

Abstract
The RNA polymerase II (Pol II) core promoter is the strategic site of convergence of the signals that lead to the initiation of DNA transcription1,2,3,4,5, but the downstream core promoter in humans has been difficult to understand1,2,3. Here we analyse the human Pol II core promoter and use machine learning to generate predictive models for the downstream core promoter region (DPR) and the TATA box. We developed a method termed HARPE (high-throughput analysis of randomized promoter elements) to create hundreds of thousands of DPR (or TATA box) variants, each with known transcriptional strength. We then analysed the HARPE data by support vector regression (SVR) to provide comprehensive models for the sequence motifs, and found that the SVR-based approach is more effective than a consensus-based method for predicting transcriptional activity. These results show that the DPR is a functionally important core promoter element that is widely used in human promoters. Notably, there appears to be a duality between the DPR and the TATA box, as many promoters contain one or the other element. More broadly, these findings show that functional DNA motifs can be identified by machine learning analysis of a comprehensive set of sequence variants.
Similar content being viewed by others


Main
The core promoter is generally considered to be the stretch of DNA that directs the initiation of transcription of a gene; it ranges from about −40 to +40 nucleotides (nt) relative to the +1 nt transcription start site (TSS)1,2,3,4,5. The core promoter comprises DNA sequence elements such as the TATA box, initiator (Inr), motif ten element (MTE), and downstream core promoter element (DPE) (Extended Data Fig. 1a). Each of these motifs is present only at a subset of core promoters. Hence, there are no universal core promoter elements. Moreover, specific core promoter motifs can be important for enhancer–promoter specificity6,7,8 and can be involved in gene networks7,9,10,11.
The key DNA sequence motifs of human core promoters remain to be clarified. In focused human promoters, in which transcription initiates at a single site or a narrow cluster of sites, the TATA box is the best known core promoter element, but most human core promoters lack a TATA box12. In Drosophila, TATA-less transcription is frequently driven by the downstream MTE and DPE motifs13,14,15,16; however, these motifs have rarely been found in human promoters and have been thought perhaps not to exist in humans1,2,3.
HARPE analysis of the downstream promoter
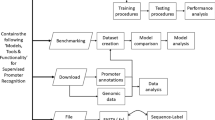
To decipher the downstream core promoter in humans, we generated and analysed an extensive library of promoters that contain randomized sequences in the region from +17 to +35 nt relative to the +1 nt TSS. This stretch, which we term the DPR, comprises the positions that correspond to the MTE and DPE (Fig. 1a, Extended Data Fig. 1a), which are overlapping elements in the downstream core promoter region in Drosophila that span multiple contact points with the transcription factor TFIID16,17,18,19. In previous studies, libraries of entire core promoter regions have been screened and characterized by using cell-based systems20,21,22,23,24. By contrast, here we have analysed specific segments of the core promoter in vitro and in cells, with the strategy of obtaining high coverage and carrying out machine learning analysis of the data.

a, Schematic of HARPE for the analysis of DNA sequence variants in the DPR. The randomized segment was generated by oligonucleotide synthesis with mixed nucleotides. ORF, open reading frame. b, Most sequence variants exhibit low transcriptional activity. The distribution of transcription strength for each of the approximately 500,000 core promoter variants is shown. c, A distinct DPR sequence motif can be seen in the nucleotide frequencies of the 0.1% most transcribed DPR sequences (top) as well as in the web logo for the top HOMER motif that is identified with these sequences (bottom). All panels show a representative experiment, n = 2 biologically independent samples.
In natural promoters, it can be difficult to elucidate the characteristics of a specific DNA element, such as the DPR, owing to the different promoter backgrounds in which the sequence motif is situated. To circumvent this problem, we adapted the survey of regulatory elements23 (SuRE) and developed the HARPE method. HARPE involves the generation of around 500,000 random DPR variants in an invariant promoter cassette followed by assessment of the transcription strength (defined as the RNA tag count divided by the DNA tag count; Methods) of each variant in vitro (Fig. 1a, Extended Data Fig. 1, Supplementary Table 1). This analysis showed that most DPR sequence variants support only a low level of transcription (Fig. 1b) and that the most active DPR sequences exhibit distinct nucleotide preferences (Extended Data Fig. 1d). Moreover, hypergeometric optimization of motif enrichment (HOMER) motif discovery analysis25 of the top 0.1% most-transcribed HARPE sequences identified a distinct motif that resembled the Drosophila DPE consensus sequence (RGWYGT from +28 to +32)14 (Fig. 1c, Extended Data Fig. 1e, f). The results of HARPE are reproducible (Extended Data Fig. 1g–i) in the absence or presence of sarkosyl, which limits transcription to a single round (Extended Data Fig. 2a–d, Supplementary Discussion 1).
HARPE is a robust and versatile method
To determine the versatility of the HARPE method, we tested the assay by varying different experimental parameters. First, we compared the results of HARPE assays that were performed with two different core promoter cassettes: SCP1m (as in Fig. 1), which is a version of the synthetic SCP1 promoter with a mutant TATA box (also known as SCP1mTATA26); and the human IRF1 core promoter, which lacks a TATA box and contains a DPE motif17. Both core promoters contain a consensus Inr sequence12, but otherwise they share no sequence similarity. With these two different core promoter cassettes, the HARPE results were nearly indistinguishable (Fig. 2a, Extended Data Figs. 1i, 2e). In addition, we observed nearly the same results with TATA-less versus TATA-box-containing promoters (Fig. 2b, Extended Data Figs. 1i, 2e). Thus, HARPE can function consistently in different core promoter backgrounds.

The top HOMER motifs obtained from the 0.1% most active sequences are shown. a, HARPE of the DPR with two different promoter cassettes: SCP1 lacking a TATA box (SCP1m) and the human IRF1 core promoter (in vitro transcription). b, HARPE of the DPR with a TATA-less promoter (SCP1m) and a TATA-box-containing promoter (SCP1) in vitro. c, HARPE of the DPR (+17 to +35 nt), DPE (+23 to +34 nt), and MTE (+18 to +29 nt) motifs with the SCP1m promoter in vitro. d, HARPE of the DPR in the SCP1m promoter transcribed in vitro or in cells. All panels show a representative experiment, n = 2 biologically independent samples.
Second, we investigated whether we would obtain consistent HARPE data if we randomized only a subset of the DPR rather than the entire DPR. To this end, we performed HARPE by randomization of only the MTE region (+18 to +29 nt) or only an extended DPE region (+23 to +34 nt) (Fig. 2c, Extended Data Figs. 1i, 2f). These experiments showed that randomization of subregions of the DPR yielded nucleotide preferences similar to those obtained by randomization of the entire DPR.
Third, we tested whether transcription of the HARPE promoter libraries in cells would yield results similar to those seen in vitro (Fig. 2d, Extended Data Fig. 2g). To this end, we carried out HARPE by transfection of the promoter libraries into HeLa cells and observed nucleotide preferences in the DPR that were nearly identical to those seen in vitro. Furthermore, we found a strong resemblance between HARPE data generated in vitro and in cells with the DPR sequence in the human IRF1 and TATA-box-containing SCP1 core promoter cassettes, as well as with the MTE and DPE sequences (Extended Data Fig. 2h–j). Therefore, HARPE appears to be a robust method that provides consistent data under a variety of different conditions.
HARPE analysis of the upstream TATA box
To enable the use of HARPE for the analysis of upstream promoter elements, we developed a modified version that includes linkage of each of the upstream randomized motifs with a corresponding downstream barcode (Extended Data Fig. 2k–p). We performed this analysis with randomized sequences in the region of the TATA box. We tested a long TATA region (−32 to −21 nt relative to the +1 nt TSS) and a short TATA region (−30 to −23 nt) (Extended Data Figs. 1a, 2k–p). The long-TATA analysis yielded an A/T-rich stretch that resembled that seen in natural human promoters. The short-TATA construct contained a TA dinucleotide at positions −32 and −31 that served to fix the phasing of the TATA sequence. Hence, with the short TATA construct, we observed a more distinct TATA-box-like sequence in a single register. Thus, HARPE can be used to analyse upstream as well as downstream promoter sequences.
Machine learning analysis of the HARPE data
HARPE analysis of the DPR yielded hundreds of thousands of sequence variants (Supplementary Table 1), each of which was associated with a specific transcription strength, and the data were therefore well suited for machine learning analysis. There are many different methods for supervised learning, and we found SVR27,28 to be an effective and straightforward approach for the analysis of the HARPE data.
In the SVR analysis of the DPR, we started with 468,069 sequence variants, each of which had a known transcriptional strength (Fig. 3a). We set aside 7,500 sequences that represented the full range of observed transcription strengths (test sequences) for later testing of the SVR. Next, we trained the SVR with 200,000 sequences (Extended Data Fig. 3a) and performed grid search and cross validation to identify optimal hyperparameter values and to establish the stability of the model (Extended Data Fig. 3b–d). The resulting SVR model that was generated from the biochemical (in vitro transcription) data was termed SVRb.

