

Abstract
Dental enamel is a principal component of teeth1, and has evolved to bear large chewing forces, resist mechanical fatigue and withstand wear over decades2. Functional impairment and loss of dental enamel, caused by developmental defects or tooth decay (caries), affect health and quality of life, with associated costs to society3. Although the past decade has seen progress in our understanding of enamel formation (amelogenesis) and the functional properties of mature enamel, attempts to repair lesions in this material or to synthesize it in vitro have had limited success4,5,6. This is partly due to the highly hierarchical structure of enamel and additional complexities arising from chemical gradients7,8,9. Here we show, using atomic-scale quantitative imaging and correlative spectroscopies, that the nanoscale crystallites of hydroxylapatite (Ca5(PO4)3(OH)), which are the fundamental building blocks of enamel, comprise two nanometric layers enriched in magnesium flanking a core rich in sodium, fluoride and carbonate ions; this sandwich core is surrounded by a shell with lower concentration of substitutional defects. A mechanical model based on density functional theory calculations and X-ray diffraction data predicts that residual stresses arise because of the chemical gradients, in agreement with preferential dissolution of the crystallite core in acidic media. Furthermore, stresses may affect the mechanical resilience of enamel. The two additional layers of hierarchy suggest a possible new model for biological control over crystal growth during amelogenesis, and hint at implications for the preservation of biomarkers during tooth development.
Similar content being viewed by others


Main
Enamel covers the entire crown of human teeth (Fig. 1a), reaching thicknesses of several millimetres (Fig. 1b). A characteristic microstructural element, the enamel rod (Fig. 1c), is composed of thousands of lath-like crystallites aligned with their crystallographic c direction approximately parallel to the long axes of the rods (Fig. 1d). Crystallites sectioned normal to their long axis appear as oblong polygons with an edge length of 20–50 nm in the short direction and 70–170 nm in the long direction (Fig. 1e, f). Characteristic length scales of the periodic hydroxylapatite (OHAp) lattice are in the subnanometre regime (Fig. 1g–i).

a, Left, length scales of enamel in a human premolar (values for a–i are indicated); right, optical image. b, Section parallel to the mid-coronal cervical plane (indicated in pink in a) with the external enamel surface (EES) and the dentine-enamel junction (DEJ) labelled. c, SEM image of keyhole-shaped cross-sections of enamel rods in lactic-acid-etched outer enamel; location of image shown boxed in b. d, SEM image of OHAp crystallites. e–g, STEM-ADF images at increasing magnification of enamel crystallites in cross-section, oriented approximately parallel to the [001] zone axis (shown in g). Arrows in f indicate the intergranular space between crystallites, and shell and core regions of crystallites. The CDL in g (arrowed) appears bright in ADF. h, Cryo-STEM-ADF lattice image of a crystallite oriented parallel to the [010] zone axis (inset, fast Fourier transform). i, Close-up of h with rendering of a 2 × 2 × 2 OHAp supercell (Ca, blue; O, red; P, green; H, white).
Enamel owes its hardness (up to about 5 GPa) to its high mineral content (approximately 96 wt%)10. Although enamel is nominally composed of OHAp, magnesium (0.2–0.6 wt%), sodium (0.2–0.9 wt%), carbonate (2.7–5 wt%) and fluoride (about 0.01 wt%) are also present11. Although the distribution of the minor constituents is known to vary over tens to hundreds of micrometres11, gradients over much shorter distances have only recently been discovered. Specifically, in rodent incisor enamel, most Mg is confined between crystallites as Mg-substituted amorphous calcium phosphate (Mg-ACP), controlling enamel dissolution and mechanical properties7. Segregation of Mg and Na ions to a 2–10-nm-thick layer between human enamel crystallites was confirmed8. However, it has not yet been shown that this layer is identical to the Mg-ACP found in rodent enamel.
Perhaps uniquely to human enamel, the centre of the crystallite seems to be more soluble, is more prone to electron-beam-induced damage, and displays a poorly understood contrast feature that is known as the central dark line (CDL)12,13. All three have generally been assumed to be related to the presence of defects in the crystallite lattice, but the exact nature of these defects is not known13. We therefore set out to test whether there are compositional gradients of minor enamel constituents across single crystallites.
In annular dark-field scanning transmission electron microscopy (STEM-ADF) images of human outer enamel, crystallites are separated by narrow regions that appear darker than the crystallite (Fig. 1e, f), consistent with expectations for a Mg-rich amorphous intergranular phase. Additionally, they exhibit a shell that appears brighter than the core (Fig. 1e, f). The sensitivity of enamel to radiation limits the tolerable electron dose and hampers high-resolution analysis of enamel crystallites (Supplementary Information section 1.3). Here, sample cooling and low-dose imaging conditions in cryogenic STEM (cryo-STEM) enable atomic-resolution imaging of ultrathin sections (20–30 nm; Supplementary Fig. 1, Supplementary Table 1), revealing a continuous atomic lattice across the entire crystallite (Figs. 1h, i, 2a; Supplementary Figs. 2, 3). Unlike the shell, the core appears as a patchwork of lighter and darker areas on either side of the CDL, and seems more prone to beam damage13.

a, Spherical-aberration (Cs)-corrected cryo-STEM-ADF lattice image of the core of a single enamel crystallite oriented to the [001] zone axis, close to the CDL (yellow arrows). White arrows indicate darker patches in the core region. Inset, fast Fourier transform. b, Mg K-edge XANES of human enamel and reference materials. Peaks at approximately 1,311 eV (A) and 1,313 eV (B) are more pronounced in spectra of human enamel and indicate the presence of Mg on OHAp lattice sites. Fit parameters are reported in Supplementary Table 3. c–e, Cryo-STEM EEL spectra obtained from a region containing several enamel crystallites, with close-ups of the P L2,3-edge (d) and the Ca L2,3, O K and C K-edges (e). f, MCR components (Comp. 1,2) contributing to feature near the Mg L2,3-edge. g, Cryo-STEM-ADF image of an enamel crystallite. h, Spatial intensity map of MCR components 1 (green) and 2 (magenta) in g. i, Average intensity profile for the region of interest indicated in g and h, in the direction of the arrow.
Although STEM in conjunction with energy-dispersive X-ray spectroscopy (STEM-EDS) reported approximately 0.4 atom % (at.%) Mg, 0.7 at.% Na and 0.6 at.% F in crystallites (Supplementary Fig. 4, Supplementary Table 2), elemental maps were largely featureless (not shown). The presence of at least some Mg on apatite sites in addition to Mg-ACP was confirmed by bulk X-ray absorption spectroscopy (XAS) at the Mg K-edge (Fig. 2b, Supplementary Fig. 7). Major components are well represented in cryo-STEM electron energy-loss spectra (EELS; Fig. 2c–e). Unfortunately, Na and F were not detected, the C K-edge is rather weak (Fig. 2e inset), and spectral component maps did not show gradients within crystallites (Supplementary Fig. 5), despite the use of a sensitive direct electron detector for EELS14.
