

Abstract
The phylogenetic relationships between hominins of the Early Pleistocene epoch in Eurasia, such as Homo antecessor, and hominins that appear later in the fossil record during the Middle Pleistocene epoch, such as Homo sapiens, are highly debated1,2,3,4,5. For the oldest remains, the molecular study of these relationships is hindered by the degradation of ancient DNA. However, recent research has demonstrated that the analysis of ancient proteins can address this challenge6,7,8. Here we present the dental enamel proteomes of H. antecessor from Atapuerca (Spain)9,10 and Homo erectus from Dmanisi (Georgia)1, two key fossil assemblages that have a central role in models of Pleistocene hominin morphology, dispersal and divergence. We provide evidence that H. antecessor is a close sister lineage to subsequent Middle and Late Pleistocene hominins, including modern humans, Neanderthals and Denisovans. This placement implies that the modern-like face of H. antecessor—that is, similar to that of modern humans—may have a considerably deep ancestry in the genus Homo, and that the cranial morphology of Neanderthals represents a derived form. By recovering AMELY-specific peptide sequences, we also conclude that the H. antecessor molar fragment from Atapuerca that we analysed belonged to a male individual. Finally, these H. antecessor and H. erectus fossils preserve evidence of enamel proteome phosphorylation and proteolytic digestion that occurred in vivo during tooth formation. Our results provide important insights into the evolutionary relationships between H. antecessor and other hominin groups, and pave the way for future studies using enamel proteomes to investigate hominin biology across the existence of the genus Homo.
Similar content being viewed by others
Main
Since 1994, over 170 human fossil remains have been recovered from level TD6 of the Gran Dolina site of the Sierra de Atapuerca10 (Burgos, Spain) (Extended Data Fig. 1, Supplementary Information). These fossils have been dated to the late Early Pleistocene epoch and exhibit a unique combination of cranial, mandibular and dental features9,11. To accommodate the variation observed in the human fossils from TD6, a new species of the genus Homo—H. antecessor—was proposed in 19979. The relationship of this species to earlier or later hominins in Eurasia—such as the H. erectus specimens from Dmanisi or Neanderthals, Denisovans and modern humans, respectively—have been the subject of considerable debate3,4,12,13. These issues remain unresolved owing to the fragmentary nature of hominin fossils at other sites, and the failure to recover ancient DNA in Eurasia that dates to the Early, and most of the Middle, Pleistocene epoch.
By contrast, recent developments in the extraction and tandem mass-spectrometric analysis of ancient proteins have made it possible to retrieve phylogenetically informative protein sequences from Early Pleistocene contexts6,8. We therefore applied ancient protein analysis to a H. antecessor molar from sublevel TD6.2 of the Gran Dolina site of the Sierra de Atapuerca (specimen ATD6-92) (Extended Data Fig. 2a). This specimen, identified as an enamel fragment of a permanent lower left first or second molar, has been directly dated to 772–949 thousand years ago (ka) using a combination of electron spin resonance and U-series dating11. In addition, we sampled dentine and enamel from an isolated H. erectus upper first molar (specimen D4163) (Extended Data Fig. 2b) from Dmanisi (Georgia) that has been dated to 1.77 million years ago (Ma)1,14,15, as amino acid racemization analysis of this specimen indicated the presence of an endogenous protein component in the intracrystalline enamel fraction of the tooth (Extended Data Fig. 3, Supplementary Information). On both specimens, we performed digestion-free peptide extraction optimized for the recovery of short, degraded protein remains6. Nanoscale liquid chromatography–tandem mass spectrometry (nanoLC–MS/MS) acquisition was replicated in two independent proteomic laboratories (Extended Data Table 1), implementing common precautions and analytical workflows to minimize protein contamination (Methods). We compared the proteomic datasets retrieved from the Pleistocene hominin tooth specimens with those generated from a positive control, a recent human premolar (Ø1952; which is from a male individual and is approximately three centuries old), as well as previously published Holocene teeth16 (Methods, Supplementary Information). Finally, to validate our enamel peptide spectrum matches, we performed machine-learning-based MS/MS spectrum intensity prediction using the wiNNer algorithm17. The results show that the wiNNer model retrained for randomly cleaved and heavily modified peptides provides a predictive performance similar to that of the wiNNer model trained on modern, trypsin-digested samples, assuring accurate sequence identification for the phylogenetically informative peptides (median Pearson correlation coefficients of ≥0.76) (Methods, Supplementary Fig. 6, Supplementary Information).
Protein recovery from the Dmanisi dentine sample was limited to sporadic collagen type I fragments, and therefore in-depth analysis of this material was not further pursued. By contrast, we recovered ancient proteomes from both hominin enamel samples. We found that the composition of these proteomes is similar to that of the recent human specimen that we processed as a positive control, as well as to previously published proteomes from ancient enamel6,16,18,19 (Extended Data Table 2, Supplementary Table 6). The enamel-specific proteins include amelogenin (both AMELX and AMELY isoforms), enamelin (ENAM), ameloblastin (AMBN), amelotin (AMTN) and the enamel-specific protease matrix metalloproteinase 20 (MMP20). Serum albumin (ALB) and collagens (COL1α1, COL1α2 and COL17α1) are also present. For the enamel-specific proteins, the peptide sequences that we retrieved cover approximately the same protein regions in all of the specimens that we analysed (Extended Data Fig. 4). Although destructive, our sampling of Pleistocene hominin teeth resulted in higher protein sequence coverage than acid-etching of Holocene enamel surfaces16,20 (Supplementary Fig. 7). The AMTN-specific peptides largely derive from a single sequence region involved in hydroxyapatite precipitation through the presence of phosphorylated serines21. Finally, the observation of the AMELY-specific peptides (which is coded on the non-recombinant portion of the Y chromosome) demonstrates that the H. antecessor molar that we studied belonged to a male individual16 (Extended Data Fig. 5).
Besides proteome composition and sequence coverage, several further lines of evidence independently support the endogenous origin of the hominin enamel proteomes. Unlike exogenous trypsin, keratins and other human-skin contaminants that we identified, the enamel proteins have high deamidation rates (Extended Data Fig. 6)—above the rate observed for the recent human specimens (Supplementary Fig. 8). Both Pleistocene hominins have average peptide lengths that are shorter than those observed for our recent human controls (Extended Data Fig. 6d). The average peptide length is shorter in the Dmanisi hominin, but longer in the younger Atapuerca hominin (Extended Data Fig. 6d). By contrast, we observe that the peptide lengths in enamel from the Dmanisi hominin are indistinguishable from those of the faunal remains from the same site. Together, our protein data are therefore consistent with theoretical and experimental6,22 expectations for samples of their relative age.