a, Summary of the SVR workflow. The HARPE dataset comprises about 500,000 DPR sequence variants, each with its associated transcription strength. A subset of these data (200,000 variants) was used to generate an SVR model for the DPR. The resulting SVR model was termed SVRb because it was trained with biochemical data. The SVR model provides a numerical score for the predicted transcription strength of any test sequence. b–d, To test the effectiveness of SVRb, the experimentally observed transcription strengths of sequence variants were compared with their predicted SVRb scores. b, Analysis of 7,500 independent test sequences in the HARPE dataset that were not used in the training of SVRb. The light grey shading (SVRb score ≥ 2) indicates predicted DPR activity (representative experiment, n = 2 biologically independent samples). c, Analysis of an independently generated HARPE dataset of a low-complexity DPR library (8,431 sequence variants) with high-confidence transcription strengths (representative experiment, n = 2 biologically independent samples). For b, c, PCC, Pearson’s correlation coefficient with two-tailed P < 2.2 × 10−16; ρ, Spearman’s rank correlation coefficient with two-tailed P < 2.2 × 10−16. d, Analysis of 16 DPR sequence variants (not in the training set) that were each tested individually by in vitro transcription and primer extension methodology (representative experiment, n = 4 biologically independent samples). PCC, Pearson’s correlation coefficient with two-tailed P = 3.4 × 10−7; ρ, Spearman’s rank correlation coefficient with two-tailed P < 2.2 × 10−16. For gel source data, see Supplementary Fig. 1.
The SVRb model was then able to provide a numerical value for the predicted transcription strength of any DNA sequence. First, we found an excellent correlation (ρ = 0.90) between the predicted SVRb scores and the observed transcription strengths of independent test sequences (Fig. 3b, Extended Data Fig. 3e). Second, we generated and analysed a separate high-quality, low-complexity HARPE dataset of DPR variants (Extended Data Fig. 3f–i), and saw an excellent correlation (ρ = 0.96) between the predicted SVRb scores and the observed transcription strengths (Fig. 3c). Third, we individually transcribed 16 promoters with a range of SVRb scores (Extended Data Fig. 4). These experiments revealed an excellent correlation (ρ = 0.89–0.95) between the predicted SVRb scores and the transcriptional activities of the individual sequences tested in vitro and in cells (Fig. 3d, Extended Data Fig. 4). It is also important to note that sequence variants with an SVRb score of two or more typically have at least sixfold-higher activity than inactive sequences (comparison of median values in the two groups; Extended Data Fig. 5a–c). Thus, an SVRb score of two or more is likely to reflect an active DPR. Last, performance assessment of SVRb revealed that it reliably predicts active DPR sequences (Extended Data Fig. 5d–r).
The data thus indicate that SVRb provides an accurate model for the DPR. Furthermore, we observed that SVRb, which was created with the SCP1m promoter cassette, correlated well with an SVRirf1 model that was generated with HARPE data for the DPR with the human IRF1 promoter cassette (ρ = 0.87) (Extended Data Fig. 6a, b). We also saw a good correlation between SVRb (for the DPR in a TATA-less background) and SVRscp1, which was generated with HARPE data for the DPR with the SCP1 (TATA-containing) promoter cassette (ρ = 0.80) (Extended Data Fig. 6c–e). Hence, the combination of HARPE and SVR analysis yields similar SVR models with different promoter backgrounds.
SVR models versus consensus sequences
To test the utility of an SVR model relative to a consensus sequence, we compared DPR sequences that were obtained by a standard consensus approach to the scores predicted by SVRb. First, we identified the DPE-like RGWYGT consensus sequence (from +28 to +33 nt) in the top 0.1% most active HARPE variants (Fig. 1c, Extended Data Fig. 6f). We then examined the transcription strengths of the variants that contained a perfect match to the consensus, and saw a wide range that varied from highly active to inactive (Extended Data Fig. 6g). These findings indicate that a perfect match to the RGWYGT consensus does not accurately predict the strength of the DPR. By contrast, we compared the SVRb scores to the observed transcription strengths of the same variants and saw an excellent correlation (ρ = 0.95) (Extended Data Fig. 6h). Thus, an SVR model is more effective than a standard consensus approach for predicting the activity of a sequence motif.
We also compared SVRb scores to the HOMER motif scores, which are based on the position-weight matrix (PWM) associated with the top HOMER consensus sequence (Extended Data Fig. 6i). These results showed that the comprehensive computational SVR model (ρ = 0.90) more accurately describes the DPR than the traditional consensus-based method (ρ = 0.51). The effectiveness of the SVR approach may be due, at least in part, to the training of the SVR with the full range of DPR sequences (that is, strong, intermediate, and weak), which is in contrast to the use of only strong variants in the generation of a consensus sequence.
Unlike a consensus-based model, the SVRb model can accurately incorporate the influence of neighbouring sequences on DPR activity (Extended Data Fig. 6j, k, Supplementary Discussion 2). We also found that SVR models can detect the function of an important sequence motif, such as a DPE-like sequence or a TATA motif, that is located at different positions within a larger region of interest (Extended Data Fig. 7a–i, Supplementary Discussion 3). In addition, SVRb uses information from a broader region of the DPR than a consensus-based model (Extended Data Fig. 7j, k, Supplementary Discussion 4). These findings thus indicate that SVR models are more effective at predicting transcription activity than consensus-based models.
SVR models from cell-based data
To test the versatility of SVR in the description of core promoter motifs, we compared SVR models created with HARPE data generated in vitro and in cells. With the DPR, we made SVRc (SVR of the DPR with cell-based data; the performance assessment of SVRc is in Extended Data Fig. 5i–m), which correlated well (ρ = 0.71) with transcription strengths in cells and was reproducible (ρ = 0.85) (Extended Data Fig. 7l, m). Moreover, SVRc correlated well (ρ = 0.77) with SVRb in predicting the transcription strengths of DPR sequences (Fig. 4a).

a, The SVR model from HARPE data in cells (SVRc) is similar to SVRb (biochemical). The SVRb and SVRc DPR scores of 7,500 test sequences (Fig. 3b) are compared. PCC, Pearson’s correlation coefficient with two-tailed P < 2.2 × 10−16; ρ, Spearman’s rank correlation coefficient with two-tailed P < 2.2 × 10−16. The light grey shading (SVRb and SVRc scores ≥ 2) indicates predicted DPR activity. b, Cumulative frequency of SVRc DPR scores in natural human promoters. Approximately 30% of 11,932 human promoters33, 17% of 100,000 random sequences (61% average G/C content, as in human core promoters), and 2.6% of 10,000 inactive sequences (randomly selected from the 50% least active sequences in the HARPE assay) have an SVRc score of at least 2 (green line), which corresponds to an active DPR (Extended Data Fig. 5b). c, Mutational analysis reveals DPR activity in different human promoters (for genes shown on x-axis) with SVRc DPR scores >2.5. In the mutant promoters, the wild-type DPR was substituted with a DNA sequence that has an SVRc DPR score of 0.3 (Extended Data Fig. 4a). The promoter sequences are shown in Extended Data Fig. 8h. Promoter activity was measured by transient transfection in cells followed by primer extension analysis of the TSSs (data shown as mean ± s.d., n = 3 or 4 biologically independent samples, indicated by points representing independent samples). All P < 0.05 (two-tailed paired Student’s t-test). For gel source data, see Supplementary Fig. 1. d, The SVRc DPR score correlates inversely with the presence of TATA-like sequences in human promoters in HeLa cells. The frequency of occurrence of Inr-like sequences, TATA-like sequences12, and TATA-box motifs (SVRtata ≥ 1) (Extended Data Fig. 5c) in human promoters that were binned according to their SVRc DPR scores (Extended Data Fig. 9a). Bins with fewer than 100 promoters are indicated with open circles and are connected by dashed lines (representative experiment, n = 2 biologically independent samples).
With the TATA box, we used HARPE data generated in vitro and in cells (Extended Data Figs. 2k–p, 8a, b) to create SVR models (with the long TATA sequence) termed SVRtata (in vitro) and SVRtata (in cells) (Extended Data Fig. 7d–f; performance assessment of SVRtata (in vitro) is shown in Extended Data Fig. 5n–r). SVRtata (in vitro) was found to correlate well (ρ = 0.86) with transcription strengths as well as with SVRtata (in cells) (ρ = 0.80) (Extended Data Fig. 7d, e). These results indicate that the use of HARPE in conjunction with SVR analysis is an effective method for the analysis of core promoter motifs. Furthermore, the extensive correlation between the in vitro and cell-based data (Figs. 2d, 4a, Extended Data Figs. 2g–j, 7d, 8a, b) provides comprehensive evidence that the mechanisms of transcription initiation in vitro are similar to those in cells.
The DPR is widely used in human promoters
To assess the role of the DPR in humans, we examined the relation between the HARPE-based DPR data and the corresponding sequences in natural human core promoters. First, we found that the relative nucleotide preferences in focused human core promoters12 are similar to those in the most active sequences in the HARPE assay in vitro and in cells (Extended Data Fig. 8c–e). It is therefore likely that data from the HARPE assay reflect the properties of the DPR in natural human promoters.
By using the SVR models, we were able to estimate the occurrence of core promoter motifs in natural human focused promoters. With SVR models for the DPR, we found that about 25–34% of human promoters in different cell lines (HeLa, MCF7 and GM12878) are predicted to have an active DPR (Fig. 4b, Extended Data Fig. 8f, g, Supplementary Discussion 5). Similarly, with the SVRtata models, we determined that about 15–23% of human promoters contain an active TATA box (Extended Data Fig. 7g–i, Supplementary Discussion 5). Thus, the DPR appears to be a widely used core promoter element. Moreover, the estimated occurrence of the DPR is comparable to that of the TATA box.