However, a feature near the expected onset of the Mg L2,3-edge at 51 eV decomposes into two principal components (with no residual) by multivariate curve resolution (MCR, Fig. 2f, Supplementary Fig. 6)15. One of these is predominant in the shell, the other in the intergranular Mg-ACP and in the core (compare Fig. 2g, h, Supplementary Fig. 6). Other elements may also have minor edges in this spectral area, whereas STEM-EDS and bulk XAS do support the presence of Mg. The two components suggest distinct local environments in the core and shell, and even gradients within the core (Fig. 2i). As susceptibility to beam damage limited our ability to further explore such gradients by cryo-STEM methods, we turned to atom probe tomography (APT, Supplementary Fig. 8)16,17 to provide additional insights.
APT spectra of untreated human enamel resemble those of rodent enamel (Supplementary Fig. 9, Supplementary Table 4)7,16. In 3D reconstructions of APT data, it takes some practice to recognize the faceted cross-sections of enamel crystallites (Supplementary Figs. 10, 11). Treatment of samples with 250 mM NaF at pH 8.4 (37 °C, 24 h), however, resulted in a marked increase of CaF+ ions (Supplementary Table 5). These ions clearly outline individual crystallites (compare Supplementary Fig. 10a–c, and d, e) and thus greatly facilitate the analysis of reconstructions (Fig. 3). Fluoride treatment further results in an increase in sodium content (+0.39 at.%, +71% relative) with moderate statistical significance (P < 0.1) that also seems to be limited to the intergranular phase (see below), but has a negligible overall impact on composition of the samples otherwise (Supplementary Tables 5, 6). These observations confirm that Na+ and F− rapidly diffuse between, but do not appreciably penetrate into, crystallites under the treatment conditions. Similar to rodent enamel, Mg is enriched in this intergranular space (mean 0.35 at.%; range 0.15–0.51 at.%); in fluoridated samples levels of Na (1.27 at.%, 0.69–1.76 at.%) and F (1.36 at.%, 1.10–1.59 at.%) are also elevated there (compare Fig. 3e versus f; see also Supplementary Figs. 12, 13). In combination with the disordered local structure around Mg observed by Mg K-edge XAS, this is robust evidence for the presence of Mg-ACP in the intergranular phase; the thickness of this region is consistent with previous observations in human enamel8.

a–d, Rendering of Mg (a), Na (b), F (c) and COH (d) positions in a 3D reconstruction of fluoridated human enamel, viewed along the long axis of the crystallites. All scale bars, 20 nm. e, Concentration profiles of F (purple), Na (green), C (teal) and Mg (magenta) along the dashed line in a. Profiles for n = 15 crystallites across 3 technical replicates are shown in Supplementary Fig. 13a–o. f, Concentration profiles in a crystallite from a sample that had not been fluoridated. Profiles for n = 5 crystallites across 2 biological replicates are shown in Supplementary Fig. 13p–t. Note that fluoridation increases the concentration of Na and F in the intergranular phase (ig, grey highlights) versus the core (co, orange highlight), due to short circuit diffusion, whereas the concentration in the shell (sh) is not affected7. Coloured arrows indicate local maxima of concentrations in the core.
In striking difference to rodent enamel, however, Mg levels are elevated not only in the intergranular Mg-ACP, but also in two distinct layers in the core (Fig. 3a; Supplementary Figs. 10, 11; Supplementary Videos 1, 2). The core is further enriched in sodium, probably as Na+, and fluorine, probably as F− (Fig. 3b, c). In addition, the carbon concentration is elevated (Fig. 3d), which is most probably due to the presence of carbonate (CO32−). APT data thus prove true the hypothesis that there exist a Mg2+- and CO32−-rich core in human enamel crystallites18,19.
Line profiles taken approximately normal to the midplane in 20 crystallites identified in 3D reconstructions of five APT sample tips (three NaF-treated, two untreated; Fig. 3e, f, Supplementary Figs. 12, 13) reveal that, on average, the two Mg-rich layers (mean 0.5 at.%; range 0.33–0.72 at.%) are also enriched in Na. However, Na levels usually peak closer to the midplane (1.2 at.%; 0.87–1.55 at.%), where F (1.4 at.%; 1.13–2.44 at.%) and C (about 0.6 at.%; 0.45–1.01 at.%) are also elevated, and Mg goes through a minimum (0.4 at.%). We note that the distributions of Na, F and C are more variable than that of Mg and can be asymmetric or show additional local maxima. Although a form of shot noise may be responsible for the latter effect, we note that contrast in STEM-ADF shows similar variation. It is therefore possible that there is some clustering of substituents. Regardless, mole fractions are always noticeably lower in the shell (on average, 0.22 at.% Mg, 0.81 at.% F, 0.52 at.% Na and 0.32 at.% C). APT therefore not only confirms the core–shell structure observed in many crystallites and over large areas by STEM, but clearly indicates that the core itself has a sandwich structure.
Driven by the size mismatch between Ca2+ and Mg2+, we expect a substantial contraction of the apatite lattice in the Mg-rich layers20,21,22,23. Lattice parameters determined by density functional theory (DFT) calculations and X-ray diffraction experiments, after correction for thermal expansion, agree within 1% and indicate a contraction in both the a and c directions with increasing level of Mg substitution (Supplementary Tables 7, 8; Supplementary Figs. 14–16)24. Carbonate substitution also results in a contraction of the lattice in the a directions25. However, there is a mild expansion of the lattice in the c direction that partially offsets the effect of Mg (Supplementary Fig. 17).
Because enamel crystallites are coherent, lattice parameter changes that result from gradients in Mg2+ and CO32− are in effect residual (eigen) strains and may therefore cause a net residual stress. Residual stresses in turn can affect the overall mechanical performance of a material, but also affect the local chemical potential and therefore the solubility. To explore these possibilities, we predicted residual stresses in an idealized crystallite (Supplementary Fig. 18) using finite-element modelling (Fig. 4a–c). In mechanical equilibrium, the core experiences a net tensile stress, with distinct maxima in the Mg-rich layers (point T in Fig. 4a). The highest compressive stress (−46.4 MPa) is found on the free surface parallel to the (001) plane (Fig. 4b), again in the Mg-rich layers. The shell of the crystallite experiences compressive residual stresses (Fig. 4c). On most of the surface, these stresses are near −39 MPa. Although absolute values of the stresses reported here will vary as real crystallites differ in shape and composition from the highly idealized model we use here, we believe that the model captures trends quite well and provides insights into how we can expect crystallites to behave on average.

a, Rendering of the scalar pressure, calculated as one-third of the trace of the stress tensor, as a measure of residual stress in a finite-element model of an enamel crystallite. Note that symmetric boundary conditions were applied to two faces (white ‘S’); values on these represent internal rather than surface stresses. b, View of a showing the free surface parallel to the (001) plane. c, Plot of the mole fractions of C (black) and Mg (magenta), and of the residual pressure (blue), against the distance from Q to R in a. d, SEM image of an acid-etched enamel section in which crystallites emerge end-on, displaying intergranular corrosion (arrowhead) and preferential dissolution of the core (white arrows).