In addition to diagenetic modifications, we observe two kinds of in vivo modification in our recent and ancient enamel proteomes. First, we detect serine (S) phosphorylation within the S-X-E motif (Fig. 1a, b). This motif, as well as the S-X-phosphorylated S motif, is recognized by the FAM20C secreted kinase, which is active in the phosphorylation of extracellular proteins23,24. The presence of phosphoserine in fossil enamel and its location in the S-X-E and/or S-X-phosphorylated S motifs has also previously been observed in other Pleistocene enamel proteomes6,25. Phosphorylation occupancy can be computed successfully for ancient and recent samples, and reveals differences in the ratios of phosphorylated peptides between samples (Fig. 1c, Supplementary Table 5). Second, the peptide populations that we retrieve primarily cover the ameloblastin, enamelin and amelogenin sequence regions, representing cleavage products deriving from in vivo activity of the proteases MMP20 and—subsequently—kallikrein 4 (KLK4) (Extended Data Fig. 4, Methods). The peptide populations are also enriched in N and C termini that correspond to known MMP20 and KLK4 cleavage sites (Extended Data Fig. 7, Supplementary Fig. 9). FAM20C phosphorylation and MMP20 and KLK4 proteolysis are the two main processes that occur in vivo during enamel biomineralization. Our observation of products deriving from both processes opens up the possibility of studying in vivo processes of hominin tooth formation across the Pleistocene epoch.

a, Phosphorylation sequence motif analysis of H. antecessor specimen ATD6-92. b, Phosphorylation sequence motif analysis of H. erectus specimen D4163. c, Phosphorylation occupancy comparison, expressed as log2-transformed summed intensity ratio of modified and unmodified peptides, for amino acid sites for which data are available for at least two specimens. y axis labels indicate the position of the phosphorylated amino acids for each protein (UniProt accession numbers Q9NP70 (AMBN), Q99217 (AMELX) and Q9NRM1 (ENAM)). SK339 denotes an archaeological specimen from a modern human, which is approximately three centuries old (see ‘Recent human control specimens’ in the Methods for details).
Homo antecessor is known only from the Gran Dolina TD6 assemblage in Atapuerca9. Its relationship with other European Middle Pleistocene fossils is heavily debated3,4,5,26,27. It remains contentious as to whether H. antecessor represents the last common ancestor of H. sapiens, Neanderthals and Denisovans9, or whether it represents a sister lineage to the last common ancestor of these species28,29. We address this issue by conducting phylogenetic analyses on the basis of our ancient protein sequences from H. antecessor (ATD6-92), a panel of present-day great ape genomes and protein sequences translated from archaic hominin genomes (Methods).
We built several phylogenetic trees using maximum likelihood and Bayesian methods (Fig. 2a, Supplementary Figs. 13–16). In these trees, the H. antecessor sequence represents a sister taxon that is closely related to, but not part of, the group composed of Late Pleistocene hominins for which molecular data are available (Fig. 2a, Supplementary Figs. 13, 15, 16). The enamel protein sequences do not resolve the relationships between H. sapiens, Neanderthals and Denisovans owing to the low number of informative single amino acid polymorphisms. However, pairwise divergence of the amino acid sequences between H. antecessor and the clade containing H. sapiens, Neanderthals and the Denisovan is larger than the divergence between the members of this clade (Fig. 2b, Supplementary Fig. 12, Supplementary Information). The concatenated gene tree may be subjected to incomplete lineage sorting, and we have too little sequence data to discard this possibility at the moment. However, if we use the concatenation of available gene trees as a best guess for the population tree, and assume that such a population tree is a good descriptor of the relationships among ancient hominins, then our results support the placement of H. antecessor as a closely related sister taxon of the last common ancestor of H. sapiens, Neanderthals and Denisovans. The phylogenetic position of H. antecessor agrees with a divergence of the H. sapiens and Neanderthal + Denisovan lineages between 550 and 765 ka30,31, as ATD6-92 has been dated to 772–949 ka11. This is further supported by recent reconsiderations of the morphology of H. antecessor in relation to Middle and Late Pleistocene hominins29.

a, Maximum credibility tree estimated using BEAST and a concatenated alignment of seven protein sequences recovered for the ancient sample. Posterior Bayesian probabilities are indicated at nodes with a probability of ≤ 1. Horizontal error bars at each node indicate the 95% highest posterior density intervals for the split time estimates. The position of H. antecessor is consistent with that obtained via maximum likelihood (Supplementary Fig. 13) and Bayesian (Supplementary Fig. 16) analyses. ERZ and HG codes in parentheses after H. sapiens refer to identifiers for data from the Simons Genome Diversity Panel33 and 1000 Genomes Project34, respectively (see ‘Comparison between the ancient protein sequences and modern reference proteins’ in the Methods for details). b, Histograms of the divergence times obtained for the split between H. antecessor and the H. sapiens, Neanderthal and Denisovan clade (HND) (red), the HND–HND split (blue), and the Pan–(HND + H. antecessor) split (grey). Divergence times in a and b are shown as a percentage of the time since the divergence of all great apes.

For each sample, the presence (green) or absence (blank) of endogenous DNA, collagens, non-collagenous proteins (NCPs) or an enamel proteome is given. Only samples for which mammalian proteomes are published are considered6,7,8,35,36,37,38. Hominin samples are indicated with squares, other mammalian samples are indicated with circles. Selected specimens have their separate molecular components joined, and are named. Xiahe refers to the Xiahe mandible7; the Thistle Creek Equus refers to a horse metapodial from the Canadian permafrost38.
Homo antecessor has tentatively been proposed as the last common ancestor of Neanderthals and modern humans9. The similarities observed between the modern-like mid-facial topography of H. antecessor and H. sapiens—including a modern pattern of coronal orientation of the infraorbital surface, the sloping and directionality of this plane, as well as the anterior flexion of the maxillary surface and arching of the zygomatic-alveolar crest—were key in this proposal9,32. Additional studies of the face of ATD6-69 have confirmed that H. antecessor exhibits the oldest known modern-like face in the fossil record12,13. The phylogenetic placement of H. antecessor implies that this modern-like face—as represented by H. antecessor—must have a considerably deep ancestry in the genus Homo. Findings made between 2003 and 2005 have shown that the H. antecessor hypodigm includes some features that were previously considered Neanderthal autapomorphies28. Our results suggest that these features appeared in Early Pleistocene hominins, and were retained by Neanderthals and lost by modern humans.
By contrast, the phylogenetic tree built with the H. erectus specimen from Dmanisi has only moderate resolution (Extended Data Fig. 8, Supplementary Fig. 11), despite deeper shotgun protein sequencing for this specimen (Extended Data Table 1). This partly inconclusive result might be due to the shorter average peptide lengths compared to the Atapuerca H. antecessor specimen (Extended Data Fig. 6d, Methods) and an absence of uniquely segregating single amino acid polymorphisms (Supplementary Table 9). Although our H. erectus data from Dmanisi demonstrate that ancient hominin proteins can be reliably obtained from the Early Pleistocene epoch, they also highlight the current limits of ancient protein analysis when applied to the phylogenetic placement of Early Pleistocene hominin remains.
