

Abstract
Isoprenoids are vital for all organisms, in which they maintain membrane stability and support core functions such as respiration1. IspH, an enzyme in the methyl erythritol phosphate pathway of isoprenoid synthesis, is essential for Gram-negative bacteria, mycobacteria and apicomplexans2,3. Its substrate, (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate (HMBPP), is not produced in metazoans, and in humans and other primates it activates cytotoxic Vγ9Vδ2 T cells at extremely low concentrations4,5,6. Here we describe a class of IspH inhibitors and refine their potency to nanomolar levels through structure-guided analogue design. After modification of these compounds into prodrugs for delivery into bacteria, we show that they kill clinical isolates of several multidrug-resistant bacteria—including those from the genera Acinetobacter, Pseudomonas, Klebsiella, Enterobacter, Vibrio, Shigella, Salmonella, Yersinia, Mycobacterium and Bacillus—yet are relatively non-toxic to mammalian cells. Proteomic analysis reveals that bacteria treated with these prodrugs resemble those after conditional IspH knockdown. Notably, these prodrugs also induce the expansion and activation of human Vγ9Vδ2 T cells in a humanized mouse model of bacterial infection. The prodrugs we describe here synergize the direct killing of bacteria with a simultaneous rapid immune response by cytotoxic γδ T cells, which may limit the increase of antibiotic-resistant bacterial populations.
Similar content being viewed by others

Main
As a first line of defence, innate immune cells—such as dendritic cells, monocytes and macrophages—phagocytose bacteria and present the bacterial antigens on their cell surface using the major histocompatibility complex7. This antigen presentation initiates the adaptive T and B cell immune response that clears the infected host cells and the bacteria within them in 6–30 days8. Antibiotics prevent bacteria from overwhelming the body of the host, while the combined immune responses clear the bacterial infection. A group of six bacteria, known as the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter species), is the leading cause of multidrug-resistant nosocomial infections9. In addition, multidrug-resistant strains of Mycobacterium tuberculosis and Plasmodium falciparum are also global public health threats10,11. Rare mutations and the acquisition of antibiotic-resistance genetic elements give rise to bacterial cells that resist antibiotics through various mechanisms, including modification of the antibiotic target, the secretion of inactivating enzymes, the use of drug efflux pumps and metabolic bypass12,13,14. It has previously been reported that natural killer and cytotoxic T cells deliver granzymes to within bacteria or protozoan parasites, where they disrupt several essential systems and induce a mechanism of programmed pathogen death known as microptosis15,16,17. Bacteria that are undergoing microptosis do not develop resistance16. However, M. tuberculosis, P. falciparum and the ESKAPE pathogens evade antigen presentation by killing antigen-presenting cells, by preventing phago-lysosomal fusion or by segregating themselves inside different compartments of antigen-presenting cells18. In addition, some antibiotics impair the function of immune cells19.
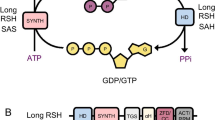
Here we introduce a double-pronged antimicrobial strategy through the use of dual-acting immuno-antibiotics (DAIAs)20,21. We focus on the methyl-d-erythritol phosphate (MEP) pathway for isoprenoid biosynthesis, which is essential for the survival of most Gram-negative bacteria and apicomplexans (malaria parasites) (Fig. 1a) but is absent in humans and other metazoans2,3. The first line of attack in the DAIA strategy targets the MEP enzyme IspH, which metabolizes HMBPP into isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP). IPP and DMAPP are building blocks for downstream terpenoids, which are essential for protein prenylation, the synthesis of peptidoglycan cell walls and the production of quinones for respiration1,22. The Escherichia coli strain CGSC 8074 (here termed ΔispH) conditionally expresses E. coli IspH in the presence of 0.5% arabinose; however, the addition of glucose to the medium shuts down IspH expression, which is lethal to this strain3 (Fig. 1b). The lack of IspH causes a build-up of HMBPP—a bacterial pathogen-associated molecular pattern—which stimulates the Vγ9Vδ2 T cells to expand and produce the cytotoxic proteins perforin, granulysin and granzymes, all of which are important for microptosis4,5,6. This forms the second line of attack in the DAIA strategy, and was demonstrated either by treating human peripheral blood mononuclear cells (PBMCs) with HMBPP + IL2 or by infecting them with CGSC 8074 in the presence of glucose. Under both conditions, a greater expansion of γδ T cells (Fig. 1c, top) and higher levels of the cytotoxic proteins perforin and granzyme A (Fig. 1c, middle) and the T cell surface activation markers CD69 and HLA-DR (Fig. 1c, bottom) were observed in comparison with PBMCs infected with wild-type (BL21) E. coli. An IspH inhibitor will therefore kill bacteria directly, as with other antibiotics, but will also kill persistent bacteria by microptosis15,16,23,24.

a, IspH is an essential enzyme of the MEP pathway (found in Gram-negative bacteria, mycobacteria and apicomplexan parasites), in which it produces IPP and DMAPP from HMBPP. IspH is absent from the mevalonate pathway (found in humans and complex metazoans). b, Left and bottom, the E. coli strain CGSC 8074 produces IspH in the presence of arabinose but not in the presence of glucose. Right, conditional knockdown of IspH, achieved by decreasing the levels of arabinose, reduces bacterial viability as measured by CFU assay (n = 3 biological and 3 technical replicates). Data are mean ± s.e.m. c, Left, human PBMCs co-infected with wild-type E. coli or CGSC 8074 (ΔispH) E. coli were analysed for expansion of CD3+Vγ9TCR+ (γδ) T cells after 24 h and compared to uninfected (UI) or HMBPP-treated PBMCs (top). Gated γδ T cell populations were analysed for the cytotoxic granule proteins granzyme A and perforin (middle) or cell surface markers of T cell activation CD69 and HLA-DR (bottom). Data are representative of 4 independent experiments (4 donors). Right, the percentage of Vγ9+ T cells from the CD3+ population and the percentage of Vγ9+ T cells with increased expression of granzyme A, perforin, CD69 and HLA-DR. Data are mean ± s.e.m. ***P < 0.001; one-way ANOVA. d, Activity of IspH in the presence of different concentrations of HMBPP, as measured by the methyl viologen assay at 30 min. Related to Extended Data Fig. 1d, e. e, Lineweaver–Burk double reciprocal plot showing the activity of E. coli IspH at different concentrations of the enzyme and its substrate HMBPP. f, Activity of 50 nM E. coli IspH over time in the presence of 1 mM HMBPP. g, The activities of purified recombinant IspH from P. falciparum, P. aeruginosa or M. tuberculosis LytB2. For d–g, n = 3 biological replicates with 8 technical replicates. Data are mean ± s.e.m.
Molecular docking and biochemical activity
We purified recombinant IspH proteins from several bacterial species—E. coli, M. tuberculosis and P. aeruginosa—and from the malaria parasite P. falciparum (Extended Data Fig. 1a, b). The activity of IspH is coupled to a system that reduces the oxidized iron–sulfur cluster25,26 [4Fe-4S]2+. In vitro, this reduction can be achieved chemically using sodium dithionite-reduced methyl viologen (Extended Data Fig. 1c), after which IspH activity is determined by the proportional change in the UV absorbance (398 nm) of oxidized methyl viologen27. Using E. coli IspH, we determined optimal concentrations of 50 nM IspH and 1 mM HMBPP and an optimal reaction time of 30 min (Fig. 1d–f, Extended Data Fig. 1d, e). We also measured the activities of purified recombinant IspH from P. falciparum, P. aeruginosa and M. tuberculosis using this methyl viologen assay (Fig. 1g).
We next performed a molecular docking study using the crystal structure of E. coli IspH (Protein Data Bank (PDB) ID 3KE8)28. The HMBPP-binding pocket was modelled (Methods) and the atomic property field established (Extended Data Fig. 2a) for the automated molecular docking of 9.6 million compounds. The 168 best-scoring compounds (Extended Data Fig. 2b) were visually compared to HMBPP. The top 24 compounds (denoted C1–24)—that is, those with lower binding energies and atomic property field scores than HMBPP (Extended Data Fig. 2c)—were further evaluated in terms of their chemical and drug-like properties; the three-dimensional conformations of the docked ligand−IspH complex were also assessed (Extended Data Fig. 3a, Supplementary Fig. 2a). Analysis by methyl viologen assay revealed C10, C17 and C23 as the best inhibitors of E. coli IspH—with half-maximal inhibitory concentrations (IC50) of 9 μM, 4 nM and 85 nM, respectively (Fig. 2a, Supplementary Table 1)—whereas the assessment of IspH activity over time showed that C17 and C23 were more stable inhibitors of E. coli, M. tuberculosis, P. aeruginosa and P. falciparum IspH than was C10 (Fig. 2b, Extended Data Fig. 3b). Although both C17 and C23 were found to be potent inhibitors of IspH from different pathogens (Fig. 2c, Supplementary Table 2), we tested several analogues of each of C10, C17 and C23 to improve their potency against purified E. coli IspH (Fig. 2d, Extended Data Fig. 3c–e, Supplementary Table 3, Supplementary Fig. 2b–d). C23 analogues showed substantial improvement (that is, lower IC50 values) in terms of E. coli IspH inhibition compared with the parent compound, whereas C10 and C17 analogues did not. Compared to the IspH substrate HMBPP, the compounds C23.20 and C23.21—the two most potent C23 analogues—show improved binding (a lower dissociation constant, K D) to purified E. coli IspH according to surface plasmon resonance analysis (Extended Data Fig. 4a). By testing different C23 analogues, we established a structure–activity relationship (Fig. 2d, Extended Data Figs. 3e, 4b) and confirmed C23.07, C23.20, C23.21, C23.28 and C23.47 as the most potent inhibitors of E. coli IspH.

a, Dose–response (nonlinear regression) curves for the inhibition of E. coli IspH by compounds C1–24, determined by the methyl viologen assay. The IC50 values (Supplementary Table 1) were calculated from the respective curves. Data are mean ± s.e.m. Associated with Extended Data Fig. 1d, e. b, Activity of E. coli IspH pre-treated with DMSO (control), C10, C17 and C23 over time. Data are mean ± s.e.m. Associated with Extended Data Fig. 3b. c, Inhibition of M. tuberculosis, P. aeruginosa and P. falciparum IspH at varying concentrations of C17 or C23. The IC50 values were calculated from the respective curves and are given in Supplementary Table 2. Data are mean ± s.e.m. d, Dose–response (nonlinear regression) curves for the inhibition of E. coli IspH by C23 analogues, as measured by the methyl viologen assay. The IC50 values were calculated from the respective curves and are provided in Supplementary Table 3. Data are mean ± s.e.m. Associated with Extended Data Fig. 3e. e, The killing of E. coli by TPP-linked analogues of the prodrug C23 (C23.07, C23.20, C23.21, C23.28; Supplementary Fig. 2e) compared to TPP-treated control, as illustrated by dynamic growth curves. The delivery of the prodrugs into bacteria and their cleavage into the active form is shown in Extended Data Fig. 4e, f. For a–d, e, n = 3 biological replicates and 8 technical replicates.
Bacterial killing by prodrugs
Because C23 and its analogues are not bacteria-permeable, we coupled them to triphenyl phosphonium to aid in the penetration of membranes29. However, because cations are efficiently effluxed out of Gram-negative bacteria by transporters—such as AcrAB–TolC in E. coli—we designed prodrugs from which the negatively charged IspH inhibitor would be released once inside the bacteria. We synthesized ester prodrugs from the C23.47 analogue by linking it to a lipophilic cation (6-hydroxyhexyl triphenylphosphonium bromide (TPP), a lipophilic alcohol (ethanol) or a basic amine (3-(dimethylamino)propan-1-ol) (Supplementary Fig. 3). Similar strategies involving the use of prodrugs with cleavable ester bonds have been shown to facilitate drug delivery into bacteria30. We found that the C23.47 + TPP ester was the most potent against E. coli, with a MIC90 value—the minimum drug concentration at which 90% bacteria are killed—of 4 μM (Extended Data Fig. 4c, d). We therefore focused on the TPP ester form of C23 analogues (Supplementary Fig. 2e). C23.20–TPP, C23.21–TPP and C23.28–TPP showed the best activity against E. coli (MIC90 < 4 μM) (Fig. 2e). Analysis of the lysates of prodrug-treated bacteria by mass spectrometry detected both the delivery of the prodrug molecule C23.28–TPP into E. coli and its subsequent cleavage into C23.28 and TPP (Extended Data Fig. 4e, f). Notably, the inhibition of E. coli IspH by C23.28 prevented the conversion of HMBPP to DMAPP and IPP, whereas treatment with TPP alone had no effect on this process (Extended Data Fig. 4g, h).
The levels of IspH in the E. coli strain CGSC 8074 can be regulated by changing the amount of arabinose in the culture medium (Extended Data Fig. 5a). Increasing IspH levels in this manner increased the dose of C23.28–TPP that was required to kill CGSC 8074 (Extended Data Fig. 5b, c). We tested several C23 derivatives on drug-resistant clinical isolates of Vibrio cholerae using the resazurin blue assay and the colony-forming unit (CFU) assay, and determined their MIC90 values (Extended Data Fig. 5d–f). Whereas TPP alone did not kill V. cholerae, the prodrugs C23.20–TPP, C23.21–TPP and C23.28–TPP had the lowest MIC90 values of 16 μM (8 μg ml−1), followed by C23.07–TPP (MIC90 = 125 μM; 63 μg ml−1) and C23.47–TPP (MIC90 = 63 μM; 31 μg ml−1). The MIC90 values for these compounds in several species of antibiotic-resistant bacteria are shown in Supplementary Table 4. In summary, the IspH inhibitor prodrugs had lower MIC90 values in multidrug-resistant clinical isolates of Enterobacter aerogenes, A. baumanii, P. aeruginosa, V. cholerae and K. pneumoniae than the current best-in-class antibiotics, including meropenem (a member of the carbapenem class), amikacin and tobramycin (aminoglycosides), ciprofloxacin (a fluoroquinolone) as well as ceftriaxone, cefepime and ceftaroline (third, fourth and fifth generation cephalosporins, respectively (Fig. 3, Supplementary Table 5).

a, b, The prodrugs C23.20–TPP, C23.21–TPP and C23.28–TPP—as well as a range of current best-in-class antibiotics—were tested against pan-resistant or multidrug-resistant clinical isolates of E. aerogenes, V. cholerae, K. pneumoniae, A. baumannii and P. aeruginosa using the resazurin blue assay (a) and CFU plating (b) after 24 h treatment (n = 3 biological replicates). For the resazurin blue assay, pink indicates bacterial growth and blue indicates no bacterial growth. TPP was used as a negative control, and uninfected culture medium was used as a positive control. Data are mean of three independent experiments ± s.e.m., with individual data points shown. *P < 0.05, **P < 0.01, ***P < 0.001, the remainder are not significant; two-tailed paired Student’s t-test. Associated with Supplementary Table 5.