For instance, at the water-accessible endcap (Fig. 4b), stresses in the core are between 4 and 40 times higher than in the shell. This is expected to increase the solubility of the core compared to the shell. Indeed, the core of outer enamel crystallites is preferentially etched, similar to the intergranular Mg-ACP (Fig. 4d, Supplementary Fig. 19), and consistent with reports for crystallites extracted from human caries lesions26,27. The core–shell architecture and associated residual stresses are thus an important aspect of disease progression, and may be used for the modelling of dissolution and re-precipitation during the progression of caries lesions. Compressive stresses in the shell may further impede crack initiation, extend the size range at which crystallites perform at their theoretical strength28, and deflect cracks, thereby increasing the tolerance of enamel29,30. On a different note, the strong modulation of stresses and the resulting strain just beneath the surface of the endcaps may be responsible for the CDL feature observed in electron microscopy. We note that although the Mg-rich layers do appear to be parallel to the CDL, proof that the CDL runs between the layers would require correlative imaging of the same crystallite by STEM and APT. Although correlative imaging of this kind is not unprecedented, it is a substantial challenge, especially for beam-sensitive materials such as enamel31,32. It would, however, be particularly rewarding, as one could also address the spatial correlation between contrast in STEM-ADF and local concentration maxima reported by APT.
Taken together, we find strong evidence that the core–shell architecture and resulting residual stresses affect the dissolution behaviour of human enamel crystallites and provide a plausible avenue for extrinsic toughening of enamel. This leads us to the question of how the gradients are created in the first place. During amelogenesis, mineral first precipitates in the organic enamel matrix as ribbons of amorphous calcium phosphate (ACP)33,34,35. ACP is tolerant of impurities, and it is conceivable that the ribbons retain Na+ and CO32− as they crystallize by an unknown mechanism. Crystallites initially grow much more slowly in thickness than in width. In human primary teeth for instance, ribbons are 3 nm thick and 29 nm wide at a distance of 25 μm from the ameloblasts, and grow to 10 nm thick and 58 nm wide by the time the ameloblasts have moved an additional 175 μm (Supplementary Fig. 20)33.
In this first phase, the fast growth direction is thus parallel to the Mg-rich layers (Fig. 5; for relative growth velocities, see Supplementary Table 9), suggesting that Mg substitution breaks the crystal symmetry. Presumably, Mg acts on its own, or in concert with organic matrix molecules, by blocking active sites for growth in the direction normal to the layer direction, through anisotropic stresses generated as it is incorporated into the crystal, or a combination of these effects. This would require Mg to be deliberately introduced into the system after the ribbon has formed, and indeed the Mg concentration in porcine enamel is known to reach a maximum in the late secretory stage36. Consistent with a regulatory role, perturbation of putative Mg transporters is known to affect amelogenesis, even though the impact on crystallite shape is not known37,38,39.

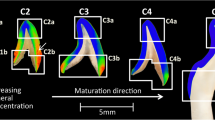
a, Schematic drawing of growth stages (time points t0–t8, shown in b) of human primary enamel crystallites (white hexagons, after ref. 33) superimposed on an idealized map of the Mg concentration based on observation of human permanent enamel crystallites reported herein. b, On the left y axis, plot of the mole fraction of Mg (blue) and carbon (present as carbonate; black, see Supplementary Fig. 21 for map) against distance along the white arrow in a. The open circles indicate the mole fractions at the interface of the growing crystallite at t0–t8. On the right y axis, plot of the ratio of the average growth velocities in the x and y directions in successive time intervals (Supplementary Table 9). Note that scaling of the time axis is unknown, and probably nonlinear. As a consequence, absolute speeds cannot be determined and may vary.
In the second phase, which is probably identical to the maturation stage of amelogenesis, growth slows down. At the same time, the ratio of the growth velocities changes, with crystals growing thicker rather than wider. In human primary enamel, this results in mature crystallites with a thickness of 26 nm and a width of 80 nm (ref. 33). The Mg-poor shell is probably formed during this period. Slow growth at low supersaturation, which may be combined with a drop in Mg and Na concentration in the enamel matrix36,40, is indeed expected to result in low rates of incorporation of impurities on the apatite lattice. Any Mg still present would accumulate ahead of the interface of the growing crystallite.
At the end of the maturation stage, as crystals start to impinge on one another, the Mg/Ca ratio in the matrix would rise rapidly, and when the threshold for Mg-ACP is exceeded23, precipitation of the amorphous intergranular phase is triggered. Although this mechanism is consistent with and provides an explanation for a number of independent observations, we note that the data we draw on come from both primary and permanent human enamel and include observations in other species. Clearly, a thorough compositional analysis of immature enamel ribbons and crystallites in one species would greatly aid in confirming the proposed sequence of events.
Regardless of whether this mechanistic proposal is accurate, what emerges is that the concentration of Mg and other minor ions at the surface of the crystallite, and therefore also the medium in its immediate proximity, varies systematically during amelogenesis of human permanent teeth. This may affect how enamel matrix proteins and their degradation products, thought to be involved in controlling enamel crystallite formation, interact with the mineral phase and each other.
The fact that ions not essential for amelogenesis, such as fluoride, are incorporated into the crystallite core has an important corollary. Enamel forms over very specific times during the development of different teeth (in humans it starts as early as the second trimester in utero and continues until the late teens), is not appreciably remodelled, and is very well preserved in remains and fossils. Crystallite cores might thus encapsulate spatially resolved biomarkers for environmental exposure, disease, or medical intervention, over an extended period of time. With APT and correlative imaging and spectroscopy, this record is now accessible and might help decipher for instance genetic predisposition to caries or the mechanism behind molar-incisor hypomineralization (MIH), a dental developmental defect of unclear aetiology that affects as many as 20% of all schoolchildren2,41.
Methods
Consumables
Unless otherwise specified, all solutions were prepared using ultra-pure water (18.2 MΩ cm) dispensed from a Barnstead Nanopure UF+UV unit (Thermo-Fisher Scientific). Lactic acid (C3H6O3; Mallinckrodt Chemicals); propionamide (98%), NaF (Sigma-Aldrich); NaH2PO4, Na2HPO4, HNO3 (65 wt%), Mg(NO3)2⋅6H2O (99%), Ca(NO3)2⋅4H2O (99%, lot no. 86432), ethanol (VWR, Radnor, PA); Ca(NO3)2⋅4H2O (99.98%, lot no. 61600281), formaldehyde (CH2O) (Alfa Aesar); (NH4)2HPO4 (99%, lot A0059707, Merck KGaA); PELCO liquid silver paint, graphite tape (Ted Pella); EPO-TEK 301 (Epoxy Technology); CarbiMet SiC grinding paper, Metadi supreme polycrystalline aqueous diamond polishing suspension, Microcloth polishing cloth (Buehler); and MM22 microtip coupons for FIB liftout (CAMECA Instruments).
