

Abstract
Class B G-protein-coupled receptors are major targets for the treatment of chronic diseases, including diabetes and obesity1. Structures of active receptors reveal peptide agonists engage deep within the receptor core, leading to an outward movement of extracellular loop 3 and the tops of transmembrane helices 6 and 7, an inward movement of transmembrane helix 1, reorganization of extracellular loop 2 and outward movement of the intracellular side of transmembrane helix 6, resulting in G-protein interaction and activation2,3,4,5,6. Here we solved the structure of a non-peptide agonist, TT-OAD2, bound to the glucagon-like peptide-1 (GLP-1) receptor. Our structure identified an unpredicted non-peptide agonist-binding pocket in which reorganization of extracellular loop 3 and transmembrane helices 6 and 7 manifests independently of direct ligand interaction within the deep transmembrane domain pocket. TT-OAD2 exhibits biased agonism, and kinetics of G-protein activation and signalling that are distinct from peptide agonists. Within the structure, TT-OAD2 protrudes beyond the receptor core to interact with the lipid or detergent, providing an explanation for the distinct activation kinetics that may contribute to the clinical efficacy of this compound series. This work alters our understanding of the events that drive the activation of class B receptors.
Similar content being viewed by others

Main
Class B peptide G-protein-coupled receptors (GPCRs) regulate the control of glucose and energy homeostasis, bone turnover, and cardiovascular development and tone1. Several peptide agonists are clinically approved for disorders of energy and bone metabolism1; however, attempts to develop non-peptide, orally available analogues have yielded only limited success. Understanding the structural basis of class B GPCR activation is crucial to the rational development of peptidic and non-peptidic drugs. Recent structural determination of full-length, active class B receptors bound to peptide agonists2,3,4,5,6 confirmed that the N terminus of the peptide ligands, required for receptor activation, binds deep within the seven-transmembrane helical bundle. This is associated with an outward movement of the tops of transmembrane helices (TM) 6 and 7 (and interconnecting extracellular loop (ECL) 3) and a large kink in the centre of TM6 that opens up the intracellular face of the receptor to allow G-protein coupling2,3,4,7,8,9,10. In parallel, a conformational reorganization of ECL2 and an inward movement of TM1 facilitates peptide interaction and receptor activation.
The GLP-1 receptor (GLP-1R) is an established therapeutic target for type 2 diabetes and obesity11. Despite their clinical success, GLP-1R peptide drugs are suboptimal owing to their route of administration and side-effect profiles, most notably nausea and vomiting that reduce patient compliance11. For many years, oral GLP-1R agonists have been pursued, with recent studies reporting promising clinical trial data for oral semaglutide—a new formulation of the approved peptide semaglutide12,13. However, it induced slightly greater severity of nausea and gastrointestinal side effects than those observed with injectable GLP-1 mimetics13. Future development of non-peptide drugs could offer more traditional small molecule absorption characteristics that may assure better long-term patient compliance with the potential for reduced gastrointestinal liability, especially for patients who are co-administering with other medications.
Several non-peptidic GLP-1R agonists have been identified14. One class form covalent interactions with C3476.36 (in which the superscript denotes the Wootten class B GPCR numbering) and are predicted to allosterically disrupt polar networks at the base of the receptor, promoting activation15, whereas other small molecule compounds bind to unknown sites at the receptor extracellular face14,16,17. However, it is assumed that these molecules may need to mimic key interactions of the peptide N terminus deep within the transmembrane core to initiate receptor activation, as is seen for short stabilized 11-mer peptides, that occupy an overlapping site to full-length peptides18.
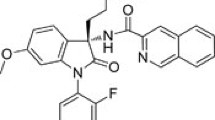
Here we investigate TT-OAD2 (Fig. 1a), a non-peptidic compound reported in the patent literature and part of the chemical series that contains the vTv Therapeutics investigational drug candidate, TTP273. TTP273, an orally administered GLP-1R agonist, successfully completed phase IIa efficacy trials for type 2 diabetes (ClinicalTrials.gov Identifier: NCT02653599), in which it met its primary endpoint, reducing levels of glycated haemoglobin in patients with type 2 diabetes, with no reported cases of nausea19, suggesting a potential clinical advantage for compounds of this series. Little has been disclosed about the molecular properties of this compound series; however, recent progression of TTP273 has been hampered by unexpected complexity in identifying optimal dosing that may be linked to a lack of understanding of its mechanism of action. Assessment of acute in vivo activity in humanized GLP-1R mice revealed that TT-OAD2 is insulinotropic and that this effect is dependent on the GLP-1R (Fig. 1b).