Specificity, mechanism and toxicity
Isoprenoids are required in Gram-negative bacteria and in M. tuberculosis for respiration and for synthesis of the cell wall1,31. Using a Seahorse XF Analyzer we found that prodrug-treated E. coli cells show a significant decrease in oxygen consumption rate (aerobic respiration) and extracellular acidification rate (glycolysis) compared with untreated cells (Extended Data Fig. 6a, b). This was accompanied by increased levels of superoxide and hydrogen peroxide16 (Extended Data Fig. 6c, d). Prodrug-treated bacteria showed a dose-dependent loss of membrane integrity (as assessed by SYTO 9 and propidium iodide staining) and of membrane potential, whereas treatment with TPP alone had no effect (Extended Data Fig. 6e–h). Scanning and transmission electron micrographs showed spherocyte formation, cell-membrane protrusions, and defects in the cell wall and in the periplasm of prodrug-treated E. coli and V. cholerae, and in conditional IspH knockdown E. coli cells (strain CGSC 8074) (Extended Data Fig. 6i, j).
The half-lives (t 1/2) of the prodrugs C23.28–TPP and C23.21–TPP were 40 and 56 min in human plasma, 218 and 245 min in pig plasma, and 20 and 21 min in mouse plasma, respectively (Extended Data Fig. 7a). Similarly, t 1/2 values in presence of liver microsomes were 27 and 48 min (human plasma), 25 and 24 min (monkey plasma), and 24 and 41 min (mouse plasma), respectively (Extended Data Fig. 7b). The disappearance of the prodrug forms coincided with the appearance of the respective parent drugs. Although our prodrugs showed low toxicity in the mammalian cell lines HepG2, RAW264.7 and Vero (Extended Data Fig. 7c), lipophilic triphenylphosphonium cations are reported to cause mitochondrial proton leak and toxicity in C2C12 myoblasts32. Furthermore, the human hERG gene (KCNH2) is a known target for lipophilic cations such as TPP33. However, our 6-hydroxyhexyl TPP carrier molecule and our prodrugs were neither toxic to C2C12 cells nor caused loss of mitochondrial membrane potential (Extended Data Fig. 7d, e). Additionally, C23.28–TPP, methyl TPP (Me-TPP) and the carrier molecule showed tenfold higher IC50 values (5–10 μM) than verapamil in hERG electrophysiological profiling experiments using an automated QPatch HTX assay (Extended Data Fig. 7f).
Notably, we found that the prodrug C23.28–TPP reduces IspH levels in E. coli and in clinical isolates of several antibiotic-resistant bacteria (Extended Data Fig. 8a, b). We next performed proteomics analysis on E. coli treated with C23.28–TPP and on CGSC 8074 (ΔispH) in the presence of glucose. Out of 2,346 proteins, 525 showed similar changes after treatment with C23.28–TPP and after IspH knockdown (Extended Data Fig. 8c, d). Among the downregulated proteins, 323 (22%) were common to drug treatment and conditional IspH knockdown (Extended Data Fig. 8e). Pathway analysis34 showed enrichment of the electron transport chain complexes, of ubiquinone, and of other pathways (Extended Data Figs. 8f–h, 9).
Dual action leads to γδ T cell response
The activation of human γδ T cells does not require epitope presentation by the major histocompatibility complex or by CD1 receptors. Instead, the butyrophilin receptors BTN2A1 and BTN3A1 on target cells act to detect phosphoantigens such as HMBPP35,36 and as a direct ligand for the Vγ9Vδ2 T cell receptor, respectively37,38. The treatment of E. coli with C23.07–TPP, similar to the conditional IspH-knockdown strain CGSC 8074, resulted in activation of Vγ9Vδ2 T cells within 24–48 h (Fig. 1c, Extended Data Fig. 10a), and the activated cells showed high levels of cytotoxic markers such as perforin and granulysin, as well as high levels of the T cell surface activation markers CD69 and HLA-DR. We observed similar results after the treatment of Mycobacterium smegmatis- or V. cholerae-infected PBMCs with C23.07–TPP (Extended Data Fig. 10b). By contrast, kanamycin-treated and TPP-treated samples did not show γδ T cell activation. Whereas E. coli and V. cholerae were resistant to kanamycin, our prodrug C23.07–TPP could effectively kill both bacteria (Extended Data Fig. 10c). To assess for resistance to IspH inhibitors, we grew clinical isolates of V. cholerae and K. pneumoniae for 18 serial passages with the prodrug C23.28–TPP in the presence or in the absence of human PBMCs. To demonstrate the critical role of Vγ9Vδ2 T cell activation and expansion in the efficacy of these prodrugs, PBMCs depleted in γδ T cells were also used in the serial passaging. The efficacy of γδ T cell depletion is reflected in the lack of Vγ9Vδ2 T cell expansion after 6 days of treatment with HMBPP and IL-15 (Extended Data Fig. 10d). In the absence of PBMCs, both V. cholerae and K. pneumoniae developed resistance to the prodrug C23.28–TPP as well as to conventional antibiotics (V. cholerae to hygromycin and K. pneumoniae to streptomycin) (Extended Data Fig. 10e, f, top). However, in the presence of human PBMCs, neither V. cholerae nor K. pneumoniae developed resistance to C23.28–TPP (Extended Data Fig. 10e, f, bottom). Passaging V. cholerae and K. pneumoniae in γδ-T-cell-depleted human PBMCs significantly diminished the dual action of the prodrug, supporting the relevance of γδ T cells in its mechanism of action. Owing to the lack of reliable in vivo γδ-depleting antibodies, we used E. coli infection in NSG mice (instead of humanized mice) to corroborate the role of Vγ9Vδ2 T cells in vivo. We injected one group of NSG mice with human PBMCs and another group with ex-vivo γδ-T-cell-depleted human PBMCs. Both groups of mice were infected with 107 E. coli cells (Fig. 4a–c) and the levels of γδ T cells were monitored by fluorescence-activated cell sorting. After infection, mice in both the γδ-T-cell-depleted and the undepleted groups were given suboptimal doses (1 mg kg−1) of C23.28–TPP, to minimize the killing of bacteria by direct antibiotic action and increase the prominence of the immune effect. Mice with γδ-T-cell-depleted PBMCs had 2–10-fold higher CFU (Fig. 4a) and significantly lower levels of γδ T cells (Fig. 4b, c) than their counterparts with undepleted PBMCs.

a, Escherichia coli load (CFU mg−1) in the organs of NSG mice injected with γδ depleted (γδ−) or undepleted (γδ+) human PBMCs, infected with E. coli and treated with 1 mg kg−1 C23.28–TPP for 3 days. b, CD3+ Vγ9TCR+ T cell expansion in γδ− or γδ+ NSG mice, four days after infection. c, Percentage of CD3+ cells that are also Vγ9+ in each organ. For a–c, n = 10 mice, 3 technical replicates. Data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, NS, not significant; two-tailed unpaired Student’s t-test, relative to γδ− mice. d–f, Humanized mice infected with E. coli were treated with C23.07–TPP (d, e, top; f, left) or C23.28–TPP (d, e, bottom; f, right) and monitored daily for survival (d), bacteraemia in terms of CFU per ml of blood (e) and E. coli load in different organs at the experimental endpoint measured as CFU mg−1 (f). g, Left, CD3+Vγ9TCR+ T cell expansion in E. coli-infected humanized mice, treated with TPP or C23.07–TPP for five days after infection. Right, the percentage of Vγ9+ T cells from CD3+ cells in each organ is shown. Associated with Extended Data Fig. 11a. For d–g, n = 6 mice, 3 technical replicates. Data are mean ± s.e.m. ***P < 0.001, **P < 0.01, *P < 0.05; two-tailed unpaired Student’s t-test, relative to TPP-treated mice. h, i, BALBc mice were infected with E. aerogenes, treated with 10 mg kg−1 TPP, C23.28–TPP or meropenem and monitored for survival (h) and i, Enterobacter load (CFU mg−1) (i) (n = 19 mice, 3 technical replicates). Data are mean ± s.e.m. ***P < 0.001, **P < 0.01, *P < 0.05, NS, not significant; one-way ANOVA, relative to TPP-treated or meropenem-treated mice.
As a final test, we assessed the direct bactericidal effects of IspH prodrugs in C57BL/6 mice infected with V. cholerae. Mice treated with C23.28–TPP showed significantly lower mortality and had a lower bacterial load in all organs tested compared to those treated with TPP alone (Extended Data Fig. 10g, h). Because mouse γδ T cells do not respond to HMBPP39,40, we used humanized mice in experiments to test the dual action of IspH prodrugs. After the injection of HMBPP into humanized mice, we saw rapid expansion of the human Vγ9Vδ2 T cells but not the αβ T cells (Extended Data Fig. 10i). Humanized mice that were infected with E. coli and treated with prodrugs treated showed lower levels of bacterial CFU in circulation as well as improved survival compared with mice that were treated with TPP alone (Fig. 4d, e). Similarly, prodrug-treated humanized mice showed significantly lower bacterial load and expansion of Vγ9Vδ2 T cells in several organs than their TPP-treated counterparts (Fig. 4f, g). We confirmed both the expansion of Vγ9Vδ2 T cells and the lower bacterial burden in the tissues of prodrug-treated humanized mice by immunofluorescence microscopy (Extended Data Fig. 11). Finally, we found that the prodrug C23.28–TPP was able to clear infection by a clinically isolated multidrug-resistant strain of E. aerogenes (UCI 15) and significantly improve the survival of infected BALBc mice, whereas meropenem—a current best-in-class carbapenem antibiotic—did not (Fig. 4h, i).
Discussion
The family of antibiotics and the antimicrobial strategy that we report here synergize direct antibiotic action with rapid immune response. This dual mechanism of action, an inherent feature of these compounds, could delay the emergence of drug resistance15,16,17 Our DAIA prodrugs are bacteria-permeable and are more effective against several species of multidrug-resistant bacteria than the current best-in-class antibiotics. They act specifically on IspH, show low toxicity to mammalian cells (specifically to myoblasts, with MIC90 values 10–100 times higher than in bacteria) and high IC50 values against hERG channels32. Unlike antibiotics derived from natural sources, no IspH inhibitors have been discovered in microorganisms, so it is less likely that resistance mechanisms—such as β-lactamases and macrolide esterases in the case of β-lactam and macrolide antibiotics—have evolved specifically against our prodrugs. Future experiments should investigate the potential mechanisms of resistance to IspH inhibitors. In addition, the synergy between γδ T cells activated by these prodrugs and other immune cells merits further study.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Molecular docking studies
The IspH–HMBPP complex with PDB ID 3KE8 was used for the virtual screening28. The protein was prepared using standard automated protocols embedded in MolSoft’s (Internal Coordinate Mechanics) ICM-Pro software41,42. Hydrogen atoms were added to the structure, and considerations were made regarding the correct orientation of Asn and Gln side chains, ligand and protein charges, histidine orientation and protonation state and any crystallographic quality flags such as high b-factors or low occupancy. All waters and heteroatoms were deleted except for the iron–sulfur complex. Virtual screening of the MolCart chemical database (http://www.molsoft.com/screening.html, v.2017, containing around 9.6 million chemicals) was undertaken using MolSoft’s ICM-VLS software43,44. The binding site was represented by five types of interactions to create a potential docking map: (i) van der Waals potential for a hydrogen atom probe; (ii) van der Waals potential for a heavy-atom probe (generic carbon of 1.7 Å radius); (iii) optimized electrostatic term; (iv) hydrophobic terms; and (v) loan-pair-based potential, which reflects directional preferences in hydrogen bonding. The energy terms are based on the all-atom vacuum force field ECEPP/3 and conformational sampling is based on the ICM biased probability Monte Carlo (BPMC) procedure42. This method randomly selects a conformation in the internal coordinate space and then makes a step to a new random position independent of the previous one but according to a predefined continuous probability distribution followed by local minimization.
A hit list of 37,849 chemicals was obtained and this was filtered down to a set of 168 chemicals recommended for experimental testing using the following criteria: (1) low van der Waals interaction energy; (2) low ICM docking score; (3) similarity between the 3D atomic property fields of the pharmacophore and the substrate45; and (4) number of hydrogen-bond acceptors in the phosphate-binding region.
Bacteria
Escherichia coli BL21(DE3) from New England Biolabs was used as a model strain. Clinical isolates of E. aerogenes (CRE) (UCI 15), K. pneumoniae 1.53 (ST147+, CTX-M15+), Salmonella enterica subsp. enterica serovar Typhimurium (LT2 – SL7207), V. cholerae (M045), A. baumannii (BC-5), A. baumannii (AB5075-UW), P. aeruginosa (PA14 and MRSN 5524), Helicobacter pylori (Hp CPY6081), Shigella flexneri (2457T), Bacillus sphaericus (CCM 2177), M. tuberculosis (M. tuberculosis H37Ra) and Yersinia pestis (KIM 10+) were obtained from BEI Resources. The conditional IspH knockdown E. coli strain CGSC 8074 (ΔispH), was obtained from the Coli Genetic Stock Center at Yale University. All strains were cultured at 37 °C in their respective medium (2.5% brain heart infusion agar, Middlebrook 7H10 with OADC, Luria Bertani (LB), tryptic soy agar, 5% blood agar, Columbia agar (BD Difco, BD 241830, BD 262710, BD 244610, BD 236950; and Fisher, R01217, R02030) based on the vendors’ recommendation. LB medium with 0.5% arabinose (Sigma, A3256) was used to culture the CGSC 8074 (ΔispH) strain. Changing arabinose and glucose concentrations (0.5–0.05%) in the LB medium enabled us to modulate IspH protein levels in CGSC 8074. For testing the antibiotic sensitivity, bacteria were grown in RPMI medium containing 10% fetal bovine serum or human serum.