Preparation of enamel sections
De-identified human premolars extracted for orthodontic reasons were kept in 10% buffered formalin at room temperature for 10 days, and at 4 °C thereafter. Before use, samples were rinsed with water and dried under a gentle stream of nitrogen gas (‘rinsed and dried’). Samples were embedded in Epo-Tek 301 epoxy, sectioned along the buccal-lingual line, ground with SiC paper (600, 800, 1,200 grit), and polished on a Buehler Trident polishing cloth with polycrystalline diamond suspensions (3 μm, 1 μm), rinsed and dried. Some sections were treated with fluoride by immersion in 50 ml of aqueous NaF (250 mM, pH 8.4) under gentle agitation using an orbital shaker, at 37 °C for 24 h, then rinsed and dried. Some sections were exposed to lactic acid (250 mM, pH 4, for ~20 s), and subsequently rinsed and dried. Some sections, oriented such that rods (and crystallites) emerge approximately perpendicular to the surface, were positioned at an angle of roughly 45° with respect to the underlying lab bench surface. The sample was etched using a steady stream of droplets of lactic acid (250 mM, pH 4) flowed across the surface for a total of 30 s. Thereafter, the tooth sections were immediately immersed in ethanol, rinsed and dried.
Unless otherwise noted, samples were affixed to an aluminium stub using carbon tape, coated with AuPd (~25 nm) using a Denton Desk IV sputter coating system (Denton Vacuum). The surface of the sample was then grounded to the stub using colloidal silver paint.
Scanning electron microscopy (SEM)
SEM was performed using a Hitachi S4800-II or a Hitachi SU8030 (Hitachi High-Tech), both equipped with a cold cathode field emission electron gun, operated at an accelerating voltage of 5 kV and an emission current of 8,600 nA. Images were acquired using secondary electron contrast.
Transmission electron microscopy (TEM)
Lamellae were prepared from ground and polished, transverse sections of outer, buccal human enamel. A dual-beam FIB/SEM (FEI Helios NanoLab or FEI Strata 400) with a gallium liquid metal ion source (LMIS) operating at an accelerating voltage of 2–30 kV was used to prepare FIB samples for TEM. A ~200-nm-thick layer of protective carbon was deposited on a 2 μm × 15 μm area of interest, either by using the electron beam (5 kV, 1.4 nA) through decomposition of a phenanthrene precursor gas (FEI Helios Nanolab), or by selecting a similar area of interest using a permanent marker deposition method42 (FEI Strata 400). On top of the carbon, a ~1-μm-thick protective platinum layer was deposited using the ion beam (30 kV, 93 pA) through decomposition of a (methylcyclopentadienyl)-trimethyl platinum precursor gas. Subsequently, two trenches were cut to allow for a roughly 2-μm-thick lamella of enamel. Next, the micromanipulator was welded onto the lamella, and the sample was cut loose from the bulk material. An in situ liftout of the sample was performed, and the lamella was welded onto a TEM half-grid. After thinning to about 40 nm in a sub-region of the lamella (5 kV, 81 pA), the section was cleaned at low voltage and current (2 kV, 28 pA) until a final thickness of 20–30 nm was achieved.
Scanning TEM (STEM) images were acquired on a JEOL JEM-2100F (JEOL USA, Peabody, MA), a JEOL GrandARM 300F, or an aberration-corrected FEI Titan Themis (FEI Co.) equipped with a monochromator, a side-entry double tilt liquid-nitrogen-cooled sample holder (Gatan 636; Gatan, Pleasanton, CA), and a cryogenically cooled anti-contamination device (for typical conditions see Supplementary Table 1). Image post-processing was performed as described in the Supplementary Information.
STEM-EDS
EDS spectrum images of regions of interest chosen in STEM-HAADF images were acquired on a windowless 100 mm2 XMaxN 100TLE Silicon Drift Detector (SDD) with a solid angle of ~0.98 sr (Oxford Instruments NanoAnalysis) with a dwell time of 5 μs per pixel.
Cryo-STEM-EELS
EELS 2D spectrum images were acquired on a Titan Themis (FEI) using a K2 Summit direct electron detector in counting mode (Gatan). This direct electron detector with high quantum efficiency (DQE up to 80%) allowed simultaneous acquisition of all relevant inner shell ionization (core loss) edges at high energy resolution and low background levels despite the low dose required to minimize beam damage14. The entrance aperture was 5 mm, the energy dispersion 0.5 eV per channel. The beam current was 4.0–8.5 pA, the dwell time 2.5 μs per pixel. STEM-ADF images were recorded in parallel. Concentration maps were extracted by fitting and subtracting the pre-edge background with a linear combination of power laws and integrating the intensity under the EELS edge of interest. MCR analysis of the Mg L2,3-edge region (Supplementary Fig. 6) was performed as described previously43.
X-ray absorption spectroscopy (XAS)
XAS measurements were performed at the Spherical Grating Monochromator (SGM, 11ID-1) at the Canadian Light Source (Saskatoon, Saskatchewan, Canada), following a literature protocol7. Briefly, enamel from de-identified human third molars was ground into a powder using an agate mortar pestle and spread on graphite tape. Spectra of the Mg K-edge (1,303 eV) were recorded from −60 eV to −12 eV in steps of 2 eV, from −12 eV to −8 eV in steps of 0.5 eV, from −8 eV to 30 eV in steps of 0.1 eV, from 30 eV to 190 eV in steps of 0.2 eV, from 190 eV to 300 eV in steps of 0.3 eV, and from 300 eV to 400 eV in steps of 0.5 eV, with a constant dwell time of 2 s per step. Monochromator energy calibration was performed by setting the first absorbance maxima of the MgO reference sample spectra to 1,309.5 eV. X-ray fluorescence intensity was measured simultaneously with four solid-state silicon drift energy dispersive X-ray detectors (Amptek). Incident flux was measured by recording the current from a gold mesh upstream. The exit slit was adjusted and the undulator detuned to reduce flux to prevent saturation of X-ray fluorescence detectors when measuring concentrated reference samples. Between 1 and 7 scans were collected for each sample and averaged. No beam-induced changes were observed when comparing sequential spectra. The Mg X-ray fluorescence intensity was isolated from the total fluorescence intensity (which contained contributions from X-ray fluorescence from other elements and the scattered incident beam) using custom written code in Mathematica (Wolfram Research). For XANES spectra, see Supplementary Fig. 7.
