repeated (Supplementary Table 2); hypothesis testing of our observed phenotypic rates as significant effects versus null-background deformity rates (Supplementary Table 3) and a summary table of how many animals were genotyped for each target site (Supplementary Table 4). This information is also described in detail below for each assay.
We have never observed the ednra, ednrb, ednra+b, ednra.L+S, ednA, ednE or edn3.L+S mutant phenotypes in WT or negative control embryos. In other words, the ednra, ednrb, ednra+b, ednra.L+S, ednA, ednE or edn3.L+S phenotypes are only seen in embryos and larvae injected with Cas9 and sgRNAs targeting these genes. Similarly, we have never observed the reduced expression patterns we report in WT or negative control embryos. However, non-specific body axis deformities (mainly incomplete yolk sac extension) can occur at a frequency of 5–8% in surviving uninjected and negative control-injected larvae. While these deformities are qualitatively different from the ednra, ednrb, ednra+b, ednra.L+S, ednA, ednE or edn3.L+S mutant phenotypes, we used this background level of developmental deformity as a proxy to estimate mutant phenotype frequencies in negative control sea lamprey larvae (Supplementary Table 3). Using the conservative estimate that one out of ten negative control (untreated) individuals will spontaneously display the observed phenotypes, we applied Fisher’s exact test to evaluate the null hypothesis that our treatments can be explained by a high ‘background’ level of developmental deformities. This null hypothesis is rejected with P values of <0.017 for all mutant phenotypes, with the majority having P values ≪ 0.000001 (see Supplementary Table 3 for individual P values).
Most in situ hybridization, immunohistochemistry and histological stains were performed on embryos and larvae displaying the consistent, severe morphological phenotype characteristic of each targeted gene. Because these specimens were non-randomly selected phenotypic mutants, statistical analysis is inappropriate. For preselected phenotypic mutants, we report the fraction of those assayed by ISH displaying disrupted gene expression patterns in Supplementary Table 2. The remaining ISH assays were performed on embryos before the mutant phenotype became apparent and severe mutants could be selected. In these cases, selected individuals were a random sample of the pool of sgRNA–Cas9 individuals and could be compared with untreated controls with Fisher’s exact test (Supplementary Table 3). For these experiments, we assumed spontaneous disruption of gene expression in 5 out of 100 of untreated, WT embryos and larvae. We view this assumption as conservative as we have never observed such variation in gene expression patterns in wild-type embryos that have been properly processed for in situ hybridization or immunohistochemistry. Under this assumption, every reported effect of ‘no expression change’ in this work is consistent with a null hypothesis of no effect or 5% background levels of gene disruption (Fisher’s exact test, all P > 0.35, see Supplementary Table 3 for individual values). For all genes we report as having discontiguous, missing, or otherwise reduced gene expression after treatment, the null hypothesis is rejected with P < 0.004 (See Supplementary Table 3 for individual values).
Genotyping
To confirm successful mutagenesis, individual severe F0 mutants were genotyped by preparing genomic DNA, PCR amplifying the target site, subcloning the amplicons and Sanger sequencing individual alleles as previously described11,48,49,50,51. In total, 86 diploid loci across 74 individuals were genotyped for this work (some animals were genotyped at multiple loci). See Supplementary Table 4 for a breakdown of individuals and target sites genotyped. Target sites and genotyping primers for each sgRNA are in Supplementary Table 1. Overall, we genotyped at least 3 severely affected individuals for each targeted gene or combination of genes (Extended Data Figs. 2, 4, 5, 7, 8, 10, Supplementary Table 2) except in the case of the P. marinus ednrb sgRNA2 target site, which probably lies immediately adjacent to an intron–exon boundary conserved across jawed vertebrates (on the 5′ end of exon 4 in zebrafish ednraa (NM_001099445.2)), and is incompletely assembled in all three publicly available genomic assemblies (including the most recent petMar327). For dlx mutants, genotyping after ISH of lightly fixed mutants was performed as previously described49,50,51.
Frequently, we found six or more unique indel alleles at a given locus in a single specimen (in X. laevis, we consider the homeologous L and S loci separately), which indicates that biallelic Cas9-driven mutagenesis is still occurring after the second cleavage event in both species. As previously reported11,52,55, when insertions of DNA fragments were discovered, these motifs often appeared on the endogenous reverse or forward strand near the target site or induced lesion (see green and purple nucleotide strings in Extended Data Figs. 2, 4, 5, 7, 10).
In situ hybridization, immunohistochemistry and histological staining
All ISH, alcian blue cartilage staining and toluidine blue staining was carried out as described previously22,56,57. The cDNA sequences used to synthesize lamprey riboprobes were dlxA22, dlxB22, dlxD22, FGFRa36, foxD-A58, hand22, ID59, lecA (GenBank: MK487484.1; see Extended Data Fig. 1 for WT expression), msxA22 (formerly referred to as msxB), myc58, phox260, soxE161, soxE262, twistA58 and soxB1b63. The cDNA sequences used to synthesize X. laevis riboprobes were phox2a64, dlx3.S56, hand2.L56 and sox9.S56. The antibody used for riboprobe detection ISH was anti-digoxygenin-alkaline phophatase, diluted 1:3,000 (Sigma SKU 11093274910). Neurofilament IHC was as described previously65 (primary antibody, Fisher 13-0700 (diluted 1:300); secondary antibody, Fisher G-21060 (diluted 1:2,000)), with the addition of 1% dimethyl sulfoxide (DMSO) to the phosphate buffer solution before the blocking step, and for X. laevis only, the secondary antibody was incubated overnight at 4 °C. For HNK-1 IHC66, digestive tracts were dissected from 2-year-old subadult frogs (see Extended Data Fig. 8f) and fixed in MEMFA overnight at 4 °C. The guts were rinsed once and washed twice for 10 min in 1× PBS at room temperature, and stored in PBS at 4 °C overnight. Thin (0.5–1 mm wide) transverse rings of the small and large intestines were cut with a razor blade. The samples were then pretreated with PBS-Triton X-100 + 1% DMSO for 1 h, followed by a 2 h block at room temperature in 10% heat-inactivated goat serum (all blocking solutions are PBS-Triton X-100 supplemented with either 5% or 10% heat-inactivated goat serum, as specified below). The HNK-1 primary antibody (Sigma SKU C6680) was diluted 1:10 in block (10% goat serum) and incubated with the samples for 1–3 days at 4 °C with high agitation. The samples were then washed with PBS-Triton X-100 at least six times over a 3-h interval before being incubated with the Alexa Fluor 488-conjugated secondary antibody (Fisher A-21042), diluted 1:100 in block (10% goat serum), for either 4 h at room temperature or overnight at 4 °C, agitated. Samples were then washed at least three times for 10 min in PBS-Triton X-100 and imaged. HuC/D IHC was performed essentially as described previously62,67, diluting the primary antibody (Fisher A-21271) 1:200, and the Alexa Fluor 488-conjugated secondary antibody 1:150 (Fisher A-11001), both in block (5% goat serum).
