

Abstract
Human immunodeficiency virus (HIV) persists indefinitely in individuals with HIV who receive antiretroviral therapy (ART) owing to a reservoir of latently infected cells that contain replication-competent virus1,2,3,4. Here, to better understand the mechanisms responsible for latency persistence and reversal, we used the interleukin-15 superagonist N-803 in conjunction with the depletion of CD8+ lymphocytes in ART-treated macaques infected with simian immunodeficiency virus (SIV). Although N-803 alone did not reactivate virus production, its administration after the depletion of CD8+ lymphocytes in conjunction with ART treatment induced robust and persistent reactivation of the virus in vivo. We found viraemia of more than 60 copies per ml in all macaques (n = 14; 100%) and in 41 out of a total of 56 samples (73.2%) that were collected each week after N-803 administration. Notably, concordant results were obtained in ART-treated HIV-infected humanized mice. In addition, we observed that co-culture with CD8+ T cells blocked the in vitro latency-reversing effect of N-803 on primary human CD4+ T cells that were latently infected with HIV. These results advance our understanding of the mechanisms responsible for latency reversal and lentivirus reactivation during ART-suppressed infection.
Similar content being viewed by others


Main
HIV remains a major global health problem, leading to approximately 1.1 million deaths worldwide annually5. Despite the major declines in morbidity and mortality associated with the use of ART, there is neither a vaccine nor a cure for HIV infection. The inability to eradicate HIV infection with the current therapies is due to the presence of latently infected cells that contain integrated replication-competent virus that persist indefinitely in HIV-infected individuals undergoing ART and contribute to rebound viraemia when therapy is discontinued (that is, the viral reservoir)1,2,3,4. A key paradigm in the field of HIV cure—referred to as ‘shock and kill’6,7—supposes that the induction of virus expression (that is, ‘virus reactivation’) in these latently infected cells (‘shock’) followed by immune-mediated clearing (‘kill’) may substantially reduce the reservoir size and possibly lead to a functional cure for HIV infection. Unfortunately, no latency-reversing agent (LRA) tested to date has successfully perturbed the viral reservoir in clinical trials in humans. In particular, histone deacetylase inhibitors have not induced either robust virus reactivation or reduction of the viral reservoir in ART-treated HIV-infected individuals8,9,10,11,12. More encouragingly, in SIV-infected ART-treated rhesus macaques (Macaca mulatta), treatment with toll-like receptor 7 (TLR7) agonists was linked to transient blips of plasma viraemia13. However, this result was not reproduced in further studies14,15. More recently, persistent remission was observed in a subset of ART-treated simian–human chimeric immunodeficiency virus (SHIV)-infected macaques that received the TLR7 agonist GS-9620 in combination with the broadly neutralizing PGT121 antibody16. In all, these published data indicate that novel, more potent approaches for latency reversal are needed to achieve a functional cure for HIV infection.
SIV and SHIV infection of rhesus macaques
Infection of rhesus macaques with SIV or SHIV is the most widely used animal model for the study of the mechanisms by which the viral reservoir is established and maintained under ART and to preclinically test interventions aimed at reducing the viral reservoir in vivo17. A previous study demonstrated that the depletion of CD8+ lymphocytes in SIV-infected ART-treated macaques was consistently followed by increased plasma viraemia, thus indicating that these cells contribute to viral suppression under ART18. Although the precise mechanisms responsible for this observation remain unclear, phylogenetic analysis of the rebounding virus suggested that silencing of virus transcription contributes to this antiviral effect. On the basis of these observations, we hypothesized that the depletion of CD8+ lymphocytes combined with LRAs may enhance virus production under ART. As shown in Fig. 1a, the IL-15 superagonist N-803 is a complex of a mutant IL-15 and a dimeric IL-15 receptor αSu/Fc fusion protein19. The engineered structure is at least 25 times more biologically potent than IL-15 as it mimics transpresentation, and the IgG–Fc component confers improved in vivo safety and bioavailability20,21. In the setting of ART-suppressed lentiviral infection, N-803 may target the residual virus pool through its ability to act in vitro as a potent LRA and to strengthen the antiviral immune responses mediated by T and natural killer (NK) cells22.

a, IL-15 superagonist N-803 structure. i.v., intravenous; TCID50, 50% tissue-culture infectious dose. b, Study design. Arrows indicate the administration of 100 μg kg−1 N-803 (green) and 50 mg kg−1 MT807R1 (blue). c, Plasma viral load pre-intervention (n = 35 macaques), including infection and initiation of ART (grey bar). Limit of detection is 60 copies of SIV RNA per ml of plasma (black bar). d, Mean peripheral CD4+ T cell (maroon), CD8+ T cell (purple) and NK cell (grey) count in the lymph node. e, Percentage of CD4+ and CD8+ T cells in the lymph node. f, Ki-67 expression in cellular subsets after intervention with N-803 (n = 7 macaques). g, Ki-67 expression in bulk CD4+ T cells after treatment with only N-803 (green, n = 7 macaques), CD8 depletion alone (blue, n = 14 macaques), and CD8 depletion combined with the administration of N-803 (red, n = 14 macaques). h, Gene-set enrichment analysis of RNA-sequencing data from bulk CD4+ T cells comparing gene sets enriched on day 3 after intervention with N-803 alone (green, n = 7 macaques), CD8 depletion alone (blue, n = 7 macaques) or CD8 depletion combined with N-803 treatment (red, n = 7 macaques). i, Heat map of the enriched genes in bulk CD8+ T cells in the IL-2 and STAT-5 signalling gene set after administration of N-803 alone (n = 7 macaques). Data are mean ± s.e.m. Two-sided Kruskal–Wallis tests (d, f) and Friedman tests (e, g) were used to compare values after the interventions to the pre-intervention baseline and approximate P-value summaries are provided.
The current study included a total of 35 SIV-infected macaques that started ART at day 56 after infection and were treated for at least 1 year before any further intervention. The macaques were divided in three groups as follows (Fig. 1b): 7 macaques were treated with 4 weekly doses of 100 μg kg−1 of N-803 (group 1, N-803 alone), 14 macaques received 1 dose of the CD8-depleting antibody, MT807R1 (anti-CD8α) at 50 mg kg−1 intravenously (group 2, CD8 depletion alone) and 14 macaques received 4 weekly doses of N-803 starting at the time of CD8 depletion (group 3, CD8 depletion with N-803). After reconstitution of CD8+ T cells (defined as more than 100 CD8+ T cells per μl of blood), 7 macaques in groups 2 and 3 received four additional weekly administrations of N-803. Peripheral blood samples, lymph node and rectal biopsies were collected at various time points before, during and after these interventions. All macaques underwent analytical treatment interruption at week 3 after either CD8+ T cell reconstitution or the last N-803 treatment. As shown in Fig. 1c, all macaques showed suppression of viraemia after 1 year of ART, with plasma viral loads below the detectable limit of our standard assay (60 copies per ml of plasma)23 at the time of the additional interventions in 33 out of 35 macaques (94.3%). We also measured residual plasma viraemia using an ultrasensitive assay (limit of detection of 3 copies per ml)24,25,26 at three monthly sampling points before the interventions. Viraemia was below 3 copies per ml in 19 out of 35 ART-treated macaques (52.3%), and 26 out of 35 macaques (74.3%) showed levels of residual viraemia of ≤10 copies per ml at the time of intervention (Extended Data Table 1). These results indicate that the level of virus suppression observed in our cohort of macaques was in most cases comparable to that of long-term ART-treated HIV-infected individuals27,28,29.
As shown in Extended Data Fig. 1a–d and consistent with previous studies18,30, treatment with anti-CD8α MT807R1, with or without N-803, depleted on average 99.1% of CD3+CD8+ T cells in peripheral blood, 97.9% in lymph nodes and 99.5% in rectal biopsies. In addition, treatment with MT807R1 alone depleted 93.2% of NK cells in peripheral blood (Extended Data Fig. 1e). As expected on the basis of previous studies, N-803 administration alone resulted in the expansion of CD8+ T cells in the blood and lymph nodes (Fig. 1d, e), as well as increased proliferation of peripheral CD8+ T cells, CD4+ T cells and NK cells (Fig. 1f). Of note, although CD8 depletion alone did not result in a rapid increase in CD4+ T cell proliferation (as measured by Ki-67 expression), the combination of CD8 depletion and treatment with N-803 led to a significant increase in CD4+ T cell proliferation (Fig. 1g and Extended Data Fig. 2r–v). The frequency of CD4+ T cells co-expressing Ki-67 and the HIV and SIV coreceptor CCR5—potential target cells for infection—was significantly increased across all groups by the third week after intervention (Extended Data Fig. 2l). Additionally, CD8 depletion with or without N-803 administration resulted in the expansion of effector memory CD4+ T cells and increased PD-1 expression on CD4+ T cells (Extended Data Fig. 2f, g).