Absorption data were normalized, background-subtracted using AUTOBK, and converted to k-space using Athena44. Edge energy (E0) was set to the maximum of the first derivative of the absorption spectra. χ(k) data were weighted by k2 and Fourier-transformed over a k-range of 2–9.5 Å−1, applying a Hanning window with a sill width of 1 Å−1. Theoretical photoelectron scattering amplitudes and phase shifts based on the crystal structures of dolomite45, huntite46, whitlockite47 and hydroxyapatite22 were calculated using FEFF648. Shell-by-shell fitting of the EXAFS data was performed in R-space using Artemis44. An energy shift parameter (ΔE0) was maintained constant for the scattering paths but allowed to vary between samples. The amplitude reduction factor (S02 = 0.8) was determined on the basis of a fit to the dolomite, huntite and whitlockite spectra with coordination numbers constrained based on their respective crystal structures. Multiple scattering in the carbonate reference samples was accounted for following Reeder and co-workers49. Enamel and ACP EXAFS spectra were fitted using a model based on the Ca[II] site of hydroxylapatite, consisting of a single Mg-O and two Mg-P scattering paths50,51. To minimize the number of fitting parameters, the coordination number and σ2 for the two Mg-P paths were constrained for each sample but allowed to vary between samples. For EXAFS spectra, see Supplementary Fig. 7. For fitted parameters, see Supplementary Table 3.
Atom probe tomography (APT)
Samples for APT were extracted ~10 μm below the external enamel surface on mid-coronal cervical sections of human premolars, using a Dual Beam SEM/FIB (Helios NanoLab; FEI), and following standard protocols52,53. Briefly, a 200-nm-thick layer of protective “FIB-Pt” was deposited using the electron beam (5 kV, 1.4 nA) on a 2 μm × 25 μm area of interest through decomposition of a (methylcyclopentadienyl) trimethyl platinum precursor gas. A thicker coating of FIB-Pt (~400 nm) was then deposited using the ion beam (30 kV, 93 pA). An angled cut was then made on either side of the Pt strap, and one end was cut free and attached to an in-situ manipulator (Omniprobe) using FIB-Pt. After cutting the final side free, 1–2 μm segments were attached to the top of silicon posts on the APT array with FIB-Pt. Tips were sharpened in the ion beam using annular mill patterns with progressively smaller inner and outer diameters (16–30 kV, 0.28−0.47 nA). The majority of contamination/gallium implantation was removed by a final cleaning step (2 kV, 0.25 nA).
APT analysis was performed using a LEAP 5000 XS (CAMECA Instruments) with a laser operating at a wavelength of 355 nm and a pulse frequency of 250 kHz, at a power of 40 pJ. The temperature in the analysis chamber was kept 25 K, the pressure <10−8 Pa. The d.c. potential on the microtip was adjusted to maintain an evaporation rate of 0.005 ions per laser pulse. 3D reconstructions of the sample tips were made using the IVAS software package (CAMECA Instruments). Standard parameters were used for all reconstructions.
For representative APT spectra, see Supplementary Fig. 9. For peak identities and integration limits, see Supplementary Table 4, and for a comparison of the composition of human and rodent samples see Supplementary Table 6. In this manuscript, we analyse data from three reconstructions of fluoridated enamel, and two reconstructions of enamel that were not fluoridated by us. Inspection revealed that the cross-sections of 8 crystallites (8 fluoridated, 0 non-fluoridated) were fully contained in the reconstructions, and that of 14 crystallites (7/5) were partially contained. For 20 crystallites (15/5) we were able to extract 1D concentration profiles approximately normal to the midplane of the crystallite, using regions of interest (ROIs) that were defined manually in IVAS (CAMECA Instruments; Supplementary Fig. 12). For the remaining crystallites, too small a part was contained in the reconstruction, or it was not possible to deduce the orientation. Consequently, 1D profiles could not be extracted. 1D profiles were corrected for a homogeneous background. Additional reconstructions are given in Supplementary Figs. 10 and 11. As a visual aid, we animated our reconstruction of a 30-nm-thick slice through one enamel crystallite, showing individual ion positions in 3D space (Supplementary Video 1) and iso-concentration surfaces (Supplementary Video 2). We used the Matlab platform (The Mathworks) to render these videos.
Hydrothermal syntheses
Hydroxylapatite (OHAp, 0 at.% Mg) was synthesized following a literature protocol54. Briefly, 5 ml of an aqueous solution of 99.98% Ca(NO3)2∙4H2O (0.1 M, 0.5 mmol) was mixed with 5 ml aqueous solution of (NH4)2HPO4 (0.06 M, 0.3 mmol). To the resulting suspension, 5 ml of an aqueous solution of propionamide (1 M, 5 mmol) was added. The pH was adjusted to 3 by addition of approximately 45 μl of aqueous HNO3 (5 M), to give a clear, transparent solution. The solution was transferred to a PTFE-lined microwave digestion vessel and treated hydrothermally (heating ramp: 30 °C min−1, final temperature of 180 °C held for 30 min), using a Milestone EthosEZ Microwave Digestion System (Milestone). The resulting precipitate was centrifuged and washed with deionized water (3 × 15 ml) and ethanol (3 × 15 ml), and dried in vacuo.
OHAp (0.22 at.% Mg) was synthesized as described above, but using 99% Ca(NO3)2∙4 H2O instead of 99.98% Ca(NO3)2∙4 H2O.
OHAp (1.15 at.% Mg) was synthesized as follows: to 475 μl of an aqueous solution of 99.98% Ca(NO3)2∙4 H2O (1 M, 0.475 mmol) was added 25 μl of an aqueous solution of Mg(NO3)2∙6 H2O (1 M, 0.025 mmol). The solution was diluted to 5 ml overall volume with deionized water. A 5 ml aqueous solution of (NH4)2HPO4 (0.06 M, 0.3 mmol) and a 5 ml aqueous solution of propionamide (1 M, 5 mmol) were added to give a suspension. The pH was adjusted to 3 by addition of approximately 45 μl aqueous HNO3 (5 M) to give a clear, transparent solution. The solution was transferred to a PTFE-lined microwave digestion vessel and treated hydrothermally as described above. The resulting precipitate was centrifuged and washed with deionized water (3 × 15 ml) and ethanol (3 × 15 ml), and dried in vacuo to give OHAp (approximately 20 wt% by powder X-ray diffraction (PXRD)) and whitlockite (approximately 80 wt% by PXRD). Needle-shaped OHAp crystals could easily be differentiated from whitlockite platelets.
Phase identity and purity for all samples were confirmed by PXRD. The magnesium mole fraction was determined using ICP-MS.
Powder X-ray diffraction
PXRD patterns of OHAp were collected at 100 K on a STOE-STADI-P powder diffractometer (STOE Corporation) equipped with an asymmetric curved germanium monochromator (Cu Kα1 radiation, λ = 1.54056 Å) and a one-dimensional silicon strip detector (MYTHEN2 1K, DECTRIS). The line-focused Cu X-ray tube was operated at 40 kV and 40 mA. Powder was packed in a polyimide capillary (0.5 mm inner diameter) and intensity data were collected over an angular range of 2θ = 10°–70°, over a period of 10 min. The instrument was calibrated against an NIST silicon standard (640d). Data were processed and Rietveld refinement was performed using MDI Jade 2010 (Materials Data). Lattice parameters are reported in Supplementary Table 7.