Notably, in sharp contrast to the DPR, a correctly positioned match to the RGWYGT DPE-like sequence14 (Fig. 1c) was found in only about 0.4–0.5% of human focused promoters (Supplementary Discussion 5). Therefore, in humans, a consensus DPE-like sequence is rare, as previously noted1,2,3, but the SVR-based DPR is relatively common. These findings further highlight the utility of machine learning relative to consensus approaches for the identification of core promoter sequence motifs.
We also tested the activities of individual DPR-like sequences in natural human promoters. To this end, we identified eight human promoters with an SVRc score of at least 2.5 and determined the activities of wild-type and mutant versions of the core promoters in cells (Fig. 4c, Extended Data Fig. 8h) and in vitro (Extended Data Fig. 8h, i). In all of the promoters that were tested, mutation of the DPR region resulted in a substantial decrease in transcriptional activity. These findings show that functionally active DPR motifs can be identified in natural promoters by using the SVR models.
Duality between the DPR and TATA box
To investigate the relation between the DPR, the TATA box, and the Inr, we examined the co-occurrence of these motifs in human promoters (Fig. 4d, Extended Data Fig. 9, Supplementary Discussion 6). We typically observed an increase in the occurrence of the Inr and Inr-like sequences with an increase in the SVR scores for the DPR. This effect is consistent with the cooperative function of the DPE and Inr motifs in Drosophila13. By contrast, the TATA motif is enriched in promoters lacking a DPR and depleted in promoters with high DPR scores. Similarly, but to a lesser extent, strong DPR motifs are more abundant in TATA-less promoters than in TATA-containing promoters (Extended Data Fig. 10). These findings suggest that some human core promoters depend predominantly on the DPR, whereas others depend mostly on the TATA box. This duality between the human DPR and TATA box suggests that they might have different biological functions and is consistent with the mutually exclusive properties of the DPE and TATA box in Drosophila7,29,30,31,32. Hence, the TATA–DPR duality is likely to reflect different mechanisms of transcription and potentially different modes of regulation of TATA-dependent versus DPR-dependent promoters in humans.
Here, we have used machine learning to decipher a promoter motif that could not be identified by the analysis of overrepresented sequences (Supplementary Discussion 7). Beyond the study of core promoters, this work describes a strategy for the machine learning analysis of functionally important DNA sequence motifs. In the future, it seems likely that machine learning models will continue to supersede consensus sequences in the characterization of DNA sequence motifs.
Methods
HARPE screening vector and promoter inserts
The HARPE screening vector (Extended Data Fig. 1b) was created by modification of the SuRE plasmid23 (a gift from J. van Arensbergen and B. van Steensel, Netherlands Cancer Institute). New features of the HARPE vector are as follows. First, to increase transcription levels, two GC-boxes (GGGGCGGGGC; binding sites for transcription factor Sp1) are located at positions −80 and −51 (the numbers indicate the positions of the upstream G of each GC-box) relative to the A+1 in the initiator (Inr) sequence of the core promoter that is to be inserted into the vector. Second, a TATA-like sequence (TTAACTATAA) upstream of the GC-boxes was mutated to CTGACTGGAC. Third, a KpnI restriction site is downstream of the −51 GC-box. Fourth, the KpnI site is followed by a spacer sequence and an AatII restriction site for insertion of core promoter sequences between the KpnI and AatII sites. Fifth, downstream of the AatII site, there is an RNA polymerase III (Pol III) terminator sequence (TTTTTTT) upstream of the transcribed sequence that is complementary to the reverse transcription primer. The Pol III terminator minimizes any potential background signal from Pol III transcription. For HARPE screening of randomized upstream sequences such as the TATA box, we used a slightly different screening vector in which the KpnI site is upstream of position −51. In this case, the downstream GC-box is included in the promoter insert rather than in the vector.
Randomized promoter inserts were generated by 5′ phosphorylation (T4 polynucleotide kinase; New England Biolabs) and annealing of partially complementary oligonucleotides (Extended Data Fig. 1c). The double-stranded DNA products were designed with 3′-overhangs for insertion between the KpnI and AatII sites of the HARPE vector. The SCP1m and human IRF1 core promoter sequences that were used are shown in Supplementary Table 2. In the analysis of the DPE region, the SCP1m region between +18 and +22 (CGAGC) was mutated to ATCCA (mutant MTE26). In the analysis of the TATA region, the SCP1m region between +28 and +34 (AGACGTG) was mutated to CTCATGT (mutant DPE6). In the IRF1 sequence, we introduced an A+11 to T substitution to eliminate a partial Pol III box A-like sequence.
HARPE library generation
The methodology for the preparation of the HARPE library was adapted from the SuRE procedure23. Annealed and phosphorylated promoter inserts were ligated into KpnI- and AatII-digested HARPE vector by using the TAKARA DNA Ligation Kit, Version 1 (Takara Bio). The resulting DNA was electroporated into DH5G CloneCatcher Gold (Genlantis) bacteria as recommended by the manufacturer, and the number of transformants was assessed by plating. Typically, a complexity of about 1,000,000 to 80,000,000 transformants was achieved. Next, a secondary downscaling step was performed to decrease the complexity of the library to about 100,000 or about 500,000 for shorter (8 to 12 bp) or longer (19 bp) randomized regions, respectively. Isolation of the DNA yielded the final HARPE DNA libraries, which were then transcribed in HeLa cells or in vitro.
Transcription of HARPE libraries in cells
HeLa cells (kind gift from the laboratory of A. Rao, La Jolla Institute for Immunology) were maintained at 37 °C under 5% CO2 in DMEM (Gibco) supplemented with 10% FBS (ATCC), 50 U/ml penicillin (Thermo Fisher Scientific), and 50 μg/ml streptomycin (Thermo Fisher Scientific). HeLa cells were not authenticated but were tested and found to be negative for mycoplasma contamination. Transfections were performed with Lipofectamine 3000 (Thermo Fisher Scientific) as recommended by the manufacturer. Typically, two 10-cm culture dishes were used per sample. During collection, one-third of the cell pellet was reserved for plasmid DNA extraction, whereas the rest of the cells were used for RNA extraction. RNA processing was then performed as described below. All HARPE experiments in cells were performed independently two times to ensure reproducibility of the data. Replicates originated from the same HARPE DNA libraries that underwent independent transfection and downstream processing.
Transcription of HARPE libraries in vitro
For each sample library, the products from 12 standard in vitro transcription reactions were combined. Standard reactions were performed as follows. DNA template (500 ng) was incubated with HeLa nuclear extract34 for preinitiation complex assembly at 30 °C for 1 h in 46 μl transcription buffer (20 mM HEPES-K+ (pH 7.6), 50 mM KCl, 6 mM MgCl2, 1.25% (w/v) polyvinyl alcohol, 1.25% (w/v) polyethylene glycol, 0.5 mM DTT, 3 mM ATP, 0.02 mM EDTA, and 2% (v/v) glycerol). rNTPs (4 μl; 0.4 mM final concentration of each rNTP) were added to initiate transcription. (Where indicated, sarkosyl was added to 0.2% (w/v) final concentration at 20 s after the addition of rNTPs.) The reaction was incubated at 30 °C for 20 min and terminated by the addition of 150 μl Stop Mix (20 mM EDTA, 200 mM NaCl, 1% (w/v) SDS, 0.3 mg/ml glycogen). Proteinase K (5 μl; 2.5 mg/ml) was added, and the mixture was incubated at 30 °C for 15 min. All in vitro transcription HARPE experiments were performed independently at least two times to ensure reproducibility of the data. Replicates originated from the same HARPE DNA libraries that underwent independent transcription and downstream processing.
RNA extraction and processing after transcription of HARPE libraries
RNA transcripts from cells or from in vitro transcription reactions were extracted with Trizol or Trizol LS (Thermo Fisher Scientific), respectively. Total RNA (40 μg for cell transfection experiments or the entire yield for in vitro experiments) was processed as follows. Contaminating plasmid DNA was removed with the TURBO DNA-free Kit—rigorous DNase treatment protocol (Thermo Fisher Scientific) as recommended by the manufacturer. The nucleic acids were precipitated with ethanol, and reverse transcription was performed with SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) with the RT primer (5′- GTGACTGGAGTTCAGACGTGT; Supplementary Table 2) as recommended by the manufacturer. The reaction products were then treated with 30 U RNase H (New England Biolabs) for 20 min at 37 °C. The nucleic acids were extracted with phenol-chloroform-isoamyl alcohol and precipitated with ethanol. The resulting cDNAs were then size-selected on a 6% polyacrylamide-8M urea gel using radiolabelled size markers (Supplementary Table 2) that enable the purification of cDNAs corresponding to transcription that initiates in the region from −5 to +6 relative to the A+1 in the Inr sequence.
Size-selected cDNAs were used as templates to generate DNA amplicons for Illumina sequencing using custom forward oligonucleotides containing the Illumina P5 and Read1-primer sequences preceding the sequence corresponding to nucleotides +1 to +16 of the promoter analysed (Supplementary Table 2). Reverse primers were selected from the NEBNext Multiplex Oligos for Illumina kits (NEB). NGS PCR amplicons were then size-selected on native 6% polyacrylamide gels before Illumina sequencing.