To better characterize the biological effects of N-803, we conducted a transcriptional analysis using RNA sequencing of sorted CD4+ T cells collected before intervention and day 3, week 2 and week 4 after the start of intervention. Regardless of concurrent CD8 depletion, N-803 induced a significant upregulation of gene sets associated with cell cycling and proliferation, activation, antiviral responses and cell signalling (Fig. 1h). In CD8+ T cells, N-803 resulted in significant enrichment of genes in the IL-2 and STAT-5 signalling gene set, which is also indicative of IL-15 signalling, as the receptor for this cytokine shares two out of three subunits with IL-2 and uses STAT-5 as the key adaptor molecule (Fig. 1i). In addition, we examined the expression of 25 genes specifically involved in the host–virus interaction during SIV infection and found that N-803 administration induced a consistent and transient upregulation of APOBEC3 in CD4+ T cells, CD8+ T cells and NK cells (Extended Data Fig. 3a–c).
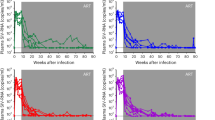
As shown in Fig. 2a and Extended Data Table 1, administration of N-803 was not associated with an increase of plasma viraemia of >60 copies per ml in any of the treated macaques, indicating that the IL-15 superagonist is not sufficient to exert an in vivo LRA effect in ART-treated SIV-infected macaques when used alone. As expected on the basis of previous studies18, macaques undergoing CD8 depletion alone showed a moderate but significant increase in virus production, with plasma viraemia of >60 copies per ml detected in 11 out of 14 macaques (78.6%) and 18 out of 56 samples (32.1%) collected weekly after CD8 depletion (Fig. 2b). Viraemia of >1,000 copies per ml was observed in 2 out of 14 macaques (14.2%) and 2 out of 56 (3.6%) of the same samples (Fig. 2b). In all cases, the level of virus production returned to below 60 copies per ml of plasma at the time of CD8+ T cell reconstitution (Fig. 2e). Overall, the level of increased viraemia observed in this study was consistent with previous studies18, even though the magnitude of virus production after CD8 depletion was slightly less marked, possibly related to the longer period of ART treatment (12 months compared with 2–8 months)18. Notably, macaques treated with N-803 during CD8 depletion showed highly robust and persistent levels of virus production, with viraemia of >60 copies per ml detected in 14 out of 14 macaques (100%) and 41 out of 56 samples (73.2%) and viraemia of >1,000 copies per ml observed in 6 out of 14 macaques (42.9%) and 13 out of 56 samples (23.2%) (Fig. 2c). We emphasize that all seven macaques with full suppression of virus production at the time of intervention with CD8 depletion and N-803 administration (that is, repeated viral load measurements <3 copies per ml of plasma) demonstrated clear virus reactivation with detectable levels in 26 out of 28 time points 1 week after each N-803 administration, and >60 copies per ml in 16 out of 28 of the same samples (Fig. 2d). Similar results were obtained in a smaller pilot study in which N-803 administration during CD8 depletion was performed in five ART-suppressed SHIVSF162P3-infected macaques (Fig. 2g). The level of viraemia observed in SIV-infected macaques treated with combined CD8 depletion and N-803 administration during long-term ART is higher and more persistent than the results of previous shock-and-kill cure strategies tested in humans and nonhuman primates8,9,10,11,12,13,31.

a–c, Plasma viral loads after intervention with N-803 alone (n = 7 macaques) (a), CD8 depletion alone (n = 14 macaques) (b) or CD8 depletion combined with N-803 (n = 14 macaques) (c). d, Longitudinal plasma viral loads for macaques with fully suppressed viral load (<3 copies per ml of plasma) before CD8 depletion combined with administration of N-803. e, f, Comparison of viral load pre-intervention (pre), post-intervention when CD8+ T cells are <100 cells per μl blood (post) and during CD8+ T cell reconstitution >100 cells per μl blood (CD8 return) in macaques that underwent CD8 depletion alone (n = 14 macaques) (e) or CD8 depletion with N-803 (n = 14 macaques) (f). Data are mean ± s.e.m. g, Plasma viral loads after CD8 depletion with N-803 administration in SHIVSF162P3-infected macaques after 6 months of ART (n = 5 macaques). Viral suppression of <60 copies per ml is shown as a black bar (a–d, g) or dashed line (e, f) and the limit of detection for these assays was 3 copies per ml of plasma. h, RNAscope determination of the percentage of SIV RNA+ lymph node cells expressing high levels (>4 copies) of viral RNA per cell 1 week after CD8 depletion combined with N-803 treatment. i–k, Plasma viral loads of HIV-infected, ART-treated humanized mice treated with N-803 alone (green, n = 7 mice) (i), CD8 depletion alone (blue, n = 8 mice) (j) or CD8 depletion with N-803 administration (red, n = 8 mice) (k). The limit of detection was 346 copies per ml. Statistical significance was calculated using a two-sided Kruskal–Wallis test (e, f) or Wilcoxon signed-rank test (h). A key for the macaque ID codes is provided in Extended Data Fig. 5.
After the last treatment with N-803, the level of viraemia rapidly decreased coincident with the reconstitution of CD8+ T cells, and all macaques returned to <60 copies per ml by week 6 after CD8 depletion and N-803 administration (Fig. 2c, f). As expected, CD8+ T cell reconstitution was faster in CD8-depleted macaques co-treated with N-803 (Extended Data Fig. 5e–g), as the IL-15 superagonist enhances CD8+ T cell proliferation (Fig. 1d–f).
We next investigated the correlates of virus reactivation in ART-treated SIV-infected macaques that underwent CD8 depletion with N-803, and observed that the post-depletion viral load (day 3 to week 6) was negatively correlated with the frequency of peripheral CD8+ T cells (Extended Data Fig. 5b). Additionally, the area under the curve of virus production during CD8 depletion and administration of N-803 was directly correlated with pre-intervention viraemia (Extended Data Fig. 5d). Of note, no correlation was found between the level of virus production after CD8 depletion and/or N-803 treatment and either the size of the peripheral blood DNA reservoir (measured as the fraction of SIV DNA+CD4+ T cells) or the level of CD4+ T cell activation (measured as Ki-67 or PD-1 expression on CD4+ T cells) (data not shown). To assess whether combined CD8 depletion with N-803 administration induced SIV production in lymphoid tissues, we next analysed the levels of SIV RNA in the lymph nodes using the RNAscope technology at pre-intervention and day 7 after intervention in 5 representative macaques that were both depleted of CD8+ lymphocytes and treated with N-803. As shown in Fig. 2h, and consistent with the measurements of plasma viraemia, we found a statistically significant increase in the percentage of SIV RNA+ cells with high levels of SIV RNA after intervention. No changes were observed in the level of SIV RNA in peripheral CD4+ T cells (Extended Data Fig. 4a–c), suggesting that lymphoid tissues are the main source of reactivated virus after the combined CD8 depletion and administration of N-803.
HIV infection of humanized mice
To confirm the virus reactivation induced by the combined depletion of CD8+ lymphocytes and administration of N-803 in SIV-infected ART-treated macaques in an in vivo model using HIV, we next conducted a similarly designed experiment using bone-marrow–liver–thymus (BLT) humanized mice infected with HIV-1JR-CSF and treated with ART. As shown in Fig. 2i–k, HIV-infected humanized mice showed markedly similar results to those obtained in SIV-infected macaques, with no plasma virus reactivation after the administration of N-803 alone, a moderate level of virus reactivation after CD8 depletion alone, and a robust level of virus reactivation in 7 out of 8 (87.5%) humanized mice that were depleted of CD8+ lymphocytes and treated with N-803. Furthermore, we also noted a statistically significant increase in the levels of cell-associated HIV RNA in the spleen and human-derived thymus of humanized mice that were depleted of CD8+ lymphocytes and treated with N-803 (Extended Data Fig. 6b).
CD8+ T cells inhibit latency reversal in vitro
The combined data obtained in SIV-infected macaques and HIV-infected humanized mice indicate that the strong virus reactivation activity attributed to N-803 is revealed only in the absence of CD8+ T cells, thus delineating a novel mechanism of latency maintenance and/or inhibition of latency reversal mediated by CD8+ lymphocytes. To recapitulate this observation in a reductionist in vitro model of HIV latency in human cells, we used the recently developed latency and reversal assay (LARA)32 to evaluate how CD8+ T cells affect the virus reactivation activity of N-803 in autologous memory CD4+ T cells latently infected with HIV89.6 (Fig. 3a). Of note, this assay was conducted using cells derived from HIV-negative donors, thus HIV-specific cytotoxic CD8+ T cells were absent. Whereas N-803 (and its biological counterpart, IL-15) reactivates HIV expression in latently infected CD4+ T cell monocultures, co-culture with activated CD8+ T cells significantly suppresses this ability (Fig. 3b). These data indicate that CD8+ T cells effectively suppress the latency-reversing activity of N-803, and therefore confirm the discovery of a previously unrecognized CD8+ T-cell-mediated activity that contributes to the maintenance of lentivirus latency in vivo in primates.