To ensure equivalent signal development in injected and WT individuals, morphologically stage-matched WT embryos, larvae or tissue samples were included in every ISH, IHC and histological staining experiment, with WT and treated larvae kept in the same tubes, with the caudal 1/4 cut off for identification when necessary. In addition, to verify that none of the disrupted expression patterns or aberrant histology of mutants could be explained by slight developmental delay, a previously reported side effect of sgRNA+Cas9 injection, WT embryos one stage younger were also used for comparisons; for example, morphological stage T26.5 mutant larvae were compared with both morphological stage T25.5 and stage T26.5 WT larvae. The number of embryos and larvae processed for each histological method, and the frequencies of aberrations, are reported in Supplementary Table 2.
Paraffin sectioning of subadult frog digestive tracts and haematoxylin and eosin (H&E) staining was performed per standard methods with some modifications. Entire frog digestive tracts were fixed in MEMFA overnight at 4 °C, rinsed once and washed twice for 10 min in 1× PBS at room temperature, and stored in PBS at 4 °C for one week. Transverse samples, ~3 mm (that is, ‘rings’ of gut tissue), were cut with a razor blade from the distalmost large intestine. Dissected gut tissue samples were washed 5 min each in 30, 50, 70% ethanol (in deionized water), then twice for 10 min in 100% ethanol, followed by 10-min washes in 50% ethanol/50% Hemo-De (Electron Microscopy Biosciences), 75% Hemo-De/25% ethanol and twice for 10 min in 100% Hemo-De. Samples were then washed in preheated 50% Hemo-De/50% paraffin wax at 67 °C for one hour, then rinsed and washed into preheated 100% paraffin wax overnight at 67 °C. Samples were then embedded, mounted, and sectioned on the transverse plane at a thickness of 7 μm using a Microm HM340E microtome (see Extended Data Fig. 8f). Slides with sectioned samples were allowed to desiccate overnight in a 37 °C dry incubator. Slides were then annealed face up for 10 min at 67 °C, allowed to cool to room temperature, and then de-waxed by washing twice for 10 min in Hemo-De, once for 10 min in 100% ethanol, once for 10 min in 75% ethanol, and 2 for 10 min in deionized water. H&E staining was performed immediately, using the following series of washes: haematoxylin solution (VWR) for 3 min, tap water twice for 20 s, bluing solution (0.1% sodium bicarbonate) for 2 min, tap water for 20 s, acid alcohol (0.32% HCl in 95% ethanol) for 20 s, tap water for 20 s, eosin solution (VWR) for 30 s, 95% ethanol twice for 2 min, 100% ethanol twice for 2 min, then Hemo-De twice for 5 min. Slides were then immediately coverslipped with Permount (Fisher), allowed to dry overnight and imaged on a compound microscope.
Dorsal root ganglia counts, submucosal ganglia quantification, and cranial ganglia size analyses
To quantify DRGs in Δedn3.L+S X. laevis, we counterstained whole-mount neurofilament IHC specimens with DAPI, flat-mounted the dissected trunks (all myomeres), and viewed them with a compound microscope. We ran a Student’s one-sided t-test on the WT versus Δedn3.L+S DRG counts from each left and right half of each animal and found no significant difference (P = 0.381). For this test, we reduced the degrees of freedom to match the number of individuals we analysed (rather than the number of left and right halves individually measured). To quantify Δednra+b P. marinus DRGs, we counted the number of HuC/D-positive clusters in the first ~10 myomeres (anterior to the yolk), left and right halves combined. This subset was chosen owing to variation in WT posterior DRG staining at stage 26. We ran a Student’s one-sided t-test on the WT versus Δednra+b DRG counts and found no significant difference (P = 0.129).
To quantify the reduction in Δedn3 submucosal ganglia, we counted the number of ganglia visible by HNK-1 IHC (Extended Data Fig. 8k–n), and divided that number by the total area of each gut fragment assayed (counted and/or measured in ImageJ68) to find the average number of submucosal ganglia per unit area in each treatment. The material used were distalmost gut pieces derived (dissected by hand with a razor blade, averaging ~1.1 mm2 in surface area per sample) from n = 4 Δedn3 and n = 3 WT frogs, for a total area analysed of 5.4 mm2 for WT, and 6.6 mm2 for Δedn3. A Student’s one-sided t-test yielded a P value of 0.0015, suggesting that these ganglia are reduced in Δedn3 frogs.
To quantify the planar lateral area occupied by different cranial ganglia in P. marinus, we size-calibrated images of n = 4 WT and n = 6 Δednra+b lampreys and used ImageJ to quantify the area of each (counting nodose 1–5 as a single field). We then tested for a difference in these raw area values using one-sided t-tests. We found significant differences only in the area of two ganglia, opV (P = 0.0076) and n1–5 (P = 0.0012), as was recently observed in soxE2 mutant lampreys62. No other ganglia tested yielded a significant difference in area (mmV P = 0.276, g/all P = 0.189, p P = 0.289, pll P = 0.212). Abbreviations of ganglia are as follows: all, anterior lateral line; g/all, geniculate/anterior lateral line (fused); mmV, maxillomandibular trigeminal; n1–5, nodose 1–5; opV, ophthalmic trigeminal; p, petrosal; pll, posterior lateral line.
Ectopic pigment analyses
To test for the presence of excessive pigment cells in Δednra lampreys, we applied equivalent contrast thresholds to whole lateral images of WT and Δednra mutant lampreys (n = 5 each) and inferred the percentage of melanin cover using an image analysis. Lampreys were fixed as described above, washed into 50% glycerol to clear them slightly, and imaged laterally on a white background with intense lighting. We traced the outline of each lamprey, applied a contrast threshold that only selected melanized tissue (see Extended Data Fig. 2f), and calculated the pixel cover within each body using ImageJ. Each of five images contained a single WT and a single Δednra lamprey, thus ensuring that threshold values were applied equivalently between WT and Δednra. A Student’s one-sided t-test supported an increase in melanin cover in Δednra (P = 0.0075), suggesting that melanophores have overproliferated and/or migrated to ectopic locations.
Using bright-field images, we also counted the number of melanophores present in WT (n = 5) and Δednra (n = 9) lamprey ventral fin folds, a region not usually heavily pigmented in WT. Using a Student’s one-sided t-test, we also found a significant increase in melanophores in this specific region (P = 0.00051). See Extended Data Fig. 2e.