Animal models
All studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH). All animal experiments were performed according to protocols approved by the Wistar Institute’s Institutional Animal Care and User Committee (IACUC). The humanized mice (Hu-mice) were generated by R. Somasundaram in the Herlyn laboratory and transferred over to the Dotiwala laboratory. NOD/LtSscidIL2Rgnull (NSG) mice were inbred at The Wistar Institute under licence from the Jackson Laboratory. For humanization, fetal liver and thymus were obtained from the same donor (18–22 weeks of gestation). Female NSG mice (6–8 weeks) received a thymus graft (1 mm3) in the sub-renal capsule 24 h after myeloablation using busulfan (30 mg kg−1, intraperitoneally (i.p.); Sigma-Aldrich, B2635). This was immediately followed by the injection of autologous liver-derived CD34+ haematopoietic stem cells (105 cells per mouse, intravenously (i.v.)) that were magnetically sorted by microbeads conjugated with anti-human CD34 (Miltenyi, 130-046-703)46. Six to eight weeks (>50 days) later, the presence of human immune cells was monitored by multi-colour flow cytometry using an 18-colour BD LSR II Analyzer (BD Biosciences)47. NSG mice with human PBMCs were generated by i.v. injection of human PBMCs or PBMCs depleted of all γδ T cells using Anti-TCRγ/δ Microbead Kit (Miltenyi, 130-050-701). About 107 cells per mouse were injected every 3 days, for a total of 3 doses per mouse and the presence of human immune cells was monitored by multi-colour flow cytometry. An equal number of male and female C57BL/6 or BALBc mice were obtained from the Jackson Laboratory and used for mouse models of Vibrio or Enterobacter infection, respectively. Mice were housed in plastic cages on an ad libitum diet and maintained on a 12-h light/12-h dark cycle at 22 °C at 60% humidity. Controls and experimental groups were age- and genotype-matched non-littermates. Both initial infection and drug treatments were administered by i.p. or i.v. routes. Infected mice were monitored twice daily for survival and distress. To monitor bacteraemia, mice were bled daily from tail nicks. At the end of the experiment mice were euthanized by CO2 inhalation and their spleens, livers, kidneys, lungs and brains were collected for CFU and flow cytometry analysis.
Human samples
Human PBMCs were obtained from the Human Immunology Core of the University of Pennsylvania (UPenn) under UPenn protocol 705906 (PI: Riley) ‘Pre-clinical studies of the Human Immune System’. The donors of the PBMCs provided informed written consent for the use of their samples. De-identified specimens were transferred to the Wistar Institute under Wistar protocol 21906321, reviewed and approved by the Wistar Institutional Review Board. PBMCs were washed in PBS counted and kept in plastic culture plates in RPMI medium containing 10% human serum. Human cell lines (HepG2, Vero, RAW264.7 and C2C12) were obtained from the American Type Culture Collection (ATCC), authenticated by short tandem repeat profiling and PCR assays with species-specific primers and were confirmed to be free of mycoplasma contamination.
Antibodies
The following antibodies were used in this study.
Antibodies for western blotting and immunohistochemistry (unless otherwise mentioned, dilution: primary antibody, 1:50; secondary antibody, 1:200): anti-E. coli antibody (Abcam, ab137967); anti-E. coli IspH rabbit polyclonal antibody (Genscript, generated in this study, dilution 1:100,000); anti-E. coli RNA Sigma 70 mouse antibody (BioLegend, 63208); secondary-biotinylated rabbit anti-rat IgG (Vector Laboratories, BA-4001); mouse IgG HRP-linked whole antibody (GE Healthcare, NA931V); rabbit IgG HRP-linked whole antibody (GE Healthcare, NA934V); biotinylated goat anti-rabbit IgG antibody (Vector Laboratories, BA-1000); donkey anti-rabbit IgG AF-488 (BioLegend, 406416).
Antibodies for fluorescence-activated cell sorting (FACS; dilution 1:100): anti-CD3-PerCP-Cy5.5 (clone UCHT1, BD Biosciences, 560835); anti-CD4-Alexa Fluor 700 (clone RPA-T4, BD Biosciences, 557922); anti-CD8a-Brilliant Violet 711 (clone RPA-T8, BioLegend, 301044); anti-TCR vg9-FITC (clone 7A5, Invitrogen, TCR2720) (or anti-TCR vd2 (clone B6, BioLegend, 331402) with anti-mouse IgG-AF647 (Invitrogen, A21236)); anti-CD107a (LAMP-1)-Brilliant Violet 510 (clone H4A3, BioLegend, 328632); anti-CD69-PE/Cy7 (clone FN50, BD Biosciences, 557745); anti-HLA-DR-Brilliant Violet 421 (clone L243, BioLegend, 307636); anti-CD38-Brilliant Violet 510 (clone HIT2, BD Biosciences, 563251); anti-CD25-Alexa Fluor 647 (clone BC96, BioLegend, 302618).
Antibodies for FACS compensation (dilution 1:200): anti CD3 mouse monoclonal PE/Dazzle 594 (BioLegend, 317346); anti CD3 mouse monoclonal APC (BioLegend, 300412); anti CD3 mouse monoclonal APC Cy7 (BioLegend, 300317); anti CD3 mouse monoclonal BV711 (BioLegend, 344838); anti CD3 mouse monoclonal PE (BioLegend, 300408); anti CD3 mouse monoclonal PE Cy7 (BioLegend, 300316).
Anti-E. coli IspH antibody generation: the control sera (2–3 ml) were collected from the ear pinna of the rabbit before the start of immunization. The 200 μg of purified E. coli IspH protein was mixed with the KLH conjugate and Freud’s complete adjuvant and injected subcutaneously into the rabbit (2–4 site per rabbit) in the animal facility at Genscript. The second immunization was performed 14 days after the first immunization with 200 μg purified protein, KLH conjugate and Freud’s incomplete adjuvant. One week after the second immunization, the test sera (first test bleed) were collected from the rabbit to test the antibody titration by ELISA and western blot. The third immunization, with 200 μg purified protein, KLH conjugate and Freud’s incomplete adjuvant was performed 14 days after the first test bleed. One week later the second test bleed was performed, and sera were purified for IgG antibodies using a protein A column. The purified IgG antibodies were used for the confirmation of anti-IspH antibody production by ELISA and western blot. After confirmation, that antibody was raised in rabbits, the production bleed was performed, the sera were separated, and antibodies were purified using a protein A column. The purified anti-E. coli IspH rabbit polyclonal antibody was validated by western blots using purified ispH protein from E. coli, P. aeruginosa, M. tuberculosis and P. falciparum. The antibody was further validated using lysates of A. baumannii, S. flexneri, S. enterica, V. cholerae and H. pylori.
Depletion of γδ T cells from human PBMCs
The γδ T cells were separated from human PBMCs using anti-TCRγ/δ Microbead Kit (Miltenyi, 130-050-701). After Ficoll separation the human PBMCs were washed and resuspended in RPMI medium containing human serum. The cells were counted, pelleted at 300g for 10 min and resuspended in 40 μl of MACS buffer for every 107 cells. The cells were incubated with 10 μl of anti-TCR γ/δ hapten-antibody per 107 cells, at 4–8 °C for 10 min. After incubation, 30 μl MACS buffer and 20 μl of MACS anti-hapten MicroBeads-FITC per 107 cells were added followed by further incubation at 4–8 °C for 15 min. The cells were washed with 1–2 ml of MACS buffer per 107 cells and centrifuged at 300g for 10 min. The supernatant was removed, and the cells resuspended in 500 μl MACS buffer per 108 cells. The sample was loaded on the MACS buffer-rinsed LS column and was kept in the magnetic field. The cells in the flow through were collected and the column washed three times with 3 ml MACS buffer. The cells in the flow through and washes were combined, pelleted and resuspended in RPMI + 10% human serum and counted for further experiments.
Mouse infection studies
In experiments with Hu-mice or NSG mice injected with human PBMCs, infection was induced by injecting 107 E. coli per mouse i.p. in 200 μl Dulbecco phosphate buffered saline (DPBS). In experiments with C57BL/6 mice, 106 V. cholerae and in experiments with BALBc mice, 5 × 104 E. aerogenes (UCI 15) were injected i.p. After 24 h, prodrugs the C23.07–TPP or C23.28–TPP (where mentioned), or just the carrier molecule TPP, (10 mg per kg per mouse) in 1% DMSO–DPBS solution were injected i.p. (or i.v. in case of E. aerogenes-infected BALBc mice) once a day for 1–2 weeks, until mice succumbed to infection or were euthanized for tissue analysis, as indicated. A group of E. aerogenes-infected mice were given meropenem (10 mg per kg per mouse) for comparison to a best-in-class antibiotic. NSG mice injected with human PBMCs were given a suboptimal (1 mg kg−1) dose of C23.28–TPP through the i.v. route, once a day for 4 days. Blood from infected mice was collected daily using tail snips and analysed for bacteremia by CFU and flow cytometry for γδ T cell expansion. After death from infection or euthanasia at the end of the experiment, the spleen, liver, lungs, brain and kidneys were collected, sectioned and studied for bacterial CFU, immunohistochemistry or flow cytometry as indicated.
Isolation of cells and bacteria from different organs
Samples of mouse spleen, liver, lung, brain and kidney were weighed and crushed in 12-well plastic tissue culture plates using a 5-ml syringe. RBCs were lysed in RBC lysis (ACK) buffer at 37 °C and for 1 min. Cells were washed 3–5 times with MACS buffer at 4 °C. Cells were then either analysed by flow cytometry or lysed in distilled, deionized water and serial dilutions of samples were plated for bacterial CFU on medium plates respective to the bacteria studied.
Ex vivo infection in human PBMCs
Human PBMCs were washed in medium (10% human serum RPMI medium supplemented with 100 U ml−1 penicillin G and 100 μg ml−1 streptomycin sulfate, 6 mM HEPES, 1.6 mM l-glutamine, 50 mM 2-mercaptoethanol) then cultured in medium without penicillin or streptomycin in 6-, 12-, 24- or 96-well Primaria plates (Fisher Scientific, 08-772). Escherichia coli, V. cholerae, K. pneumoniae or M. tuberculosis ex vivo infections were induced at a multiplicity of infection of 1:0.1, 1:1, 1:10 or 1:100. Various dilutions of 100 mM stock solutions of prodrugs C23.07, C23.28–TPP or TPP (control) were added to sample wells to give a final working concentration range from 500 μM to 4 μM. Infected PBMC samples were analysed at 24, 48 or 72 h by flow cytometry or lysed in distilled water at different time points where indicated and the lysates were used for CFU analysis. The Vγ9Vδ2 T cells in uninfected PBMCs show low initial levels of perforin, probably because of the length of time spent in culture (up to 72 h).
CFU analysis
Bacterial cultures treated with different prodrugs or antibiotics, or lysates from infected mouse blood, tissues or infected ex vivo human PBMCs were serially diluted and 50 μl was plated on bacterial culture plates. The plates were incubated at 37 °C and counted after overnight incubation (after 20 days for M. tuberculosis colonies). The CFU were normalized per ml for blood or per mg weight for tissues. For all experiments, at least three independent experiments were performed, with 3–8 technical replicates in each experiment.
Recombinant IspH cloning and expression
IspH gene sequences from E. coli, Pseudomonas, Plasmodium and M. tuberculosis (LytB2) were optimized for expression in E. coli and synthesized by Genscript. These sequences were cloned in a pET24a-KAN vector and co-expressed with iron–sulfur cluster (isc) proteins (encoded in the pACYC184 plasmid) in Nico (DE3) cells (NEB, C2529H)48. Transformed Nico (DE3) cells were grown at 37 °C in Terrific Broth (12 g tryptone, 24 g yeast extract, 5 ml glycerol per litre of broth) supplemented with sterile monopotassium phosphate (23.1 g l−1), dipotassium phosphate (125.4 g l−1), ferric ammonium citrate (35 mg l−1), l-cysteine (1 mM) and the antibiotics kanamycin (50 mg l−1) and chloramphenicol (35 mg l−1). At an optical density at 600 nm (OD600) of 0.6–0.7, IspH production was induced by adding IPTG at 1 mM concentration before overnight incubation at 25 °C.
IspH purification
After IspH induction, bacteria were spun down at 6,000g and washed three times with 50 ml degassed PBS. All subsequent steps were performed in an anaerobic glove box at 0.5 ppm O2. After the final wash, the bacteria were resuspended in 20 ml degassed lysis buffer (25 mM Tris, 1M KCl, 5% glycerol, cOmplete protease inhibitor cocktail (Sigma, 4693132001), 5 mM sodium dithionite, pH 7.5). The rest of the procedure was carried out under anaerobic conditions (<0.5 ppm O2) in an mBraun glovebox. Bacteria were lysed by freeze-thawing five or six times in liquid nitrogen. Nucleic acids were eliminated by incubating with 500 units of Benzonase (Sigma E1014) at room temperature for 30 min. The lysate was spun down at 6,000g and filtered through a 0.45-μm filter under anaerobic conditions (<0.5 ppm O2). The lysate was incubated for 2–3 h at room temperature with 3–5 ml Ni-NTA resin (Qiagen, 30230) that had been equilibrated in lysis buffer. The Ni-NTA resin was washed with 3 column volumes of wash buffer 1 (25 mM Tris, 1 M KCl, 5% glycerol, cOmplete protease inhibitor cocktail, 30 mM imidazole, pH 7.5) and 1 column volume of wash buffer 2 (25 mM Tris, 0.1 M KCl, 5% glycerol, cOmplete protease inhibitor cocktail, 30 mM imidazole, pH 7.5). The protein was eluted from Ni-NTA using 15 ml elution buffer (25 mM Tris, 0.1 M KCl, 5% glycerol, cOmplete protease inhibitor cocktail, 300 mM imidazole, pH 7.5). The eluted protein was passed through a 5-ml bed of chitin resin to remove contaminating proteins and then passed in tandem through Sepharose SP (GE Healthcare, 17072910) and Sepharose Q (GE Healthcare, 17051010) resin beds. The protein was eluted from the Q column using the Q column elution buffer (25 mM Tris, 1 M KCl, 5% glycerol, pH 7.5) desalted using Econo-Pac 10DG (Bio-Rad, 732-2010) desalting columns and concentrated using Amicon Ultra 10k spin columns.
Methyl viologen assay
All solutions were degassed by boiling before use and the assays were performed under <0.5 ppm O2 in a glove box. To monitor the activity of IspH, methyl viologen was used as the reducing agent. The oxidation of methyl viologen (blue to colourless) was followed by measuring the loss of absorption at 398 nm. The assay solution contained 50 mM Tris–HCl (pH 8), 1 mM methyl viologen and 0.5 mM sodium dithionite in a total volume of 100 μl in 96-well flat bottom plastic plates. Varying concentrations of IspH (0–5 μM) and HMBPP (0–1.25 mM) were titrated and optimal concentrations of 50 nM IspH and 1 mM HMBPP were used for subsequent experiments. After reduction of methyl viologen with sodium dithionite an approximate absorbance of 3 was reached. The reactions were initiated by the addition of IspH. For inhibition studies, varying concentrations of candidate drugs (1 nM–250 μM) or DMSO (negative control) were added. The plates were sealed by Parafilm, incubated at 37 °C and the absorbance at 398 nm was read every 5 min in a Biotek Synergy 2 plate reader. The activity was expressed as micromoles of HMBPP consumed per second, as measured by the decrease in absorbance at 398 nm. Samples that lacked HMBPP served as a baseline negative control. The assay is linear with respect to time and protein concentration.