a, Schematic of HIV latency model. Memory CD4+ T cells (mCD4+) were enriched on day 0, infected in vitro on day 3 with HIV89.6 and maintained in saquinavir. On day 6, TGFβ, IL-7, conditioned medium from the H-80 feeder cell line and additional antiretroviral drugs were added to the culture. Cryopreserved autologous peripheral-blood mononuclear cells (PBMCs) were thawed on day 8 and rested overnight before enriching for total CD8+ (tCD8+) cells and then TCR-activated for 3 days. On day 12, HIV-infected mCD4+ and TCR-activated total CD8+ cells were co-cultured in a 1:1 or 1:5 ratio in the presence of anti-CD3/CD28 antibody, N-803 or recombinant IL-15 until day 15. b, HIV Gag+CD4+ T cell frequency was quantified using flow cytometry and the frequency in co-cultures was calculated as a fold change compared to CD4 T cell monoculture. Each colour represents a unique donor (n = 8 biologically independent samples) and data are mean ± s.e.m. (indicated by the grey bars). Statistical significance was measured using a matched one-way analysis of variance (ANOVA).
Virus sequence analysis
To investigate the viral dynamics associated with reactivation after CD8 depletion and administration of N-803 in SIV-infected ART-treated macaques, we performed a longitudinal sequence analysis of plasma virus using single-genome PCR amplification of the SIVmac239-derived env genes. The viral env was sequenced at three pivotal time points: day 7 after infection (at peak viraemia), day 56 after infection—that is, immediately before ART initiation (pre-ART)—and during peak virus reactivation. We conducted this analysis in six macaques that showed robust virus reactivation (plasma viraemia of >800 copies per ml) (Fig. 2c). Extended Data Figure 7 shows phylogenetic analysis of the translated Env amino acid sequences of the circulating viruses. The peak viral load Env sequences were homogeneous and nearly identical to the input SIVmac239 sequence; however, the diversity and number of informative sites at subsequent time points were limited, such that sequences could not be clustered on the basis of time point with significant bootstrap support. The diversity at each time point, although limited, was quantified by determining the number of amino acid differences from the input SIVmac239 sequence. Extended Data Figure 8a shows that, for all macaques, the peak viraemia Env sequences are the least different from the input virus, as expected, and increased divergence was observed at the pre-ART time point and after reactivation. We next calculated the average number of amino acid differences from SIVmac239 at each time point, and compared these with contemporaneous plasma viraemia for each macaque in a correlation matrix. The only significant association was a direct relationship between Env divergence and plasma viraemia during reactivation (Extended Data Fig. 8b). Finally, Extended Data Fig. 9 shows the location of sequence changes at each time point using highlighter plots of the Env amino acid sequences. Overall, this analysis supports the hypothesis that CD8 depletion and administration of N-803 induces robust reactivation of a diverse population of viral variants. As no signs of virus evolution emerged from the longitudinal sequence analysis during high levels of latency reversal, it is unlikely that the rebounding virus is a product of de novo viral replication. Supporting this hypothesis, the combined CD8 depletion and administration of N-803 did not increase the levels of two-long-terminal repeat circles—which are considered a marker of recent lentivirus infection—in peripheral-blood mononuclear cells (data not shown).
Analytic treatment interruption
To determine whether the interventions used induced a decrease in the virus reservoir, we first longitudinally measured the level of cell-associated SIV DNA in blood and lymph nodes. As shown in Fig. 4a–f, none of the experimental groups showed significant changes in either the total fraction of circulating CD4+ T cells or the calculated fraction of lymph-node-derived cells that contained SIV DNA. To functionally assess the effect of the treatment regimens on the reservoir size, we performed an analytical treatment interruption in all macaques three weeks after either CD8 reconstitution and/or the last N-803 treatment. As shown in Fig. 4g–i, all macaques rebounded within three weeks of ART interruption and most macaques sustained high viral loads until the time of necropsy. It should be noted that in the current study, ART was initiated at day 56 after infection, thus substantially later than in other published macaque studies that included analytical treatment interruption, and thus in the setting of a larger and more-disseminated reservoir16,33. The rapid rebound after ART interruption was therefore not unexpected as the experimental design was focused on assessing the shock effect of CD8 depletion combined with N-803 administration, with no anticipated effect on the reservoir size in the absence of an intervention aimed at clearing the cells that have reactivated SIV production (kill phase of the shock-and-kill approach). The absence of a decrease in the level of SIV DNA+ cells after the combined CD8 depletion and administration of N-803 may be due to the lack of CD8+ T-cell-mediated clearance of cells that have reactivated virus expression and/or the N-803-mediated proliferative expansion of infected CD4+ T cells that have survived the events of virus reactivation.

a–f, Copies of cell-associated SIV DNA per 106 cells was determined in peripheral CD4+ T cells (a–c) and frozen bulk lymph node cells (d–f) in macaques treated with N-803 alone (n = 7 (a) and n = 3 (d) macaques), CD8 depletion alone (n = 14 (b) and n = 5 (e) macaques), and CD8 depletion with N-803 treatment (n = 14 (c) and n = 3 (f) macaques). Two-sided Friedman tests (a–c) and Wilcoxon signed-rank tests (d–f) were used to determine statistical significance between pre-treatment and post-treatment time points. For all comparisons, P > 0.05. Sample mean ± s.e.m. are indicated by grey bars. g–i, Viral rebound after interruption of ART (indicated by arrowheads) in macaques that received N-803 alone (n = 7 macaques) (g), CD8 depletion alone (n = 13 macaques) (h) or CD8 depletion with N-803 (n = 14 macaques) (i). ATI, analytical treatment interruption. The limit of detection was <3 copies of SIV RNA per ml plasma. The black bar indicates viral loads <60 copies per ml and the grey box indicates ART.
Discussion
The current paradigm for shock-and-kill interventions for an HIV cure suggests that reactivation of virus transcription in latently infected cells is the first essential step to eliminate the persistent reservoir of replication-competent virus in ART-treated HIV-infected individuals. In this study, we have shown that the administration of the IL-15 superagonist N-803 in both SIV-infected macaques and HIV-infected humanized mice induces a highly robust and persistent reversal of latency only in the setting of CD8+ lymphocyte depletion, thus suggesting a substantial role for CD8+ lymphocytes in suppressing the LRA effect of N-803. Notably, this previously undescribed role of CD8+ lymphocytes in maintaining virus latency was fully reproduced in an in vitro experimental approach that involved the co-culture of activated, unprimed CD8+ T cells with autologous, latently HIV-infected human primary CD4+ T cells. Here we use a novel approach to manipulate latently infected cells and have independently confirmed this method in the two best-validated and most widely used in vivo models for HIV cure interventions and then recapitulated the results in an in vitro experimental system of virus latency. In addition to this conceptual advance, this study defines a robust shock approach that could provide a key experimental system to directly compare and contrast the efficacy of different kill interventions in vivo, including those that may act in a CD8+ T-cell-independent manner (such as neutralizing antibodies, CD4 mimetics or immunotoxins). Further studies that aim to identify the specific molecular pathways used by CD8+ T cells to promote latency may allow the suppression of this activity, and therefore enable the full use of the virus-reactivating potential of N-803 or other LRAs in the clinical setting without depleting CD8+ lymphocytes.
Methods
Rhesus macaques, SIV-infection, ART, CD8 depletion and administration of N-803
This study was conducted using a cohort of 35 Indian-origin rhesus macaques housed at Yerkes National Primate Research Center (male and female, 2–3 years of age at the start of the study). All macaques were Mamu*B07− and Mamu*B17−; the following macaques were Mamu*A01+: 77_13, RFr15, 208_13, RPb16, RJt15, RHv15, RAu15, RNa16, RNz15, ROr15, RRb16, RSt15, RAk16, RUs15, RYe16, RVz15, RBn16 and RRn16. All procedures were approved by the Emory University Institutional Animal Care and Use Committee (IACUC) and animal care facilities are accredited by the US Department of Agriculture and the Association for Assessment and Accreditation of Laboratory Animal Care International.
Rhesus macaques were infected intravenously with 3,000 TCID50 of SIVmac239 (nef open). SIVmac239 stock was titrated in vitro for viral infectivity by standard end-point titration on CEMx174 cells. The TCID50 was calculated using a previously published method34. All macaques were put on a three-drug ART regimen at 8 weeks after SIV infection. Tenofovir disoproxil fumarate (5.1 mg kg−1 per day or tenofovir, 20 mg kg−1 per day) and emtricitabine (40 mg kg−1 per day) were both provided by Gilead Pharmaceuticals. Dolutegravir (2.5 mg kg−1 per day) was provided by ViiV Pharmaceuticals. Drugs were administered daily by subcutaneous injection.