Processing of plasmid DNA for Illumina sequencing
For in vitro experiments, the starting material used was the HARPE DNA libraries. For cell transfection experiments, post-transfection plasmid DNA extraction was performed as described23. In brief, cells were treated with trypsin, washed with PBS, and then incubated in 500 μl nuclear extraction buffer (10 mM NaCl, 2 mM MgCl2, 10 mM Tris-HCl (pH 7.8), 5 mM DTT, 0.5% NP40) on ice for 5 min. Nuclei were pelleted at 7,000g and washed twice with 1 ml nuclear extraction buffer. DNA was then extracted with ZymoPURE Plasmid Miniprep Kit (Zymo Research). Plasmid DNA samples were used as a template for the generation of DNA amplicons for Illumina sequencing. The forward oligonucleotides contain the Illumina P5 and Read1-primer sequences followed by a promoter-specific sequence (Supplementary Table 2) that comprises nucleotides +1 through +16 (relative to the +1 TSS) for accurate DNA count assessment. Reverse primers were selected from the NEBNext Multiplex Oligos for Illumina kits (New England Biolabs), which match the Illumina Read2-primer sequence present on the HARPE plasmid. NGS PCR amplicons were then size-selected on native 6% polyacrylamide gels before Illumina sequencing.
Illumina sequencing
Illumina sequencing of NGS PCR amplicons was carried out on a HiSeq 4000 or Novaseq 6000 at the IGM Genomics Center, University of California, San Diego, La Jolla, CA (Moores Cancer Center, supported by NIH grant P30 CA023100 and NIH SIG grant S10 OD026929).
Transcription of individual test sequences and candidate human promoters
The plasmids used for testing individual clones were constructed with the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) as recommended by the manufacturer. These constructs include core promoter sequences12 from −36 to +50 nt relative to the +1 TSS of the specified genes.
For testing transcription activity in vitro, nucleic acids resulting from single standard reactions were isolated by phenol-chloroform-isoamyl alcohol extraction and ethanol precipitation, and subjected to primer extension analysis with 5′-32P-labelled RT primer. For testing transcription activity in cells, HeLa cells were transfected, and RNA was extracted with Trizol (Thermo Fisher Scientific). Total RNA (15 μg) was subjected to primer extension analysis with 5′-32P-labelled RT primer.
Primer extension products were resolved on 6% polyacrylamide-8M urea gels and quantified by using a Typhoon imager (GE Health Sciences) and the associated Amersham Typhoon control software v1.1. Quantification of radiolabelled samples was measured with Fiji v1.52i. All experiments for individual clones were performed independently at least three times to ensure reproducibility of the data.
NGS data processing
Single-read sequences (SR75) were screened according to the following criteria: a perfect match to the 10 nt directly upstream of the randomized region followed by the exact nucleotide count within the randomized region and a perfect match to the 10 nt directly downstream of the randomized region. (For the analysis of the TATA box (long version), the SR75 sequencing reads only allowed for 8 nt following the barcode; thus, the criteria that we employed were as follows: perfect match to the 12 nt directly upstream of the barcode; exact size of randomized barcode; and perfect match to the 8 nt directly downstream of the barcode.) All reads containing a match to the selection pattern were deemed usable and trimmed for sequences outside the randomized region. When present, highly abundant reads in the randomized box that correspond to the original promoter sequence or to invariant sequences from other constructs were discarded, as they are likely to have originated from inaccurate indexing of other multiplexed samples. Read counts for each variant were then computed and yielded a plasmid DNA dataset (DNA dataset) and a cDNA dataset (RNA dataset) for each sample.
For each DNA dataset, we used only sequences with a minimum read count of 10 and a minimum relative count of 0.75 reads per million (RPM) so that low-confidence variants would not be included in the analysis. RNA dataset sequences were then matched to the corresponding DNA dataset, which was used as a reference. For each HARPE experiment, transcription strength was then defined as RNA tag count (in RPMs) divided by DNA tag count (in RPMs). Total read counts, number of variants, coverage values, and required DNA read counts are in Supplementary Table 1.
HARPE targeting the TATA box
HARPE libraries for the analysis of the TATA-box region were prepared using the same methodology as for the other HARPE libraries, except that a second randomized ‘barcode’ box was added between +53 and +63 nt (short TATA version) or +53 and +67 nt (long TATA version). The SCP1m region between +28 and +34 nt (AGACGTG) was also mutated to CTCATGT (mutant DPE13). Conversion tables from barcode to TATA-box variant were built by paired-end sequencing of amplicons from the starting plasmid libraries. Sequencing reads were screened as described above and clusters for which both read 1 and read 2 passed the screening criteria were used to compute read counts. A minimum read count threshold was set so that ≥98% of barcodes were associated with a single TATA-box variant. Pairs that did not reach the threshold and the remaining 2% of unassigned barcodes were discarded. DNA datasets and RNA datasets for all TATA-box HARPE experiments were matched to their corresponding barcode-to-TATA conversion tables. All non-matching barcodes were not included. TATA variants associated with multiple barcodes were combined, and their transcription strengths were computed as the average transcription strength across the multiple barcodes.
Low-complexity, high-confidence HARPE dataset
Low-complexity libraries were generated by limiting the randomization of the DPR (that is, setting nucleotides +17 to +35 to TCGKYYKSSYWKKRMRTGC, which yields a maximum complexity of 8,192) as well as by adding a randomized 3-nt tag from +55 to +57 nt. The final library contained about 130,000 DPR-tag pairs, which resulted in a median value of 13 out of 64 possible 3-nt tags per DPR variant. The transcription strength for each DPR variant was computed by determining the average of the RNA tag count/DNA tag count values for all of the DPR–tag pairs for that variant.
Motif discovery
Motif discovery was performed using HOMER25. findMotifs.pl was used to search the 0.1% most transcribed HARPE sequences in the region of interest. Variants randomly selected from all tested sequences were used as background. We looked for 19-nt motifs in the DPR datasets and 12-nt motifs in the DPE only and MTE only datasets. Because the TATA box is not constrained to a single position, we did not specify a motif length for the TATA-box datasets. The homer2 find tool was used to retrieve the sequences matching the top motif as well as to compute position-weight-matrix-based HOMER motif scores. These sequences were then used to generate the sequence logo using WebLogo 335,36.
Data processing, statistics and graphical displays
All calculations (including Pearson’s correlation coefficients, Spearman’s rank correlation coefficients, P values, means, and standard deviations) were performed in the R environment (version 3.6.1) in Rstudio v1.1.463 with R packages ggplot2 v3.2.1, tidyr v1.0.0, dplyr v0.8.3 and rlist v0.4.6.1, or with Microsoft Excel. All replicate measurements were taken from distinct samples. Adobe Illustrator CS v11.0.0 was used to build figures.
Training of SVR models
Machine learning analyses were performed using functions of the R package e1071 (D. Meyer, E. Dimitriadou, K. Hornik, A. Weingessel and F. Leisch (2019). e1071: Misc Functions of the Department of Statistics, Probability Theory Group (formerly: E1071), TU Wien. R package version 1.7-2. https://CRAN.R-project.org/package=e1071). For SVR training, we used the default radial basis function (RBF) kernel, which yielded the best results among those tested. Grid search was performed for hyperparameters C (cost) and gamma, and cross validation was done by using two independent sets of sequences that were not used for the training (Extended Data Fig. 3b–d). Nucleotide variables for HARPE variants were computed as four categories (A, C, G and T), known as factors in R. To build the SVR model, we used the nucleotide variables as the input features and transcription strength as the output variable. For SVRb (or SVRc), we set aside 7,500 (or 6,500) test sequences (with the full range of transcription strengths) and trained the SVR with 200,000 of the remaining sequences (Extended Data Fig. 3a). For SVRtata, we set aside 5,000 test sequences (with the full range of transcription strengths) and trained the SVR with all remaining (232,713) sequence variants.
Use of the SVR models to predict transcription strength
The SVR models described in this study can be used to predict transcription strength with R by using the predict() function included in CRAN package e1071. Models are imported with readRDS(). Query sequence data must be formatted as follows. The variable names are V1 to V12 for SVRtata (corresponding to positions −32 to −21) and V1 to V19 for SVRc and SVRb (corresponding to positions from +17 to +35). Query sequences are split with one nucleotide per column and one sequence per row. Each column must have at least one A, one C, one G and one T to ensure that all variables are read as four categories (A, C, G, T). Prediction using an SVR model and a query sequence will return an output ‘SVR score’ that is related to the transcription strength and set on an arbitrary scale.
To streamline use of the models, we also provide an R script named SVRpredict.R (requires R with CRAN packages e1071 and docopt). SVRpredict.R inputs a model file as well as a sequence file (12- or 19-letter words/sequences, one sequence per line), and outputs a new file with each sequence and its associated predicted transcription strength in an added column (SVR_score).
Position index
To assess the effect of each sequence position on the SVR score, we used the position index (Extended Data Fig. 7j, k), which is the maximal SVR score increase that can be attained by a single nucleotide substitution at each position of the DPR. Because the positional contribution is affected by the sequence context (that is, the nucleotides at other positions within the DPR), the average positional contribution in 200 DPR contexts (that is, sequences in 200 different natural human promoters) was used to determine the position index.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The HARPE data are available from Gene Expression Omnibus (GEO; accession number, GSE139635). We obtained 5′-GRO-seq files (GSE6387233 and GSE9003512) and GRO-cap files (GSM1480321)37 from the Gene Expression Omnibus website (https://www.ncbi.nlm.nih.gov/geo/). Source data are provided with this paper.