Our dataset provides a unique molecular resource of hominin biomolecular sequences from Early and Middle Pleistocene hominins, and represents—to our knowledge—the oldest ancient hominin proteomes presented to date. Comparison of hominin and fauna proteomes from different skeletal tissues reveals that the dental enamel proteome outlasts dentine and bone proteome preservation (Fig. 3). Here the prolonged survival of hominin enamel proteomes is exploited to show that H. antecessor represents a hominin taxon closely related to the last common ancestor of H. sapiens, Neanderthals and Denisovans. In addition, our datasets demonstrate that in vivo proteome modifications, such as serine phosphorylation, survive over time scales of hundreds of thousands of years. Current research therefore suggests that dental enamel, the hardest tissue in the mammalian skeleton, is the material of choice for the analysis of hominin evolution in deep time.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
Site location and specimen selection
Recent human control specimens
We analysed Ø1952, a human premolar recovered in an archaeological excavation in Copenhagen (Almindeligt Hospital Kirkegård, excavated in 1952, from kisse ‘2’). The tooth is approximately three centuries old, as the cemetery was in use from approximately ad 1600 to approximately ad 1800, and originates from a male individual. We also re-analysed previously published data16 related to specimens that are dated to between approximately 5,700 and 200 years ago; of these specimens, we took SK339 as a recent example in our comparative figures (a male individual from Fewston (UK) dated to the nineteenth century ad).
Atapuerca
One fragmentary permanent lower left first or second molar (ATD6-92; field number and museum accession number at CENIEH) was used for ancient protein analysis (Extended Data Fig. 2a, Supplementary Information). ATD6-92 originates from sublevel TD6.2 of the Gran Dolina cave site. Sublevel TD6.2 contains a large number of faunal remains, about 170 hominin fossils and about 830 archaeological artefacts. All hominin specimens from sublevel TD6.2, including ATD6-92, are attributed to H. antecessor9. ATD6-92 has recently been directly dated through electron spin resonance, laser-ablation inductively coupled plasma mass spectrometry U-series and bulk U-series dating11. Together with previous chronological research at the site, these analyses constrain the age of ATD6-92 to 772–949 thousand years old11.
Dmanisi
One fragmentary permanent upper first molar (D4163; field number and museum accession number at the Georgian National Museum) was used for ancient protein analysis (Extended Data Fig. 2b, Supplementary Information). D4163 derives from layer B1 in excavation block M6 (Dmanisi). Layer B1 at Dmanisi contains one of the richest palaeontological assemblages attributed to the Eurasian Early Pleistocene epoch, including several hominin crania. Below, we refer to these specimens as H. erectus (Dmanisi). They represent the earliest hominin fossils outside Africa, and are dated to 1.77 Ma14. Faunal material from the site previously demonstrated ancient protein survival for most specimens, but a total absence of ancient DNA6 (Fig. 3).
Amino acid racemization
Chiral amino acid analysis was undertaken on one Pleistocene sample from the hominin tooth (D4163) to test the endogeneity of the enamel protein through its degradation patterns. The tooth chip was separated into the enamel and dentine portions, and each was powdered with an agate pestle and mortar. All samples were prepared using previously published procedures39, modified to be optimized for enamel, using a bleach time of 72 h to isolate the intracrystalline protein, demineralization in HCl, KOH neutralization and formation of a biphasic solution through centrifugation40. Two subsamples were analysed from each portion: one fraction was directly demineralized and the free amino acids analysed, and the second was treated to release the peptide-bound amino acids, thus yielding the total hydrolysable amino acid fraction. Samples were analysed in duplicate by reversed-phase high-performance liquid chromatography, with standards and blanks analysed alongside samples. During preparative hydrolysis, both asparagine (Asn) and glutamine (Gln) undergo rapid irreversible deamidation to aspartic acid (Asp) and glutamic acid (Glu), respectively41. It is therefore not possible to distinguish between the acidic amino acids and their derivatives, and they are reported together as Asx and Glx, respectively. Additional descriptions of the methods, as well as additional results, are given in the Supplementary Information.
Proteomic extraction and nanoLC–MS/MS
Protein extraction
Protein extraction was conducted on enamel samples (from the Atapuerca H. antecessor, Dmanisi H. erectus and Ø1952) and a dentine sample (Dmanisi), using one of three protocols. In brief, the first extraction method used HCl for demineralization, but included no subsequent reduction, alkylation or digestion. The second extraction method used a more standard approach, in which the pellet left from the demineralization in extraction one was reduced, alkylated and digested with LysC and trypsin. The third extraction method used TFA for demineralization, and had no subsequent reduction, alkylation or digestion. The first and third extraction approaches provided more extensive peptide recovery in ancient enamel proteomes6 compared to the second extraction approach42. Further details can be found in the Supplementary Information and a previous publication6. Ø1952 was processed using extraction methods one and three. No proteinase and phosphatase inhibitors were used during extraction, as we assumed that catalytically active enzymes were not present in our specimens and the high acidic conditions during our extraction would have irreversibly denatured any proteases possibly present as contaminants in our reagents. Extended Data Table 1 provides a breakdown of the use of specific extraction methods, hominin samples and hominin tissues.
NanoLC–MS/MS analysis
Shotgun proteomic data were obtained on peptide extracts of both hominins at separate facilities at the Novo Nordisk Centre for Protein Research (University of Copenhagen) and the Proteomics Unit (Centre for Genomic Regulation, Barcelona Institute of Science and Technology). Full peptide elutions were injected, in some cases across replicate runs in both Copenhagen and Barcelona. In brief, samples processed in Copenhagen were suspended in 0.1% trifluoroacetic acid, 5% acetonitrile, and analysed on a Q-Exactive HF or HF-X mass spectrometer (Thermo Fisher Scientific) coupled to an EASY-nLC 1200 (Thermo Fisher Scientific). The HF or HF-X mass spectrometer was operated in positive ion mode with a nanospray voltage of 2 kV and a source temperature of 275 °C. Data-dependent acquisition mode was used for all mass spectrometric measurements. Full mass spectrometry scans were done at a resolution of 120,000 with a mass range of m/z 300–1,750 and 350–1,400 for the HF and HF-X mass spectrometers, respectively, with detection in the Orbitrap mass analyser. Fragment ion spectra were produced at a resolution of 60,000 via high-energy collision dissociation (HCD) at a normalized collision energy of 28% and acquired in the Orbitrap mass analyser. In addition, test runs for the Dmanisi sample were performed at a shorter gradient (Supplementary Information). In Barcelona, samples were dissolved in 0.1% formic acid and analysed on a LTQ-Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) coupled to an EASY-nLC 1000. The mass spectrometer was operated similarly to the parameters stated for the HF and HF-X mass spectrometers in Copenhagen, except the nanospray voltage was 2.4 kV and full mass spectrometry scans with 1 micro scan were used over a mass range of m/z 350–1,500. Further details of the LC–MS/MS analysis can be found in the Supplementary Information.