After over 12 months of ART, macaques were assigned to intervention groups. Age, weight, sex, A01 status, peak post-infection viral load and time to suppression after ART were all controlled for when allocating macaques to intervention groups. Group sizes were determined using a power analysis based on previous studies. No blinding was performed in this study. One dose of the anti-CD8α-depleting antibody, MT807R1 (50 mg kg−1), was administered to 28 macaques. The initial 15 macaques that received the depleting antibody received the administration intravenously. Owing to safety concerns, the administration was changed to subcutaneous for the remaining 13 macaques. There was no observable effect of the different administration routes on the efficacy of depletion.
At the start of the intervention, 21 macaques received a dose of N-803 either in addition to CD8 depletion (n = 14) or as a single treatment (n = 7). N-803 was administered subcutaneously in a cycle of 100 μg kg−1 once a week for 4 consecutive weeks.
The study design included a subsequent four-dose administration of N-803 in seven macaques of groups 1 and 2 that was carried out at the time of CD8+ T cell reconstitution to potentially accelerate the recovery of these cells and improve their antiviral cytotoxic potential. As expected, this second cycle of N-803 induced a faster recovery of CD8+ T cells (data not shown) and was associated with an increase in T cell activation and proliferation that was similar to the increased activation and proliferation observed after the first N-803 cycle (data not shown). The late administration did not result in an increase in plasma viraemia.
Macaques were interrupted of ART 3 weeks after either the last dose of N-803 or 3 weeks after the reconstitution of CD8+ T cells (whichever occurred first). Plasma viral loads were monitored for about 6 months until necropsy was performed.
CD8 depletion in combination with N-803 treatment in SHIV-infected ART-treated macaques (pilot study)
An additional five Indian-origin rhesus macaques were included in this study as part of a follow-up pilot study using a SHIV model of infection. These macaques were also housed at the Yerkes National Primate Research Center and all procedures were approved by the Emory University IACUC. Macaques were infected intrarectally with high-dose SHIVSF162P3, administered as a 1:50 dilution of a 2,032 TCID50 per ml, 109 RNA copies per ml, 182 ng ml−1 P27 stock. All macaques were placed on the same tenofovir, emtricitabine and dolutegravir ART regimen 12 weeks after SHIV infection. After 6 months of ART, macaques received one dose of MT-807R1 at 50 mg kg−1 subcutaneously. N-803 was administered subcutaneously in a cycle of 100 μg kg−1 once a week for 4 consecutive weeks starting at the time of CD8 depletion.
Collection and processing of tissues from macaques
Blood, lymph nodes and rectal biopsies were collected longitudinally including at the time of necropsy and processed for further analyses as previously described18.
Immunophenotypes of macaques by flow cytometry
Multiparametric flow cytometry was performed according to a standard protocol on PBMCs and lymph node mononuclear cells using fluorescently labelled monoclonal antibodies cross-reactive in rhesus macaques. The following antibodies were used at 37 °C for 30 min: CCR5 APC (BD Biosciences, 560748, clone 3A9) and CCR7 FITC (BD Biosciences, 561271, clone 150503), in addition to LIVE/DEAD aqua viability dye (ThermoFisher, L35957) . The following antibodies were subsequently used at room temperature for 30 min: CD3 APC-Cy7 (BD Biosciences, 557757, clone SP34-2), CD4 BV650 (Biolegend, 317436, clone OKT4), CD8α BV711 (Biolegend, 301044, clone RPA-T8), pure CD8β (Thermo, 14-5273-82, clone SIDI8BEE) conjugated to Pe-Cy5 via a kit (Innova Biosciences, 760-0010), CD45RA Pe-Cy7 (BD Biosciences, 561216, clone 5H9), CD62L PE (BD Biosciences, 341012, clone SK11), CD95 BV605 (Biolegend, 305628, clone DX2), PD-1 BV421 (Biolegend, 329920, clone EH12.2H7), CD16 BV421 (BD Biosciences, 562874, clone 3G8), CD20 PE-Cy5 (BD Biosciences, 555624, clone 2H7), CD14 PE-Cy7 (BD Biosciences, 557742, clone M5E2), NKG2A (also known as CD159) PE (Beckman Coulter, IM3291U, clone Z199), CD28 PE-Cy5.5 or ECD (Beckman Coulter, B24027 and 6607111, respectively, clone CD28.2), CD56 FITC (BD Biosciences, 340723, clone NCAM16.2). Additional panels included CD69 Pe-Cy5 (BD Biosciences, 555532, clone FN50) and CD25 APC (BD Biosciences, 555434, clone M-A251). Cells stained for Ki-67 were fixed and permeabilized with Perm II kit (BD Biosciences) before staining at room temperature for 30 min with Ki-67 AF700 (BD Biosciences, 561277, clone B56).
All flow cytometry specimens were acquired on an LSR II (BD Biosciences) equipped with fluorescence-activated cell sorting (FACS) software (FACS Diva), and analysis of the acquired data was performed using FlowJo software (TreeStar).
Determination of plasma SIV RNA, and cell-associated RNA and DNA
For pre-intervention time points, quantitative PCR with reverse transcription (RT–qPCR) was performed to determine SIV plasma viral load as previously described23 with a sensitivity of 60 copies per ml. For the three time points before intervention and all post-intervention time points, plasma SIV gag RNA levels were measured high-sensitivity assay formats24,25,26. Quantification of total cell-associated SIVmac239 gag DNA was performed as previously described35. The number of gag DNA copies per 106 CD4+ T cells was calculated by dividing the number of gag DNA copies per 106 PBMCs by the percentage of CD3+CD8−CD4+ cells in the PBMC population. CD4+ T cells were isolated from PBMCs using a CD4+ T cell isolation kit (Miltenyi) and cell-associated RNA was measured as previously described36.
In situ RNA analysis and quantification
Viral RNA (vRNA) detection using RNAscope and quantitative image analysis was performed on formaldehyde-fixed, paraffin-embedded tissue sections (5 μm) as previously published37, with the following minor modifications: heat-induced epitope retrieval was performed by boiling slides in 1× target retrieval (322000; ACD) for 30 min, followed by incubation at 40 °C with a 1:10 dilution of protease III (322337; ACD) in 1× PBS for 20 min. Slides were incubated with the target probe SIVmac239 (312811; ACD) for 2 h at 40 °C and amplification was performed with RNAscope 2.5 HD Detection kits (322360; ACD) according to the manufacturer’s instructions, with 0.5× wash buffer (310091; ACD) used between steps. The resulting signal was detected with Warp Red chromogen (WR806M; Biocare Medical). Slides were counterstained with CAT haematoxylin (CATHE-GL; Biocare Medical), mounted with Clearmount (17885-15; EMS) until dry, after which a coverslip was added and sealed using Permount (SP15-100; Fisher Scientific). Slides were scanned at 40× magnification on an Aperio AT2 (Leica Biosystems). RNAscope images were analysed for the total number of vRNA+ cells per 105 total cells (quantitative) and the relative amount of vRNA present (semiquantitative) using the ISH module (v.2.2) within the Halo software (v.2.3.2089.27; Indica Labs). The relative amount of vRNA within a single cell was first estimated by quantifying the total area of the vRNA signal spot size (μm2). As the signal spot size is a function of several steps in the experimental procedures, module settings were established on concomitantly assayed, acutely infected SIV+ control slides. To estimate the signal spot size of a single vRNA molecule, we measured the signal area (minimum, mean and maximum) of more than 10 identifiable individual virions within B cell follicles, which corresponds to two copies of vRNA, and multiplied this by 0.5. We set the vRNA minimum signal spot size within the analysis module to exclude detection of a single vRNA molecule and/or integrated viral DNA. Relative vRNA copy numbers that were present within vRNA+ cells were calculated as (signal spot size within vRNA+ cell (μm2))/(0.5 × mean signal size for a virion).
FACS of live cells from macaques
Mononuclear cells isolated from blood were stained with LIVE/DEAD aqua viability dye (ThermoFisher, L35957), CD3 AF700 (BD Biosciences, 557917, clone SP34-2), CD4 BV650 (Biolegend, 317436, clone OKT4), CD8 APC-Cy7 (BD Biosciences, 557834, clone SK1), CD14 PB (Biolegend, 301828, clone M5E2), CD20 PB (Biolegend, 302328, clone 2H7) and CD16 BV421 (Biolegend, 302032, clone 3G8) for 30 min at room temperature. Aliquots of 50,000 CD4+ T cells (live CD3+CD20−CD14−CD16−CD8−CD4+) and CD8+ T cells (live CD3+CD20−CD14−CD16−CD4−CD8+) were then sorted using a FACS Aria II (BD Biosciences). Mononuclear cells were separately stained with Live/Dead stain, CD3 AF700 (BD Biosciences, 557917, clone SP34-2), CD4 BV650 (Biolegend, 317436, clone OKT4), CD8 APC-Cy7 (BD Biosciences, 557834, clone SK1), CD14 PB (Biolegend, 301828, clone M5E2), CD20 PB (Biolegend, 302328, clone 2H7) and NKG2A PE (Beckman Coulter, IM3291U, clone Z199) to sort aliquots of 50,000 NK cells (live CD3−CD20−CD14−CD4−CD8+NKG2A+).