Code availability
All computational analyses were performed by using R version 3.6.1 and previously described packages, as noted in the Methods.
References
Sandelin, A. et al. Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat. Rev. Genet. 8, 424–436 (2007).
Vo ngoc, L., Wang, Y.-L., Kassavetis, G. A. & Kadonaga, J. T. The punctilious RNA polymerase II core promoter. Genes Dev. 31, 1289–1301 (2017).
Haberle, V. & Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 19, 621–637 (2018).
Meylan, P., Dreos, R., Ambrosini, G., Groux, R. & Bucher, P. EPD in 2020: enhanced data visualization and extension to ncRNA promoters. Nucleic Acids Res. 48 (D1), D65–D69 (2020).
Roeder, R. G. 50+ years of eukaryotic transcription: an expanding universe of factors and mechanisms. Nat. Struct. Mol. Biol. 26, 783–791 (2019).
Butler, J. E. & Kadonaga, J. T. Enhancer-promoter specificity mediated by DPE or TATA core promoter motifs. Genes Dev. 15, 2515–2519 (2001).
Juven-Gershon, T., Hsu, J. Y. & Kadonaga, J. T. Caudal, a key developmental regulator, is a DPE-specific transcriptional factor. Genes Dev. 22, 2823–2830 (2008).
Zabidi, M. A. et al. Enhancer-core-promoter specificity separates developmental and housekeeping gene regulation. Nature 518, 556–559 (2015).
Parry, T. J. et al. The TCT motif, a key component of an RNA polymerase II transcription system for the translational machinery. Genes Dev. 24, 2013–2018 (2010).
Wang, Y. L. et al. TRF2, but not TBP, mediates the transcription of ribosomal protein genes. Genes Dev. 28, 1550–1555 (2014).
Duttke, S. H. C., Doolittle, R. F., Wang, Y.-L. & Kadonaga, J. T. TRF2 and the evolution of the bilateria. Genes Dev. 28, 2071–2076 (2014).
Vo Ngoc, L., Cassidy, C. J., Huang, C. Y., Duttke, S. H. & Kadonaga, J. T. The human initiator is a distinct and abundant element that is precisely positioned in focused core promoters. Genes Dev. 31, 6–11 (2017).
Burke, T. W. & Kadonaga, J. T. Drosophila TFIID binds to a conserved downstream basal promoter element that is present in many TATA-box-deficient promoters. Genes Dev. 10, 711–724 (1996).
Kutach, A. K. & Kadonaga, J. T. The downstream promoter element DPE appears to be as widely used as the TATA box in Drosophila core promoters. Mol. Cell. Biol. 20, 4754–4764 (2000).
Lim, C. Y. et al. The MTE, a new core promoter element for transcription by RNA polymerase II. Genes Dev. 18, 1606–1617 (2004).
Theisen, J. W. M., Lim, C. Y. & Kadonaga, J. T. Three key subregions contribute to the function of the downstream RNA polymerase II core promoter. Mol. Cell. Biol. 30, 3471–3479 (2010).
Burke, T. W. & Kadonaga, J. T. The downstream core promoter element, DPE, is conserved from Drosophila to humans and is recognized by TAFII60 of Drosophila. Genes Dev. 11, 3020–3031 (1997).
Louder, R. K. et al. Structure of promoter-bound TFIID and model of human pre-initiation complex assembly. Nature 531, 604–609 (2016).
Patel, A. B. et al. Structure of human TFIID and mechanism of TBP loading onto promoter DNA. Science 362, eaau8872 (2018).
Patwardhan, R. P. et al. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 27, 1173–1175 (2009).
Lubliner, S. et al. Core promoter sequence in yeast is a major determinant of expression level. Genome Res. 25, 1008–1017 (2015).
Arnold, C. D. et al. Genome-wide assessment of sequence-intrinsic enhancer responsiveness at single-base-pair resolution. Nat. Biotechnol. 35, 136–144 (2017).
van Arensbergen, J. et al. Genome-wide mapping of autonomous promoter activity in human cells. Nat. Biotechnol. 35, 145–153 (2017).
Weingarten-Gabbay, S. et al. Systematic interrogation of human promoters. Genome Res. 29, 171–183 (2019).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Juven-Gershon, T., Cheng, S. & Kadonaga, J. T. Rational design of a super core promoter that enhances gene expression. Nat. Methods 3, 917–922 (2006).
Cortes, C. & Vapnik, V. Support-vector networks. Mach. Learn. 20, 273–297 (1995).
Vapnik, V. N. The Nature of Statistical Learning Theory (Springer, 1995).
Willy, P. J., Kobayashi, R. & Kadonaga, J. T. A basal transcription factor that activates or represses transcription. Science 290, 982–985 (2000).
Hsu, J. Y. et al. TBP, Mot1, and NC2 establish a regulatory circuit that controls DPE-dependent versus TATA-dependent transcription. Genes Dev. 22, 2353–2358 (2008).
Chen, K. et al. A global change in RNA polymerase II pausing during the Drosophila midblastula transition. eLife 2, e00861 (2013).
Kedmi, A. et al. Drosophila TRF2 is a preferential core promoter regulator. Genes Dev. 28, 2163–2174 (2014).
Duttke, S. H. C. et al. Human promoters are intrinsically directional. Mol. Cell 57, 674–684 (2015).
Dignam, J. D., Lebovitz, R. M. & Roeder, R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 (1983).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 (2004).
Schneider, T. D. & Stephens, R. M. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100 (1990).
Core, L. J. et al. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 46, 1311–1320 (2014).
Acknowledgements
We thank E. P. Geiduschek, T. Juven-Gershon, G. Kassavetis, B. Delatte, J. Fei, G. Cruz-Becerra, and S. Chen for critical reading of the manuscript; J. van Arensbergen and B. van Steensel for the SuRE plasmid and protocols; B. Grant and C. Benner for advice; A. Rao for the HeLa cells; and the DNA sequencing facility at the Moores Cancer Center at UCSD (supported by NIH grant P30 CA023100 and NIH SIG grant S10 OD026929). L.V.n. received a UCSD Molecular Biology Cancer Fellowship. J.T.K. is the Amylin Chair in the Life Sciences. This work was supported by funding from NIH/NIGMS (R35 GM118060) to J.T.K.
Author information
Authors and Affiliations
Contributions
L.V.n., C.Y.H. and J.T.K. oversaw the overall design and execution of the project. The experiments were performed mostly by L.V.n. and C.Y.H. The analysis of the natural promoters was carried out by C.M. The computational analyses were performed by L.V.n., C.J.C. and C.Y.H. L.V.n. and J.T.K. were primarily responsible for writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Design and initial characterization of the HARPE assay.
a, RNA polymerase II core promoter elements that were examined in this study. This diagram shows the positions of the TATA box, initiator (Inr), motif ten element (MTE), downstream core promoter element (DPE), and downstream core promoter region (DPR) relative to the A+1 nucleotide in the Inr consensus sequence. The Inr and MTE function together with a strict spacing requirement between the two motifs. The Inr and DPE similarly act together with a strict spacing requirement between the motifs. The Figure is drawn roughly to scale. The sequences that were randomized in the HARPE experiments are also indicated. b, c, Preparation of the HARPE library. b, HARPE constructs have two GC-boxes (Sp1 binding sites) upstream of the core promoter. The core promoters used in this study (SCP1m and IRF1) are TATA-less (mTATA = mutant TATA box), initiator (Inr)-containing promoters. An RNA polymerase III (Pol III) terminator prevents transcription by Pol III. The open reading frame of green fluorescent protein (ORF) and the polyadenylation signal (PAS) promote the synthesis of mature and stable transcripts. For the study of the DPR, the randomized region is from +17 to +35 relative to the +1 TSS. c, The fragments containing randomized elements are produced by annealing oligonucleotides that give protruding ends matching the KpnI and AatII sticky ends on the pre-digested plasmid. A high-complexity library of ~1M to 80M variants is typically obtained after bacterial transformation. If required, the level of complexity is decreased to ~100k to ~500k variants with a subset of the transformants. d, Nucleotide preferences can be observed in the most active DPR sequences. The nucleotide frequencies at each position of the DPR in the top 50% to the top 0.1% of the most transcribed sequences are indicated. All sequences (100%) are included as a reference. e, f, DPR motifs identified by HOMER. e, HOMER motifs found in the top 0.1% of HARPE DPR variants. f, Position-weight matrix for the top HOMER motif. P-values associated with hypergeometric tests (one tailed, no adjustment). All panels show a representative experiment (n = 2 biologically independent samples). g–i, HARPE is highly reproducible. g, Most variants are present and detectable in biological replicates. The intersection comprises variants detected in both biological replicates (exact sequence match). PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16. h, Reproducibility of the DNA and RNA tag counts, and the resulting transcription strength value, for variants detected in both biological replicates. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16. i, Reproducibility of the MTE, DPE, IRF1, and SCP1 (with TATA box) datasets, for variants detected in both biological replicates. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16.