Single crystal X-ray diffraction
Diffraction data for OHAp (0.22 at.% Mg) and OHAp (1.15 at.% Mg) were collected at a set temperature of 100 K using a Bruker Kappa APEX2 diffractometer (Bruker AXS) equipped with a Mo Kα (λ = 0.71073 Å) source. Single crystals (50 μm × 5 μm × 5 μm) were picked from powders and mounted with Paratone N on a cryo-loop. Diffraction patterns were indexed, refined, and integrated using SAINT of the APEX2 package (Bruker AXS). Using Olex255, the structure was solved with XT and refined with the ShelXL package using least squares minimization (Supplementary Fig. 14)56. Lattice parameters are reported in Supplementary Table 7 and Supplementary Fig. 15.
Inductively coupled plasma mass spectrometry (ICP-MS)
ICP-MS was carried out on a Thermo iCAP Q ICP-MS (Thermo Fisher Scientific). For phase-pure samples, powders were used as-received. For the sample containing whitlockite, the single crystal that was analysed by X-ray diffraction was used. Samples were dissolved in trace-metal-grade HNO3 solution (0.1 M) in a metal-free tube. Trace-metal-grade HCl solution was used as a blank.
Density functional theory (DFT) calculations
DFT calculations were performed within the generalized gradient approximation (GGA) using the GGA-PBEsol (Perdew–Burke–Ernzerhof revised for solids) exchange-correlation functional57 with the planewave pseudopotential code, Quantum ESPRESSO58. We used the ultrasoft pseudopotentials59 taken from the PSLibrary60. A plane-wave cutoff of 60 Ry was used during the ionic and electronic relaxation steps. For the simulation of Mg-doped OHAp solid solutions, we employed a 2 × 2 × 1 supercell (with 352 atoms) in the monoclinic crystal structure (P21/c symmetry). Our initial simulation performed on a pristine Ca10(PO4)6(OH)2 supercell serves as the reference. Additional simulations were performed, whereby 1, 2, 3 and 4 Ca atoms were substituted with Mg atoms. The atomic positions and the cell volume were relaxed until the Hellmann–Feynman forces were less than 2 meV Å–1 and components of the stress tensor were less than 0.1 kbar. The Brillouin zone integration was performed using a 1 × 1 × 1 Monkhorst–Pack k-point mesh (Γ-point calculation). Lattice parameters as calculated (at 0 K) and corrected to 298 K using the coefficient of thermal expansion reported by Babushkin and co-workers61 are reported in Supplementary Fig. 15.
Lattice strain as a function of composition
Lattice parameters as a function of magnesium mole fraction were determined by XRD and DFT as outlined above (Supplementary Fig. 15). Lattice parameters as a function of carbonate weight fraction were obtained from Demier and co-workers25 using WebPlotDigitizer62. Carbonate weight fractions were converted to mole fractions (XC) using the stoichiometric models postulated by Deymier and co-workers25. The concentration-dependent lattice strain in the a and c directions was calculated from the lattice parameters and fitted with a linear model:
where \({\varepsilon }_{i}^{j}\) denotes the strain in the i direction due to substitution with species j, Xj is the mole fraction of species j, \({n}_{i}^{j}\) is the slope, and \({b}_{i}^{j}\) is the intercept (Supplementary Figs. 16 and 17). Fit parameters are reported in Supplementary Table 8.
Finite-element modelling
Enamel crystallites were idealized as slabs with rectangular cross-section, oriented with the [001] direction parallel to the z axis and the [100] parallel to the x axis. Mg and carbonate concentrations were modelled as continuous, 2D distributions (Supplementary Fig. 18), chosen to represent experimental 1D concentrations profiles in the x direction (Supplementary Fig. 13). For simplicity, the contributions of fluoride and of sodium ions in excess of those needed to charge-balance carbonate ions were ignored, as were contributions from surface free energies. All modelling was performed using COMSOL Multiphysics.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Code availability
This manuscript primarily made use of commercial (IVAS, Origin, Matlab, MDI Jade, APEX2, Thermo Scientific Qtegra ISDS, COMSOL Multiphysics, TEM Imaging and Analysis, DigitalMicrograph, AZtec, Adobe Illustrator) and freely available (DEMETER, OLEX2, SHELX, Quantum ESPRESSO, Cornell Spectrum Imager, ImageJ) software packages for acquisition, processing and visualization of data. MCR was performed using custom code using the Matlab mcr.m package from the Eigenvector Research PLS_toolbox, as described elsewhere43. In addition, custom code written for the Mathematica and Matlab environments was used for file conversions, plotting and visualization. This code is available from the corresponding author upon reasonable request.
Change history
21 July 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41586-020-2544-x
References
Nanci, A. Ten Cate’s Oral Histology: Development, Structure, and Function 7th edn (Mosby Elsevier, 2008).
Chai, H., Lee, J. J. W., Constantino, P. J., Lucas, P. W. & Lawn, B. R. Remarkable resilience of teeth. Proc. Natl Acad. Sci. USA 106, 7289–7293 (2009).
Klein, O. D. et al. Meeting report: a hard look at the state of enamel research. Int. J. Oral Sci. 9, e3 (2017).
Moradian-Oldak, J. Protein-mediated enamel mineralization. Front. Biosci. 17, 1996–2023 (2012).
Lacruz, R. S., Habelitz, S., Wright, J. T. & Paine, M. L. Dental enamel formation and implications for oral health and disease. Physiol. Rev. 97, 939–993 (2017).
Robinson, C. Enamel maturation: a brief background with implications for some enamel dysplasias. Front. Physiol. 5, 388 (2014).
Gordon, L. M. et al. Amorphous intergranular phases control the properties of rodent tooth enamel. Science 347, 746–750 (2015).
La Fontaine, A. et al. Atomic-scale compositional mapping reveals Mg-rich amorphous calcium phosphate in human dental enamel. Sci. Adv. 2, e1601145 (2016).
Gordon, L. M. & Joester, D. Mapping residual organics and carbonate at grain boundaries and in the amorphous interphase in mouse incisor enamel. Front. Physiol. 6, 57 (2015).
Zhang, Y.-R., Du, W., Zhou, X.-D. & Yu, H.-Y. Review of research on the mechanical properties of the human tooth. Int. J. Oral Sci. 6, 61–69 (2014).
Robinson, C. et al. The chemistry of enamel caries. Crit. Rev. Oral Biol. Med. 11, 481–495 (2000).
Yanagisawa, T. & Miake, Y. High-resolution electron microscopy of enamel-crystal demineralization and remineralization in carious lesions. J. Electron Microsc. 52, 605–613 (2003).
Reyes-Gasga, J., Hemmerle, J. & Bres, E. F. Aberration-corrected transmission electron microscopy study of the central dark line defect in human tooth enamel crystals. Microsc. Microanal. 22, 1047–1055 (2016).