Surface plasmon resonance
Approximately 30,000 RU of purified recombinant His-tagged E. coli IspH was immobilized onto a Ni-NTA surface plasmon resonance (SPR) chip activated by N-(3-dimethylaminopropyl)-N′-ethyl carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS). The remaining binding sites were blocked with 1 M ethanolamine at pH 8.5. Test compounds C23.20, C23.21 and HMBPP were serially diluted to 1:3.16 starting at 100 μM final concentration in running buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% Tween20, 5% DMSO) and run on a Biacore T200 instrument at a flow rate of 50 μl min−1, to reduce the mass transport limitation effects.
General chemistry
All reactions were conducted under an inert gas atmosphere (nitrogen or argon) using a Teflon-coated magnetic stir bar at the temperature indicated. Commercial reagents and anhydrous solvents were used without further purification. Solvents were removed using a rotary evaporator, and residual solvent was removed from non-volatile compounds using a vacuum manifold maintained at approximately 1 Torr. All yields reported are isolated yields. Preparative reversed-phase high pressure liquid chromatography (RP-HPLC) was performed using a Gilson GX-271 semi-prep HPLC, eluting with a binary solvent system A and B using a gradient elution (A, H2O with 0.1% trifluoroacetic acid (TFA); B, CH3CN with 0.1% TFA) with UV detection at 220 nm. Low-resolution mass spectral (MS) data were obtained on a Waters ACQUITY QDa LC–MS mass spectrometer with UV detection at 254 nm. Proton nuclear magnetic resonance (1H NMR) spectra were obtained on a Bruker Avance II 400 (400 MHz) spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) relative to residual undeuterated solvent as an internal reference. The following abbreviations are used for the multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; dt, doublet of triplets; td, triplet of doublets; tt, triplet of triplets; ddd, doublet of doublet of doublets; m, multiplet; br, broad.
Synthesis of (6-hydroxyhexyl)triphenylphosphonium bromide (TPP)
To a stirred solution of 6-bromohexan-1-ol (5.0 g, 27.61 mmol) in 70 ml of acetonitrile at room temperature was added triphenylphosphine (7.967 g, 30.37 mmol), and the reaction mixture was heated under reflux for 48 h under a nitrogen atmosphere. Completion of the reaction was confirmed by thin layer chromatography (TLC). The solvent was evaporated under reduced pressure, the crude product was washed with ethanol (2 × 30 ml), and the solid was dried under high vacuum without further purification to afford the title compound (0.95 mmol) as a white solid. The product was confirmed by 1H NMR and liquid chromatography coupled to mass spectrometry (LC–MS). 1H NMR (400 MHz, CDCl3) δ 7.92–7.75 (m, 9H), 7.71 (td, J = 7.5, 3.4 Hz, 6H), 3.87–3.71 (m, 2H), 3.63 (t, J = 5.4 Hz, 2H), 1.77–1.56 (m, 4H), 1.51 (d, J = 2.9 Hz, 4H). Mass spectrometry: m/z: calcd for [C24H28OP]+ ([M]+), 363.19; found, 363.16 (Supplementary Fig. 3a).
Synthesis of (6-hydroxyhexyl)triphenylphosphonium bromide esters
4-(Naphthalen-2-yl)-4-oxobutanoic acid, 4-(naphthalen-1-yl)-4-oxobutanoic acid and 4-(2,5-dimethylphenyl)-4-oxobutanoic acid were used for the synthesis of (6-(4-(naphthalen-2-yl)-4-oxobutanoyloxy)hexyl)triphenylphosphonium bromide (C23.20–TPP), (6-(4-(naphthalen-1-yl)-4-oxobutanoyloxy)hexyl)triphenylphosphonium bromide (C23.21–TPP) and (6-(4-(2,5-dimethylphenyl)-4-oxobutanoyloxy)hexyl)triphenylphosphonium bromide (C23.28–TPP) respectively (Supplementary Fig. 3b–d). To a stirred solution of the respective aryl-4-oxobutanoic acid (about 0.3 g, 1.31 mmol), (6-hydroxyhexyl)triphenylphosphonium bromide (0.583 g, 1.31 mmol) and N,N-dimethylpyridin-4-amine (DMAP; 0.176 g, 1.58 mmol) in anhydrous CH2Cl2 (15 ml) at 0 °C was added dicyclohexylcarbodiimide (0.271 g, 1.45 mmol) under a nitrogen atmosphere. Then the reaction mixture was brought to room temperature and stirred for 16 h. Completion of the reaction was confirmed by TLC. The reaction mixture was then cooled to −10 °C and the insoluble material was filtered off. The solid was washed with cold (−10 °C) CH2Cl2. The combined organic layer was then washed with aqueous 1 M HCl (15 ml), water (15 ml), saturated aqueous NaHCO3 (15 ml) and saturated aqueous NaCl (15 ml), and then dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by silica gel flash column chromatography using 5–10% MeOH in CH2Cl2 to afford the title compound, (about 0.687 g, 1.05 mmol). The products were confirmed by 1H NMR and LC–MS as follows (Supplementary Fig. 3b–d):
(6-(4-(Naphthalen-2-yl)-4-oxobutanoyloxy)hexyl)triphenylphosphonium bromide (C23.20–TPP): 1H NMR (400 MHz, CDCl3) δ 8.50 (s, 1H), 8.03–7.93 (m, 2H), 7.87 (ddd, J = 12.6, 5.5, 3.3 Hz, 7H), 7.81–7.73 (m, 3H), 7.73–7.65 (m, 5H), 7.64–7.49 (m, 2H), 4.12–4.00 (m, 2H), 3.99–3.84 (m, 2H), 3.44 (t, J = 6.6 Hz, 2H), 2.79 (t, J = 6.6 Hz, 2H), 1.72–1.49 (m, 6H), 1.36 (dt, J = 15.0, 7.5 Hz, 2H). Mass spectrometry: m/z: calcd for [C38H38O3P]+ ([M]+), 573.26; found, 573.21.
(6-(4-(Naphthalen-1-yl)-4-oxobutanoyloxy)hexyl)triphenylphosphonium bromide (C23.21–TPP): 1H NMR (400 MHz, CDCl3) δ 8.55–8.48 (m, 1H), 8.02–7.90 (m, 2H), 7.89–7.80 (m, 7H), 7.75 (tt, J = 12.0, 5.3 Hz, 3H), 7.71–7.61 (m, 6H), 7.56–7.45 (m, 3H), 4.13–4.01 (m, 2H), 3.97–3.83 (m, 2H), 3.40–3.30 (m, 2H), 2.86–2.75 (m, 2H), 1.73–1.51 (m, 6H), 1.36 (dt, J = 15.0, 7.5 Hz, 2H). Mass spectrometry: m/z: calcd for [C38H38O3P]+ ([M]+), 573.26; found, 573.31.
(6-(4-(2,5-Dimethylphenyl)-4-oxobutanoyloxy)hexyl)triphenylphosphonium bromide (C23.28–TPP): 1H NMR (400 MHz, CDCl3) δ 7.87 (ddd, J = 12.6, 5.2, 3.3 Hz, 6H), 7.81–7.74 (m, 3H), 7.73–7.63 (m, 6H), 7.48 (s, 1H), 7.17 (dd, J = 7.8, 1.2 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 4.04 (t, J = 6.5 Hz, 2H), 3.97 – 3.84 (m, 2H), 3.18 (t, J = 6.5 Hz, 2H), 2.69 (dd, J = 11.8, 5.4 Hz, 2H), 2.38 (s, 3H), 2.35 (s, 3H), 1.76–1.50 (m, 6H), 1.35 (dt, J = 15.0, 7.6 Hz, 2H). Mass spectrometry: m/z: calcd for [C36H40O3P]+ ([M]+), 551.27; found, 551.21.
2,4-Dioxo-4-phenylbutanoic acid and 4-(naphthalen-2-yl)-2,4-dioxobutanoic acid were used for the synthesis of (6-(2,4-dioxo-4-phenylbutanoyloxy)hexyl)triphenylphosphonium bromide (C23.07–TPP) and (6-(4-(naphthalen-2-yl)-2,4-dioxobutanoyloxy)hexyl)triphenylphosphonium bromide (C23.47–TPP), respectively (Supplementary Fig. 3e, f). To a stirred solution of the respective aryl-2,4-dioxobutanoic acid (about 200 mg, 1.04 mmol) and (6-hydroxyhexyl)triphenylphosphonium bromide (461 mg, 1.04 mmol) in anhydrous CH2Cl2 (15 ml) at 0 °C was added triethylamine (316 mg, 3.12 mmol), DMAP (165 mg, 1.35 mmol) and 2-chloro-1-methylpyridinium iodide (319 mg, 1.25 mmol) and stirred for 2 h at 0 °C. Completion of the reaction was confirmed by TLC. The reaction mixture was diluted with cold water and the product was extracted with CH2Cl2 (20 ml × 2). The combined organic layer was washed with aqueous 1 M HCl (15 ml), aqueous NaHCO3 (15 ml) and brine (15 ml), and was then dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was purified by silica gel flash chromatography (ethyl acetate/hexane) to afford the title compound (about 321 mg, 0.5 mmol) as a thick liquid. The product was confirmed by NMR and LC–MS as follows (Supplementary Fig. 3b-d):
(6-(2,4-Dioxo-4-phenylbutanoyloxy)hexyl)triphenylphosphonium bromide (C23.07–TPP): 1H NMR (400 MHz, CDCl3) δ 15.29 (s, 1H), 8.06–7.96 (m, 2H), 7.93–7.82 (m, 6H), 7.78 (dt, J = 7.3, 3.6 Hz, 2H), 7.73–7.65 (m, 6H), 7.62 (dd, J = 10.5, 4.2 Hz, 1H), 7.52 (t, J = 7.6 Hz, 2H), 7.06 (s, 1H), 4.33–4.23 (m, 2H), 4.01–3.88 (m, 2H), 1.84–1.53 (m, 6H), 1.49–1.33 (m, 2H). Mass spectrometry: m/z: calcd for [C34H34O4P]+ ([M]+), 537.22; found, 537.31.
(6-(4-(Naphthalen-2-yl)-2,4-dioxobutanoyloxy)hexyl)triphenylphosphonium bromide (C23.47–TPP): 1H NMR (400 MHz, CDCl3) δ 15.32 (s, 1H), 8.55 (s, 1H), 8.05–7.97 (m, 2H), 7.92 (dd, J = 16.7, 8.4 Hz, 2H), 7.85–7.74 (m, 3H), 7.74–7.64 (m, 11H), 7.64–7.53 (m, 2H), 7.21 (s, 1H), 4.32 (t, J = 6.5 Hz, 2H), 3.35 (dd, J = 12.5, 7.4 Hz, 2H), 1.81–1.69 (m, 2H), 1.65 (d, J = 3.8 Hz, 4H), 1.50–1.36 (m, 2H). Mass spectrometry: m/z: calcd for [C38H36O4P]+ ([M]+), 587.23; found, 587.32.
Synthesis of 4-(naphthalen-2-yl)-2,4-dioxobutanoic acid (C23.47)
To a stirred solution of ethyl 4-(naphthalen-2-yl)-2,4-dioxobutanoate (504 mg, 1.86 mmol) in methanol (10 ml), tetrahydrofuran (10 ml) and water (2 ml) at room temperature was added lithium hydroxide monohydrate (235 mg, 5.59 mmol) and the reaction mixture was stirred for 6 h at room temperature. Completion of the reaction was confirmed by TLC. The volatile compounds were evaporated under reduced pressure to yield the crude product, which was acidified with aqueous 1 M HCl (20 ml), and the product was then extracted with ethyl acetate (30 ml × 2). The combined organic layers were washed with brine (10 ml), dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The resulting crude product was purified by silica gel flash chromatography (ethyl acetate/hexane) to afford the title compound (406 mg, 1.68 mmol) as a white solid. The product was confirmed by NMR and LC–MS as follows (Supplementary Fig. 3g): 1H NMR (400 MHz, DMSO) δ 14.33 (s, 2H), 8.82 (s, 1H), 8.20 (d, J = 8.0 Hz, 1H), 8.10–7.93 (m, 3H), 7.76–7.58 (m, 2H), 7.23 (s, 1H). Mass spectrometry: m/z: calcd for [C14H11O4]+ ([M+H]+), 243.07; found, 243.14.
Synthesis of 3-(dimethylamino)propyl-4-(naphthalen-2-yl)-2,4-dioxobutanoate (C23.47–DAP)
To a stirred solution of 4-(naphthalen-2-yl)-2,4-dioxobutanoic acid (100 mg, 0.41 mmol) in anhydrous CH2Cl2 (7 ml) at 0 °C was added 3-(dimethylamino)propan-1-ol (0.62 mg, 64 mmol), triethylamine (125 mg, 1.24 mmol), DMAP (65 mg, 0.54 mmol) and 2-chloro-1-methylpyridinium iodide (127 mg, 0.49 mmol), and stirred for 1 h at 0 °C. Completion of the reaction was confirmed by TLC. The reaction mixture was diluted with cold water and the product was extracted with CH2Cl2 (10 ml × 2). The combined organic layers were washed with aqueous 1 M HCl (10 ml), aqueous NaHCO3 (10 ml) and brine (10 ml), and then dried over anhydrous Na2SO4, The solvent was evaporated under reduced pressure and the crude product was purified by silica gel flash chromatography (ethyl acetate/hexane) to afford the title compound (81 mg, 0.25 mmol) as a white solid. The product confirmed by NMR and LC–MS as follows (Supplementary Fig. 3h): 1H NMR (400 MHz, CDCl3) δ 12.15 (s, 1H), 8.51 (d, J = 53.1 Hz, 1H), 8.12–7.79 (m, 4H), 7.73–7.46 (m, 2H), 7.25 (s, 1H), 4.46 (t, J = 5.9 Hz, 2H), 3.26 (dd, J = 21.8, 14.1 Hz, 2H), 2.92 (s, 6H), 2.47–2.20 (m, 2H). Mass spectrometry: m/z: calcd for [C19H22NO4]+ ([M+H]+), 328.38; found, 328.15.