Extended Data Fig. 2 Further characterization of the HARPE assay and modification of the HARPE assay to include the analysis of the upstream TATA box element.
a–d, Relative promoter strengths in HARPE experiments performed in the absence versus the presence of sarkosyl. In vitro transcription reactions were performed in the absence or presence of 0.2% (w/v) sarkosyl (added immediately after transcription initiation). a, HARPE datasets with reactions performed in the presence of sarkosyl are reproducible. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16. b, Relative promoter strength does not appear to be affected by the addition of sarkosyl. Comparison of HARPE data from reactions carried out in the absence (Control) or the presence of sarkosyl. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16. c, The top 0.1% most highly transcribed promoter variants show similar nucleotide preferences in the absence (Control) or the presence of sarkosyl (representative experiment, n = 2 biologically independent samples). d, The individual analysis of 16 independent promoter variants shows that the relative promoter strengths are approximately the same in the absence (Control) or the presence of sarkosyl. PCC, Pearson’s correlation coefficient with two-tailed P-value = 7.1 × 10−11 (replicate 1) or 1.7 × 10−11 (replicate 2). For gel source data, see Supplementary Fig. 1. e–g, HARPE yields consistent data under different conditions. The nucleotide frequencies of the top 0.1% most active sequences are shown. e, HARPE analysis (in vitro) of the DPR with three different promoter cassettes: SCP1 lacking a TATA box (SCP1m), the human IRF1 core promoter (IRF1), and SCP1 containing a TATA box (SCP1). f, HARPE of the DPR (+17 to +35), DPE (+23 to +34), and MTE (+18 to +29) motifs with the SCP1m promoter in vitro. g, HARPE of the DPR in the SCP1m promoter transcribed in vitro or in cells. All panels show a representative experiment, n = 2 biologically independent samples. h–j, HARPE data generated in cells are similar to the corresponding in vitro data. h, The nucleotide frequencies of the top 0.1% most active DPR sequences obtained in cells are consistent with their in vitro counterparts. These HARPE experiments were performed with the human IRF1 core promoter. i, The nucleotide frequencies of the top 0.1% most active MTE and DPE sequences obtained in cells are consistent with their in vitro counterparts. These experiments examined either the MTE region or the DPE region in cells or in vitro. j, The nucleotide frequencies of the top 0.1% most active DPR sequences obtained in cells are consistent with their in vitro counterparts. These HARPE experiments were performed with the TATA-box-containing SCP1 core promoter. All panels show a representative experiment (n = 2 biologically independent samples). k–p, HARPE can be used to analyse regions upstream of the TSS. k, Design of a HARPE experiment targeting the upstream TATA-box region. Sequencing of the DNA constructs provides a correspondence between each TATA-box variant and a downstream barcode. Analysis of the barcode sequence in each transcript thus identifies its associated TATA-box variant sequence. l, HARPE was performed with a randomized region from −32 to −21 (long TATA) relative to the +1 TSS. The reproducibility of two independent experiments is shown. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16. m, HARPE was carried with a randomized region from −30 to −23 (short TATA) with an upstream TA dinucleotide at positions −32 and −31. The upstream TA sequence directs the formation of the TATA box in a single phase. The reproducibility of two independent experiments is also shown. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16. n, The nucleotide frequencies and top 8-nt and 12-nt HOMER motifs for the top 0.1% most transcribed variants are shown for HARPE data with the long TATA (−32 to −21) randomized sequence. The upstream T of the 8-nt TATA box motif was found to be located at position −32, −31, or −30 (representative experiment, n = 2 biologically independent samples). o, The nucleotide frequencies and top 8-nt HOMER motif for the top 0.1% most transcribed variants are shown for HARPE data with the short TATA (−30 to −23) randomized sequence. In the short TATA analysis, the upstream T of the TATA box is fixed at position −32, and thus, a distinct TATA-box sequence can be seen in the HOMER analysis (representative experiment, n = 2 biologically independent samples). p, The nucleotide frequencies in natural human focused promoters12 are similar to those in the long TATA dataset (n), particularly with the A and T nucleotides.
Extended Data Fig. 3 Initial characterization and optimization of the SVR models and the creation of a low complexity HARPE library for further SVR analysis of the DPR.
a, Selection of sequences for training of the SVR. Different numbers of training sequences were selected either randomly (blue line) or by using a combination of the most transcribed (Best) variants and Non-Best variants (that is, those variants that are not in the Best category) at a 1:1 ratio of Best:Non-Best (orange line). The resulting SVR models were used to predict the transcriptional activity of the Test Sequences in Fig. 3b, and the correlations between the predicted versus observed transcriptional activities are shown on the Y axis. In our studies, we used the SVR model (Selected variants) that was built on the training set that consists of the 100,000 most transcribed (Best) variants and randomly selected 100,000 Non-Best variants (representative experiment n = 2 biologically independent samples). The models in this figure were built by using default parameters for SVR training. b–d, Grid search cross validation for the SVR models. Grid search results with different values for the cost of misclassification (cost) and individual training example influence (gamma) for (b) SVRb, (c) SVRc, and (d) SVRtata. Shown are Spearman’s rank correlation coefficient (rho) between the prediction of each model and the observed transcription strength with two independent datasets (validation and test sets, which are separate halves of the test sequences described in Fig. 3b) that were not used in the training of the models. SVR models were trained as described in Methods. Undefined (UD) correlation is observed when the prediction of a model is constant regardless of the sequence. The hyperparameter values that were selected in this study are as follows: SVRb (c = 10 and gamma = 0.1); SVRc (c = 1, gamma = 0.02); and SVRtata (c = 100, gamma = 0.1). e, Concordance between the predicted and observed activities of DPR sequence variants, as shown with a logarithmic scale. Analysis of 7500 independent test sequences in the HARPE dataset that were not used in the training of SVRb. This figure presents the data shown in Fig. 3b with a log scale for the x- and y-axes. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16. f–i, Design and use of a low complexity HARPE library that provides high-quality data on 8,431 unique DPR variants. f, Design of a low complexity library with multiple DNA sequence tags for each DPR variant. A restricted library was built with 8,431 unique DPR variants. Each variant was associated with about 15 downstream DNA sequence tags that enable multiple measurements of transcription strength for the same variant within the same experiment. g, To restrict the complexity of the library, the randomized region was shortened to 13 nucleotides, and each position contained one of only two different bases. h, The number of tags per variant. The median value is 13 (representative experiment, n = 2 biologically independent samples). i, The observed transcription strength for each of the DPR variants. There are multiple different sequence tags for each DPR variant. The plot shows the average (black) ± standard deviation (designated in grey) for each of the variants (representative experiment, n = 2 biologically independent samples).
Extended Data Fig. 4 Individual assessment of the transcription activity of 16 independent variants that are not present in the SVR training set.
a, The 16 variants, which include the original SCP1m sequence, represent a wide range of SVR scores. Nucleotides that differ from the SCP1m sequence are indicated in red type. b, The 16 promoter sequences were inserted into plasmids and subjected to in vitro transcription and primer extension analysis (n = 4 biologically independent samples). The plots show the predicted SVRb scores and the observed transcription strengths. Replicate 1 is shown in Fig. 3d. PCC, Pearson’s correlation coefficient with two-tailed P-values <1.7 × 10−6; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16. For gel source data, see Supplementary Fig. 1. c, The 16 promoters were subjected to transient transfection and primer extension analysis (n = 4 biologically independent samples). The plots show the predicted SVRb scores and the observed transcription strengths. PCC, Pearson’s correlation coefficient with two-tailed P-value <3.9 × 10−6; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 5 Use of the SVR models to identify active sequence elements and performance assessment of the SVR models.