Proteomic data analysis
Protein sequence database construction
We constructed an initial Hominidae sequence database containing protein sequences of all major and minor enamel proteins derived from all extant great apes, a hylobatid (Nomascus leucogenys) and a macaque (Macaca mulatta). Additionally, we added protein sequences translated from extinct Late Pleistocene hominins30,43, and sequences from Gorilla beringei, Pongo pygmaeus and Pongo tapanuliensis44,45,46. For each protein, we reconstructed the protein sequence of ancestral nodes in the Hominidae family through PhyloBot47 to minimize cross-species proteomic effects48, and added missing isoform variation on the basis of the isoforms present for each protein in the human proteome as given by UniProt (Supplementary Information). Furthermore, we downloaded the entire human reference proteome from UniProt (4 September 2018) for a single separate search to allow matches to proteins previously not encountered in enamel proteomes. To each constructed database, we added a set of known or possible laboratory contaminants to allow for the identification of possible protein contaminants49.
Proteomic software, settings and false-discovery rate
Raw mass spectrometry data were searched for each specimen and tissue separately in either PEAKS50 (v.7.5) or MaxQuant51 (v.1.5.3.30). No fixed modifications were specified in any search. For PEAKS, variable post-translational modifications were set to include proline hydroxylation, glutamine and asparagine deamidation, oxidation (M), phosphorylation (STY), carbamidomethylation (C) and pyroglutamic acid (from Q and E). For MaxQuant, the following variable post-translation modifications were additionally included: ornithine formation (R), oxidation (W), dioxidation (MW), histidine to aspartic acid (H>D), and histidine to hydroxyglutamate. Searches were conducted with unspecific digestion. For PEAKS, precursor mass tolerance was set to 10 ppm and fragment mass tolerance to 0.05 Da, and the false-discovery rate of peptide spectrum matches was set to equal ≤1.0%. For MaxQuant, default settings of 20 ppm for the first search and 4.5 ppm for the final search were used, a fragment mass tolerance of 20 ppm, and peptide spectrum match (PSM) and protein false-discovery rate was set to 1.0%, with a minimum required Andromeda score of 40 for all peptides. Protein matches were accepted with a minimum of two unique peptide matches in either the PEAKS or MaxQuant search. Proteins that conform to these criteria are detailed in Extended Data Table 2. Example MS/MS spectra from the MaxQuant search and overlapping sites of phylogenetic interest (single amino acid polymorphisms) are included as Supplementary Data 1.
Data search iterations
For both the proteomes of Dmanisi and Atapuerca specimens, we conducted two separate initial searches. First, we conducted a search in PEAKS against the entire human proteome. Only standard enamel proteins were identified in these searches, allowing us to continue with more specific searches. For the Dmanisi dentine sample, this first search resulted in a small number of peptides matching to collagen type I only. On the basis of the limited amount of sequence data, no further analysis of the Dmanisi dentine data was therefore conducted. Second, for the enamel data, we conducted a search in PEAKS and MaxQuant against the entire enamel proteome database of all extant and extinct Hominidae. This search was used to observe single amino acid polymorphisms outside the known sequence variation in PEAKS and MaxQuant through the de novo, error-tolerant and/or dependent peptide approaches implemented in each of these search engines. These initial searches indicate overall good protein preservation in both samples and the presence of peptide matches to Pan- and Homo-derived proteins only.
On the basis of these two initial searches, a novel protein sequence database was used that only includes sequences from the genus Pan, the genus Homo, their predicted ancestral sequences and novel protein sequences observed for both the Dmanisi or Atapuerca samples. Final searches and subsequent data analysis were conducted against this database using the above search and post-translational modification settings. Positions supported by insufficient spectral data were replaced by ‘X’, in resulting peptide alignments before phylogenetic analysis.
Data analysis of Ø1952 and the previously published16 dataset was conducted only in MaxQuant against a database restricted to H. sapiens. All other search settings and database restrictions were similar between these two recent human controls and the ancient hominin proteomes.
Peptide sequence and single amino acid polymorphism validation
To validate the PSMs covering single amino acid polymorphisms of interest, we performed peptide spectrum intensity prediction and validation on our dataset using wiNNer17. Data from the ancient specimens (Dmanisi H. erectus and Atapuerca H. antecessor) were divided into a subset that contained phylogenetically informative peptide sequences and a larger subset that did not contained these peptides. A training dataset was prepared by taking a subset of the latter peptides, and adding a previously published dataset of enamel proteomes from Dmanisi fauna6. We built two models, one for HCD +2 spectra and one for HCD +3 spectra. We took into account the large number of variable modifications observed in our ancient enamel proteomes, and split the retained data for each model into subsets for training, validation and testing (80:10:10). We then obtained Pearson correlation coefficients for the predicted and true fragment intensities in the test dataset and the phylogenetically informative spectra. The architecture of wiNNer was built using Keras (version 2.0.8; https://keras.io) and Tensorflow (version 1.3.0). The wiNNer analysis indicated close correspondence between predicted and true fragment ion intensities (Pearson correlation coefficient medians between 0.85 and 0.76 for different subsets of the data), indicating adequate peptide sequence identification for all our peptides, including phylogenetically informative positions and the variable post-translational modifications. The wiNNer model can be accessed on GitHub (https://github.com/cox-labs/wiNNer.git). Additional methodological details of the wiNNer architecture are given in the Supplementary Information.
Protein damage analysis
Ancient proteins can be modified diagenetically in a variety of ways compared to their modern counterparts. We quantify glutamine and asparagine deamidation following a previously publication42 for MaxQuant output, based on MS1 spectral intensities and protein-based bootstrapping (1,000 bootstraps). Further details can be found in the previous publication42. We observe that both glutamines and asparagines are almost all deamidated to glutamic acid and aspartic acid, respectively (Extended Data Fig. 6a–c). In addition, peptide length distributions were obtained for datasets presented here and elsewhere6,8, demonstrating a shortening of average peptide length and overall peptide length distributions for older samples (Extended Data Fig. 6d).
Protein in vivo modification analysis
The existing literature on enamel and enamel proteome biomineralization describes three processes that are key to the maturation of the enamel proteome: protein hydrolysis by MMP20 and KLK452,53,54,55, in vivo phosphorylation of serine residues6,8,23 and expression of different isoforms of AMELX, AMBN and AMTN52,55,56. We sought to explore the presence of both in vivo protein hydrolysis and serine phosphorylation modifications in our Pleistocene hominin proteomes.