Synthesis of ethyl esters
The synthetic steps were identical to those for the synthesis of (6-hydroxyhexyl)triphenylphosphonium bromide esters described above. Ethanol was used for esterification in place of (6-hydroxyhexyl)triphenylphosphonium bromide. To a stirred solution of the respective aryl-2,4-dioxobutanoic acid (about 100 mg, 0.52 mmol) in anhydrous CH2Cl2 (8 ml) at 0 °C was added ethanol (72 mg, 1.56 mmol), triethylamine (158 mg, 1.56 mmol), N,N-dimethylpyridin-4-amine (DMAP; 83 mg, 0.68 mmol) and 2-chloro-1-methylpyridinium iodide (159 mg, 0.62 mmol) and stirred for 1 h at 0 °C. Completion of the reaction was confirmed by TLC. The reaction mixture was diluted with cold water and the product was extracted with CH2Cl2 (10 ml × 2). The combined organic layers were washed with aqueous 1 M HCl (10 ml), aqueous NaHCO3 (10 ml) and brine (10 ml), and then dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by silica gel flash chromatography (ethyl acetate/hexane) to afford the title compound (80 mg, 0.36 mmol) as a white solid. The product was confirmed by NMR and LC–MS as follows (Supplementary Fig. 3i–l).
Ethyl 4-(naphthalen-2-yl)-2,4-dioxobutanoate (C23.20–EA): 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H), 8.03 (dt, J = 15.2, 7.6 Hz, 1H), 8.00–7.93 (m, 1H), 7.88 (t, J = 8.3 Hz, 2H), 7.65–7.46 (m, 2H), 4.18 (q, J = 7.1 Hz, 2H), 3.45 (t, J = 6.7 Hz, 2H), 2.82 (t, J = 6.7 Hz, 2H), 1.28 (t, J = 7.1 Hz, 3H). Mass spectrometry: m/z: calcd for [C16H17O3]+ ([M+H]+), 257.12; found, 257.14.
Ethyl 4-(naphthalen-1-yl)-4-oxobutanoate (C23.21–EA): 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H), 8.04 (dd, J = 8.6, 1.7 Hz, 1H), 7.96 (t, J = 8.4 Hz, 1H), 7.89 (t, J = 8.4 Hz, 2H), 7.65–7.48 (m, 2H), 4.18 (q, J = 7.1 Hz, 2H), 3.46 (t, J = 6.7 Hz, 2H), 2.83 (q, J = 6.6 Hz, 2H), 1.28 (t, J = 7.1 Hz, 3H). Mass spectrometry: m/z: calcd for [C16H17O3]+ ([M+H]+), 257.12; found, 257.14.
Ethyl 4-(2,5-dimethylphenyl)-4-oxobutanoate (C23.28–EA): 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 7.16 (dt, J = 23.2, 4.5 Hz, 2H), 4.16 (q, J = 7.1 Hz, 2H), 3.20 (dd, J = 8.8, 4.4 Hz, 2H), 2.81–2.64 (m, 2H), 2.44 (s, 3H), 2.36 (s, 3H), 1.27 (td, J = 7.1, 2.3 Hz, 3H). Mass spectrometry: m/z: calcd for [C14H19O3]+ ([M+H]+), 235.13; found, 235.24.
Ethyl 2,4-dioxo-4-phenylbutanoate (C23.07–EA): 1H NMR (400 MHz, CDCl3): δ 15.30 (s, 1H), 8.06–7.96 (m, 2H), 7.66–7.57 (m, 1H), 7.55–7.46 (m, 2H), 7.08 (s, 1H), 4.41 (q, J = 7.1 Hz, 2H), 1.42 (t, J = 7.1 Hz, 3H). Mass spectrometry: m/z: calcd for [C12H13O4]+ ([M+H]+), 221.08; found, 221.14.
Prodrug uptake and cleavage
Escherichia coli (108 cells) were treated with different concentrations (10–5,000 nM) of the prodrug C23.28–TPP for 30 min. The bacteria were washed in DPBS, lysed by freeze-thawing 5 times in liquid nitrogen and the lysate treated with acetonitrile to a final concentration of 50%. Lysates were spun down at 5,000g, passed through 0.45-μm filters and analysed by LC–MS.
Conversion of HMBPP to DMAPP and IPP
Escherichia coli IspH was incubated with varying concentrations (10–5,000 nM) of TPP (control) or the IspH inhibitor C23.28 for 10 min. A methyl viologen assay as described above was performed with final concentrations of IspH and HMBPP of 50 nM and 1 mM, respectively. At 30 min the reaction was stopped by the addition of acetonitrile to a final concentration of 50%. Purified HMBPP and DMAPP/IPP were used as benchmarks and to obtain a dilution curve. Samples were analysed by LC–MS for the presence of HMBPP and DMAPP/IPP.
Plasma stability of prodrugs
The in vitro stabilities of the prodrugs C23.20–TPP, C23.21–TPP and C23.28–TPP were measured in human (Sigma, P9523) mouse (Sigma, P9275) and pig (Sigma, P2891) plasma. The lyophilized plasma was reconstituted with the recommended volume of 0.05 M PBS (pH 7.4) to a concentration of 100% and prewarmed at 37 °C. The reactions were initiated by the addition of the prodrugs to preheated plasma solution to yield a final concentration of 100 μM. A positive control solution without the addition of plasma was also included to monitor compound stability over the course of the experiment. The assays were incubated at 37 °C and shaken at 200 rpm. Samples (50 μl) were taken at 0, 15, 30, 45, 60 and 120 min and added to 200 μl acetonitrile to deproteinize the plasma. The samples were vortexed for 1 min and centrifuged at 4 °C for 15 min at 20,000g. The clear supernatants were transferred to LC–MS vials for analysis.
Liver microsome stability of prodrugs
The in vitro stabilities of the prodrugs C23.20–TPP, C23.21–TPP and C23.28–TPP were measured in human (Sigma, M0317) mouse (Sigma, M9441) and monkey (Sigma, M8816) liver microsomes. A stock solution of the prodrug was added to a solution of 0.1 M PBS (pH 7.4) containing 1 mM NADPH to a final concentration of 100 μM. This solution was incubated at 37 °C for 5 min at which time microsomes were added at a final concentration of 1.0 mg ml−1, incubated at 37 °C and shaken at 200 rpm. A positive control solution without the addition of microsomes was also included to monitor compound stability over the course of the experiment. Aliquots were removed at 0, 15, 30, 60, 90, 120 min and 10× volume of acetonitrile was added to stop the reaction and deproteinate the sample. Samples were centrifuged at 20,000g for 5 min at 4 °C, and the supernatant was transferred to LC–MS vials for analysis.
LC–MS quantification of small molecules
LC–MS analysis was performed on a Thermo Fisher Scientific Q Exactive HF-X mass spectrometer equipped with a HESI II probe and coupled to a Thermo Fisher Scientific Vanquish Horizon UHPLC system. IPP/DMAPP and HMBPP were analysed by hydrophilic interaction chromatography (HILIC) on a ZIC-pHILIC 2.1-mm i.d × 150 mm column (EMD Millipore). The HILIC mobile phase A was 20 mM ammonium carbonate, 0.1% ammonium hydroxide, pH 9.2, and mobile phase B was acetonitrile. Prodrug compounds were analysed by reversed phase (RP) chromatography on a Synergi 4 mm Polar-RP 2-mm i.d × 100 mm column (Phenomenex). The RP mobile phase A was 0.1% formic acid in MilliQ water, and mobile phase B was 0.1% formic acid in acetonitrile. Peak areas for each compound were integrated using TraceFinder 4.1 software (Thermo Fisher Scientific).
Determination of prodrug stability
The calibration curves used to determine prodrug and drug concentrations ranged from 50 μM to 0.012 μM with twofold serial dilutions (13 points in duplicate) and were generated from LC–MS quantifications using TraceFinder 4.1 software (Thermo Fisher Scientific). Data points were plotted in GraphPad and respective half-lives (t 1/2) were calculated using the expression t 1/2 = 0.693/k where k is the rate constant. Relevant supporting information can be found in the Source Data file.
Bacterial viability and prodrug treatment
Escherichia coli or clinical isolates of E. aerogenes (CRE) (UCI 15), K. pneumoniae 1.53 (ST147+, CTX-M15+), S. enterica subsp. enterica serovar Typhimurium (LT2 – SL7207), V. cholerae (M045), A. baumannii (BC-5), A. baumannii (AB5075-UW), P. aeruginosa (PA14), P. aeruginosa (MSRN 5524), H. pylori (Hp CPY6081), B. sphaericus (CCM 2177), M. tuberculosis (M. tuberculosis H37Ra) and Y. pestis (KIM 10+) were cultured to late log phase (108 cells per ml) in their respective culture medium and quantified by measuring the optical density at 600 nm (OD600) for 3 serial dilutions. The bacteria were spun down, resuspended in RPMI medium supplemented with 10% FBS or human serum at a concentration of 105 cells per ml and aliquoted at 100 μl per well into a 96-well plate. Varying concentrations of candidate prodrugs (4–500 μM final concentration) were added and incubated for 1–4 h (4 days for M. tuberculosis) at 37 °C. Bacterial viability from each sample was tested by CFU, resazurin blue (colorimetric and fluorescence) and growth curve assays. For proteomics and electron microscopy the bacteria were treated with the respective prodrugs for 8 and 24 h. The following antibiotics were used to compare bacterial killing potency with our prodrugs: meropenem (Sigma, 1392454), amikacin (Sigma, A0365900), ceftriaxone (Sigma, C0691000), cefepime (Sigma, 1097636), ciprofloxacin (Sigma, 17850), tobramycin (Sigma, T4014), ceftaroline (Bocsci, B0084-459128), kanamycin (Sigma, B5264), chloramphenicol (Sigma, C0378), ampicillin (Sigma, A9518), doxycycline (Sigma, D3447), gentamicin (Sigma, G1264) and streptomycin (Sigma, S6501).
Resazurin blue assay
Control or prodrug-treated bacterial samples were treated with resazurin sodium salt (Sigma R7017) at a final concentration of 0.02% and incubated for 4 h (overnight for M. tuberculosis) at 37 °C in a Biotek Synergy 2 plate reader. Changes in fluorescence were measured every 20 min for 16 h (3 days for M. tuberculosis), with discontinuous shaking, using excitation filter range 530–570 nm and emission filter range 590–620 nm. An increase in fluorescence intensity corresponds to bacterial growth and is quantified by comparison with untreated bacterial control samples. The ratio of (T threshold (untreated)/T threshold (prodrug-treated)) (where T = time) was used to quantify the change in bacterial growth. To minimize inter-experimental variations, all T threshold times were corrected by subtracting the time taken for untreated control cultures to reach minimum detectable fluorescence. At the end of the experiment, wells were visualized for changes in colour from blue (inviable bacteria) to pink (viable bacteria) or by measuring the fluorescence at the aforementioned excitation and emission wavelengths.
Measurement of bacterial membrane integrity by SYTO 9/propidium iodide assay
Escherichia coli cells grown to late log phase (108 cells per ml) were treated with TPP (control) or DAIA prodrugs at varying concentrations in RPMI + 10% FBS. Bacteria were spun down and washed three times in Tris buffered saline (TBS) (pH 7.5). Component A (SYTO 9 dye) (1.5 μl ml−1) and component B (propidium iodide) from the BacLight Live/Dead kit (Life Tech, L7012) were added to the bacterial samples and incubated for 15 min. An aliquot was run for flow cytometry on a BD LSR II instrument (BD Biosciences). With the excitation wavelength centred at about 485 nm, the fluorescence intensities at 530 nm (green) and 630 nm (red) were measured and the data analysed using FlowJo software. TPP- or isopropanol-treated bacteria served as negative or positive controls, respectively, and their flow plots were used to gate the prodrug-treated samples. As bacteria lose their membrane integrity the green SYTO 9 dye is displaced by the red propidium iodide dye. The remaining samples were spun down at 5,000g for 10 min, resuspended in 10 μl of TBS, spread on glass microscopy slides and dried. The samples were mounted using Cytoseal 60 or Mounting Medium (Electron Microscopy Sciences). Specimens were documented photographically using an 80i upright microscope and analysed with the NIS-Elements Basic Research software.
Measuring respiration using a Seahorse XF Analyzer
On the day before the assay, the sensor cartridges from the Seahorse XFe96 FluxPaks (Agilent, 102416) were calibrated according to the manufacturer’s instructions using pre-warmed Seahorse XF Calibrant. Escherichia coli cells were grown in LB medium overnight to an OD600 of 0.3, washed in PBS and resuspended in Seahorse XF RPMI medium, pH 7.4 (Agilent, 103576) supplemented with 1% glucose. Bacteria (105, 106 or 107) were added to XF Cell Culture Microplates (Agilent, 101085-004) precoated with poly-d-lysine and spun down at 2,000g for 10 min to attach them to the plate. The wells in the plate were divided to include bacteria treated with TPP (negative control) and 3 concentrations (500, 100 and 20 μM) of C23.28–TPP; 8 technical replicates were used for each condition. Fresh medium (90 μl) was added to each well and 90 μl of TPP or prodrug solution was added to each injection port A. The baseline oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured for 12 min, after which the TPP or prodrug solution was injected into each sample. Readings were obtained as pmol min−1 (OCR) and mpH min−1 (ECAR) every 6 min for up to 90 min. The mean of the 8 technical replicates was plotted for each treatment condition and changes in OCR and ECAR were compared to the control samples.
Detection of superoxide and H2O2
Superoxide anion was measured in prodrug- and TPP-treated bacteria by diluting them 1/50 into PBS containing 2 μM dihydroethidium (Sigma, D7008) just before flow cytometry (excitation, 535 nm; emission, 610 nm). H2O2 production was measured in similar bacterial samples using the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Thermo Fisher, A22188). Fluorescence measurements were calibrated by comparison to calibration curves for wells containing H2O2 in a final concentration ranging between 0.1 to 100 μM. Fluorescence was measured using the 540/620 nm wavelength pair in a Biotek Synergy 2 plate reader.
Staining for bacterial membrane potential
The procedure for studying the changes in membrane potential in prodrug-treated E. coli was identical to that used for the Live/Dead assay with the following exceptions. The BacLight Bacterial Membrane Potential Kit (Lifetech, B34950) was used in this case. Component A (10 μl) (3 mM 3,3′-diethyloxacarbocyanine (DiOC2)) was used to stain the bacterial samples for 30 min at room temperature. TPP- or component B (carbonyl cyanide m-chlorophenyl hydrazine (CCCP))-treated bacteria were used as negative or positive controls, respectively, and to gate prodrug-treated samples. When the membrane potential is intact, the DiOC2 dye forms tetramers within bacteria that fluoresce at 630 nm (red). Loss of membrane potential leads to the formation of dimers that fluoresce at 530 nm (green). Bacteria were analysed by both flow cytometry and microscopy.