Ventral hand/hand2.L domain size ratio analysis
To quantify any difference in hand (P. marinus) or hand2.L (X. laevis) expression domain sizes in the ventral pharynx in Δednra+b P. marinus and Δednra.L+S X. laevis, we quantified the ratio of lateral X/Y plane hand/hand2.L domain size to head ratios on both the left and right side of. Using right and left lateral images of WT and ednra mutant P. marinus and X. laevis, we used ImageJ to outline the head (from the anterior end to the back of the pharyngeal skeleton) and the hand or hand2.L expression domain. We did this for n = 8 Δednra+b P. marinus versus n = 6 WT P. marinus, and n = 8 Δednra.L+S X. laevis versus n = 4 WT X. laevis. Dividing the hand orthologue expression domain size by the overall head size yielded hand domain:head size ratios for each species, which are graphed in Extended Data Figs. 4h, 5h (for X. laevis and P. marinus, respectively). To test for significant differences in the WT versus mutant groups, we ran t-tests to characterize any consistent difference between WT and ednra mutants. For these tests, we grouped the hand domain:head size ratio value from each image by treatment, and reduced the degrees of freedom to match the number of individuals we analysed (rather than the number of images measured). We found that in X. laevis, the hand2.L expression domain did significantly decrease in size (Student’s one-sided t-test P = 0.000833), as expected from work in other model vertebrates (see text). However, unexpectedly, the P. marinus hand expression domain does not appear to be proportionally reduced, and trended towards an increase in its proportional size (Student’s two-sided t-test P = 0.0647).
Synteny and phylogenetic analysis
For the Ednrs and ligands, we looked at synteny at each locus where possible (Extended Data Fig. 11). The synteny analysis was performed by finding the coding sequences of all ednr and edn ortholologs in the 2017 P. marinus genome27 via the UCSC genome browser (https://genome.ucsc.edu/) and comparing the neighbouring genes to that of chicken (Gallus gallus) and/or human (Homo sapiens) as published previously5. For the Edns, synteny information alone was ambiguous and amino acid similarity across large phylogenetic distances is poor (other than in the 21-amino-acid secreted peptide sequence, which is highly conserved). We thus used the conceptual gene products (both separately and concatemerized) of the closely linked hivep and phactr genes to deduce the likely evolutionary history of these gene families6 (Extended Data Fig. 10c–e). For the Ednrs, we repeated an amino acid similarity analysis according to the same methods as we used previously21, but with a subset of sequences. All phylogenetic and molecular evolutionary analyses were conducted using MEGA version 669. See Supplementary Table 5 for all accession numbers associated with these analyses.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All data generated or analysed, and all methods used during this study are summarized in the Article (and its Supplementary Information). The raw data and images are available from the first and second authors upon reasonable request.
References
Meulemans, D. & Bronner-Fraser, M. Gene-regulatory interactions in neural crest evolution and development. Dev. Cell 7, 291–299 (2004).
Martik, M. L. et al. Evolution of the new head by gradual acquisition of neural crest regulatory circuits. Nature 574, 675–678 (2019).
Martinez-Morales, J. R., Henrich, T., Ramialison, M. & Wittbrodt, J. New genes in the evolution of the neural crest differentiation program. Genome Biol. 8, R36 (2007).
Ohno, S. Evolution by Gene Duplication (Springer–Verlag, 1970).
Braasch, I. & Schartl, M. Evolution of endothelin receptors in vertebrates. Gen. Comp. Endocrinol. 209, 21–34 (2014).
Braasch, I., Volff, J. N. & Schartl, M. The endothelin system: evolution of vertebrate-specific ligand-receptor interactions by three rounds of genome duplication. Mol. Biol. Evol. 26, 783–799 (2009).
Miller, C. T., Schilling, T. F., Lee, K., Parker, J. & Kimmel, C. B. sucker encodes a zebrafish Endothelin-1 required for ventral pharyngeal arch development. Development 127, 3815–3828 (2000).
Miller, C. T., Yelon, D., Stainier, D. Y. & Kimmel, C. B. Two endothelin 1 effectors, hand2 and bapx1, pattern ventral pharyngeal cartilage and the jaw joint. Development 130, 1353–1365 (2003).
Krauss, J. et al. Endothelin signalling in iridophore development and stripe pattern formation of zebrafish. Biol. Open 3, 503–509 (2014).
Baynash, A. G. et al. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell 79, 1277–1285 (1994).
Square, T. et al. CRISPR/Cas9-mediated mutagenesis in the sea lamprey Petromyzon marinus: a powerful tool for understanding ancestral gene functions in vertebrates. Development 142, 4180–4187 (2015).
Stock, D. W. & Whitt, G. S. Evidence from 18S ribosomal RNA sequences that lampreys and hagfishes form a natural group. Science 257, 787–789 (1992).
Clouthier, D. E. et al. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development 125, 813–824 (1998).
Ruest, L. B., Xiang, X., Lim, K. C., Levi, G. & Clouthier, D. E. Endothelin-A receptor-dependent and -independent signaling pathways in establishing mandibular identity. Development 131, 4413–4422 (2004).
Tavares, A. L. P. et al. Ectodermal-derived Endothelin1 is required for patterning the distal and intermediate domains of the mouse mandibular arch. Dev. Biol. 371, 47–56 (2012).
Charité, J. et al. Role of Dlx6 in regulation of an endothelin-1-dependent, dHAND branchial arch enhancer. Genes Dev. 15, 3039–3049 (2001).
Parichy, D. M. et al. Mutational analysis of endothelin receptor b1 (rose) during neural crest and pigment pattern development in the zebrafish Danio rerio. Dev. Biol. 227, 294–306 (2000).
Kawasaki-Nishihara, A., Nishihara, D., Nakamura, H. & Yamamoto, H. ET3/Ednrb2 signaling is critically involved in regulating melanophore migration in Xenopus. Dev. Dyn. 240, 1454–1466 (2011).
Metallinos, D. L., Bowling, A. T. & Rine, J. A missense mutation in the endothelin-B receptor gene is associated with lethal white foal syndrome: an equine version of Hirschsprung disease. Mamm. Genome 9, 426–431 (1998).
Sánchez-Mejías, A., Fernández, R. M., López-Alonso, M., Antiñolo, G. & Borrego, S. New roles of EDNRB and EDN3 in the pathogenesis of Hirschsprung disease. Genet. Med. 12, 39–43 (2010).
Square, T., Jandzik, D., Cattell, M., Hansen, A. & Medeiros, D. M. Embryonic expression of endothelins and their receptors in lamprey and frog reveals stem vertebrate origins of complex Endothelin signaling. Sci. Rep. 6, 34282 (2016).
Cerny, R. et al. Evidence for the prepattern/cooption model of vertebrate jaw evolution. Proc. Natl Acad. Sci. USA 107, 17262–17267 (2010).
Kuraku, S., Takio, Y., Sugahara, F., Takechi, M. & Kuratani, S. Evolution of oropharyngeal patterning mechanisms involving Dlx and endothelins in vertebrates. Dev. Biol. 341, 315–323 (2010).