Hart, J. L. et al. Direct detection electron energy-loss spectroscopy: a method to push the limits of resolution and sensitivity. Sci. Rep. 7, 8243 (2017).
de Juan, A. & Tauler, R. Multivariate curve resolution (MCR) from 2000: progress in concepts and applications. Crit. Rev. Anal. Chem. 36, 163–176 (2006).
Gordon, L. M., Tran, L. & Joester, D. Atom probe tomography of apatites and bone-type mineralized tissues. ACS Nano 6, 10667–10675 (2012).
Gault, B., Moody, M. P., Cairney, J. M. & Ringer, S. P. Atom Probe Microscopy (Springer, 2012).
Robinson, C., Kirkham, J., Brookes, S. J., Bonass, W. A. & Shore, R. C. Int. J. Dev. Biol. The chemistry of enamel development. 39, 145–152 (1995).
Robinson, C., Weatherell, J. A. & Hallsworth, A. S. Distribution of magnesium in mature human enamel. Caries Res. 15, 70–77 (1981).
Laurencin, D. et al. Magnesium incorporation into hydroxyapatite. Biomaterials 32, 1826–1837 (2011).
Shannon, R. D. & Prewitt, C. T. Effective ionic radii in oxides and fluorides. Acta Crystallogr. B 25, 925–946 (1969).
Hughes, J. M., Cameron, M. & Crowley, K. D. Structural variations in natural F, OH, and Cl apatites. Am. Mineral. 74, 870–876 (1989).
LeGeros, R. Z., Sakae, T., Bautista, C., Retino, M. & LeGeros, J. P. Magnesium and carbonate in enamel and synthetic apatites. Adv. Dent. Res. 10, 225–231 (1996).
Ben Abdelkader, S., Khattech, I., Rey, C. & Jemal, M. Synthése, caractérisation et thermochimie d’apatites calco-magnésiennes hydroxylées et fluorées. Thermochim. Acta 376, 25–36 (2001).
Deymier, A. C. et al. Protein-free formation of bone-like apatite: new insights into the key role of carbonation. Biomaterials 127, 75–88 (2017).
Voegel, J. C. & Frank, R. M. Stages in dissolution of human enamel crystals in dental caries. Calcif. Tissue Res. 24, 19–27 (1977).
Tohda, H., Takuma, S. & Tanaka, N. Intercrystalline structure of enamel crystals affected by caries. J. Dent. Res. 66, 1647–1653 (1987).
Gao, H. J., Ji, B. H., Jager, I. L., Arzt, E. & Fratzl, P. Materials become insensitive to flaws at nanoscale: lessons from nature. Proc. Natl Acad. Sci. USA 100, 5597–5600 (2003).
Yahyazadehfar, M. et al. On the mechanics of fatigue and fracture in teeth. Appl. Mech. Rev. 66, 030803 (2014).
Yilmaz, E. D., Schneider, G. A. & Swain, M. V. Influence of structural hierarchy on the fracture behaviour of tooth enamel. Phil. Trans. R. Soc. A 373, 1–20 (2015).
Langelier, B., Wang, X. & Grandfield, K. Atomic scale chemical tomography of human bone. Sci. Rep. 7, 39958 (2017).
Stoffers, A. et. al. Correlating atom probe tomography with atomic-resolved scanning transmission electron microscopy: example of segregation at silicon grain boundaries. Microsc. Microanal. 23, 291–299 (2017).
Daculsi, G. & Kerebel, B. High-resolution electron-microscope study of human enamel crystallites — size, shape, and growth. J. Ultrastruct. Res. 65, 163–172 (1978).
Beniash, E., Metzler, R. A., Lam, R. S. & Gilbert, P. U. Transient amorphous calcium phosphate in forming enamel. J. Struct. Biol. 166, 133–143 (2009).
Robinson, C., Fuchs, P. & Weatherell, J. A. The appearance of developing rat incisor enamel using a freeze fracturing technique. J. Cryst. Growth 53, 160–165 (1981).
Kirkham, J. et al. Self-assembling peptide scaffolds promote enamel remineralization. J. Dent. Res. 86, 426–430 (2007).
Luder, H. U., Gerth-Kahlert, C., Ostertag-Benzinger, S. & Schorderet, D. F. Dental phenotype in Jalili syndrome due to a c.1312 dupC homozygous mutation in the CNNM4 gene. PLoS ONE 8, e78529 (2013).
Yamazaki, D. et al. Basolateral Mg2+ extrusion via CNNM4 mediates transcellular Mg2+ transport across epithelia: a mouse model. PLoS Genet. 9, e1003983 (2013).
Nakano, Y. et al. A critical role of TRPM7 as an ion channel protein in mediating the mineralization of the craniofacial hard tissues. Front. Physiol. 7, 258 (2016).
Aoba, T., Shimoda, S. & Moreno, E. C. Labile or surface pools of magnesium, sodium, and potassium in developing porcine enamel mineral. J. Dent. Res. 71, 1826–1831 (1992).
Hubbard, M. J., Mangum, J. E., Perez, V. A., Nervo, G. J. & Hall, R. K. Molar hypomineralisation: a call to arms for enamel researchers. Front. Physiol. 8, 546 (2017).
Park, Y. C. et al. Use of permanent marker to deposit a protection layer against FIB damage in TEM specimen preparation. J. Microsc. 255, 180–187 (2014).
Zachman, M. J., Tu, Z. Y., Choudhury, S., Archer, L. A. & Kourkoutis, L. F. Cryo-STEM mapping of solid-liquid interfaces and dendrites in lithium-metal batteries. Nature 560, 345–349 (2018).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Antao, S. M., Mulder, W. H., Hassan, I., Crichton, W. A. & Parise, J. B. Cation disorder in dolomite, CaMg(CO3)2, and its influence on the aragonite + magnesite ↔ dolomite reaction boundary. Am. Mineral. 89, 1142–1147 (2004).
Dollase, W. A. & Reeder, R. J. Crystal-structure refinement of huntite, CaMg3(CO3)4, with X-ray powder data. Am. Mineral. 71, 163–166 (1986).
Calvo, C. & Gopal, R. Crystal structure of whitlockite from Palermo Quarry. Am. Mineral. 60, 120–133 (1975).
Rehr, J. J. & Albers, R. C. Theoretical approaches to X-ray absorption fine structure. Rev. Mod. Phys. 72, 621–654 (2000).
Reeder, R. J., Lamble, G. M. & Northrup, P. A. XAFS study of the coordination and local relaxation around Co2+, Zn2+, Pb2+, and Ba2+ trace elements. Am. Mineral. 84, 1049–1060 (1999).
Holt, C. et al. Preparation of amorphous calcium-magnesium phosphates at pH 7 and characterization by x-ray absorption and fourier transform infrared spectroscopy. J. Cryst. Growth 92, 239–252 (1988).
Harries, J. E., Hukins, D. W. L., Holt, C. & Hasnain, S. S. Conversion of amorphous calcium phosphate into hydroxyapatite investigated by EXAFS spectroscopy. J. Cryst. Growth 84, 563–570 (1987).