Transmission electron microscopy
Bacteria (E. coli or V. cholerae) were treated with TPP or with the prodrug C23.28–TPP in RPMI medium with 10% FBS for 0, 8 or 24 h. The ΔispH conditional knockdown E. coli was cultured similarly in the presence of 1% dextrose for 8 or 24 h to inhibit IspH expression. At respective time points the samples were fixed in 2.5% glutaraldehyde, 2% paraformaldehyde at 4 °C in 100 mM cacodylate buffer (pH 7.0) containing 2 mM CaCl2 and 0.2% picric acid. Samples were briefly washed and treated for 2 h at 4 °C with 1% osmium tetroxide in 100 mM cacodylate buffer (pH 7.0). After washing with distilled water 3–5 times, samples were dehydrated using increasing ethanol concentrations and embedded in Epon resin (Sigma-Aldrich). Ultrathin sections of the embedded samples were cut and loaded onto grids and stained further with Reynold’s lead citrate (Sigma-Aldrich) for 3–15 min. Grids were dried overnight and observed using a JEOL 1010 transmission electron microscope equipped with an AMT 2k CCD camera.
Scanning electron microscopy
Scanning electron microscope experiments were carried out at the CDB Microscopy Core (Perelman School of Medicine, University of Pennsylvania). Bacterial samples were washed three times with 50 mM Na-cacodylate buffer, fixed for 2–3 h with 2% glutaraldehyde in 50 mM Na-cacodylate buffer (pH 7.3), spun down over 0.22-μm filter membranes and dehydrated in an increasing ethanol concentration over a period of 1.5 h. Dehydration in 100% ethanol was performed three times. Dehydrated samples were incubated for 20 min in 50% hexamethyldisilane (HMDS; Sigma-Aldrich) in ethanol followed by 100% HMDS (refreshed three times) and then overnight air-drying as described previously49. Samples were then mounted on stubs and sputter-coated with gold–palladium. Specimens were observed and photographed using a Quanta 250 FEG scanning electron microscope (FEI) at 10 kV accelerating voltage.
Toxicity assays in mammalian cell lines
The cytotoxicity of prodrugs to C2C12, HepG2, Raw 264.7 and Vero cells was estimated using an LDH-GloTM cytotoxic assay kit (Promega, J2381). Cells were grown, counted, aliquoted in 96-well plates at a cell density of 105 cells per well and allowed to adhere to the bottom of the wells for 1 day at 37 °C and 5% CO2. The cells were treated with prodrugs at different concentrations (1–5,000 μM). Cells treated with 2% DMSO served as negative control whereas cells treated with 0.2% Triton X-100 served as positive control for cytotoxicity. Eight replicates were performed for each condition. Supernatant medium samples were taken from each well at intervals of 24, 48 and 72 h, diluted 300-fold in PBS, added to the lactate dehydrogenase (LDH) assay reagent in a 1:1 ratio (20 μl:20 μl) in white opaque 96-well plates and further incubated at room temperature for 1 h in the dark. Luminescence was measured using a Biotek Synergy 2 plate reader with integration time 1 s per well. Cytotoxicity was calculated using the following equation: % cytotoxicity = 100 × (experimental LDH release − medium background)/(maximum LDH release control − medium background).
Measurement of mitochondrial membrane potential
To quantify the effect of IspH prodrugs on the mitochondrial membrane potential of C2C12 myoblasts (ATCC, CRL-1772), cells were grown in DMEM + 10% FBS up to 90% confluence and suspended by trypsinization. Cells were washed and pelleted at 500g for 5 min and resuspended in DMEM consisting of 100 nM of tetramethyl rhodamine methyl ester (Thermo Fisher, I34361) for 30 min at 37 °C with slow shaking. Myoblasts were pelleted down and resuspended in PBS. One million cells were incubated with 1, 10 or 100 μM concentrations of TPP, IspH prodrugs or CCCP (Invitrogen, B34950) for 10 min at room temperature. CCCP is an ox-phos uncoupler that causes loss of mitochondrial membrane potential and is used as a positive control. After 10 min cells were analysed by flow cytometry according to the manufacturer’s instructions and the plots were gated using negative control (unstained cells).
Profiling the effect of the TPP carrier molecule on hERG channel
Compound profiling against hERG—to evaluate the potential cardiac liability of 6-hydroxyhexyl TPP, methyl TPP and our prodrug C23.28–TPP—was carried out at Reaction Biology using the QPatch HTX fully automated patch-clamp platform that enables the testing of up to 48 cells in parallel. Electrophysiological profiling was done in the presence of verapamil (positive control), DMSO (vehicle control) and the TPP compounds at a concentration range of 10 nM to 10 μM (n = 3 cells per sample concentration × 6 concentrations). Exemplar hERG traces were elicited from a holding potential of −80 mV followed by steps from −60 to +50 mV in 10 mV increments; tail currents were elicited by a step to −50 mV. Response data obtained were normalized to peak current at 0.1% DMSO. Nonlinear regression curve fits were used to calculate the IC50 of each compound.
Protein isolation and western blot analysis
Bacterial samples were washed with PBS and treated with 10 mg ml−1 lysozyme in 20 mM Tris-HCl, pH 8.0; 2 mM EDTA at 37 °C for 30 min. Lysates are prepared by freeze-thawing in Ripa lysis buffer (10 mM Tris-Cl pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl) supplemented with protease inhibitors at 4 °C. Whole-cell lysates (100 μg per reaction) were mixed with an equal volume of 2× SDS–PAGE sample buffer supplemented with 10% 2-mercaptoethanol and heated for 5 min at 100 °C. Protein samples were size-fractionated on 4–20% Tris-glycine gradient gels (Lonza) or lab-made 12.5% Tris-glycine gels using constant voltage at room temperature, transferred overnight onto Immuno-Blot PVDF membranes (Bio-Rad, 162-0177) at 4 °C and subjected to protein blotting using the mouse anti-E.coli RNA Sigma 70 antibody (BioLegend, 663208) or rabbit anti-E. coli IspH antibody (generated in this work). Both primary antibodies show cross-reactivity across several bacterial species. Secondary antibodies conjugated to horseradish peroxidase were used at a dilution of 1:10,000 (GE Healthcare, NA931V, NA934V). The immunoblots were scanned using Image Quant LAS 4000. Uncropped western blots with molecular weight markers are shown in Supplementary Fig. 1.
Proteomics
Triplicate samples of C23.28–TPP-treated or ΔispH E. coli lysates at 0, 8 and 24 h (a total of 18 samples) were processed. Protein samples were concentrated (up to eightfold) by lyophilization and 25 μg from each sample was separated by SDS–PAGE for a distance of 0.5 cm into the gel. The entire lanes were excised, digested with trypsin and analysed by LC–MS/MS on a Q Exactive HF mass spectrometer using a 240 min LC gradient. Tandem mass spectrometry (MS/MS) spectra were searched with full tryptic specificity against the UniProt E. coli database (https://www.uniprot.org; accessed 7 December 2019) using the MaxQuant 1.6.3.3 program. The ‘match between runs’ feature was used to help to transfer identifications across experiments to minimize missing values. Protein quantification was performed using razor and unique peptides. False discovery rates (FDRs) for protein and peptide identifications were set at 1%. A total of 2,346 protein groups were identified, including proteins identified by a single razor and unique peptide. Label-free quantitation (LFQ) intensity was used for protein quantification50. The LFQ intensity levels were log2-transformed and undetected intensities were floored to a minimum detected intensity across all proteins or a minimum across 4 samples in the case that both replicates were undetected.
Bioinformatics analysis
Unpaired t-tests were performed to estimate the significance of difference between conditions, and false discovery rate was estimated using a procedure described previously51. Proteins that passed the P < 0.05 threshold were considered significant (all passed FDR <5% threshold). A total of 525 proteins changed in both the IspH prodrug treatment and the ΔispH conditional knockdown systems. Proteins showing more than 2-fold up- or downregulation under both conditions and at both 8- and 24-h time points were analysed using Venny52. Enrichment analysis of proteins common to both conditions and time points was performed using Search Tool for the Retrieval of Interacting Genes/Proteins (STRING)34. Functions with at least 5 differentially expressed proteins enriched at the P < 0.001 threshold were considered.
Antibiotic-resistance assays with DAIAs
For this study, K. pneumoniae and V. cholerae clinical strains mentioned in the section ‘Bacteria’ were cultured to exponential phase in RPMI medium containing 10% FBS. The bacteria were washed in DBPS and quantified by OD600. The bacteria were aliquoted into identical samples containing 105 CFU. These aliquots were used to start fresh cultures in RPMI medium containing 5% human serum, in the presence or absence of 106 human PBMCs, and for each condition in the presence or absence of the prodrug C23.28–TPP. Hygromycin and streptomycin were used as control antibiotics for Vibrio and Klebsiella, respectively. After 8 h of incubation at 37 °C and 5% CO2, 50 μl of each sample was plated by serial dilution for the measurement of CFU, and the rest of the cultures were allowed to grow. After 24 h of incubation, bacteria from each sample were washed and quantified by OD600. Bacteria (105) from the respective samples in the first passage were used to start the next passage (cycle of selection by antibiotic) under the same conditions. For samples co-incubated with human PBMCs, fresh PBMCs from the same donor were used for every passage. Bacterial growth in each passage was measured up to the 18th passage. Bacterial growth (CFU) in the absence of antibiotics was considered as 100% growth and resistance to an antibiotic in each passage was defined as the percentage of bacterial growth that occurred in the presence of that antibiotic.
Flow cytometry
Cells were washed with 2 ml of 1× PBS at 1,500 rpm for 5 min and then stained with 1 ul of Zombie Yellow (BioLegend, 423103) for 20 min at room temperature to check the viability. The cells were stained for cell surface markers with a combination of (where indicated) CD3- PerCP-Cy5.5 (clone UCHT1, BD Biosciences, 560835), CD4-Alexa Fluor 700 (clone RPA-T4, BD Biosciences, 557922), CD8a-Brilliant Violet 711 (clone RPA-T8, BioLegend, 301044), TCR Vγ9-FITC (clone 7A5, Invitrogen, TCR2720), CD69- PE/Cy7 (clone FN50, BD Biosciences, 557745), HLA-DR-Brilliant Violet 421 (clone L243, BioLegend, 307636), for 20 min in FACS buffer (1% FBS in PBS) at room temperature. Next the cells were washed with PBS, fixed and permeabilized using a Fixation/Permeabilization Kit (BD Biosciences, 554714) for 15 min at 4 °C. After washing with 1 ml of 1× permeabilization buffer, intracellular proteins were stained using Perforin-Brilliant Violet 421 (clone dG9, BioLegend, 308122), Granulysin-Alexa Fluor 647 (clone DH2, Bio Legend, 348006), Granzyme A-PE/Cy7 (clone CB9, Bio Legend, 507222). Cells were washed with 1× permeabilization buffer twice. The cells were resuspended in 300 μl of 1% paraformaldehyde fixation buffer (BioLegend, B244799) in PBS. Samples were run on BD LSR II (BD Biosciences) and the data analysed using FlowJo software. Cells were first gated for lymphocytes (forward scatter/side scatter (FSC/SSC)) then singlets (FSC-A vs FSC-H). The singlets were further analysed for their uptake of the Live/Dead Aqua or Zombie Yellow stain to determine live and dead cells. Live cells were gated for CD3+ cells then gated for their identifying surface markers—CD4, CD8 and Vγ9 (γδ T lymphocytes)—followed by their respective cytotoxic markers perforin, granulysin and granzyme A or cell surface markers of T cell activation such as HLA-DR, and CD69. Gating strategy for every FACS plot is shown in the Source Data.
Validating the anti-Vδ2-TCR antibody for immunofluorescence
Human PBMCs from one donor were split into two aliquots; one sample was treated with 10 μM HMBPP and 50 ng ml−1 IL-15 to expand Vγ9Vδ2 T cells and the other sample was depleted of all γδ T cells using Anti-TCRγ/δ Microbead Kit (Miltenyi, 130-050-701). HepG2 and Vero cells served as negative control. Cells (106 of each type) were collected in Eppendorf tubes and washed with 1× PBS 3 times. The cell pellet was resuspended in PBS and fixed for 20 min by adding formaldehyde to a final concentration of 4%. Fixed cells were washed with 1× PBS, pelleted and embedded in 100 μl of 4% agar (Fisher Scientific, BP14232). The agar block was then treated with 70% ethanol before paraffin embedding and sectioning at the Wistar Histotechnology Facility. For immunofluorescence studies, sections were deparaffinized in xylene, then rehydrated in ethanol (100–95% to 80–70%) and distilled water. The endogenous peroxidase activity was eliminated by treating the sections with 0.5% hydrogen peroxide in methanol for 10 min. The slides were washed under tap water for 5 min before simmering them in Tris-EDTA buffer. The slides were washed with PBS before blocking them in 5% BSA blocking solution for 1 h. The tissue sections were subsequently incubated with primary anti Vδ2-TCR primary antibody (BioLegend, 331402) 1:50 in 5% BSA overnight at 4 °C, washed next day with 1× PBS and incubated with AF647 (Invitrogen, A21236) secondary antibody 1:200 for 45 min. DAPI was added for 5 min and the sections mounted using Cytoseal 60 or Mounting Medium (Electron Microscopy Sciences). Specimens were documented photographically using a Leica TCS SP5 Scanning Confocal Microscope and analysed with the NIS-Elements Basic Research software. The validation images are shown in Extended Data Fig. 11b, c.
Tissue staining and immunofluorescence
Tissues were collected and fixed in Formalde-Fresh Solution overnight at 4 °C, washed with 1× PBS and transferred to 70% ethanol before paraffin embedding and sectioning. Tissue embedding and sectioning were performed by the Histotechnology Facility at The Wistar Institute. For immunohistochemistry studies, tissue sections were deparaffinized in xylene, rehydrated in ethanol (100–95% to 80–70%) and distilled water. The endogenous peroxidase activity was quenched with 0.5% hydrogen peroxide in methanol for 10 min. The slides were washed under tap water for 5 min, simmered in Tris-EDTA buffer, and washed with PBS before blocking in 5% BSA blocking solution for 1 h. The tissue sections were subsequently incubated with anti Vδ2-TCR primary antibody (BioLegend, 331402) and anti-E. coli antibody (Abcam, ab137967) 1:50 in 5% BSA overnight at 4 °C, washed the next day with 1× PBS and incubated with AF647 (Invitrogen, A21236) and AF488 (BioLegend, 406416) secondary antibodies 1:200 for 45 min. DAPI (1:5,000) was added for 5 min and the samples mounted using Cytoseal 60 or Mounting Medium (Electron Microscopy Sciences). Specimens were photographed using an 80i upright microscope and analysed with the NIS-Elements Basic Research software.
Software used for data collection
NIS-Elements Basic Research (Nikon) v.4.60.00; FlowJo (FlowJo LLC) v.10; Internal Coordinate Mechanics software (ICM) (MolSoft) v.3.7-2a; Virtual Ligand Screening (VLS) (MolSoft) v.3.7-2a; Seahorse Wave controller software v.2.4.2 (Agilent).