Johnels, A. G. On the development and morphology of the skeleton of the head of Petromyzon. Acta Zool. 29, 139–277 (1948).
Green, S. A., Uy, B. R. & Bronner, M. E. Ancient evolutionary origin of vertebrate enteric neurons from trunk-derived neural crest. Nature 544, 88–91 (2017).
Kuratani, S. Evolution of the vertebrate jaw from developmental perspectives. Evol. Dev. 14, 76–92 (2012).
Smith, J. J. et al. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat. Genet. 50, 270–277 (2018).
Camargo Sosa, K. et al. Endothelin receptor Aa regulates proliferation and differentiation of Erb-dependant pigment progenitors in zebrafish. PLOS Genet. 15, e1007941 (2019).
Yao, T., Ohtani, K., Kuratani, S. & Wada, H. Development of lamprey mucocartilage and its dorsal–ventral patterning by endothelin signaling, with insight into vertebrate jaw evolution. J. Exp. Zoolog. B 316, 339–346 (2011).
Clouthier, D. E., Garcia, E. & Schilling, T. F. Regulation of facial morphogenesis by endothelin signaling: insights from mice and fish. Am. J. Med. Genet. A. 152A, 2962–2973 (2010).
Fujimoto, S., Oisi, Y., Kuraku, S., Ota, K. G. & Kuratani, S. Non-parsimonious evolution of hagfish Dlx genes. BMC Evol. Biol. 13, 15 (2013).
Tahara, Y. Normal stages of development in the lamprey Lampetra reissneri (Dybowski). Zool. Sci. 5, 109–118 (1988).
Nair, S., Li, W., Cornell, R. & Schilling, T. F. Requirements for Endothelin type-A receptors and Endothelin-1 signaling in the facial ectoderm for the patterning of skeletogenic neural crest cells in zebrafish. Development 134, 335–245 (2007).
Bonano, M. et al. A new role for the Endothelin-1/Endothelin-A receptor signaling during early neural crest specification. Dev. Biol. 323, 114–129 (2008).
Asai, R. et al. Endothelin receptor type A expression defines a distinct cardiac subdomain within the heart field and is later implicated in chamber myocardium formation. Development 137, 3823–3833 (2010).
Jandzik, D. et al. Roles for FGF in lamprey pharyngeal pouch formation and skeletogenesis highlight ancestral functions in the vertebrate head. Development 141, 629–638 (2014).
Bondurand, N., Dufour, S. & Pingault, V. News from the endothelin-3/EDNRB signaling pathway: role during enteric nervous system development and involvement in neural crest-associated disorders. Dev. Biol. 444 (Suppl 1), S156–S169 (2018).
Higashiyama, H. et al. On the vagal cardiac nerves, with special reference to the early evolution of the head-trunk interface. J. Morphol. 277, 1146–1158 (2016).
Thiagarajah, J. R. et al. Altered goblet cell differentiation and surface mucus properties in Hirschsprung disease. PLoS ONE 9, e99944 (2014).
von Boyen, G. B. et al. Abnormalities of the enteric nervous system in heterozygous endothelin B receptor deficient (spotting lethal) rats resembling intestinal neuronal dysplasia. Gut 51, 414–419 (2002).
Karne, S., Jayawickreme, C. K. & Lerner, M. R. Cloning and characterization of an endothelin-3 specific receptor (ETC receptor) from Xenopus laevis dermal melanophores. J. Biol. Chem. 268, 19126–19133 (1993).
Spiewak, J. E. et al. Evolution of Endothelin signaling and diversification of adult pigment pattern in Danio fishes. PLoS Genet. 14, e1007538 (2018).
Woodcock, M. R. et al. Identification of mutant genes and introgressed tiger salamander DNA in the laboratory axolotl, Ambystoma mexicanum. Sci. Rep. 7, 6 (2017).
Simakov, O. et al. Deeply conserved synteny resolves early events in vertebrate evolution. Nat. Ecol. Evol. 4, 820–830 (2020).
Yanagisawa, H. et al. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development 125, 825–836 (1998).
Sive, H. L., Grainger, R. M. & Harland, R. M. Early development of Xenopus laevis: A Laboratory Manual (Cold Spring Harbor Laboratory Press, 2000).
Nieuwkoop, P. D. & Faber, J. Normal Table of Xenopus laevis (Daudin): A Systematical and Chronological Survey of the Development from the Fertilized Egg Till the End of Metamorphosis (Garland, 1994).
Zu, Y. et al. Biallelic editing of a lamprey genome using the CRISPR/Cas9 system. Sci. Rep. 6, 23496 (2016).
York, J. R., Yuan, T., Lakiza, O. & McCauley, D. W. An ancestral role for Semaphorin3F-Neuropilin signaling in patterning neural crest within the new vertebrate head. Development 145, dev164780 (2018).
York, J. R., Yuan, T., Zehnder, K. & McCauley, D. W. Lamprey neural crest migration is Snail-dependent and occurs without a differential shift in cadherin expression. Dev. Biol. 428, 176–187 (2017).
Yuan, T., York, J. R. & McCauley, D. W. Gliogenesis in lampreys shares gene regulatory interactions with oligodendrocyte development in jawed vertebrates. Dev. Biol. 441, 176–190 (2018).
Wang, F. et al. Targeted gene disruption in Xenopus laevis using CRISPR/Cas9. Cell Biosci. 5, 15 (2015).
Rosen, J. N., Sweeney, M. F. & Mably, J. D. Microinjection of zebrafish embryos to analyze gene function. J. Vis. Exp. 25,1115 (2009).
Session, A. M. et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538, 336–343 (2016).
Flowers, G. P., Timberlake, A. T., McLean, K. C., Monaghan, J. R. & Crews, C. M. Highly efficient targeted mutagenesis in axolotl using Cas9 RNA-guided nuclease. Development 141, 2165–2171 (2014).
Square, T. et al. A gene expression map of the larval Xenopus laevis head reveals developmental changes underlying the evolution of new skeletal elements. Dev. Biol. 397, 293–304 (2015).
Aigler, S. R., Jandzik, D., Hatta, K., Uesugi, K. & Stock, D. W. Selection and constraint underlie irreversibility of tooth loss in cypriniform fishes. Proc. Natl Acad. Sci. USA 111, 7707–7712 (2014).
Sauka-Spengler, T., Meulemans, D., Jones, M. & Bronner-Fraser, M. Ancient evolutionary origin of the neural crest gene regulatory network. Dev. Cell 13, 405–420 (2007).
Meulemans, D., McCauley, D. & Bronner-Fraser, M. Id expression in amphioxus and lamprey highlights the role of gene cooption during neural crest evolution. Dev. Biol. 264, 430–442 (2003).