a–c, The relationship between SVR scores and transcription strength. Box-plot diagrams are shown for (a) SVRb, (b) SVRc, and (c) SVRtata with all of their corresponding HARPE sequence variants that are placed in bins of the indicated SVR score ranges. Sequence variants with SVRb score ≥ 2, SVRc score ≥ 2, and SVRtata score ≥ 1 are typically at least about 6 times more active than an inactive sequence (light blue shaded regions), and are thus designated as “active”. The thick horizontal lines are the medians, and the lower and upper hinges are the first and third quartiles, respectively. Each upper (or lower) whisker extends from the upper (or lower) hinge to the largest (or lowest) value no further than 1.5 * IQR from the hinge. Data beyond the end of the whiskers (outlying points) are omitted from the box plot. Sequence variants with transcription strength = 0 were removed to allow log-scale display of the diagrams. The horizontal dashed grey lines denote the transcription strengths of the median inactive sequences. d–h, Performance assessment of SVRb. All panels show a representative experiment (n = 2 biologically independent samples). d, Selection of HARPE variants used in performance assessment. The top 10% sequence variants were designated as active/positive for transcription, and an equal (randomly selected) number of the bottom 50% of sequence variants were designated as inactive/negative for transcription. These sequences were then used in the performance assessment. Intermediate variants that were between the top and bottom groups were not included. The transcription strengths of all selected sequences are shown. e, Receiver operating characteristic (ROC) curve. f, Precision-recall (PR) curve. g, Performance measures relative to the minimum SVRb score required for a positive prediction. Performance was computed by counting true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN). Accuracy [(TP+TN) / (TP+FP+TN+FN)] reflects how often SVRb predictions are correct. Precision [TP / (TP + FP)] is the proportion of positive predictions that are correct. Sensitivity or recall or true positive rate [TP / (TP + FN)] is the proportion of transcriptionally active variants that are correctly predicted as positives. h, False positive and false negative rates. The false positive rate [FP / (FP + TN)] is the probability for an inactive sequence to be incorrectly predicted as positive. The false negative rate [FN / (FN + TP)] = (1 − Sensitivity) is the probability for an active sequence to be incorrectly predicted as negative. Performance values are shown for selected minimum SVRb scores (1.5 and 2). All panels show a representative experiment (n = 2 biologically independent samples). i–m, Performance assessment of SVRc. i, Selection of HARPE variants used in performance assessment. The top 10% sequence variants were designated as active/positive for transcription, and an equal (randomly selected) number of the bottom 50% of sequence variants were designated as inactive/negative for transcription. These sequences were then used in the performance assessment. Intermediate variants that were between the top and bottom groups were not included. The transcription strengths of all selected sequences are shown. j, Receiver operating characteristic (ROC) curve. k, Precision-recall (PR) curve. l, Performance measures relative to the minimum SVRc score required for a positive prediction. Performance was computed by counting true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN). Accuracy [(TP+TN) / (TP+FP+TN+FN)] reflects how often SVRc predictions are correct. Precision [TP / (TP + FP)] is the proportion of positive predictions that are correct. Sensitivity [TP / (TP + FN)] is the proportion of transcriptionally active variants that are correctly predicted as positives. m, False positive and false negative rates. The false positive rate [FP / (FP + TN)] is the probability for an inactive sequence to be incorrectly predicted as positive. The false negative rate [FN / (FN + TP)] = (1 − Sensitivity) is the probability for an active sequence to be incorrectly predicted as negative. Performance values are shown for selected minimum SVRc scores (1.5 and 2). All panels show a representative experiment (n = 2 biologically independent samples). n–r, Performance assessment of SVRtata. n, Selection of HARPE variants used in performance assessment. The top 10% sequence variants were designated as active/positive for transcription, and an equal (randomly selected) number of the bottom 50% of sequence variants were designated as inactive/negative for transcription. These sequences were then used in the performance assessment. Intermediate variants that were between the top and bottom groups were not included. The transcription strengths of all selected sequences are shown. One outlier variant with an exceptionally high transcription level was omitted in the graph, but was included in the performance analysis. o, Receiver operating characteristic (ROC) curve. p, Precision-recall (PR) curve. q, Performance measures relative to the minimum SVRtata score required for a positive prediction. Performance was computed by counting true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN). Accuracy [(TP+TN) / (TP+FP+TN+FN)] reflects how often SVRtata predictions are correct. Precision [TP / (TP + FP)] is the proportion of positive predictions that are correct. Sensitivity [TP / (TP + FN)] is the proportion of transcriptionally active variants that are correctly predicted as positives. r, False positive and false negative rates. The false positive rate [FP / (FP + TN)] is the probability for an inactive sequence to be incorrectly predicted as positive. The false negative rate [FN / (FN + TP)] = (1 − Sensitivity) is the probability for an active sequence to be incorrectly predicted as negative. Performance values are shown for minimum SVRtata scores = 1.0. All panels show a representative experiment (n = 2 biologically independent samples).
Extended Data Fig. 6 Further analysis of the SVR models and their relation to consensus sequence-based approaches.
a–e, SVR models based on HARPE data with different promoter backgrounds are consistent. SVR models were tested with the 7500 DPR sequence variants used in Fig. 3b. a, SVRirf1 models trained with HARPE data for the DPR with the IRF1 promoter cassette (promoter background) are reproducible. b, SVRb based on HARPE data for the DPR with the SCP1m promoter cassette (promoter background) is similar to the SVRirf1 model trained with HARPE data for the DPR in the IRF1 background. c, SVRscp1 models trained with HARPE data for the DPR with the SCP1 (TATA-containing) promoter cassette (promoter background) are reproducible. d, SVRb for the DPR in the TATA-less SCP1m promoter cassette (promoter background) is similar to the SVRscp1 model for the DPR in the TATA-containing SCP1 promoter cassette. e, SVRb and SVRscp1 exhibit similar DNA sequence preferences. This figure shows the web logos for the top HOMER motifs identified with the top 0.1% DPR sequences (in 500,000 random sequences), as assessed with either SVRb or SVRscp1. f–h, SVR analysis incorporates information that is not encapsulated in a consensus of enriched sequences in the most active variants. f, Web logo for the top HOMER motif identified with the 0.1% most transcribed DPR sequences. This panel is adapted from Fig. 1c and shows the DPE-like RGWYGT consensus of enriched sequences from +28 to +33. In contrast, the SVR model is generated from strong, intermediate, and weak variants of the entire DPR region. g, HARPE variants with a perfect match to the RGWYGT consensus exhibit transcription strengths that range from highly active to inactive. h, SVRb accurately predicts the transcription strengths of different HARPE variants with a perfect match to the RGWYGT consensus. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16. i, An SVR-based approach provides a more accurate prediction of DPR activity than a consensus sequence-based method. The plots show the correlation between the observed transcription strength (in vitro) and the predicted scores of the DPR, as assessed with either SVRb (upper; adapted from Fig. 3b) or a consensus sequence/position-weight matrix-based method (HOMER; lower). The HOMER consensus/position-weight matrix (Fig. 1c, Extended Data Fig. 1e, f) is based on the top 0.1% most transcribed DPR sequences. The DPR variants are the 7500 Test Sequences shown in Fig. 3. The coloured density scale is identical for both plots (representative experiment, n = 2 biologically independent samples). PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16. j, k, SVRb scores are influenced by DNA sequence context (that is, flanking nucleotides), whereas PWM-based HOMER scores treat individual nucleotide positions independently. j, Box-plot diagrams of the changes in the HOMER motif scores (top) and the SVRb scores (bottom) due to an A-to-G substitution at each of the indicated positions. The values were generated with 200 different DPR sequences in randomly-selected natural human promoters. The thick horizontal lines are the medians, and the lower and upper hinges are the first and third quartiles, respectively. Each upper (or lower) whisker extends from the upper (or lower) hinge to the largest (or lowest) value no further than 1.5 * IQR from the hinge. Data beyond the end of the whiskers (outlying points) are omitted from the box plot. A representative experiment is shown (n = 2 biologically independent samples). k, The influence of sequence context is accurately captured by the SVR model. Shown are the changes in SVRb score and transcription strength for 4,081 DPR variants when A is mutated to G at positions +30 (left) or +32 (right). The transcription data of the sequence variants were from the Low Complexity Library (Fig. 3c). PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16.
Extended Data Fig. 7 Characterization of the properties of the SVR models and the generation of SVRtata for the TATA box and SVRc for the DPR with cell-based data.
a–c, SVR models capture the preferred distances between the TSS and the DPR. a, The most significantly enriched 8-nt HOMER motif found in the top 0.1% of HARPE DPR variants (top) and its associated position-weight matrix (bottom). P-value associated with hypergeometric tests (one tailed). This 8-nt DPE-like motif closely resembles the Drosophila DPE consensus sequence2,14. Importantly, the DPE-like sequence is shorter than the DPR region and is therefore not at a fixed position. b, Positional preference analysis of the 8-nt motif in the top 0.1% HARPE DPR variants shows a preferred major position (74%) as well as a minor position (17%) that is 1 nt upstream of the major position. c, SVRb accurately predicts the transcription strength of sequence variants in all positions. This figure shows box-plot diagrams of the transcription strength for all variants within the HARPE dataset that contain the 8-nt motif at each position. The quality of the prediction at each position is indicated by Spearman’s rank correlation coefficient (rho) between the observed transcription strength and SVRb score, HOMER motif score with the 19-nt DPR motif (shown in Extended Data Fig. 1e, f), or HOMER motif score with the 8-nt DPR motif (shown in a). The thick horizontal lines are the medians, and the lower and upper hinges are the first and third quartiles, respectively. Each upper (or lower) whisker extends from the upper (or lower) hinge to the largest (or lowest) value no further than 1.5 * IQR from the hinge. Data beyond the end of the whiskers (outlying points) are omitted from the box plot. All panels show a representative experiment (n = 2 biologically independent samples). d–i, Machine learning analysis of the HARPE TATA-box data yields an SVRtata model for the TATA box. The HARPE data for the long TATA-box region (−32 to −21; Extended Data Figs. 1a, 2k–p, 8a, b) were subjected to SVR analysis. The resulting SVR models (derived from data generated in vitro or in cells) were termed SVRtata. d, The SVRtata model from HARPE data in cells is similar to that from HARPE data in vitro. The SVRtata (in vitro) and SVRtata (in cells) scores are compared by using 5000 independent test sequences that were not used in the training of the SVR. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16. e, Comparison of SVRtata scores and the observed transcription strengths of 5000 independent test sequences. These results are based on in vitro data. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16. f, Comparison of HOMER motif scores and the observed transcription strengths of the same 5000 test sequences used in e. The position-weight matrices of the top 12-nt (left) or 8-nt (right) HOMER motifs (Extended Data Fig. 2n) were used to determine HOMER motif scores. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16. g, Cumulative frequency of SVRtata scores of natural human promoters in HeLa cells. Approximately 23% of 11,932 human promoters and 4% of 100,000 random sequences (61% average G/C content, as in human core promoters) have an SVRtata (in vitro) score of at least 1 (marked with a green line), which corresponds to an active TATA box (Extended Data Fig. 5c). h, Cumulative frequency of SVRtata scores of natural human promoters in MCF7 cells. Focused promoters identified in ref. 12 were used. Approximately 18% of 7,678 MCF7 promoters and 4% of 100,000 random sequences (61% average G/C content, as in human core promoters) have an SVRtata (in vitro) score of at least 1 (marked with a green line), which corresponds to an active TATA box. i, Cumulative frequency of SVRtata scores of natural human promoters in GM12878 cells. Focused promoters were identified as described in ref. 12 by using GRO-cap data in human GM12878 cells from ref. 37. Approximately 15% of 30,643 GM12878 promoters and 4% of 100,000 random sequences (61% average G/C content, as in human core promoters) have an SVRtata (in vitro) score of at least 1 (marked with a green line), which corresponds to an active TATA box. All panels show a representative experiment (n = 2 biologically independent samples). j, k, Most positions within the DPR have a moderate impact upon the overall SVR score. The influence of each position in the DPR on the model prediction score is shown by the value of the Position Index. The Position Index at position X is the average of the maximal magnitude of variation in (j) the SVR score or (k) the HOMER motif score with A, C, G or T at position X with 200 different DPR sequences that were randomly selected from natural human promoters. As a reference, the Web Logo for the top HOMER motif identified with the 0.1% most transcribed DPR sequences is also shown. l, m, SVRc model of the DPR with HARPE data generated in cells. l, HARPE libraries were transfected in cells, and normalized RNA tags were obtained. The SVRc (SVR from cell-based data) scores derived from these data correlate with measured transcription strengths in cells (with data that are independent of the SVRc training data) (representative experiment, n = 2 biologically independent samples). PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16. m, The SVRc models obtained from cells are reproducible. PCC, Pearson’s correlation coefficient with two-tailed P-value <2.2 × 10−16; rho, Spearman’s rank correlation coefficient with two-tailed P-value <2.2 × 10−16.