Larson, D. J., Prosa, T. J., Ulfig, R. M., Geiser, B. P. & Kelly, T. F. Local Electrode Atom Probe Tomography: A User’s Guide (Springer Science and Business Media, 2013).
Thompson, K. et al. In situ site-specific specimen preparation for atom probe tomography. Ultramicroscopy 107, 131–139 (2007).
Qi, M.-l., Xiao, G.-y. & Lu, Y.-p. Rapid hydrothermal synthesis of submillimeter ultralong flexible hydroxyapatite fiber using different pH regulators. Acta Metall. Sinica Engl. Lett. 29, 609–613 (2016).
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 42, 339–341 (2009).
Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 71, 3–8 (2015).
Perdew, J. P. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (2008).
Giannozzi, P. et al. QUANTUM EXPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892–7895 (1990).
Dal Corso, A. Pseudopotentials periodic table: from H to Pu. Comput. Mater. Sci 95, 337–350 (2014).
Babushkin, O., Lindbäck, T., Holmgren, A., Li, J. & Hermansson, L. Thermal expansion of hot isostatically pressed hydroxyapatite. J. Mater. Chem. 4, 413–415 (1994).
Rohatgi, A. WebPlotDigitizer Version 4.2 https://automeris.io/WebPlotDigitizer (2019).
Acknowledgements
This work was supported in part by the National Institute of Health–National Institute of Dental and Craniofacial Research (NIH-NIDCR R03 DE025303-01, R01 DE025702-01), the National Science Foundation (DMR-1508399), the NSF Platform for the Accelerated Realization, Analysis and Discovery of Interface Materials (PARADIM) under Cooperative Agreement no. DMR-1539918, and the University of Virginia. K.A.D. was in part supported by a 3M fellowship. The Canadian National Sciences and Engineering Research Council in part supported L.M.G. K.A.D. and M.J.C. were supported in part by the Northwestern University Graduate School Cluster in Biotechnology, Systems and Synthetic Biology, which is affiliated with the Biotechnology Training Program. L.S. was supported by a Deutsche Forschungsgemeinschaft research fellowship (STE2689/1-1). This work made use of the following core facilities operated by Northwestern University: NUCAPT, which received support from NSF (DMR-0420532), ONR (N00014-0400798, N00014-0610539, N00014-0910781 and N00014-1712870), and the Initiative for Sustainability and Energy at Northwestern University (ISEN); MatCI; NUANCE and EPIC, which received support from the International Institute for Nanotechnology (IIN), the Keck Foundation, and the State of Illinois, through the IIN; IMSERC; the Jerome B. Cohen X-Ray Diffraction Facility; QBIC, which received support from NASA Ames Research Center (NNA06CB93G). NUCAPT, MatCI, NUANCE and EPIC were further supported by the MRSEC programme (NSF DMR-1720139) at the Materials Research Center; NUCAPT, NUANCE, EPIC and IMSERC were also supported by the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205). This work made use of the Cornell Center for Materials Research (CCMR) Shared Facilities supported through the NSF MRSEC Program (no. DMR-1719875). The Titan Themis 300 was acquired through NSF-MRI-1429155, with additional support from Cornell University, the Weill Institute, and the Kavli Institute at Cornell. This work made use of the Rivanna cluster maintained by the Advanced Research Computing Services at the University of Virginia. Portions of this work were performed at the Canadian Light Source (CLS), which received support from The Natural Sciences and Engineering Research Council of Canada, the National Research Council of Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan. The authors thank P. Akers, M. Stohle and G. Borden for providing de-identified human premolars, and C. Malliakas, K. MacRenaris, M. Thomas and especially K. Rice for technical support.
Author information
Authors and Affiliations
Contributions
The experiments and simulations were designed by K.A.D., P.J.M.S., L.S., P.V.B., J.M.R., D.J. STEM experiments were performed by P.J.M.S., B.H.G. and M.J.Z., with additional help in analysis and simulations provided by M.J.Z. and L.F.K. APT data were collected and analysed by K.A.D., with D.J. assisting the analysis. The finite-element model was developed by K.A.D. and D.J. using DFT calculations performed by P.V.B., and synthetic Mg-rich OHAp crystals made and analysed by L.S. XAS data were collected and analysed by L.M.G. and M.J.C. K.A.D., P.J.M.S., B.H.G., P.V.B., M.J.Z., L.F.K., J.M.R. and D.J. were all involved in preparing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Tables, Discussion, and References, organized by experimental technique (1: STEM; 2: XAS; 3: APT; 4: FEM; 5: SEM) with an additional chapter that details the proposed model for amelogenesis in greater detail. This section contains 21 Supplementary Figures, 9 Supplementary Tables, and 40 references.
Supplementary Data
This zipped file contains source data for Supplementary Figures 4–7 and 14–17.
Video 1
Ion positions in a human enamel crystallite. All 24Mg2+ (magenta), 40Ca19F+ (blue), 23Na+ (turquoise), and COxHy+ ions (red) are rendered for a 30 nm thick slice through a human enamel crystallite (crystallite D in Supplementary Fig. S10a). Note that dataset was rotated such that view direction (x axis) is approximately parallel to the Mg layers. The animation shows rotation about the z axis, which is normal to the Mg layers. This visualization was scripted using the Matlab R2019b platform (The MathWorks).
Video 2
Core-shell and sandwich structure of human enamel crystallite. The reconstruction was rotated as in Video 1, voxelized (1 nm isotropic voxels) and smoothed. Iso-concentration surfaces and iso-caps for 24Mg2+ (magenta, ciso = 0.1 nm−3), 23Na+ (turquoise, 0.3 nm−3), and COxHy+ ions (red, 0.12 nm−3) were rendered together with that for 40Ca19F+ (blue, 0.07 nm−3) for the same slice as in Video 1, but ions outside the central crystallite are not shown. The animation shows one revolution about the z axis, followed by an oscillatory tilt series about the y axis. This visualization was scripted using the Matlab R2019b platform (The MathWorks).
Rights and permissions
About this article

Cite this article
DeRocher, K.A., Smeets, P.J.M., Goodge, B.H. et al. Chemical gradients in human enamel crystallites. Nature 583, 66–71 (2020). https://doi.org/10.1038/s41586-020-2433-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2433-3
- Springer Nature Limited
This article is cited by
-
The circadian clock in enamel development
International Journal of Oral Science (2024)
-
Effect of Fluoride on the Ion-association of Calcium Phosphate and Crystallization of Hydroxyapatite
Journal of Wuhan University of Technology-Mater. Sci. Ed. (2024)
-
A strong and tough hydroxyapatite-based fiber with enamel-inspired hierarchical structure
Science China Materials (2024)
-
Research progress of biomimetic materials in oral medicine
Journal of Biological Engineering (2023)
-
Effect of the prism-interprisms three-dimension spatial microstructure on the enamel bond strength
BMC Oral Health (2023)