Haming, D. et al. Expression of sympathetic nervous system genes in lamprey suggests their recruitment for specification of a new vertebrate feature. PLoS ONE 6, 0026543 (2011).
McCauley, D. W. & Bronner-Fraser, M. Importance of SoxE in neural crest development and the evolution of the pharynx. Nature 441, 750–752 (2006).
Yuan, T., York, J. R. & McCauley, D. W. Neural crest and placode roles in formation and patterning of cranial sensory ganglia in lamprey. Genesis 58, e23356 (2020).
Cattell, M. V., Garnett, A. T., Klymkowsky, M. W. & Medeiros, D. M. A maternally established SoxB1/SoxF axis is a conserved feature of chordate germ layer patterning. Evol. Dev. 14, 104–115 (2012).
Talikka, M., Stefani, G., Brivanlou, A. H. & Zimmerman, K. Characterization of Xenopus Phox2a and Phox2b defines expression domains within the embryonic nervous system and early heart field. Gene Expr. Patterns 4, 601–607 (2004).
McCauley, D. W. & Bronner-Fraser, M. Conservation of Pax gene expression in ectodermal placodes of the lamprey. Gene 287, 129–139 (2002).
Ware, M., Dupé, V. & Schubert, F. R. Evolutionary conservation of the early axon scaffold in the vertebrate brain. Dev. Dyn. 244, 1202–1214 (2015).
Modrell, M. S. et al. A fate-map for cranial sensory ganglia in the sea lamprey. Dev. Biol. 385, 405–416 (2014).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Acknowledgements
We thank S. Miehls at the USGS Hammond Bay Biological Station and B. Laflamme at the Holyoke Dam for providing adult sea lampreys; B. Birsoy, J. Shi and M. Klymkowsky for assistance with X. laevis fertilizations; Z. Root for assistance with X. laevis and sea lamprey injections and husbandry; C. Altier for assistance with X. laevis husbandry; S. Schwikert for pro bono statistics consultation; C. Miller for his input on the manuscript; D. McCauley and J. York for providing HuC/D primary antibody and the IHC protocol; and R. Harland for providing secondary antibodies and staining advice for X. laevis. D.M.M., T.A.S., D.J., M.R., J.L.M. and M.V.C. were supported by National Science Foundation grants IOS 1656843, IOS 1257040 and IOS 0920751 to D.M.M. H.P.S. and A.W.H. were supported by the University of Colorado, Boulder Undergraduate Research Opportunities Program. D.J. was supported by the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 751066 and by the Scientific Grant Agency of the Slovak Republic VEGA grant no. 1/0415/17.
Author information
Authors and Affiliations
Contributions
D.M.M. conceived the project. D.M.M., T.A.S., D.J. and M.R. designed the experiments. All authors performed experiments and collected data. D.M.M., T.A.S. and D.J. wrote the manuscript. All authors discussed and provided input on the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Ingo Braasch and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Petromyzon marinus wild-type and mutant larval alcian blue stained head skeletons and lecticanA expression.
Anterior to left in all panels. For detailed quantification information, see Supplementary Table 2 and Methods section ‘Statistics and reproducibility’. a, WT ventral view at st. T30. b, WT lateral view of the same specimen, with skeletal elements and cartilage types labelled in b′ and b″, respectively. In b′, regions of the oral skeleton are delineated. In b″, Epitrematic indicates the epitrematic processes on PAs 3 and 4, though these also exist on all branchial arches. Hypotrematic indicates the hypotrematic processes of PAs 3 and 4, though these also exist on all branchial arches. Hypobranchial indicates the hypobrancial cartilage connecting PAs 4, 5, and 6, but exists between all branchial arches. Subchordal indicates the subchordal cartilage connecting PAs 4, 5, and 6, but exists between all branchial arches. c–f, Alcian blue reveals Δednra, Δednrb, Δednra+b, ΔednA head skeleton phenotypes at st. T30 (n = 16/16, n = 5/18, n = 19/19, and n = 12/15 individuals for each gene, respectively). Red arrowheads highlight some regions where cartilages are missing or separated. g–k, lecticanA WT expression summary in P. marinus. This gene is homologous to gnathostome lectican genes (such as aggrecan and versican) and like those genes it is expressed in neural crest-derived mesenchyme before (for example, arrows in h and i) and during chondrogenesis (for example, expression in j and k). ll, lower lip; lmp, lateral mouth plate; mvs, medial velar skeleton; 1-9, pharyngeal arches (numbered individually); tr, trabecular; ul, upper lip. The scale bar in a is 500 μm and applies to images in a–f.
Extended Data Fig. 2 Petromyzon marinus Δednra phenotype and genotype summary.
For detailed quantification information, see Supplementary Tables 1, 2, 4, and Methods section ‘Statistics and reproducibility’. a, Phenotypic series of ednra sgRNA4 hypomorphs at st. T30 (n = 264/325 injected individuals displayed a similarly severe phenotype, here labelled ‘head skeleton + heart + pigment’). Left lateral views. Scale bar represents 500 μm. b, An example of a genotyped animal from injected with ednra sgRNA3 (n = 113/154 injected individuals displayed a similarly severe phenotype). Left lateral view. Scale bar represents 500 μm. Target site is shown in orange with a red PAM. Green nucleotide string indicates an insertion with sequence that is also observed at a nearby endogenous locus near the lesion on the reverse strand (underlined nucleotide string). This insertion is stacked inside of the lesion on the 5′ end. c, A staging series of Δednra illustrating the typical manifestation of the severe phenotype shown in a and b. Left lateral views in all panels. At st. T24, a slight heart oedema is usually apparent (arrow). From this stage, the reduction in head skeleton size becomes progressively more dramatic through development relative to WT (brackets mark the anterior and posterior boundaries of the skeletogenic mesenchyme). Scale bar represents 100 μm and applies to all images in this panel. d, A ventral view of the WT and Δednra heart, showing the most prominent aggregation of ectopic melanophores. Anterior to top, bracket indicates the heart in WT and the heart oedema in Δednra. e, Melanophore number is increased in the ventral fin fold in Δednra relative to WT (n = 5 WT versus n = 5 Δednra individuals, Student’s one-sided t-test P = 0.000255, Cohen’s d = −1.53, df = 13). Images at right illustrate the pigment cells counted in (green arrowheads). The median fin fold is outlined. f, External melanin cover is also increased in Δednra relative to WT (n = 5 WT versus n = 5 Δednra individuals, Student’s one-sided t-test P = 0.00749, Cohen’s d = −1.74, df = 9). Image at right is an example threshold image quantified in the analysis, see Methods. Box plots show all points and delineate all quartile thresholds; medians are indicated with a horizontal line.
Extended Data Fig. 3 Petromyzon marinus dlxA, -D, -B, hand, ID, lecticanA (lecA), myc, msxA, phox2, soxE1, soxE2, and twistA expression in Δednr lampreys at st. T26.5.