Software used for data analysis
Microsoft Office 2016; Prism 7 v.7.04 (GraphPad); MaxQuant v.16.3.3; Max Planck Institute Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) v.11; Venny v.2.1; TraceFinder v.4.1; ICM Browser v.3.7-2a (Molsoft); Seahorse Wave analysis software (Agilent) v.2.4.2; ChemDraw v.19.1.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Molecular docking studies were performed using the E. coli IspH structure 3KE8 from the Protein Data Bank. The atomic field property of the IspH binding pocket was mapped using the Internal Coordinate Mechanics (ICM) software (http://www.molsoft.com/icm_pro.html) from Molsoft and the molecular docking of 10 million compounds from the MolCart library (https://www.molsoft.com/molcart.html) was carried out using theVirtual Ligand Screening software (https://molsoft.com/vls.html) from Molsoft. Owing to the lack of a suitable online repository, all docking data are available upon request as an .icb file, viewable using the free ICM browser (http://www.molsoft.com/icm_browser.html). LC–MS/MS spectra were searched against the UniProt E. coli (BL21-DE3) database (https://www.uniprot.org/proteomes/UP000002032). The proteomics data are available on MassIVE (https://massive.ucsd.edu/) using the accession number MSV000086359, or they can be downloaded from ftp://massive.ucsd.edu/MSV000086359/. All reagents used or generated and all data that support the findings of this study are available from the authors on reasonable request, see author contributions for contacts for specific datasets. Source data are provided with this paper.
Change history
27 September 2021
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41586-021-03961-x
References
Odom, A. R. Five questions about non-mevalonate isoprenoid biosynthesis. PLoS Pathog. 7, e1002323 (2011).
Jomaa, H. et al. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 285, 1573–1576 (1999).
McAteer, S., Coulson, A., McLennan, N. & Masters, M. The lytB gene of Escherichia coli is essential and specifies a product needed for isoprenoid biosynthesis. J. Bacteriol. 183, 7403–7407 (2001).
Rhodes, D. A. et al. Activation of human γδ T cells by cytosolic interactions of BTN3A1 with soluble phosphoantigens and the cytoskeletal adaptor periplakin. J. Immunol. 194, 2390–2398 (2015).
Chien, Y. H., Meyer, C. & Bonneville, M. γδ T cells: first line of defense and beyond. Annu. Rev. Immunol. 32, 121–155 (2014).
Chen, Z. W. Multifunctional immune responses of HMBPP-specific Vγ2Vδ2 T cells in M. tuberculosis and other infections. Cell. Mol. Immunol. 10, 58–64 (2013).
Alberts, B. et al. Molecular Biology of the Cell 4th edn (Garland Science, 2002).
Lieberman, J. in Fundamental Immunology 7th edn (ed. Paul, W. E.) Ch. 37 (Lippincott, Williams & Wilkins, 2012).
Magill, S. S. et al. Multistate point-prevalence survey of health care-associated infections. N. Engl. J. Med. 370, 1198–1208 (2014).
WHO. Tuberculosis https://www.who.int/en/news-room/fact-sheets/detail/tuberculosis (World Health Organization, 2020).
WHO. Artemisinin resistance and artemisinin-based combination therapy efficacy https://www.who.int/docs/default-source/documents/publications/gmp/who-cds-gmp-2019-17-eng.pdf?ua=1 (World Health Organization, 2019).
Wright, G. D. Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv. Drug Deliv. Rev. 57, 1451–1470 (2005).
Li, X. Z., Plésiat, P. & Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 28, 337–418 (2015).
Wilson, D. N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 12, 35–48 (2014).
Dotiwala, F. et al. Granzyme B disrupts central metabolism and protein synthesis in bacteria to promote an immune cell death program. Cell 171, 1125–1137.e11 (2017).
Walch, M. et al. Cytotoxic cells kill intracellular bacteria through granulysin-mediated delivery of granzymes. Cell 157, 1309–1323 (2014).
Dotiwala, F. et al. Killer lymphocytes use granulysin, perforin and granzymes to kill intracellular parasites. Nat. Med. 22, 210–216 (2016).
Finlay, B. B. & McFadden, G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124, 767–782 (2006).
Yang, J. H. et al. Antibiotic-induced changes to the host metabolic environment inhibit drug efficacy and alter immune function. Cell Host Microbe 22, 757–765.e3 (2017).
Chiang, C. Y. et al. Mitigating the impact of antibacterial drug resistance through host-directed therapies: current progress, outlook, and challenges. MBio 9, e01932-17 (2018).
Oldfield, E. & Feng, X. Resistance-resistant antibiotics. Trends Pharmacol. Sci. 35, 664–674 (2014).
Marakasova, E. S. et al. Prenylation: from bacteria to eukaryotes. Mol. Biol. (Mosk.) 47, 717–730 (2013).
Workalemahu, G. et al. Metabolic engineering of Salmonella vaccine bacteria to boost human Vγ2Vδ2 T cell immunity. J. Immunol. 193, 708–721 (2014).
Dieli, F. et al. Granulysin-dependent killing of intracellular and extracellular Mycobacterium tuberculosis by Vγ9/Vδ2 T lymphocytes. J. Infect. Dis. 184, 1082–1085 (2001).
Wolff, M. et al. Isoprenoid biosynthesis via the methylerythritol phosphate pathway: the (E)-4-hydroxy-3-methylbut-2-enyl diphosphate reductase (LytB/IspH) from Escherichia coli is a [4Fe-4S] protein. FEBS Lett. 541, 115–120 (2003).
Rohdich, F. et al. The deoxyxylulose phosphate pathway of isoprenoid biosynthesis: studies on the mechanisms of the reactions catalyzed by IspG and IspH protein. Proc. Natl Acad. Sci. USA 100, 1586–1591 (2003).
Xiao, Y., Chu, L., Sanakis, Y. & Liu, P. Revisiting the IspH catalytic system in the deoxyxylulose phosphate pathway: achieving high activity. J. Am. Chem. Soc. 131, 9931–9933 (2009).
Gräwert, T. et al. Probing the reaction mechanism of IspH protein by X-ray structure analysis. Proc. Natl Acad. Sci. USA 107, 1077–1081 (2010).
Nazarov, P. A. et al. Mitochondria-targeted antioxidants as highly effective antibiotics. Sci. Rep. 7, 1394 (2017).
Ortmann, R. et al. Acyloxyalkyl ester prodrugs of FR900098 with improved in vivo anti-malarial activity. Bioorg. Med. Chem. Lett. 13, 2163–2166 (2003).
Søballe, B. & Poole, R. K. Microbial ubiquinones: multiple roles in respiration, gene regulation and oxidative stress management. Microbiology 145, 1817–1830 (1999).
Trnka, J., Elkalaf, M. & Anděl, M. Lipophilic triphenylphosphonium cations inhibit mitochondrial electron transport chain and induce mitochondrial proton leak. PLoS ONE 10, e0121837 (2015).
Wingrove, D. E. & Gunter, T. E. Kinetics of mitochondrial calcium transport. II. A kinetic description of the sodium-dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. J. Biol. Chem. 261, 15166–15171 (1986).
Schwartz, A. S., Yu, J., Gardenour, K. R., Finley, R. L., Jr & Ideker, T. Cost-effective strategies for completing the interactome. Nat. Methods 6, 55–61 (2009).
Wang, H. et al. Butyrophilin 3A1 plays an essential role in prenyl pyrophosphate stimulation of human Vγ2Vδ2 T cells. J. Immunol. 191, 1029–1042 (2013).
Sandstrom, A. et al. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vγ9Vδ2 T cells. Immunity 40, 490–500 (2014).
Rigau, M. et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science 367, eaay5516 (2020).
Karunakaran, M. M. et al. Butyrophilin-2A1 directly binds germline-encoded regions of the Vγ9Vδ2 TCR and is essential for phosphoantigen sensing. Immunity 52, 487–498.e6 (2020).
Wei, H. et al. Definition of APC presentation of phosphoantigen (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate to Vγ2Vδ2 TCR. J. Immunol. 181, 4798–4806 (2008).
Mogues, T., Goodrich, M. E., Ryan, L., LaCourse, R. & North, R. J. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 193, 271–280 (2001).
Abagyan, R., Totrov, M. & Kuznetsov, D. ICM—a new method for protein modeling and design - applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 15, 488–506 (1994).
Abagyan, R. & Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235, 983–1002 (1994).
Lam, P. C., Abagyan, R. & Totrov, M. Hybrid receptor structure/ligand-based docking and activity prediction in ICM: development and evaluation in D3R Grand Challenge 3. J. Comput. Aided Mol. Des. 33, 35–46 (2019).
Lam, P. C., Abagyan, R. & Totrov, M. Ligand-biased ensemble receptor docking (LigBEnD): a hybrid ligand/receptor structure-based approach. J. Comput. Aided Mol. Des. 32, 187–198 (2018).
Totrov, M. Atomic property fields: generalized 3D pharmacophoric potential for automated ligand superposition, pharmacophore elucidation and 3D QSAR. Chem. Biol. Drug Des. 71, 15–27 (2008).
Chen, Q. & Chen, J. Isolation of CD34+ cells from human fetal liver and cord blood. Bio Protoc. 3, e991 (2013).
Somasundaram, R. et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat. Commun. 8, 607 (2017).
Span, I. et al. Insights into the binding of pyridines to the iron–sulfur enzyme IspH. J. Am. Chem. Soc. 136, 7926–7932 (2014).
Braet, F., De Zanger, R. & Wisse, E. Drying cells for SEM, AFM and TEM by hexamethyldisilazane: a study on hepatic endothelial cells. J. Microsc. 186, 84–87 (1997).
Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteomics 13, 2513–2526 (2014).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA 100, 9440–9445 (2003).
Oliveros, J. C. Venny v.2.1 (BioinfoGP, 2007).
Acknowledgements
This research was supported by the G. Harold and Leila Y. Mathers Charitable Foundation, Commonwealth Universal Research Enhancement Program (CURE – Pennsylvania Department of Health) and the Wistar Science Discovery Fund (F.D.). F.D. was supported by a Wistar Institute recruitment grant from The Pew Charitable Trusts. R.S.S. and M.H. were funded by the Adelson Medical Research Foundation and DOD for Hu-mice generation. We thank D. Speicher from the Proteomics Facility at the Wistar Institute, S. Molugu from the Electron Microscopy Resource Laboratory and Y. Velich from the Cell and Developmental Biology Microscopy core at UPenn. Support for the Wistar Institute Proteomics and Metabolomics and Genomics Shared Resources was provided by Cancer Center Support Grant P30 CA010815 and National Institutes of Health instrument grant S10 OD023586. We thank M. Groll, E. Oldfield, A. Odom John and C. Morita for advice on the bacterial isoprenoid synthesis pathway, IspH and γδ T cell fields.
Author information
Authors and Affiliations
Contributions
F.D. conceived the study and planned the experiments. M.T. set up the atomic field property of the IspH catalytic site and performed molecular docking experiments. K.S.S., R. Sharma, P.V. and A.S. purified the proteins, performed the biochemical activity assays, bacterial killing experiments, mouse infection studies and contributed to the preparation of the manuscript. P.V. performed flow cytometry and microscopy studies. K.S.S. performed the electron microscopy studies with assistance from the UPenn electron microscopy core. A.R.G. and H.-Y.T. ran the samples for proteomics and small-molecule studies. A.K. and R. Sharma performed the bioinformatics and pathway analysis on proteomics data and helped to illustrate it in a figure. J.C. performed the surface plasmon resonance studies. J.M.S. planned the synthesis of DAIA prodrugs and P.A.N.R. synthesized them. H.C., K.M., R. Somasundaram and M.H. provided Hu-mice. M.G. and M.E.M. performed the seahorse experiments. F.D. and J.M.S. analysed the data. F.D. generated the figures and drafted the manuscript. J.M.S. and P.A.N.R. provided reagents and expertise. All authors provided critical revisions to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Herman Sintim, Ben Willcox and Gerry Wright for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail:https://doi.org/10.1038/s41586-021-03961-x
Extended data figures and tables
Extended Data Fig. 1 Purification of recombinant IspH proteins from multiple microbial species and measurement of their biochemical activity by methyl viologen assay.
a, Coomassie-stained gels showing IPTG induction of recombinant 6His-tagged P. falciparum (Pf), E. coli (Ec), P. aeruginosa (Pa) and M. tuberculosis (MTB) IspH. b, Anti-His-tag immunoblots showing the respective purified IspH proteins. Images in a, b are representative of 3 independent purification attempts. c, IspH uses methyl viologen (MV) as an electron donor for the reductive dehydroxylation of HMBPP. Colourless oxidized methyl viologen is restored to its reduced blue form by sodium dithionite. In the absence or inhibition of IspH activity, methyl viologen stays blue. d, e, Methyl viologen assays measuring IspH activity using different concentrations of E. coli IspH at 10 min in the presence of 1 mM HMBPP (d) and different concentrations of HMBPP (e) at 30 min in the presence of 50 nM E. coli IspH. For d, e, data are mean of 3 independent experiments ± s.e.m. ***P < 0.001, **P < 0.01, *P < 0.05, ns, not significant; two-tailed unpaired Student’s t-test, relative to 0 nM IspH in d or 0 μM HMBPP in e.
Extended Data Fig. 2 In silico molecular docking with the active pocket of E. coli IspH.
a, Crystal structure of E. coli IspH (PDB: 3KE8)28 (top left) was used to generate the atomic property field (bottom left) and mimic HMBPP interactions in the active binding pocket (right). b, Automated virtual ligand screening (Molsoft) identified 168 out of 9.6 million compounds on the basis of predicted binding at the active site. c, The top 24 compounds were compared with HMBPP visually and on the basis of their predicted number of hydrogen bonds formed, hydrogen-bond energy, Van der Waals interaction energy and other interactions as mentioned. In silico docking for C1–24 is shown in Extended Data Fig. 3a.
Extended Data Fig. 3 In silico molecular docking of compounds C1–24 and their inhibitory activity on E. coli IspH.
a, Chemical structures and in silico docking of the top 24 candidate IspH inhibitors at the E. coli IspH active pocket rendered by Molsoft. Structures are shown in Supplementary Fig. 2a. b, Activity of P. aeruginosa (Pa), M. tuberculosis (MTB) and P. falciparum (Pf) IspH pretreated with DMSO (control), C10, C17 and C23 over time. Data are mean of 4 independent experiments ± s.e.m. c–e, Inhibition of E. coli IspH by analogues of C10 (c), C17 (d) or C23 (e). Structures are shown in Supplementary Fig. 2. For analogues with better activity than the parent compound, ***P < 0.001, **P < 0.01, *P < 0.05; two-tailed unpaired Student’s t-test, relative to C23 (n = 8 technical replicates). Data are mean ± s.e.m.