RNA sequencing and data analysis
Bulk CD4+ T cells were sorted from fresh PBMCs before intervention, and on day 3, week 2 and week 4 post-intervention. In brief, RNA from sorted cells was collected and extracted, and the DNA was digested. Libraries were prepared and normalized, pooled and clustered on a flow cell for sequencing. RNA-sequencing data were aligned to the MacaM v.7.8 assembly of the Indian rhesus macaque genome. To identify pathways that were differentially modulated, gene-set enrichment analysis38 was performed on the ranked transcript lists using 1,000 phenotype permutations and random seeding. Gene sets used included the MSigDB H (hallmark) gene sets39. Normalized enrichment scores for select upregulated gene sets were used, for which the normalization was group-specific. A normalized enrichment score cut-off of greater than 1.35 for upregulated gene sets with a false-discovery rate of less than 0.2 was used, as per the recommendations for gene-set enrichment analysis. For the generation for heat maps, colours represent log2-transformed library-size-normalized read counts scaled to unit variance across transcript vectors and normalized to the baseline median sample value of each transcript.
Single-genome PCR amplification of SIVmac239 env sequences
cDNA synthesis and 384-well single-genome PCR amplification (SGA) were performed using an approach similar to previously described methods40,41,42. In brief, RNA was extracted from cryopreserved plasma samples using the QIAmp viral RNA kit (Qiagen, 52906) and reverse transcription was performed using the SuperScript III kit (Invitrogen, 18080-044) with reverse primer SM-ER1 (5′-CTATCACTGTAATAAATCCCTTCCAGTCCC-3′). cDNA was diluted to result in less than 30% positive wells for SGA. First-round PCR was performed in a 15-μl volume using the Phusion Hotstart II High Fidelity DNA Polymerase (Thermo Scientific, F537S) with forward primer H2SM-EF1 (5′-CCCTTGAAGGMGCMRGAGAGCTCATTA-3′) and SM-ER1. Cycling conditions were as follows: 98 °C for 2 min; 10 cycles of 95 °C for 15 s, 54 °C for 60 s and 68 °C for 4 min; 25 cycles of 95 °C for 15 s, 54 °C for 60 s and 68 °C for 4 min, adding 5 s to the extension per cycle; 72 °C for 30 min; and 4 °C hold. Second-round PCR was performed with the same enzyme in a 10-μl volume with 1 μl of the first-round PCR reaction as template and primers H2SM-EF2 (5′-CACCTAAAAARTGYTGCTAYCATTGCCAG-3′) and SM-ER2 (5′-ATAAAATGAGACATGTCTATTGCCAATTTG-3′). Cycling conditions were as follows: 95 °C for 2 min; 30 cycles of 95 °C for 15 s, 54 °C for 60 s and 72 °C for 2.5 min; 72 °C for 10 min; and 4 °C hold. PCR amplicons were purified using a Qiaquick PCR Purification Kit (Qiagen 28106).
Sequencing of env amplicons
On average, 26 SGA PCR amplicons per time point (range, 20–30) were sequenced by Eurofins Genomic DNA Sanger sequencing using the following primers: SIVmac251seqF1 5′-GGGATATGTTATGAGCAGTCACG-3′; SIVmac251seqF2 5′-ATCCAAGAGTCTTGTGACAAGC-3′; SIVmac251seqF3 5′-AAGAGAGGGAGACCTCACG-3′; SIVmac251seqF4 5′-AGGCCAGTGTTCTCTTCC-3′; SIVmac251seqR1 5′-CTTGTTCCAAGCCTGTGC-3′; SIVmac251seqR2 5′-CCTCTGCAATTTGTCCACATG-3′; SIVmac251seqR3 5′-TCCAAGAAGTCAACCTTTCGC-3′; SIVmac251seqR4 5′-AGCTGGGTTTCTCCATGG-3′18. Sequencher v.5.1 was used to generate nucleotide sequence contigs and sequences with mixed peaks in the chromatogram were excluded from further analysis.
Sequence analysis of SIVmac239 contigs
Geneious v.9.1.7 was used to translate nucleotide sequences into amino acids and generate alignments. Amino acid alignments were exported from Geneious in FASTA format and used to generate highlighter plots to visualize amino acid mismatches (http://www.hiv.lanl.gov/content/sequence/HIGHLIGHT/highlighter_top.html). Phylogenetic neighbour-joining consensus trees (Jukes–Cantor, resampling with 100 bootstrap replicates) were created in Geneious using amino acid alignments, and were exported in NEXUS format into Figtree v.1.4.4 for further modification (A. Rambaut, http://tree.bio.ed.ac.uk/). Phylogenetic trees were presented as unrooted or were rooted on the midpoint. Bootstrap values that were higher than 80% are considered significant. Pairwise differences between the infecting SIVmac239 clone and each SGA-derived Env amino acid sequence were determined using Geneious.
Experimental design of the BLT humanized mouse model
The BLT humanized model of HIV infection was used to determine the efficacy of CD8 depletion alone, CD8 depletion in combination with N-803 or N-803 alone as a LRA. BLT mice (15–19 weeks after humanization surgery) were exposed to HIV-1JR-CSF intravenously. ART was initiated 4–5 weeks later. Viraemia was durably suppressed by ART for 4 weeks. A single dose of N-803, CD8-depleting antibody or the combination of N-803 and CD8-depleting antibody was administered to HIV-infected and suppressed mice as indicated below. HIV RNA induction was measured on days 4 and 7.
Generation of BLT humanized mice
BLT humanized mice were prepared as previously reported43,44,45,46. In brief, a 1–2-mm piece of human liver tissue was sandwiched between two pieces of autologous thymus tissue (Advanced Bioscience Resources) under the kidney capsule of sublethally irradiated (200 cGy) 12–15-week-old NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG; The Jackson Laboratory) mice. After implantation, mice were transplanted intravenously with haematopoietic CD34+ stem cells isolated from autologous human liver tissue. Human immune cell reconstitution was monitored in the peripheral blood of BLT mice by flow cytometry every 3–4 weeks as previously described43,44,45,46. Mice were maintained under specific-pathogen-free conditions by the Division of Comparative Medicine at the University of North Carolina, Chapel Hill. Mouse experiments were conducted in accordance with NIH guidelines for the housing and care of laboratory animals and in accordance with protocols reviewed and approved by the IACUC at the University of North Carolina, Chapel Hill.
Production of HIV and infection of BLT mice
Stocks of HIV-1JR-CSF were prepared as previously reported43,44,45. The proviral clone was transfected into human embryonic kidney (HEK)293T cells using Lipofectamine 2000 (Invitrogen, 11668030) following the manufacturer’s protocol. Viral supernatants were collected 48 h after transfection and titred onto TZM-bl indicator cells in triplicate to determine the tissue-culture infectious units (TCIU) per ml. At least two different titre determinations were performed for each virus stock. BLT mice were exposed to 3 × 104 TCIU HIV-1JR-CSF by tail-vein injection.
Analysis of HIV infection in BLT mice
The peripheral-blood plasma viral load was monitored longitudinally by quantitative real-time PCR using a TaqMan RNA-to-Ct 1-step kit (Applied Biosystems, 4392656). The sequences of the forward and reverse primers and the TaqMan probe for PCR amplification and detection of HIV gag RNA were: 5′-CATGTTTTCAGCATTATCAGAAGGA-3′, 5′-TGCTTGATGTCCCCCCACT-3′ and 5′-FAM-CCACCCCACAAGATTTAAACACCATGCTAAQ-3′, respectively. Known quantities of HIV gag RNA were run in parallel, creating a standard curve for HIV gag and sample RNA levels were quantified by extrapolation from the standard curve. All samples were run and analysed on an ABI 7500 Fast Real-time PCR System (Applied Biosystems).
HIV DNA levels were measured in tissue cells collected at the time of collection and cryopreserved in cryopreservation medium (10% DMSO:90% fetal bovine serum). Cells were thawed slowly, counted by Trypan exclusion, aliquoted and pelleted. DNA was extracted from cell pellets using the QIAamp DNA Blood Mini Kit (Qiagen) according to the manufacturer’s instructions. RT–PCR was performed with a TaqMan Fast Universal PCR Master Mix (Applied Biosystems). The sequences of the forward primer, reverse primer and the TaqMan probe for amplification and detection of HIV gag DNA were: 5′-CATGTTTTCAGCATTATCAGAAGGA-3′, 5′-TGCTTGATGTCCCCCCACT-3′ and 5′-FAM CCACCCCACAAGATTTAAACACCATGCTAA Q -3′, respectively. As a control, Homo sapiens haemoglobin subunit gamma-2 (HBG2) was included to quantify the presence of human DNA in each sample. The sequences of the forward primer, reverse primer and the TaqMan probe for amplification and detection of HBG2 were 5′-CGCTTCTGGAACGTCTGAGATT-3′, 5′-CCTTGTCCTCCTCTGTGAAATGA-3′ and 5′-FAM-TCAATAAGCTCCTAGTCCAGAC-3′, respectively. All samples were run and analysed on an ABI 7500 Fast Real-time PCR System (Applied Biosystems).