Extended Data Fig. 8 Analysis of the HARPE TATA data as well as the DPR in natural human promoters.
a, b, The nucleotide preferences of the top 0.1% most active TATA-box sequences in cells are similar to those of their in vitro counterparts. a, Long randomized TATA-box region (-32 to -21 relative to the +1 TSS). b, Short randomized TATA-box region (-30 to -23 relative to the +1 TSS). All panels show a representative experiment (n = 2 biologically independent samples). c, Distinct nucleotide preferences can be seen at the DPR in focused human promoters, which were identified as described in ref. 12 by using 5′GRO-seq data in HeLa cells33. d, The top ~2.5% (11,932) most active DPR sequences in cells, as assessed by HARPE, have nucleotide preferences that are similar to those seen in natural human core promoters in HeLa cells (representative experiment, n = 2 biologically independent samples). e–g, Relationship between natural human promoter sequences and HARPE data in vitro. e, The top ~2.5% (11,932) most active DPR sequences in vitro, as assessed by HARPE, have nucleotide preferences that are similar to those seen in natural human core promoters in HeLa cells. f, Cumulative frequency of SVRb DPR scores of natural human promoters. Approximately 26% of 11,932 human promoters (HeLa cells), 12% of 100,000 random sequences (61% average G/C content, as in human core promoters), and 0.4% of 10,000 inactive sequences (randomly selected from the 50% least active sequences in the HARPE assay; not used in the training of the SVR) have an SVRb score of at least 2 (marked with a green line), which corresponds to an active DPR (Extended Data Fig. 5a). g, Cumulative frequency of SVRc and SVRb DPR scores of natural human promoters in MCF7 and GM12878 cells. Approximately 34% of 7,678 MCF7 promoters, 34% of 30,643 GM12878 promoters, 17% of 100,000 random sequences (61% average G/C content, as in human core promoters), and 2.6% of 10,000 inactive sequences (randomly selected from the 50% least active sequences in the HARPE assay; not used in the training of the SVR) have an SVRc score of at least 2 (marked with a green line), which corresponds to an active DPR (Extended Data Fig. 5b). Approximately 26% of 7,678 MCF7 promoters, 25% of 30,643 GM12878 promoters, 12% of 100,000 random sequences (61% average G/C content, as in human core promoters), and 0.4% of 10,000 inactive sequences (randomly selected from the 50% least active sequences in the HARPE assay; not used in the training of the SVR) have an SVRb score of at least 2 (marked with a green line), which corresponds to an active DPR (Extended Data Fig. 5a). All panels show a representative experiment (n = 2 biologically independent samples). h, i, Analysis of the DPR in natural human promoters. h, Sequences of natural human promoters that contain DPR motifs with an SVRb score >6 and an SVRc score >2.5. The mutant DPR sequence has an SVRb score = 0.3 and an SVRc score = 0.3. i, Mutational analysis reveals DPR activity in different human promoters with SVRb DPR scores >6. In each of the mutant promoters, the wild-type DPR was substituted with a DNA sequence that has an SVRb DPR score of 0.3 (data are depicted as the mean with error bars denoting standard deviation, n = 3 or 4 biologically independent samples, as indicated by the points representing independent samples on the graph). The sequences of the tested promoters are shown in f. Promoter activity was measured by in vitro transcription followed by primer extension analysis of the TSSs. All P-values <0.01 (Student’s t-test, two-tailed, paired). For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 9 Analysis of the DPR and its relationship to the Inr and TATA box in active human promoters in different human cell lines.
a–e Analysis of the DPR and its relationship to the Inr and TATA box in active human promoters in HeLa cells. a, Distribution of focused human promoters derived from HeLa cells in increasing SVRc DPR score bins. Bins 9 and 10 have less than 100 promoters. b, The frequencies of occurrence of the Inr and Inr-like sequences in different bins of promoters with increasing SVRc DPR scores. The Inr-like sequence is as defined previously12. c, The frequencies of occurrence of the TATA box and TATA-like sequences decrease as the SVRc DPR score increases. d, Distribution of focused human promoters in increasing SVRb DPR score bins. Promoters with SVRb scores between 4.24 and 17 were combined together in bin 11. e, The frequencies of occurrence of Inr-like sequences, TATA-like sequences, and TATA-box motifs (as assessed with SVRtata ≥ 1; Extended Data Fig. 5c) in different bins of promoters with increasing SVRb DPR scores. The Inr-like and TATA-like sequences are as defined previously12. In b and c, bins with less than 100 promoters are indicated with open circles and are connected by dashed lines. In e, bin 11 is shown in black circles connected by dashed black lines. All panels show a representative experiment (n = 2 biologically independent samples). f, g, Analysis of the DPR and its relationship to the Inr and TATA box in active human promoters in MCF7 and GM12878 cells. f, Distribution of focused human promoters in increasing SVRc DPR score bins. For each cell line, bin 10 has less than 100 promoters. MCF7 focused promoters are described in ref. 12. GM12878 focused promoters were identified as described in ref. 12 by using GRO-cap data in human GM12878 cells from ref. 37. g, The frequencies of occurrence of Inr-like sequences, TATA-like sequences, and TATA-box motifs (as assessed with SVRtata ≥ 1; Extended Data Fig. 5c) in different bins of promoters with increasing SVRc DPR scores. The Inr-like and TATA-like sequences are as defined previously12. Bins with less than 100 promoters are indicated with open circles and are connected by dashed lines. All panels show a representative experiment (n = 2 biologically independent samples).
Extended Data Fig. 10 Distribution of SVR DPR scores for human promoters in relation to their SVRtata scores.
Human promoters were divided into four groups according to their SVRtata score. For each TATA box category, the distribution of SVR DPR scores is shown for each of five classes of promoters (no DPR, weak DPR, intermediate DPR, good DPR, and strong DPR). a, Human focused promoters obtained from HeLa cells12,33 analysed with SVRtata and SVRc. b, Human focused promoters obtained from HeLa cells analysed with SVRtata and SVRb. c, Human focused promoters obtained from MCF7 cells12 analysed with SVRtata and SVRc. d, Human focused promoters obtained from GM12878 cells37 analysed with SVRtata and SVRc. Focused promoters were identified as described in ref. 12 by using GRO-cap data in human GM12878 cells from ref. 37. All panels show a representative experiment (n = 2 biologically independent samples).
Supplementary information
Supplementary Information
This file contains Supplementary Discussions 1 to 7, Supplementary References, Supplementary Tables 1 and 2, and Supplementary Fig. 1.
Source data
Rights and permissions
About this article

Cite this article
Vo ngoc, L., Huang, C.Y., Cassidy, C.J. et al. Identification of the human DPR core promoter element using machine learning. Nature 585, 459–463 (2020). https://doi.org/10.1038/s41586-020-2689-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2689-7
- Springer Nature Limited
This article is cited by
-
Quantitative analysis of transcription start site selection reveals control by DNA sequence, RNA polymerase II activity and NTP levels
Nature Structural & Molecular Biology (2024)
-
Superior electroadhesion force with permittivity-engineered bilayer films using electrostatic simulation and machine learning approaches
Scientific Reports (2024)
-
A 3-Gene Random Forest Model to Diagnose Non-obstructive Azoospermia Based on Transcription Factor-Related Henes
Reproductive Sciences (2023)
-
Plant synthetic epigenomic engineering for crop improvement
Science China Life Sciences (2022)
-
The TFIID pivot of preinitiation complex
Science China Life Sciences (2021)