For protein hydrolysis by MMP20 and KLK4, we made use of the Atapuerca digestion-free dataset and the described locations of AMBN, AMELX and AMELY, and ENAM cleavage by MMP20 and KLK452,53,54,55. We compared the experimentally observed cleavage sites to a random cleavage model of each protein separately and tested whether the cleavage sites are present in a larger portion of PSMs in the ancient sample. Here we can indeed show an increased presence of PSMs with termini at, or close to, known MMP20 and KLK4 cleavage locations (Extended Data Fig. 7). This corresponds with our observation that protein regions with continuous sequence coverage correspond to known proteolytic fragments after MMP20 and KLK4 activity (Extended Data Fig. 4).
Phosphorylation of serines (S), threonines (T) and tyrosines (Y) was assessed using Icelogo57 sequence motif analysis. This analysis was based on the MaxQuant results, from which only identified phosphorylation sites with a localization probability of ≥0.95 were selected. STY sites with no phosphorylation or localization probabilities ≤ 0.95 were taken as the non-phosphorylated background, and a sequence motif window of 7 amino acids on either side of the STY was selected. Sequence motif analysis indicates a strong preference for the phosphorylation of S with a glutamic acid (E) on the +2 position (S-X-E motif) (Fig. 1a, b) in both hominin enamel proteomes. This substrate motif and the S-X-phosphorylated S motif are recognized by the kinase FAM20C, which is known to be active in vivo on extracellular proteins involved in biomineralization23, and has previously been reported for ancient, non-hominin enamel proteomes as well6,8.
To compare phosphorylation occupancy between the Dmanisi and Atapuerca enamel proteomes, we performed a separate MaxQuant database search (Supplementary Information) and restricted our analyses to amino acid positions covered by phosphorylated and non-phosphorylated peptides, observed in both hominins and quantified through label-free quantification.
Phylogenetic analysis
Comparison between the ancient protein sequences and modern reference proteins
We compared the reconstructed ancient protein sequences from the Dmanisi H. erectus and Atapuerca H. antecessor with protein sequences from great apes44,46, three Neanderthals31,43,58, a Denisovan59 and a panel of present-day humans, including 256 samples from the Simons Genome Diversity Panel33 and 41 high-coverage individuals from the 1000 Genomes Project34. Altogether, our reference data represent worldwide human and great ape variation (Supplementary Tables 7, 8). Additionally, we included protein sequences from macaque (M. mulatta) and gibbon (N. leucogenys) to root phylogenetic trees. The protein sequences were retrieved from the UniProt database or reconstructed from the reference whole-genome sequences as described in Supplementary Methods.
The ancient and reference protein sequences were aligned using mafft60. We aligned the sequences of each protein separately and obtained an alignment for each of the ancient individuals independently (Supplementary Table 9). The isobaric amino acids leucine (L) and isoleucine (I) cannot be distinguished with the experimental procedure used for this study. Therefore, we have to take the following precautions to avoid unintentional sequence differences. If either I or L was present at a specific amino acid position in the reference protein sequences, we replaced all corresponding amino acids in the ancient protein sequences with the amino acid that is present. Alternatively, if both amino acids are present in the reference protein sequence, we replace all I to L for all sequences. We used sequence information for seven proteins (ALB, AMBN, AMELX, AMELY, COL17α1, ENAM and MMP20) for the H. antecessor individual and six proteins for the H. erectus individual (ALB, AMBN, AMELX, COL17α1, ENAM and MMP20) with a total of 22.08% and 22.14% non-missing sites, respectively (Supplementary Table 9). We were able to recover a unique single amino acid polymorphism for H. antecessor; however, for H. erectus no unique single amino acid polymorphism was detected (Supplementary Tables 9–11, Supplementary Figs. 10–12).
Phylogenetic reconstruction
We built phylogenetic trees using our protein sequence alignments following three approaches: a maximum likelihood approach using PhyML v.361, and two Bayesian approaches using mrBayes62 and BEAST63.
For the maximum likelihood approach, we built maximum likelihood trees for each protein independently and for a concatenated alignment consisting of all of the available protein sequences for each of the ancient samples (Supplementary Figs. 13, 14). We used PhyML v.3 and the parameters described in the Supplementary Information section 2.3.5a to build and optimize the tree topologies, branch length and substitutions rates for each of the alignments. Support for each bipartition was obtained based on 100 non-parametric bootstrap replicates. We evaluated the effect of significant missingness in the ancient samples on the inferred topology. Finally, we looked at the effect of varying which of the subset of present-day human samples was included in the tree (Supplementary Information section 2.3.5b, c).
For the Bayesian approach using mrBayes, to assess the robustness of the maximum likelihood inference results, we performed Bayesian phylogenetic inference on the basis of the concatenated alignments using mrBayes 3.2 and the parameters described in Supplementary Information section 2.3.5d (Extended Data Fig. 8, Supplementary Fig. 16). Bayesian inference was performed using the CIPRES Science Gateway64.
For the Bayesian approach using BEAST, we used BEAST 2.5 to obtain a time calibrated tree for the seven proteins used for H. antecessor. For this analysis, we used concatenated alignments including the Neanderthals, the Denisovan, seven randomly chosen H. sapiens individuals and a single individual per great ape species. The alignment was partitioned by gene and a coalescent constant population model was used for the tree prior. The dates of the ancient samples included in the analysis (Vindija Neanderthal, 52 ka58; Altai Neanderthal, 112 ka31; Denisovan, 72 ka59 and H. antecessor, 860.5 ka11) were used as tip dates for calibration. For each partition, we used the Jones–Taylor–Thornton substitution model with four categories for the gamma parameter, for which we allowed the Markov chain Monte Carlo chain to sample the shape of the gamma distribution (with an exponentially distributed prior) and assigned independent clock models. Additionally, we set a prior for the divergence time of great apes to 23.85 ± 2.5 Ma (normally distributed)65, and rooted the tree using the macaque (M. mulatta). The overall topology of the tree was estimated for the seven partitions jointly. The convergence of the algorithm was assessed using Tracer v.1.7.066. Finally, we repeated this analysis with 100 alignments, each of them consisting of 7 present-day humans chosen randomly. Although the topology within the clade consisting of present-day humans, Neanderthals and Denisovan was not consistent across the replicates, 99 of the replicates consistently place the H. antecessor sequence as an outgroup to this clade (Fig. 2a).
Further details on phylogenetic analysis and results can be found in the Supplementary Information. Example MS/MS spectra from the MaxQuant search and overlapping sites of phylogenetic interest (single amino acid polymorphisms) for both hominins are included as Supplementary Data 1.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD014342. Generated ancient protein consensus sequences used for phylogenetic analysis for H. antecessor (Atapuerca) and H. erectus (Dmanisi) hominins can be found in the Supplementary Data 2, which is formatted as a .fasta file. Full protein sequence alignments used during phylogenetic analysis can be accessed via Figshare (https://doi.org/10.6084/m9.figshare.9927074). Amino acid racemization data are available online through the NOAA database. The wiNNer model can be accessed on GitHub (https://github.com/cox-labs/wiNNer.git).