ART administration in BLT mice
ART was administered to BLT humanized mice as previously described47,48,49, via 12.7-mm pellets of irradiated Teklad chow containing emtricitabine (1,500 mg kg−1), tenofovir disoproxil fumarate (1,560 mg kg−1) and raltegravir (600 mg kg−1) (Research Diets).
N-803 and MT807R1 administration in BLT mice
N-803 (0.2 mg kg−1 in PBS) and the control vehicle (PBS) were administered to mice intravenously in a total volume of 200 μl. MT807R1 (3 mg kg−1 in PBS) and the control vehicle (PBS) were also administered intravenously in a total volume of 200 μl.
Immunophenotypic analysis of BLT mice
Immunophenotyping was performed on peripheral-blood samples longitudinally and at the time of collection on blood and mononuclear cells isolated from the tissues of BLT mice. All flow cytometry data were collected on a BD FACSCanto instrument using BD FACSDiva software (v.6.1.3) and data were analysed with FlowJo Software (v.10.5.0). Antibodies for the analysis of human immune cell levels included: CD45 APC (BD 555485, clone HI30), CD3 FITC (BD Biosciences, 555339, clone HIT3a), CD4 APC-Cy7 (BD Biosciences, 560158, clone RPA-T4), CD33 PE (BD Biosciences, 340679, clone P67.6); CD19 PE-Cy7 (BD Biosciences, 557835, clone SJ25C1) and CD8 PerCP (BD Biosciences, 347314, clone SK1). Flow cytometry gating for the expression of lineage-specific antigens on human leukocytes was performed as follows. Step 1, forward and side scatter were used to set a live-cell gate. Step 2, live cells were then analysed for the expression of the human pan-leukocyte marker CD45. Step 3, human leukocytes were then analysed for human CD3+ T cells and CD19+ B cells. Step 4, T cells were analysed for human CD4 and CD8 expression. The following flow cytometry antibody panel was also used to analyse HLA-DR, CD38 and CD25 expression: CD3 BV421 (BD Biosciences, 562426, clone UCTH1), CD4 BV605 (BD Biosciences, 562658, clone RPA-T4), CD45 FITC (BD Biosciences, 347463, clone 2D1), HLA-DR PerCP (BD Biosciences, 347364, clone L243), CD69 PE (BD Biosciences, 555531, clone FN50), anti-CD38 PE-Cy7 (BD Biosciences, 335790, clone HB7), CD25 APC (BD Biosciences, 340938, clone 2A3), CD8 APC-Cy7 (BD Biosciences, 557834, clone SK1) and AQUA (ThermoFisher, L35957). Flow cytometry gating was performed as follows. Step 1, forward-scatter height and forward-scatter area were used to eliminate doublets. Step 2, side-scatter area and forward-scatter area were used to distinguish leukocytes based on morphology. Step 3, the viability dye AQUA was use to discriminate live cells from dead cells. Step 4, live cells were analysed for the expression of the human pan-leukocyte marker CD45. Step 5, human leukocytes were then assessed for human CD3 expression to identify T cells. Step 6, T cells were evaluated for the expression of human CD4 and CD8. Step 7, human CD4+ or CD8+ T cells were examined for expression of HLA-DR and/or CD38, or CD25. Gates were set with fluorescence-minus-one controls. Non-specific binding was assessed with isotype controls.
Experimental design CD8 in vitro suppression assay
In vitro latently infected memory CD4+ T cells were generated using the LARA method as previously described32 with the following modifications. On day 0, after PBMCs were isolated from buffy coats of HIV-naive individuals (New York Blood Center) using SepMate density-gradient centrifugation (StemCell Technologies, 85460), a portion of PBMCs from each HIV-naive donor was cryopreserved in fetal bovine serum (VWR Life Science Seradigm, 97068-085) and 10% DMSO, and stored in liquid nitrogen. On day 8, PBMCs were thawed and rested overnight in RPMI 1640 medium (Fisher Scientific, SH3002701.01) supplemented with 10% fetal bovine serum, 1% penicillin–streptomycin (Corning, 45000-650), and 1% HEPES (Gibco, 15630080); cRPMI) before total CD8+ T-cell-positive enrichment on day 9 (Miltenyi, 130-045-201). CD8+ T cells were stimulated with anti-CD3/CD28 beads (Dynabeads, 11141D) at a 1:1 ratio plus 30 U ml−1 IL-2 (R&D Systems, 202IL050CF) for 3 days. On day 12 of LARA, CD4+ and CD8+ were prepared for co-culture. HIV latently infected memory CD4+ T cells were washed, counted and plated for latency reversal in cRPMI in the presence of ART (100 nM efavirenz, 200 nM raltegravir and 5 μM saquinavir). Activated total CD8+ T cells were removed from the anti-CD3/CD28 beads, washed and resuspended in cRPMI plus ART. CD4+ and CD8+ T cells were co-cultured at a 1:1 or 1:5 ratio at a final density of 1 × 106 cells per ml. CD4+ monocultures were also maintained in parallel. Mono- and co-cultures were then left unstimulated, TCR-activated with 1 μg ml−1 plate-bound OKT3 and 1 μg ml−1 soluble CD28 (Biolegend, 302933), or treated with 14 nM N-803 (provided by NantKwest) or 500 ng ml−1 IL-15 (R&D Systems, 247-ILB). Cells were collected after 72 h and analysed by flow cytometry and qPCR.
Multicolour flow analysis of cell-surface and intracellular marker expression was performed with a BD FACSymphony flow cytometer. Between 200,000 and 600,000 events were acquired for each sample using the live-cell gate. The data were analysed with FlowJo (v.10).
Antibodies used in in vitro suppression assays include: CD3 AF700 (BD Biosciences, 557943, clone UCHT1), CD8 BUV737 (BD Biosciences, 564629, clone SK1), HIV-1 core antigen-FITC (Beckman Coulter, 6604665, clone KC57), CD4 BV421 (BD Horizon, 565997, clone SK3), CD45RA APC-eFluor780 (Invitrogen, 47045842, clone HI100), CD27 BV650 (Biolegend, 302828, clone O323), CCR7 Pe-Cy7 (BD Pharmingen, 557648, clone 3D12) and Fixable Viability Dye eFluor 506 (Invitrogen eBioscience, 65-0866-18).
Statistics and reproducibility
Statistical analyses, including two-way Kruskal–Wallis tests, two-way Friedman tests, one-way ANOVA and Spearman r correlations, were performed using GraphPad Prism v.7.0 or v.8.0. Data are mean ± s.e.m. as indicated.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Illumina sequencing reads for RNA-sequencing experiments were submitted to the NCBI SRA repository (accession number SRP188630). RNA-sequencing datasets were submitted to the NCBI GEO repository (accession number GSE128415). env nucleotide sequences have been deposited in the GenBank (accession numbers MK922999–MK923550).
Change history
04 February 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41586-020-2002-9
References
Chun, T.-W. et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl Acad. Sci. USA 94, 13193–13197 (1997).
Finzi, D. et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300 (1997).
Wong, J. K. et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278, 1291–1295 (1997).
Siliciano, J. D. et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9, 727–728 (2003).
UNAIDS. UNAIDS Data 2017. (2017).
The International AIDS Society Scientific Working Group on HIV Cure. Towards an HIV cure: a global scientific strategy. Nat. Rev. Immunol. 12, 607–614 (2012).
Archin, N. M. et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487, 482–485 (2012).
Archin, N. M. et al. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. J. Infect. Dis. 210, 728–735 (2014).
Rasmussen, T. A. et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV 1, e13–e21 (2014).
Søgaard, O. S. et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. 11, e1005142 (2015).
Elliott, J. H. et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 10, e1004473 (2014).
Elliott, J. H. et al. Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study. Lancet HIV 2, e520–e529 (2015).
Lim, S.-Y. et al. TLR7 agonists induce transient viremia and reduce the viral reservoir in SIV-infected rhesus macaques on antiretroviral therapy. Sci. Transl. Med. 10, eaao4521 (2018).
Del Prete, G. Q. et al. TLR7 agonist administration to SIV-infected macaques receiving early initiated cART does not induce plasma viremia. JCI Insight 4, e127717 (2019).
Bekerman, E. et al. PD-1 blockade and TLR7 activation lack therapeutic benefit in chronic simian immunodeficiency virus-infected macaques on antiretroviral therapy. Antimicrob. Agents Chemother. 63, e01163-19 (2019).