Extended Data Fig. 4 Drug binding assays, structure–activity relationship, testing prodrug potency with different carrier molecules and determining prodrug cleavage and E. coli IspH inhibition by LC–MS.
a, SPR signals (resonance units (RU)) from different concentrations of HMBPP, C23.20 and C23.21 run on E. coli IspH crosslinked NTA chip, plotted against concentrations to calculate K D and R max (the amount of ligand (in RU) to be immobilized) values (n = 3 biological and 2 technical replicates). b, Structure–activity guided analogue design reduced the IC50 values of multiple C23 analogues compared with the parent compound. Structures are shown in Supplementary Fig. 2. c, d, Prodrug ester forms of analogue C23.47 obtained by linking ethanol, TPP or dimethylaminopropanol (synthetic reactions are shown in Supplementary Fig. 3) were tested for E. coli killing by dynamic growth curves (c) and by resazurin blue assay (d). For c, d, n = 3 biological and 8 technical replicates. e, Escherichia coli cells treated with 5 μM C23.28–TPP for 30 min were lysed and the lysates analysed by LC–MS to quantify the relative abundance of C23.28–TPP (prodrug), TPP (carrier molecule) and C23.28 (active drug). Respective molecules were identified by their respective retention times (RT) and mass:charge (m/z) ratios. Area under the respective peaks is measured in arbitrary units (AU) and is directly proportional to the abundance of the molecules. f, Relative abundances of C23.28–TPP (prodrug), TPP (carrier molecule) and C23.28 (active drug) found within E. coli treated with different concentrations (10–5,000 nM) of C23.28–TPP (n = 3 technical and 2 biological replicates). g, Methyl viologen assay performed by treating 1 mM HMBPP with 50 nM E. coli IspH pre-treated with 5 μM C23.28 or TPP for 30 min. Samples analysed by LC–MS to quantify relative conversion of HMBPP (IspH substrate) to DMAPP and IPP (IspH products). Respective molecules were identified by their respective retention times and mass:charge (m/z) ratios. Area under the respective peaks is measured in AU and is directly proportional to the abundance of the molecules. h, Conversion of 1 mM HMBPP (black) to DMAPP and IPP (grey) in 30 min by 50 nM E. coli IspH in the presence of different concentrations (10–5,000 nM) of TPP (dotted lines) or C23.28 (solid lines) (n = 3 technical and 2 biological replicates). For f, h, data are mean of 3 independent experiments ± s.e.m.
Extended Data Fig. 5 C23 prodrugs specifically act on IspH and kill multidrug-resistant clinical isolates of V. cholerae.
a, Immunoblot shows modulation of IspH levels in CGSC 8074 (E. coli) achieved by altering arabinose levels in culture medium. RpoD, loading control; representative of 3 independent experiments. b, c, Sensitivity of the CGSC 8074 strain to C23.28–TPP decreases with increasing IspH levels, as shown by resazurin blue assay (b) and dynamic growth curves (c). Data are mean of 3 independent experiments ± s.e.m. ***P < 0.001, **P < 0.01, *P < 0.05, the remainder are not significant; two-tailed paired Student’s t-test. d–f, TPP-linked prodrug esters of C23 analogues C23.7, C23.20, C23.21, C23.28 and C23.47 (d) were tested for ability to kill V. cholerae (strain M045) by dynamic growth curves and resazurin blue assay (e; n = 3 biological and 8 technical replicates) or by CFU plating after 24 or 48 h treatment (f; n = 3 biological replicates with 3 serial dilutions). The MIC90 values for prodrug analogues tested on drug-resistant clinical isolates of different pathogenic bacteria are shown in Extended Data Fig. 8a. Data are mean of 3 independent experiments ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, the remainder are not significant; two-tailed unpaired Student’s t-test.
Extended Data Fig. 6 DAIA prodrugs increase oxidative stress and cause defects in bacterial respiration, membrane integrity and cell-wall architecture.
a, b, Respiratory changes in E. coli treated with TPP or with the indicated concentration of the DAIA prodrug C23.28–TPP were compared by measuring OCR (for aerobic respiration) (a) and ECAR (for glycolysis) (b). ***P < 0.001; two-tailed unpaired Student’s t-test, relative to TPP-treated control. c, d, Superoxide (solid line, 2 h after treatment; dotted line, 4 h after treatment) (c) and hydrogen peroxide (d) levels were simultaneously measured by dihydroethidium (DHE) and Amplex red fluorescence respectively. n = 8 biological replicates; data are mean ± s.e.m. e, f, Changes in E. coli membrane integrity, upon TPP or prodrug treatment, measured by Live/Dead (SYTO 9/propidium iodide) assay using flow cytometry (e) or fluorescence microscopy (f). n = 3 biological replicates. Scale bar, 2 μm. g, h, Loss of E. coli membrane potential upon treatment with TPP or prodrug measured by BacLight (DiOC2) assay using flow cytometry (g) or fluorescence microscopy (h). n = 3 biological replicates. Scale bar, 2 μm. i, Scanning electron micrographs (SEM; left) and transmission electron micrographs (TEM; right) compare the morphology of E. coli after 8 h of TPP or prodrug treatment to that of the conditional ispH knockdown E. coli strain CGSC 8074 (ΔispH) kept for 8 h in 1% glucose medium. Red arrows indicate membrane blebbing. j, SEM (top) and TEM (bottom) compare the morphology of V. cholerae after 8 h of TPP or prodrug (C23.28–TPP) treatment. In i, j, images are representative of 20 fields from 3 technical replicates. Scale bar, 400 nm.
Extended Data Fig. 7 DAIA prodrugs are stable in plasma and liver microsomes, non-toxic to mammalian cells, do not disrupt mitochondrial membrane potential in C2C12 myoblasts and do not disrupt hERG function.
a, b, Nonlinear regression curves for degradation of prodrugs C23.28–TPP and C23.21–TPP and the appearance of the parent drugs C23.28 and C23.21 in the presence of human, pig and mouse plasma (a) or human, monkey and mouse liver microsomes (b). Drug and prodrug concentration measured by LC–MS and normalized on a standard curve. The half-lives (t 1/2) are calculated from respective curves. Data are mean ± s.e.m. of 3 independent experiments. c, d, Cytotoxicity of prodrug analogues on HepG2, RAW264.7 and Vero cells (c) and C2C12 myoblasts (d) measured at 24, 48 and 72 h by LDH release (n = 3 biological and 4 technical replicates). e, Effect of TPP and prodrugs C23.28–TPP and C23.47–TPP on mitochondrial membrane potential of C2C12 myoblasts, measured by tetramethyl rhodamine methyl ester (TMRM) fluorescence. CCCP, positive control (n = 3 biological and 4 technical replicates). f, Toxicity of C23.28–TPP, the carrier (6-hydroxyhexyl)triphenylphosphonium bromide and Me-TPP to hERG channel measured by automated Q patch assay; the normalized current response is plotted using nonlinear regression curves and the IC50 of respective compounds is calculated. Data are mean of 3 independent experiments ± s.e.m. Verapamil was used as the positive control and DMSO as the negative control.
Extended Data Fig. 8 Treating E. coli with an IspH inhibitor prodrug disrupts the levels of IspH and several proteins in essential bacterial metabolic and synthesis pathways.
a, Immunoblots measure relative levels of E. coli IspH at 8 and 24 h after C23.28–TPP treatment or after conditional knockdown in CGSC 8074 (ΔispH) grown on 1% glucose. b, Immunoblots measure relative levels of IspH in clinical isolates of several pathogenic bacteria at 8 and 24 h after C23.28–TPP treatment. For a, b, RpoD immunoblot serves as loading control and the blots are representative of 3 technical replicates. c, Unsupervised hierarchical clustering of 2,346 proteins resolved indicates that the 3 biological replicates for each condition clustered together. A total of 525 proteins were either up- or downregulated both after C23.28–TPP treatment or after conditional knockdown in CGSC 8074 (ΔispH). d, Functions or pathways that are significantly enriched 8 and 24 h after C23.28–TPP treatment. Bars indicate the −log10(P value) with the number of proteins identified in each category next to the respective bar. The bars are colour-coded for the percentage of proteins in the pathway that are up- or downregulated. e, Venn diagram comparing the overlap in downregulated (>2-fold) proteins at 8 or 24 h after C23.28–TPP treatment or after conditional knockdown in CGSC 8074 (ΔispH). f, Proteins important for lipid synthesis, ribosome modification, respiration, cell division, tRNA aminoacylation, DNA/RNA synthesis, DNA repair, amino acid (AA) synthesis and lipopolysaccharide (LPS) cell-wall synthesis pathways are among those significantly downregulated. Associated with Extended Data Fig. 9a. P < 0.05 and FDR < 5%. g, Venn diagram comparing the overlap in upregulated (>2-fold) proteins at 8 or 24 h after C23.28–TPP treatment or after conditional knockdown in CGSC 8074 (ΔispH). h, Ribosome component proteins or proteins important for multidrug efflux and oxidative defence pathways are among those significantly upregulated. Associated with Extended Data Fig. 9b. P < 0.05 and FDR <5%.
Extended Data Fig. 9 Escherichia coli metabolic pathways up- or downregulated after IspH inhibition.
Extended Data Fig. 10 By dual action, IspH prodrugs expand and activate Vγ9Vδ2 T cells and reduce the emergence of antibiotic resistant bacteria.
a, Top, uninfected (UI) human PBMCs or those co-infected with E. coli analysed for expansion of CD3+Vγ9TCR+ (γδ) T cells and compared to untreated (UT) or TPP-, prodrug (C23.07–TPP)- or kanamycin (Kan)-treated PBMCs. Middle, bottom, gated γδ T cell populations analysed for cytotoxic granule proteins granulysin (GNLY) and perforin (middle) or cell surface markers of T cell activation CD69 and HLA-DR (bottom). Representative of 4 independent experiments (4 donors). Percentage of Vγ9+ T cells from CD3+ population and the percentage of Vγ9+ T cells with increased expression of granulysin, perforin, CD69 and HLA-DR were plotted in the respective graphs. Data are mean ± s.e.m. **P < 0.05, ***P < 0.001, the remainder are not significant; one-way ANOVA relative to untreated sample. b, Uninfected human PBMCs or those co-infected with V. cholerae (top) or M. smegmatis (bottom) were analysed for expansion of CD3+Vγ9TCR+ (γδ) T cells and compared to untreated or TPP-, prodrug (C23.07–TPP)- or kanamycin-treated PBMCs (n = 4 biological replicates). Percentage of Vγ9+ T cells from the CD3+ population were plotted in the respective graphs. Data are mean ± s.e.m. ***P < 0.001, rest not significant, calculated by one-way ANOVA relative to untreated sample. c, Human PBMCs co-infected with kanamycin-resistant E. coli or V. cholerae can kill neither on their own. Addition of C23.07–TPP kills both V. cholerae and E. coli (n = 2 biological and 3 technical replicates). Data are mean ± s.e.m. ***P < 0.001, ns, not significant; unpaired Student’s t-test relative to untreated samples. d, γδ T cell depletion from human PBMCs is verified by treating depleted (γδ−) and undepleted human PBMCs treated with 10 μM HMBPP and 50 ng ml−1 IL-15. Representative of 4 independent experiments (4 donors). Percentage of Vγ9+ T cells from the CD3+ population on different days were plotted in the respective graphs. Data are mean ± s.e.m. ***P < 0.001 comparing γδ depleted and undepleted PBMCs calculated by unpaired t-test. e, f, Multidrug-resistant clinical isolates of Vibrio (e) and Klebsiella (f) grown for 18 serial passages in media (RPMI+ 10% human serum) containing DAIA prodrug (C23.28–TPP) or conventional antibiotics (hygromycin (Hyg) or streptomycin (Strep)) gradually develop resistance when measured by CFU (top). Similar serial passages in presence of human PBMC inhibit the development of resistance against the DAIA prodrug but not against hygromycin or streptomycin. Passages in γδ depleted (γδ−) PBMCs show higher antibiotic resistance against the DAIA prodrug. (n = 3 technical replicates). Data are mean ± s.e.m. ***P < 0.001, NS, not significant; unpaired Student’s t-test. g, C57BL/6 mice infected with V. cholerae are treated with TPP or the DAIA prodrug C23.28–TPP and monitored daily from day 2 post-infection for survival (n = 10 mice per group). h, Vibrio load in different organs at the experimental endpoint measured as CFU mg−1 (n = 10 mice with 3 technical replicates), comparing changes in bacterial CFU in C57BL/6b mice after C23.28–TPP treatment. Data are mean ± s.e.m. ***P < 0.001; unpaired Student’s t-test, relative to TPP-treated mice. i, Hu-mice injected i.p. with HMBPP at different concentrations show dose-dependent expansion of γδ T cells but not αβ T cells in blood taken every day for a week (n = 2 mice per group).
Extended Data Fig. 11 γδ T cells expand in the tissues of prodrug-treated, E. coli-infected humanized mice.
a, Hu-mice infected with E. coli (green) and treated with TPP or the prodrug C23.07–TPP are compared for expansion of Vδ2 TCR+ T cells (red) in multiple organs at day 5 post-infection. DAPI, blue. Scale bar, 100 μm (representative of samples tested from 5–6 Hu-mice). b, Vδ2 antibody validated for immunofluorescence staining of formalin-fixed human PBMCs that are γδ expanded (HMBPP + IL15) or γδ depleted. HepG2 and Vero cells serve as negative controls. c, Anti-E. coli antibody validated for immunofluorescence staining of formalin-fixed HepG2 cells co-infected with E. coli BL21 strain. HepG2 without E. coli serves as a negative control.
Supplementary information
Supplementary Figures
This file contains Supplementary Figures 1-3.
Supplementary Tables
This file contains Supplementary Tables 1-5.
Source data
About this article

Cite this article
Singh, K.S., Sharma, R., Reddy, P.A.N. et al. RETRACTED ARTICLE: IspH inhibitors kill Gram-negative bacteria and mobilize immune clearance. Nature 589, 597–602 (2021). https://doi.org/10.1038/s41586-020-03074-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-03074-x
- Springer Nature Limited
This article is cited by
-
Cryosphere: a frozen home of microbes and a potential source for drug discovery
Archives of Microbiology (2024)
-
Unrealized targets in the discovery of antibiotics for Gram-negative bacterial infections
Nature Reviews Drug Discovery (2023)
-
Synthesis, anti-inflammatory activity, and acute toxicity of the substituted 2-[2-(4-nitrobenzoyl)hydrazono]-4-oxobutanoates
Russian Chemical Bulletin (2023)
-
Cave Microbes as a Potential Source of Drugs Development in the Modern Era
Microbial Ecology (2022)
-
Unconventional T cells and kidney disease
Nature Reviews Nephrology (2021)