Change history
29 July 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41586-020-2580-6
References
Gabunia, L. et al. Earliest Pleistocene hominid cranial remains from Dmanisi, Republic of Georgia: taxonomy, geological setting, and age. Science 288, 1019–1025 (2000).
Zhu, Z. et al. Hominin occupation of the Chinese Loess Plateau since about 2.1 million years ago. Nature 559, 608–612 (2018).
Stringer, C. The origin and evolution of Homo sapiens. Phil. Trans. R. Soc. Lond. B 371, 20150237 (2016).
Hublin, J. J. The origin of Neandertals. Proc. Natl Acad. Sci. USA 106, 16022–16027 (2009).
Rightmire, G. Human evolution in the Middle Pleistocene: the role of Homo heidelbergensis. Evol. Anthropol. 6, 218–227 (1998).
Cappellini, E. et al. Early Pleistocene enamel proteome from Dmanisi resolves Stephanorhinus phylogeny. Nature 574, 103–107 (2019).
Chen, F. et al. A late Middle Pleistocene Denisovan mandible from the Tibetan Plateau. Nature 569, 409–412 (2019).
Welker, F. et al. Enamel proteome shows that Gigantopithecus was an early diverging pongine. Nature 576, 262–265 (2019).
Bermúdez de Castro, J. M. et al. A hominid from the lower Pleistocene of Atapuerca, Spain: possible ancestor to Neandertals and modern humans. Science 276, 1392–1395 (1997).
Carbonell, E. et al. Lower Pleistocene hominids and artifacts from Atapuerca-TD6 (Spain). Science 269, 826–830 (1995).
Duval, M. et al. The first direct ESR dating of a hominin tooth from Atapuerca Gran Dolina TD-6 (Spain) supports the antiquity of Homo antecessor. Quat. Geochronol. 47, 120–137 (2018).
Freidline, S. E., Gunz, P., Harvati, K. & Hublin, J.-J. Evaluating developmental shape changes in Homo antecessor subadult facial morphology. J. Hum. Evol. 65, 404–423 (2013).
Lacruz, R. S. et al. Facial morphogenesis of the earliest Europeans. PLoS One 8, e65199 (2013).
Ferring, R. et al. Earliest human occupations at Dmanisi (Georgian Caucasus) dated to 1.85–1.78 Ma. Proc. Natl Acad. Sci. USA 108, 10432–10436 (2011).
Lordkipanidze, D. et al. A complete skull from Dmanisi, Georgia, and the evolutionary biology of early Homo. Science 342, 326–331 (2013).
Stewart, N. A., Gerlach, R. F., Gowland, R. L., Gron, K. J. & Montgomery, J. Sex determination of human remains from peptides in tooth enamel. Proc. Natl Acad. Sci. USA 114, 13649–13654 (2017).
Tiwary, S. et al. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods 16, 519–525 (2019).
Castiblanco, G. A. et al. Identification of proteins from human permanent erupted enamel. Eur. J. Oral Sci. 123, 390–395 (2015).
Asaka, T. et al. Type XVII collagen is a key player in tooth enamel formation. Am. J. Pathol. 174, 91–100 (2009).
Porto, I. M., Laure, H. J., de Sousa, F. B., Rosa, J. C. & Gerlach, R. F. New techniques for the recovery of small amounts of mature enamel proteins. J. Archaeol. Sci. 38, 3596–3604 (2011).
Gasse, B., Chiari, Y., Silvent, J., Davit-Béal, T. & Sire, J.-Y. Amelotin: an enamel matrix protein that experienced distinct evolutionary histories in amphibians, sauropsids and mammals. BMC Evol. Biol. 15, 47 (2015).
Demarchi, B. et al. Protein sequences bound to mineral surfaces persist into deep time. eLife 5, e17092 (2016).
Tagliabracci, V. S. et al. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science 336, 1150–1153 (2012).
Hu, J. C. C., Yamakoshi, Y., Yamakoshi, F., Krebsbach, P. H. & Simmer, J. P. Proteomics and genetics of dental enamel. Cells Tissues Organs 181, 219–231 (2005).
Glimcher, M. J., Cohen-Solal, L., Kossiva, D. & de Ricqles, A. Biochemical analyses of fossil enamel and dentin. Paleobiology 16, 219–232 (1990).
Wagner, G. A. et al. Radiometric dating of the type-site for Homo heidelbergensis at Mauer, Germany. Proc. Natl Acad. Sci. USA 107, 19726–19730 (2010).
Martinón-Torres, M. et al. Dental evidence on the hominin dispersals during the Pleistocene. Proc. Natl Acad. Sci. USA 104, 13279–13282 (2007).
Bermúdez de Castro, J. M., Martinón-Torres, M., Arsuaga, J. L. & Carbonell, E. Twentieth anniversary of Homo antecessor (1997–2017): a review. Evol. Anthropol. 26, 157–171 (2017).
Gómez-Robles, A., Bermúdez de Castro, J. M., Arsuaga, J.-L., Carbonell, E. & Polly, P. D. No known hominin species matches the expected dental morphology of the last common ancestor of Neanderthals and modern humans. Proc. Natl Acad. Sci. USA 110, 18196–18201 (2013).
Meyer, M. et al. Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature 531, 504–507 (2016).
Prüfer, K. et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49 (2014).
Lacruz, R. S. et al. The evolutionary history of the human face. Nat. Ecol. Evol. 3, 726–736 (2019).
Mallick, S. et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016).
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Welker, F. et al. Middle Pleistocene protein sequences from the rhinoceros genus Stephanorhinus and the phylogeny of extant and extinct Middle/Late Pleistocene Rhinocerotidae. PeerJ 5, e3033 (2017).
Hill, R. C. et al. Preserved proteins from extinct Bison latifrons identified by tandem mass spectrometry; hydroxylysine glycosides are a common feature of ancient collagen. Mol. Cell. Proteomics 14, 1946–1958 (2015).
Wadsworth, C. & Buckley, M. Proteome degradation in fossils: investigating the longevity of protein survival in ancient bone. Rapid Commun. Mass Spectrom. 28, 605–615 (2014).
Orlando, L. et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 499, 74–78 (2013).
Penkman, K. E. H., Kaufman, D. S., Maddy, D. & Collins, M. J. Closed-system behaviour of the intra-crystalline fraction of amino acids in mollusc shells. Quat. Geochronol. 3, 2–25 (2008).
Dickinson, M., Lister, A. M. & Penkman, K. E. H. A new method for enamel amino acid racemization dating: a closed system approach. Quat. Geochronol. 50, 29–46 (2019).
Hill, R. L. Hydrolysis of proteins. Adv. Protein Chem. 20, 37–107 (1965).
Mackie, M. et al. Palaeoproteomic profiling of conservation layers on a 14th century italian wall painting. Angew. Chem. Int. Ed. Engl. 57, 7369–7374 (2018).