Borducchi, E. N. et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature 563, 360–364 (2018).
Nixon, C. C., Mavigner, M., Silvestri, G. & Garcia, J. V. In vivo models of human immunodeficiency virus persistence and cure strategies. J. Infect. Dis. 215, S142–S151 (2017).
Cartwright, E. K. et al. CD8+ lymphocytes are required for maintaining viral suppression in SIV-infected macaques treated with short-term antiretroviral therapy. Immunity 45, 656–668 (2016).
Xu, W. et al. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: interleukin-15 receptor αSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res. 73, 3075–3086 (2013).
Han, K. P. et al. IL-15:IL-15 receptor alpha superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 56, 804–810 (2011).
Rhode, P. R. et al. Comparison of the superagonist complex, ALT-803, to IL15 as cancer immunotherapeutics in animal models. Cancer Immunol. Res. 4, 49–60 (2016).
Jones, R. B. et al. A subset of latency-reversing agents expose HIV-infected resting CD4+ T-cells to recognition by cytotoxic T-lymphocytes. PLoS Pathog. 12, e1005545 (2016).
Hofmann-Lehmann, R. et al. Sensitive and robust one-tube real-time reverse transcriptase-polymerase chain reaction to quantify SIV RNA load: comparison of one- versus two-enzyme systems. AIDS Res. Hum. Retroviruses 16, 1247–1257 (2010).
Del Prete, G. Q. et al. Effect of suberoylanilide hydroxamic acid (SAHA) administration on the residual virus pool in a model of combination antiretroviral therapy-mediated suppression in SIVmac239-infected Indian rhesus macaques. Antimicrob. Agents Chemother. 58, 6790–6806 (2014).
Hansen, S. G. et al. Immune clearance of highly pathogenic SIV infection. Nature 502, 100–104 (2013).
Li, H. et al. Envelope residue 375 substitutions in simian–human immunodeficiency viruses enhance CD4 binding and replication in rhesus macaques. Proc. Natl Acad. Sci. USA 113, E3413–E3422 (2016).
Dornadula, G. et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. J. Am. Med. Assoc. 282, 1627–1632 (1999).
Maldarelli, F. et al. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 3, e46 (2007).
Chun, T. W. et al. Relationship between residual plasma viremia and the size of HIV proviral DNA reservoirs in infected individuals receiving effective antiretroviral therapy. J. Infect. Dis. 204, 135–138 (2011).
Chowdhury, A. et al. Differential impact of in vivo CD8+ T lymphocyte depletion in controller versus progressor simian immunodeficiency virus-infected macaques. J. Virol. 89, 8677–8686 (2015).
Spivak, A. M. et al. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clin. Infect. Dis. 58, 883–890 (2014).
Kulpa, D. A. et al. Differentiation to an effector memory phenotype potentiates HIV-l latency reversal in CD4+ T cells. J. Virol. 93, e00969-19 (2019).
Okoye, A. A. et al. Early antiretroviral therapy limits SIV reservoir establishment to delay or prevent post-treatment viral rebound. Nat. Med. 24, 1430–1440 (2018).
Reed, L. J. & Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27, 493–497 (1938).
Chahroudi, A. et al. Target cell availability, rather than breast milk factors, dictates mother-to-infant transmission of SIV in sooty mangabeys and rhesus macaques. PLoS Pathog. 10, e1003958 (2014).
Kumar, N. A. et al. Antibody-mediated CD4 depletion induces homeostatic CD4+ T cell proliferation without detectable virus reactivation in antiretroviral therapy-treated simian immunodeficiency virus-infected macaques. J. Virol. 92, e01235-18 (2018).
Deleage, C. et al. Defining HIV and SIV reservoirs in lymphoid tissues. Pathog. Immun. 1, 68–106 (2016).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Smith, S. A. et al. Diversification in the HIV-1 envelope hyper-variable domains V2, V4, and V5 and higher probability of transmitted/founder envelope glycosylation favor the development of heterologous neutralization breadth. PLoS Pathog. 12, e1005989 (2016).
Smith, S. A. et al. Signatures in simian immunodeficiency virus SIVsmE660 envelope gp120 are associated with mucosal transmission but not vaccination breakthrough in rhesus macaques. J. Virol. 90, 1880–1887 (2016).
Burton, S. L. et al. Breakthrough of SIV strain smE660 challenge in SIV strain mac239-vaccinated rhesus macaques despite potent autologous neutralizing antibody responses. Proc. Natl Acad. Sci. USA 112, 10780–10785 (2015).
Denton, P. W. et al. Antiretroviral pre-exposure prophylaxis prevents vaginal transmission of HIV-1 in humanized BLT mice. PLoS Medicine 5, e16 (2008).
Denton, P. W. et al. Generation of HIV latency in humanized BLT mice. J. Virol. 86, 630–634 (2011).
Denton, P. W. et al. One percent tenofovir applied topically to humanized BLT mice and used according to the CAPRISA 004 experimental design demonstrates partial protection from vaginal HIV infection, validating the BLT model for evaluation of new microbicide candidates. J. Virol. 85, 7582–7593 (2011).
Melkus, M. W. et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat. Med. 12, 1316–1322 (2006).
Honeycutt, J. B. et al. T cells establish and maintain CNS viral infection in HIV-infected humanized mice. J. Clin. Invest. 128, 2862–2876 (2018).
Kessing, C. F. et al. In vivo suppression of HIV rebound by didehydro-cortistatin A, a “block-and-lock” strategy for HIV-1 treatment. Cell Rep. 21, 600–611 (2017).
Tsai, P. et al. In vivo analysis of the effect of panobinostat on cell-associated HIV RNA and DNA levels and latent HIV infection. Retrovirology 13, 36 (2016).
Acknowledgements
This work was supported by NIH grants R01-AI125064 and UM1-AI124436 (to G.S. and A.C.); R01-AI143414 (to G.S. and D.A.K.); R01-MH108179 and R01-AI111899 (to J.V.G.); UM1-AI126619 (to D.M.M.); R01-AI123010 (to A.W.); P30 AI050409 (Emory Center for AIDS Research); P51 OD011092 (Oregon National Primate Research Center base grant); the National Institutes of Health’s Office of the Director, Office of Research Infrastructure Programs P51OD011132 (Yerkes National Primate Research Center base grant); and supported in part by federal funds from the National Cancer Institute, National Institutes of Health, under contracts HHSN261200800001E and 75N91019D00024 (J.D.L.). We thank B. Jones, S. O’Connor and J. Sacha for discussions; S. Ehnert, S. Jean and all of the animal care and veterinary staff at the Yerkes National Primate Research Center; B. Cervasi and K. Gill at the Emory University Flow Cytometry Core; Emory and Pediatric’s/Winship Flow Cytometry Core; the Translational Virology and Reservoir Cores of the Emory CFAR, the Emory Nonhuman Primate Genomics Core for RNA sequencing and analysis and the Quantitative Molecular Diagnostics Core of the AIDS and Cancer Virus Program, Frederick National Laboratory, for high-sensitivity plasma viral-load testing; NantKwest for providing N-803, K. Reimann and the NHP Reagent Resources for the MT807R1 antibody, R. Geleziunas and Gilead Pharmaceuticals for providing tenofovir and emtricitabine, D. Hazuda and B. Howell from Merck for providing raltegravir and J. Demarest and ViiV Healthcare for providing dolutegravir for this study.
Author information
Authors and Affiliations
Contributions
J.B.M., A.C., M.P. and G.S. designed the experiments. J.B.M., M.M., E.W. and D.G.C. performed the experiments. S.A.S. performed single-genome PCR and sequencing of SIV RNA. S.A.S. and C.A.D. performed the sequence-based analyses and wrote the relevant portions of the manuscript. J.D.L. performed ultrasensitive viral-load analyses. B.C. performed FACS of live cells. T.H.V. measured viral load, and cell-associated DNA and RNA. J.D.E. and K.B.-S. performed RNAscope analysis. S.E.B., G.K.T. and H.W. performed RNA-sequencing analysis. M.K., C.R.A.-S. and W.O.T. constructed BLT humanized mice. M.K. performed the HIV infection and ART suppression of BLT humanized mice. R.A.S. performed the viral load measurements, the isolation of nucleic acids and the analysis of tissue RNA levels for BLT humanized mice. C.R.A.-S. and W.O.T. designed and performed the N-803 and CD8 T cell depletion experiments in BLT humanized mice and analysed the data. A.W. supervised the data collection, analysis, figure preparation and reporting of all samples from the BLT humanized mice. J.V.G. designed, coordinated and supervised all of the BLT experimental work. C.R.A.-S., J.V.G., A.W. and W.O.T. wrote and revised the BLT humanized mice portions of the manuscript. L.F. and D.A.K. performed the in vitro studies and wrote the relevant portions of the manuscript. D.M.M., J.T.S. and J.H.L. provided technical support. J.B.M., A.C. and G.S. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Mathias Lichterfeld and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 MT807R1 effectively depletes CD8+ T cells in peripheral blood, lymph node and rectum of macaques in addition to depleting NK cells from the blood at day 7.