For detailed quantification information, see Supplementary Tables 1, 2, 4, and Methods section ‘Statistics and reproducibility’. a, b, Left lateral views in all panels. Treatment and gene assayed are indicated in the figure. Scale bar represents 100 μm and applies to all images (that is, all ISH images are to scale with each other). Gray and white dotted lines in f and g are examples of the head size and hand domain size measurements (respectively) used for the comparison in c. Δednra n = 7/21, n = 5/15, n = 2/5, and n = 0/8 individuals with perturbed dlxA, dlxD, dlxB and hand ISH signals, respectively. Δednra+b n = 20/24, n = 7/8, n = 4/6, and n = 0/9 individuals with perturbed dlxA, dlxD, dlxB, and hand ISH signals, respectively. Δednrb n = 0/8, n = 0/6, n = 0/5, and n = 0/7 individuals with perturbed dlxA, dlxD, dlxB, and hand ISH signals, respectively. c, Quantifying the X/Y lateral size of the hand ventral domain to head size ratio reveals a loosely supported trend upwards in Δednra+b, suggesting that the ventral hand domain is only nominally affected in these mutants (see Methods; left and right images of n = 6 WT and n = 8 Δednra+b individuals, Student’s two-sided t-test P = 0.0647, df = 13). Box plots show all points and delineate all quartile thresholds; medians are indicated with a horizonal line. d, Lateral views with anterior to left in all panels. Treatment and gene assayed are indicated in the figure. Overall, Δednra and Δednrb are most consistent in their gene expression patterns, despite all domains being shrunken in Δednra. Conversely, Δednra+b displays some disruptions in gene expression not observed in either single receptor perturbation (red arrowhead, white stars). ISH result numbers are as follows, all numbers indicate independent biological samples (individual lamprey larvae) that produced a reduced, discontiguous, or abrogated ISH signal: Δednra ID n = 0/5, lecA n = 16/16, myc n = 0/6, msxA n = 0/12, phox2 n = 0/8, soxE1 n = 0/4, soxE2 n = 15/21, twistA n = 0/6; Δednrb ID n = 0/10, lecA n = 4/8, myc n = 0/7, msxA n = 0/7, phox2 n = 0/8, soxE1 n = 0/7, soxE2 n = 4/8, twistA n = 0/9; Δednra+b ID n = 3/5, lecA n = 8/9, myc n = 0/4, msxA n = 0/8, phox2 n = 0/11, soxE1 n = 9/11, soxE2 n = 9/10, twistA n = 4/4.
Extended Data Fig. 4 Xenopus laevis Δednra and Δedn1 head skeleton defects and genotyping.
For detailed quantification information, see Supplementary Tables 1, 2, 4, and Methods section ‘Statistics and reproducibility’. a–c, Δedn1.L+S (b) and Δednra.L+S (c) show hypomorphic head skeleton elements relative to WT (a) at st. N.F.48 (n = 20/47 and n = 37/71 injected individuals for each gene, respectively). Δedn1.L+S is typically less severe, with a discernible Meckel’s cartilage (labelled mc) present but fused to the palatoquadrate (pq), thus lacking a primary jaw joint (black arrowheads in a′, red arrowheads in b′). However in Δednra.L+S, Meckel’s cartilage is highly reduced, and frequently unrecognizable, only visible as a bump on the palatoquadrate in most cases. The infrarostral (ir) is always detectable, and no fusion of this element to Meckel’s cartilage was ever observed (that is, the intramandibular joint appears unaffected by a loss of edn1 or ednra, arrows in a′ and b′). The ceratohyal (ch) was highly reduced in both perturbations. The branchial arch skeleton (comprising pharyngeal arches 3-6), though slightly reduced, maintained its overall shape and structure more robustly relative to PA1 and PA2 derivatives (for example, Meckel’s and the ceratohyal). Ventral views with anterior to top in a–c, dissected views in a′ and b′, red dotted line in a′ and b′ indicates a cut made during dissection through the palatoquadrate. Scale bar in a represents 500 μm and applies to b and c. a′ and b′ are not to scale with each other. d–g, dlx3.S and hand2.L expression are reduced in ΔednraL+S (red arrowheads; outline) at st. N.F.33 (n = 3/7 and n = 5/8 injected individuals for each gene, respectively). Lateral views with anterior to left. Scale bar in d represents 100 μm and also applies to e–g, Gray and white dotted lines in f and g are examples of the head size and hand domain size measurements (respectively) graphed in h. h, Quantifying hand2.L ventral expression domain to head size ratio (see panels f and g for examples) reveals a significant decrease in the relative X/Y lateral size of the hand2.L domain (n = 8 and n = 16 left/right halves of 4 WT and 8 Δedn3 individuals, respectively, Student’s one-sided t-test P = 0.000803, Cohen’s d = 1.378, df = 11 [adjusted to match the number of animals]; see Methods). Box plots show all points and delineate all quartile thresholds; medians are indicated with a horizontal line. i–l, Genotyping examples of Δedn1.L+S (j, l [top]) and Δednra.L+S (k, l [bottom]) larvae. The alleles shown in l are derived from the animals pictured in j and k (for each gene, respectively). Target sites are shown in orange with a red PAM. i–k show ventral views with anterior to the left. Pink and purple nucleotide strings indicate inserted sequences that are also observed at the endogenous locus near the lesion on the forward strand (underlined nucleotide strings). Red nucleotide strings represent insertions without an obvious source. Insertions are stacked inside of each lesion on the 5′ end.
Extended Data Fig. 5 Petromyzon marinus Δednrb and Δednra+b phenotypes and genotyping.