Castellano, S. et al. Patterns of coding variation in the complete exomes of three Neandertals. Proc. Natl Acad. Sci. USA 111, 6666–6671 (2014).
de Manuel, M. et al. Chimpanzee genomic diversity reveals ancient admixture with bonobos. Science 354, 477–481 (2016).
Nater, A. et al. Morphometric, behavioral, and genomic evidence for a new orangutan species. Curr. Biol. 27, 3487–3498.e10 (2017).
Prado-Martinez, J. et al. Great ape genetic diversity and population history. Nature 499, 471–475 (2013).
Hanson-Smith, V. & Johnson, A. PhyloBot: a web portal for automated phylogenetics, ancestral sequence reconstruction, and exploration of mutational trajectories. PLOS Comput. Biol. 12, e1004976 (2016).
Welker, F. Elucidation of cross-species proteomic effects in human and hominin bone proteome identification through a bioinformatics experiment. BMC Evol. Biol. 18, 23 (2018).
Hendy, J. et al. A guide to ancient protein studies. Nat. Ecol. Evol. 2, 791–799 (2018).
Zhang, J. et al. PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 11, M111.010587 (2012).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Chun, Y. H. P. et al. Cleavage site specificity of MMP-20 for secretory-stage ameloblastin. J. Dent. Res. 89, 785–790 (2010).
Yamakoshi, Y., Hu, J. C. C., Fukae, M., Yamakoshi, F. & Simmer, J. P. How do enamelysin and kallikrein 4 process the 32-kDa enamelin? Eur. J. Oral Sci. 114 (Suppl 1), 45–51, 93–95, 379–380 (2006).
Iwata, T. et al. Processing of ameloblastin by MMP-20. J. Dent. Res. 86, 153–157 (2007).
Nagano, T. et al. Mmp-20 and Klk4 cleavage site preferences for amelogenin sequences. J. Dent. Res. 88, 823–828 (2009).
Fukae, M. et al. Primary structure of the porcine 89-kDa enamelin. Adv. Dent. Res. 10, 111–118 (1996).
Colaert, N., Helsens, K., Martens, L., Vandekerckhove, J. & Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 (2009).
Prüfer, K. et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 358, 655–658 (2017).
Meyer, M. et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Bouckaert, R. et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLOS Comput. Biol. 15, e1006650 (2019).
Miller, M. A., Pfeiffer, W. & Schwartz, T. in Gateway Computing Environments Workshop (GCE) 1–8 (New Orleans, 2010).
Besenbacher, S., Hvilsom, C., Marques-Bonet, T., Mailund, T. & Schierup, M. H. Direct estimation of mutations in great apes reconciles phylogenetic dating. Nat. Ecol. Evol. 3, 286–292 (2019).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Acknowledgements
F.W. is supported by a Marie Skłodowska Curie Individual Fellowship (no. 795569). E. Cappellini was supported by VILLUM FONDEN (no. 17649). E.W. is supported by the Lundbeck Foundation, the Danish National Research Foundation, the Novo Nordisk Foundation, the Carlsberg Foundation, KU2016 and the Wellcome Trust. Without the effort of the members of the Atapuerca research team during fieldwork, this work would have not been possible; we make a special mention of J. Rosell, who supervises the excavation of the TD6 level. The research of the Atapuerca project has been supported by the Dirección General de Investigación of the Ministerio de Ciencia, Innovación y Universidades (grant numbers PGC2018-093925-B-C31, C32, and C33); field seasons are supported by the Consejería de Cultura y Turismo of the Junta de Castilla y León and the Fundación Atapuerca. We acknowledge The Leakey Foundation through the personal support of G. Getty (2013) and D. Crook (2014–2016, 2018, and 2019) to M.M.-T., as well as F.W. (2017). Restoration and conservation work on the material have been carried out by P. Fernández-Colón and E. Lacasa from the Conservation and Restoration Area of CENIEH-ICTS and L. López-Polín from IPHES. The picture of the specimen ATD6-92 was made by M. Modesto-Mata. E. Cappellini, J.C., J.V.O. and P. Gutenbrunner are supported by the Marie Skłodowska-Curie European Training Network (ETN) TEMPERA, a project funded by the European Union’s Framework Program for Research and Innovation Horizon 2020 (grant agreement no. 722606). Amino acid analyses were undertaken thanks to the Leverhulme Trust (PLP-2012-116) and NERC (NE/K500987/1). T.M.-B. is supported by BFU2017-86471-P (MINECO/FEDER, UE), U01 MH106874 grant, Howard Hughes International Early Career, Obra Social ‘La Caixa’ and Secretaria d’Universitats i Recerca and CERCA Programme del Departament d’Economia i Coneixement de la Generalitat de Catalunya (GRC 2017 SGR 880). C.L.-F. is supported by a FEDER-MINECO grant (PGC2018-095931-B-100). M.K. was supported by the Postdoctoral Junior Leader Fellowship Programme from ‘la Caixa’ Banking Foundation (LCF/BQ/PR19/11700002). M.M. is supported by the Danish National Research Foundation award PROTEIOS (DNRF128). Work at the Novo Nordisk Foundation Center for Protein Research is funded in part by a donation from the Novo Nordisk Foundation (grant number NNF14CC0001). The CRG/UPF Proteomics Unit is part of the Spanish Infrastructure for Omics Technologies (ICTS OmicsTech) and it is a member of the ProteoRed PRB3 consortium, which is supported by grant PT17/0019 of the PE I+D+i 2013-2016 from the Instituto de Salud Carlos III (ISCIII) and ERDF. We acknowledge support from the Spanish Ministry of Science, Innovation and Universities, ‘Centro de Excelencia Severo Ochoa 2013-2017’, SEV-2012-0208, and ‘Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement de la Generalitat de Catalunya’ (2017SGR595). D.L. and A.M. are supported by the John Templeton Foundation (no. 52935) and by the Shota Rustaveli National Science Foundation of Georgia (no. FR-18-27262). We thank M. L. Schjellerup Jørkov for providing specimen Ø1952.
Author information
Authors and Affiliations
Contributions
E. Cappellini, E.W., J.M.B.d.C., D.L., C.L.-F. and F.W. designed the study. E. Cappellini, M.M., F.W., J.R.-M., R.R.J.-C., M.R.D., C.C. and M.d.M. performed experiments. E. Cappellini, A.M., J.L.A., E. Carbonell, P. Gelabert, E.S., J.C., J.V.O., T.M.-B. and D.L. provided material, reagents or research infrastructure. F.W., J.R.-M., P. Gutenbrunner, S.T., E. Cappellini, F.R., M.M.-T., J.M.B.d.C., M.K., M.R.D., C.L.-F. and K.P. analysed data. F.W., E. Cappellini and J.M.B.d.C. wrote the manuscript with input from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
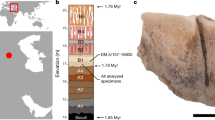
Extended Data Fig. 1 Location and stratigraphy of the hominin fossils studied.
a, Geographic location of Gran Dolina and Dmanisi. Base map was generated using public domain data from www.naturalearthdata.com. b, Summarized stratigraphic profile of Gran Dolina, including the location of hominin fossils in layers ‘Pep’ and ‘Jordi’ of sublevel TD6.2.