The percentage of CD8+ cells in the CD3+ population 7 days after depletion was compared to the levels before depletion. a–c, Sample flow cytometry shows the absence of CD8β+ cells as part of the CD3+ T cell population after depletion in the peripheral blood (a), rectum (b) and lymph nodes (c) of macaques. Similar results were found across all CD8-depleted macaques (n = 28 biologically independent samples). d, The percentage of CD8β+ cells compared to pre-depletion baseline was calculated in all CD8-depleted macaques (treated with or without N-803, n = 28 macaques) across blood and tissue samples (no differences in CD8+ T cell depletion were observed between groups 2 and 3 on day 7). A two-sided Friedman test was used to calculate statistically significant changes compared to baseline across tissues. e, Depletion of NK cells in the peripheral blood was assessed 1 week after CD8 depletion alone (n = 14 macaques) compared to baseline. Statistical significance was calculated using Wilcoxon signed-rank test. Data are mean ± s.e.m.
Extended Data Fig. 2 Phenotypic changes to CD4+ T cells after intervention.
Longitudinal flow cytometry analysis after treatment with only N-803 (green, n = 7 macaques), only CD8 depletion (blue, n = 14 macaques) and after CD8 depletion and treatment with N-803 (red, n = 14 macaques). a, CD4+ T cell frequency. b–f, Percentage of naive (b), stem cell memory (SCM) (c), central memory (CM) (d), transitional memory (TM) (e) and effector memory (EM) (f) CD4+ T cells. g–l, Percentage of bulk CD4+ T cells that express PD-1 (g), CD25 (h), CD69 (i), HLA-DR (j), CCR5 (k) and both CCR5 and Ki-67 (l). m–v, CCR5 (m–q) and Ki-67 (r–v) expression in different subsets of CD4+ T cells. Data are mean ± s.e.m. Two-sided Kruskal–Wallis tests were used to compare values after the intervention to the pre-intervention baseline and approximate P-value summaries are provided.
Extended Data Fig. 3 SIV-associated genes and IL-15 subunit genes show a transient change in expression after treatment with N-803 alone.
RNA was extracted from sorted peripheral bulk CD4+ T cells (CD3+CD4+CD8−CD20−CD14−) (left), bulk CD8+ T cells (CD3+CD4−CD8+CD20−CD14−) (middle) and NK cells (CD3−CD20−CD14−NKG2A+) (right) and libraries were prepared, normalized, pooled and clustered on flow cells for sequencing. RNA-sequencing data were aligned to the MacaM v.7.8 assembly of the Indian rhesus macaque genome. Transcripts were analysed for alignment against a custom gene set with SIV host restriction factors, PPIA (capsid folding protein), SIV receptors, SIV receptor agonists, NF-κB subunits (involved in mediating the transcription of long-terminal repeat regions), IL-15 receptor subunits and NFAT subunits. D, day; W, week.
Extended Data Fig. 4 Quantification of levels of cell-associated SIV RNA in peripheral CD4+ T cells before and after interventions.
a–c, Changes in expression of SIV RNA in relation to the number of copies of CD4 after intervention with N-803 alone (n = 7 macaques) (a), CD8 depletion alone (n = 7 macaques) (b) or CD8 depletion combined with N-803 (n = 7 macaques) (c). Data are mean ± s.e.m. Two-sided Wilcoxon signed-rank tests were used to compare values after the intervention to the pre-intervention baseline.
Extended Data Fig. 5 Level of virus reactivation correlated with the absence of CD8+ T cells.
a, b, Correlation between CD8+ T cell counts and viral load (SIV RNA copies per ml of plasma) on day 0, day 3, and weekly up to week 6 after interventions. a, CD8 depletion alone (n = 103 samples from 14 macaques). b, CD8 depletion combined with N-803 treatment (n = 112 samples from 14 macaques). c, d, The area under the curve (AUC) and the average pre-intervention viral load after CD8 depletion alone (c; n = 14 macaques) or CD8 depletion with N-803 treatment (d; n = 14 macaques). Correlation coefficients are calculated using the Spearman’s rank-order correlation (two-tailed, no adjustments). e–g, Longitudinal viral loads (top) and CD8+ T cell counts (bottom) after N-803 treatment alone (e; n = 7 macaques), CD8 depletion alone (f; n = 14 macaques) or CD8 depletion combined with N-803 treatment (g; n = 14 macaques). Colour keys along the bottom indicate animal IDs.
Extended Data Fig. 6 HIV DNA, HIV RNA and human T cell activation levels in HIV-infected, ART-suppressed BLT humanized mice treated with N-803, CD8 depletion alone or combined CD8 depletion with N-803 .
a, b, HIV-infected, ART-suppressed BLT humanized mice were treated with N-803 (green, n = 4 BLT humanized mice), CD8 depletion alone (blue, n = 4), or treated with CD8 depletion with N-803 (red, n = 4). After 7 days, total HIV DNA (a) and cell-associated HIV RNA (b) were extracted from mononuclear cells isolated from the spleen, human-derived thymus (huThy) and lymph node (LN; HIV RNA only). c–f, Percentages of HLA-DR+, CD38+, CD25+ or HLA-DR+CD38+ cells were measured in human CD4+ (c, e) or CD8+ (d, f) T cells isolated from the spleen (c, d) or human-derived thymus (e, f) of HIV-infected, ART-suppressed BLT humanized mice 7 days after treatment with N-803 (green, n = 4), CD8 depletion alone (blue, n = 4) or CD8 depletion combined with N-803 treatment (red, n = 4). Treatment groups were compared using a two-tailed Student’s t-test (a) or a Kruskal–Wallis test with a false-discovery rate correction (b–f). Data are mean ± s.e.m.
Extended Data Fig. 7 Phylogenetic trees of longitudinal SGA-derived Env amino acid sequences.
Phylogenetic trees were generated for six macaques that received CD8 depletion with N-803 using Env sequences from the peak viral load (red), pre-ART (blue) and reactivation (green) time points. The Env sequence of the SIVmac239 clone used for infection is included in each tree (black). The horizontal bar below each tree indicates the genetic distance. Sequence clusters that are supported by bootstraps greater than 80% are indicated by an asterisk. Env sequences that contain a stop codon are indicated by an arrow.
Extended Data Fig. 8 Longitudinal Env amino acid divergence from the input virus and relationship with viral load in the plasma.
The number of amino acid differences between the infecting viral clone SIVmac239 and each SGA amplicon was determined using Geneious. a, Violin plots show the frequency distribution of the number of amino acid differences between sequences at each time point in each macaque. The solid line indicates the median number of amino acid differences for each individual Env sequence; the dotted lines indicate the quartiles. Peak viral load (VL) (red), pre-ART (blue) and reactivation (green) time points are shown. The animal ID and the three time points are indicated on the x axis. Statistical differences between time points for each macaque were determined by performing multiple comparisons using a Kruskal–Wallis test with Dunn’s correction. b, The average number of sequence differences for each macaque at the reactivation time point is plotted on the y axis, and the corresponding plasma viral loads are plotted on the x axis on a log10 scale. Correlation coefficients are calculated using the Spearman’s rank-order correlation (two-tailed, no adjustments).
Extended Data Fig. 9 Highlighter plots of longitudinal SGA-derived Env amino acid sequences.
Highlighter plots were generated for six representative macaques that were depleted of CD8 and treated with N-803 using Env sequences from peak VL (red box), pre-ART (blue box) and reactivation (green box) time points. The Env sequence of the SIVmac239 clone used for infection is included as the master (reference) sequence in each plot. The position of N-linked glycosylation sites on the master sequence are indicated by pink circles. Each tick represents an amino acid difference from the master sequence, as indicated in the key. Blue diamonds indicate the loss of an N-linked glycosylation site.
Supplementary information
Rights and permissions
About this article

Cite this article
McBrien, J.B., Mavigner, M., Franchitti, L. et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature 578, 154–159 (2020). https://doi.org/10.1038/s41586-020-1946-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-1946-0
- Springer Nature Limited
This article is cited by
-
In situ analysis of neuronal injury and neuroinflammation during HIV-1 infection
Retrovirology (2024)
-
Suppression of viral rebound by a Rev-dependent lentiviral particle in SIV-infected rhesus macaques
Gene Therapy (2024)
-
A germ-free humanized mouse model shows the contribution of resident microbiota to human-specific pathogen infection
Nature Biotechnology (2024)
-
Multimeric immunotherapeutic complexes activating natural killer cells towards HIV-1 cure
Journal of Translational Medicine (2023)
-
Trispecific antibody targeting HIV-1 and T cells activates and eliminates latently-infected cells in HIV/SHIV infections
Nature Communications (2023)