For detailed quantification information, see Supplementary Tables 1, 2, 4, and Methods section ‘Statistics and reproducibility’. a, Left lateral images of Δednrb at st. T30. n = 40/42 and n = 177/403 injected individuals for sgRNA2 and sgRNA3, respectively. 100% mutant alleles were returned for the indicated individual. Target site for sgRNA3 is shown in yellow with a purple PAM. Four example alleles are shown from the indicated individual. An insertion from the reverse strand is shown in green; its endogenous ‘source’ is underlined. The insertion is stacked inside of the lesion on the 5′ end. Both scale bars represent 500 μm. b, Alcian blue staining reveals slight skeletal disruptions in Δednrb at st. T30 (red arrowheads) n = 5/18 Δednrb individuals. c, Genotyping examples of Δednra+b at st. T30 that were all found to harbour a majority of mutant alleles (>75%), n = 32/44 injected animals displayed a phenotype similar to those specimens pictured. A summary of the alleles found in the third individual are shown, which returned 100% mutant alleles. Target sites are shown in orange with a red PAM. Purple (forward) and green (reverse) nucleotide strings indicate insertions of sequences that are also observed at endogenous loci near the lesion (underlined nucleotide strings). Red nucleotide represents an insertion without a single obvious source. Insertions are stacked inside of each the lesions on the 5′ end. Scale bar in c represents 500 μm. d, HuC/D immunohistochemistry reveals only some slight defects in specific cranial ganglia, namely the opV and epibranchial ganglia (n1-5). The white dotted boxes in the top left lateral images are shown in greater detail below, as indicated for each treatment. Abbreviations: all, anterior lateral line; g/all, geniculate/anterior lateral line (fused); mmV, maxillomandibular trigeminal; n1-5, nodose 1-5; opV, ophthalmic trigeminal; p, petrosal; pll, posterior lateral line. Scale bar represents 20 μm and applies to both the WT and Δednra+b enlargements. n = 6/6 Δednra+b individuals showed a similar phenotype. e, foxD-A (left lateral, st T24) and soxB1b (dorsal view, anterior to right) ISHs show DRGs still express these genes at larval stages in Δednra+b (arrowheads). n = 0/14 and n = 0/9 Δednra+b individuals showed missing or discontiguous ISH signal for foxD-A and soxB1b, respectively. f, A size analysis of WT (n = 4) versus Δednra+b (n = 6) individuals’ left side cranial ganglia confirmed a decrease in the lateral surface area of the opV (Student’s one-sided t-test P = 0.00762, Cohen’s d = 1.42, df = 9) and n1-5 (Student’s one-sided t-test P = 0.00120, Cohen’s d = 1.625, df = 9), but not the mmV (Student’s one-sided t-test P = 0.276, df = 9), g/all (Student’s one-sided t-test P = 0.189, df = 9), p (Student’s one-sided t-test P = 0.289, df = 9), nor the pll (Student’s one-sided t-test P = 0.212, df = 9). g, Counting the number of anterior dorsal root ganglia (as visualized by HuC/D IHC, from the first somite to the anterior boundary of the yolk) in WT (n = 7) versus Δednra+b (n = 7) individuals revealed no significant change in their number (Student’s one-sided t-test P = 0.129, df = 13). Box plots show all points and delineate all quartile thresholds; medians are indicated with a horizontal line.
Extended Data Fig. 6 Petromyzon marinus Δdlx genotyping post-ISH and alcian blue staining.
For detailed quantification information, see Supplementary Tables 2, 4, and Methods section ‘Statistics and reproducibility’. a, Examples of post-ISH genotyping for each dlx locus targeted for mutagenesis at st. T26.5. ΔdlxA n = 16/51, ΔdlxD n = 16/50, ΔdlxA+C+D n = 14/17 individuals produced perturbed lecticanA (lecA) ISH signals (as pictured). Gene, target site, and measured frequency of indel alleles are all indicated in the figure for each individual. Forward strand target sites are shown in yellow with a purple PAM, reverse strand target sites are shown in orange with a red PAM. Purple and green nucleotide strings represent insertions that reflect endogenous sequence in the forward and reverse orientation, respectively, near the lesion (underlined nucleotide strings). Red nucleotide strings represent insertions without an obvious source. Insertions are stacked inside of each the lesions on the 5′ end. All ISH images are to scale with each other. b, Alcian blue staining at st. T30 reveals truncations and gaps in the intermediate head skeleton (arrowheads), and occasional fusions of branchial arches (arrows) in Δdlx lampreys. ΔdlxA n = 4/61, ΔdlxD n = 5/34, ΔdlxA+C+D n = 11/35 individuals displayed missing, discontiguous, and/or fused cartilages in the pharynx (as pictured). Scale bar in b represents 100 μm.
Extended Data Fig. 7 Xenopus laevis Δedn3 pigmentation phenotype and genotype summary.
For detailed quantification information, see Supplementary Tables 1, 4, and Methods section ‘Statistics and reproducibility’. a–h, Δedn3.L+S (a, c, e–h) have reduced neural crest-derived pigment cells (including at least melanophores and iridophores) relative to WT (b, d), n = 31/71 injected individuals displayed a > 50% reduction in pigmentation, or in the case of a, e–g, The uninjected half of the specimen (these animals were injected unilaterally at the 2 blastomere stage, a′ shows the lineage tracer GFP [coinjected as mRNA], a″ shows an overlay of a and a′), n = 27/69 animals displayed a > 50% reduction in pigment on the injected half of the animal. As expected, pigmentation loss in the eye was never observed (because eye pigmentation is not derived from the neural crest), and the black coloration of the claws always remains, which is also observed in Xenopus tyrosinase mutants (Yonglong Chen, personal communication). All images show dorsal views, anterior to top in a, d–h, and to the right in b and c. Scale bar in a represents 1mm and applies to a′ and a″. Scale bar in b represents 5 mm and applies to c. Scale bar in d also represents 5 mm and applies to e–h. i, genotyping of a leucistic tadpole revealed a high rate of mutant alleles across both the ‘long’ and ‘short’ homeologues. Target sites are shown in yellow with a purple PAM site.
Extended Data Fig. 8 Xenopus laevis Δedn3 peripheral nervous system in larvae and subadult frogs.