Extended Data Fig. 2 Hominin specimens studied.
a, ATD6-92 in buccal view. The fragment represents a portion of a permanent lower left first or second molar. b, D4163 in occlusal view. The specimen is a fragmented right upper first molar. Scale bar differs between a and b.
Extended Data Fig. 3 Amino acid racemization of D4163.
a, b, The extent of intracrystalline racemization in enamel for the free amino acid (FAA) (x axis) fraction and the total hydrolysable amino acids (THAA) (y axis) fraction for aspartic acid plus asparagine (here denoted Asx) (a), and glutamic acid plus glutamine (here denoted Glx) (b), demonstrates endogenous amino acids breaking down within a closed system. The hominin value is displayed in relation to values for enamel samples from other fauna from Dmanisi6 (blue squares) and a range of previously obtained Pleistocene and Pliocene Proboscidea from the UK40 (grey diamonds). Fauna are shown for comparison, but different rates in their protein breakdown mean that they will show different extents of racemization. The x and y axis are on different scales.
Extended Data Fig. 4 Sequence coverage for five enamel-specific proteins across Pleistocene samples and recent human controls.
For each protein, the bars span protein positions covered, with positions remapped to the human reference proteome. The top row indicates the position of a selection of known MMP20 and KLK4 cleavage products of the enamel-specific proteins AMELX55, AMBN52 and ENAM56. Several in vivo proteolytic degradation fragments of ENAM share the same N terminus, but have unknown C termini53. Dotted line for AMBN indicates a putative cleavage product based on known MMP20 (squares) and KLK4 (circles) in vivo cleavage positions. For AMTN, serines (S) at positions 115 and 116 (indicated by asterisks) are conserved among vertebrates and involved in mineral-binding21. Additional cleavage products as well as MMP20 and KLK4 cleavage sites are known in all enamel-specific proteins. SK33916 and Ø1952 are two recent human control samples (Methods). AA, amino acids; Steph., Stephanorhinus6; TRAP, tyrosine-rich amelogenin polypeptide.
Extended Data Fig. 5 Homo antecessor specimen ATD6-92 represents a male hominin.
a, Mass spectrum of an AMELY-specific peptide from the recent human control Ø1952. b, Mass spectrum of the same AMELY-specific peptide from H. antecessor. c, Alignment of a selection of AMELY- and AMELX-specific peptide fragment ion series deriving from H. antecessor. The alignment stretches along human AMELX isoform 1, positions 37 to 52 only (Uniprot accession numbers Q99217 (AMELX), Q99218 (AMELY)). See Supplementary Fig. 5 for another example of an AMELY-specific MS2 spectrum.
Extended Data Fig. 6 Enamel proteome damage.
a, b, Glutamine (Q) and asparagine (N) deamidation of enamel-specific proteins from H. antecessor (Atapuerca) (a), and H. erectus (Dmanisi) (b). Values are based on 1,000 bootstrap replications of protein deamidation. c, Relationship between mean asparagine (N) and glutamine (Q) deamidation for all proteins in both the Atapuerca and Dmanisi hominin datasets. Error bars represent 95% confidence interval window of 1,000 bootstrap replications of protein deamidation. Dashed line is x = y. d, Peptide length distribution of H. antecessor (Atapuerca), H. erectus (Dmanisi), four previously published enamel proteomes6,8,16 and one additional human Medieval control sample (Ø1952). For a, b and d, the number of peptides (n) is given for each violin plot. The box plots within the violin plots define the range of the data (whiskers extend to 1.5× the interquartile range), outliers (black dots, beyond 1.5× the interquartile range), 25th and 75th percentiles (boxes), and medians (centre lines). P values of two-sided t-tests conducted between sample pairs are indicated. No independent replication of these experiments was performed.
Extended Data Fig. 7 Survival of in vivo MMP20 and KLK4 cleavage sites in the Atapuerca enamel proteome.
a, Experimentally observed cleavage matrices for ameloblastin (AMBN), enamelin (ENAM) and amelogenin (AMELX and AMELY) (Methods). Fold differences are colour-coded by comparing observed PSM cleavage frequencies to a random cleavage matrix for each protein separately7. b, Fold differences for all observed cleavage pairs per protein. Red filled circles represent MMP20, KLK4 and signal peptide cleavage sites mentioned in the literature53,54,55,56. Red open circles indicate cleavage sites located up to two amino acid positions away from such sites. c, PSM coverage for each protein. The signal peptide (thick horizontal bar labelled ‘sig’), known MMP20 and KLK4 cleavage sites (vertical bars), and O- and N-linked glycosylation sites (asterisks) are also indicated. For AMELX, peptide positions for all three known isoforms were remapped to the coordinates of isoform 3, which represents the longest isoform (UniProt accession Q99217-3). The x and y axes differ between the three panels of c.
Extended Data Fig. 8 Phylogenetic position of D4163 through Bayesian analysis.
Nomascus leucogenys and M. mulatta were used as outgroups.
Supplementary information
Supplementary Information
Supplementary Information containing supplementary information on the archaeological sites of Gran Dolina, Atapuerca, and Dmanisi, amino acid racemization, proteomic data extraction, data generation, and data analysis, as well as phylogenetic analysis of recovered ancient hominin proteomes. The Supplementary Information contains 11 SI tables and 16 SI figures.
Supplementary Data
Supplementary information file containing annotated MS/MS spectra of phylogenetic interest.
Supplementary Data
File in .fasta format containing recovered Homo antecessor (Atapuerca) and Homo erectus (Dmanisi) protein sequences. Sequence coverage obtained after MaxQuant and PEAKS analysis is combined.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Welker, F., Ramos-Madrigal, J., Gutenbrunner, P. et al. The dental proteome of Homo antecessor. Nature 580, 235–238 (2020). https://doi.org/10.1038/s41586-020-2153-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2153-8
- Springer Nature Limited
This article is cited by
-
A label-free quantification method for assessing sex from modern and ancient bovine tooth enamel
Scientific Reports (2024)
-
Deep-time phylogenetic inference by paleoproteomic analysis of dental enamel
Nature Protocols (2024)
-
Oldest genetic data from a human relative found in 2-million-year-old teeth
Nature (2023)
-
Comparing extraction method efficiency for high-throughput palaeoproteomic bone species identification
Scientific Reports (2023)
-
Why the geosciences are becoming increasingly vital to the interpretation of the human evolutionary record
Nature Ecology & Evolution (2023)