For detailed quantification information, see Supplementary Tables 1, 2, 4, and Methods section ‘Statistics and reproducibility’. a, b, phox2a expression at st. N.F.41 in WT (a) and Δedn3 (b) larvae. Δedn3 larvae have no consistent defects or reductions in the epibranchial ganglia (arrows) or presumptive enteric nervous system precursors (regions within dashed boxes are shown enlarged and with enhanced contrast in a′ and b′), n = 0/4 Δedn3 individuals displayed a severely reduced ISH signal. Scale bar in a represents 200 μm and applies to b. c, d, Neurofilament immunohistochemistry at st. N.F.48 on WT (c) Δedn3 (d) larvae. Despite severe reductions in pigment cells on the gut and in the skin, Δedn3 larvae show no obvious defects in the cranial nerves (CNs), dorsal root ganglia (DRG), posterior lateral line (PLL), or vagal nerve (VN) n = 0/8 Δedn3 individuals displayed missing or overtly mis-patterned cranial nerves. Scale bar in c represents 500 μm and applies to d. e, Despite heavy pigmentation loss, Δedn3 tadpoles show no significant change in the number of dorsal root ganglia present at st. N.F.48 (as visualized by DAPI counterstain on Neurofilament IHC specimens, see Methods), n = 6 and n = 8 left/right halves of 3 WT and 4 Δedn3 individuals, respectively, Student’s one-sided t-test P = 0.381, df = 6 (adjusted to match the number of animals). f–p, Images of dissected and prepared subadult frog guts. f, A brightfield image of a dissected WT frog gut, illustrating the approximate locations of the assays pictured in panels g–p (boxes not to scale). A white dotted line indicates the boundary between the small and large intestine. Scale bar represents 1 mm. g, h, Cross sections of small intestines dissected from WT (g, g′) and Δedn3 (h, h′) subadult frog guts. g′ and h′ show HNK-1 immunohistochemistry. Though they lack pigmentation within the mucosa (compare g to h, lumen labelled lu), Δedn3 frogs have no overt defects in the myenteric (my) or submucosal (sm) plexuses of the small intestine (compare g′ to h′) n = 0/4 tissue pieces derived from 4 subadult frogs showed HNK-1 signal reduction, while n = 4/4 showed a reduction in pigmentation. Scale bar in g represents 100 μm and applies to h. i, j, Brightfield images of the external surface of the dissected large intestine of WT (h) and Δedn3 (i) subadult frogs. Δedn3 frogs lack pigmentation on this part of the gut (both melanophores and iridophores), n = 4/4 subadult frog guts displayed a >50% reduction in pigmentation. Anterior to top. Scale bar in i represents 200 μm and applies to j. k–n, Optical Z plane sections on flat-mounted large intestines from WT (k, m) and Δedn3 (l, n) frogs after HNK-1 immunohistochemistry, visualized at the plane of the myenteric (k, l) and submucosal (m, n) plexuses. The myenteric plexuses of Δedn3 frogs is largely normal (compare k to l), while the submucosal plexuses lack the ganglia seen in WT frogs (arrowheads, compare m to n), n = 4/4 subadult frogs displayed regions with low submucosal plexus density, see panel q for quantification. Scale bar in k represents 50 μm and applies to k–n. o, p, Haematoxylin and eosin (H&E) staining on transverse 7 μm-thick paraffin sections of the large intestine of WT (o) and Δedn3 (p) frogs. Δedn3 frogs have an excess of goblet cells (arrowheads) compared to WT as seen in mammalian Edn3/Ednrb mutants39, n = 3/3 subadult frogs showed dense increases in goblet cells. Lumen to top. q, Comparing n = 4 WT and n = 7 pieces of dissected lower intestines from 3 and 4 WT and Δedn3 frogs, respectively, reveals a significant reduction in the number of submucosal ganglia per mm2 in Δedn3 (Student’s one-sided t-test P = 0.00646, Cohen’s d = 1.47, df = 6 [adjusted to match the number of animals]). Box plots show all points and delineate all quartile thresholds; medians are indicated with a horizontal line.
Extended Data Fig. 9 Petromyzon marinus ΔednA and ΔednE phenotype and genotype summary.
For detailed quantification information, see Supplementary Tables 1, 4, and Methods section ‘Statistics and reproducibility’. a–d, Examples of genotyped material and the loci returned for each specimen. Gene, target site, and measured frequency of indel alleles are all indicated in the figure for each individual. ednA sgRNA1 n = 22/67, ednA sgRNA3 n = 29/38, ednE sgRNA2 n = 70/73, ednE sgRNA3 n = 38/51 injected individuals showed similarly severe phenotypes as those pictured for each gene. Forward strand target sites are shown in yellow with a purple PAM, reverse strand target sites are shown in orange with a red PAM. Green nucleotide string indicates an insertion that is observed at the endogenous locus in the forward orientation near the lesion (underlined nucleotide string). Red nucleotide is an insertion without a single obvious source. Insertions are stacked inside of each the lesions on the 5′ end. Scale bars represent 500 μm. e, ednA, -C, and -E sgRNAs were coinjected, and the material was subjected to soxE2 ISH at st. T26.5.
Extended Data Fig. 10 ednr and edn synteny and phylogeny.
a, Synteny of ednr genes. All ednr loci are shown for chicken (G. gallus), human (H. sapiens), and sea lamprey (P. marinus). Red and blue boxes indicate genes or groups of genes that were only observed at the ednra or ednrb loci, respectively, across species. Chicken and human information is after Braasch and Schartl5. Sea lamprey genomic information is derived from the germline genome27. b, Amino acid tree made using the Maximum Likelihood method on a ClustalW alignment, derived from a subset of sequences used in Square et al.21. Bootstrap scores (n = 100) are indicated at each node. See Supplementary Table 5 for accession numbers. c, Synteny analysis of edn loci and their surrounding hivep and phactr genes. Most vertebrate edn genes are located between hivep and phactr paralogues, which were previously used by Braasch et al.6 to determine the relationships of jawed vertebrate edn genes. We found that sea lamprey edn genes are also associated with hivep and/or phactr genes in most cases, and used the sequence similarity of their predicted gene products to infer the phylogeny of the edn locus. d, e, Hivep (b) and Phactr (c) phylogenies were created by applying the Maximum Likelihood tree building method to ClustalW alignments of amino acid sequences. Concatenated Hivep and Phactr sequences were also used to generate a phylogenetic tree but the results were largely the same as the Phactr tree, but with lower confidence values at some nodes. Bootstrap scores (n = 100) are indicated at each node. See Supplementary Table 5 for sequence information and accession numbers.
Extended Data Fig. 11 Phenotypes of larvae injected with 22 different negative control sgRNAs.
Embryos injected with an untargeted negative control sgRNA + Cas9 had normal morphology at st. T30. Embryos injected with 21 different sgRNAs targeting other developmental regulators expressed in the larval lamprey head resulted in a range of different phenotypes at st. T30 and st. T26.5. Heart oedema was not a feature of the mutant phenotype of most sgRNAs. See figure for larvae numbers. These results confirm that the phenotypes described by this work are not non-specific side effects of the CRISPR/Cas9 method in the sea lamprey.
Supplementary information
Supplementary Table
Supplementary Table 1 | sgRNA and genotyping primer sequences and frequencies of severe mutant phenotypes for genes mutagenized in this study.
Supplementary Table
Supplementary Table 2 | Numbers of sgRNA/Cas9-injected embryos and larvae displaying abnormalities revealed by ISH, IHC, and histological staining.
Supplementary Table
Supplementary Table 3 | Statistical analyses of mutant phenotypes observed in embryos and larvae injected with different sgRNAs.
Supplementary Table
Supplementary Table 4 | Numbers of Indel alleles sequenced in genotyped severe mutant individuals.
Supplementary Table
Supplementary Table 5 | GenBank accession numbers and UCSC Petromyzon marinus Genome Browser intervals used to derive amino acid sequences used for phylogenetic analyses.
Rights and permissions
About this article

Cite this article
Square, T.A., Jandzik, D., Massey, J.L. et al. Evolution of the endothelin pathway drove neural crest cell diversification. Nature 585, 563–568 (2020). https://doi.org/10.1038/s41586-020-2720-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2720-z
- Springer Nature Limited
This article is cited by
-
Shared features of blastula and neural crest stem cells evolved at the base of vertebrates
Nature Ecology & Evolution (2024)
-
Endothelin-1 axes in the framework of predictive, preventive and personalised (3P) medicine
EPMA Journal (2021)