a, Chemical structure of TT-OAD2. b, Plasma insulin induced by GLP-1 (10 μg kg−1), TT-OAD2 (3 mg kg−1) or gastric inhibitory polypeptide (GIP; 25 μg kg−1) in an acute IVGTT on humanized GLP-1R knock-in (KI) and GLP-1R knockout (KO) mice. c, Whole-cell binding assays showing the ability of GLP-1 and TT-OAD2 to displace 125I-exendin(9-39). d, cAMP accumulation, intracellular calcium mobilization, β-arrestin-1 recruitment and ERK1/2 phosphorylation (pERK1/2). e, Agonist-induced changes in trimeric Gs conformation in cell plasma membrane preparations for GLP-1 (left) and TT-OAD2 (middle). Rates (top right) and plateau (bottom right) at saturating concentrations (1 μM GLP-1, 10 μM TT-OAD2) were quantified by applying a one-phase association curve. f, Kinetics of cAMP production measured by an EPAC biosensor for GLP-1 (left) and TT-OAD2 (middle). Rates were quantified using approximate EC50 and Emax concentrations (1 nM and 0.1 μM for GLP-1, 0.1 μM and 10 μM for TT-OAD2) by applying a one-phase association curve. In e and f, arrows refer to the time at which ligand or vehicle was added. Parameters derived from kinetic data are represented as scatter plots with each individual experiment shown by black circles. All experiments were performed in GLP-1R expressing HEK293A cells. Data in b are mean + s.e.m. from 4–5 mice per treatment, representative of 3 independent experiments. Data in c–f are mean + s.e.m. of 4–5 independent experiments (in duplicate or triplicate). *P < 0.05, Student’s paired t-test.
TT-OAD2 is a biased agonist with slow kinetics
In HEK293 cells that overexpress GLP-1R, TT-OAD2 only partially displaced the orthosteric probes 125I-exendin(9–39) and ROX-exendin-4 (Fig. 1c, Extended Data Fig. 1a), consistent with an allosteric mode of interaction16. Although GLP-1R signals to several cellular pathways, TT-OAD2 activated only a subset of these responses; it was a low-potency partial agonist for cAMP accumulation, with only weak responses detected for mobilization of intracellular Ca2+ and phosphorylation of ERK1/2 at very high concentrations (100 μM) (Fig. 1d) and no detectable recruitment of β-arrestin-1. These data are indicative of bias towards cAMP and away from these other pathways relative to endogenous GLP-1. There is considerable interest in exploiting biased agonism at GPCRs to maximize the beneficial effects of receptor activation, while minimizing on-target side-effect profiles.
CRISPR-engineered HEK293 cells in which Gs/olf or Gi/o/z proteins were deleted revealed that Gs was essential for the production of cAMP; however, this response, for both ligands, was also dependent on the presence of Gi/o/z proteins. (Extended Data Fig. 1b). Assessment of proximal activation of Gs and Gi transducers using split luciferase NanoBit G-protein sensors (Extended Data Fig. 1c) determined GLP-1-decreased luminescence in a bi-phasic, concentration-dependent, manner for both G proteins with similar potencies in each phase. For TT-OAD2, the Gi sensor gave a similar decrease in luminescence to GLP-1; however, enhanced luminescence was observed for the Gs sensor, which suggests a different mechanism of Gs activation. To probe these differences further, we used membrane-based assays of bioluminescence resonance energy transfer (BRET) G-protein sensors to assess the rate and nature of the Gs conformational change. In contrast to the rates of change in the conformation of Gi, which were similar for both ligands (Extended Data Fig. 1), there was a marked distinction in kinetics for Gs coupling. GLP-1 promoted a rapid conformational change in Gs protein, whereas for TT-OAD2 this was very slow (Fig. 1e). However, both agonists induced a similar plateau of the measured response (Fig. 1e) that was reversed by excess GTP (Extended Data Fig. 1d), indicative of a similar overall conformational rearrangement. Together, this suggests that slower Gs conformational transitions, required for the exchange of GDP for GTP and Gs activation, would result in lower turnover of G protein and rate of cAMP production by TT-OAD2. Direct kinetic measurements of cAMP production validated this hypothesis (Fig. 1f, Extended Data Fig. 1e). Overall, these data revealed TT-OAD2 as a biased agonist that can only activate a subset of pathways with limited efficacy and with distinct activation kinetics relative to peptide agonists.
TT-OAD2 has an unexpected binding mode
To understand how TT-OAD2 binds and activates the GLP-1R, we determined the GLP-1R structure bound to TT-OAD2 and the transducer heterotrimeric Gs protein (Fig. 2). Complex formation was initiated in Tni insect cells by stimulation with 50 μM TT-OAD2, and complexes were then solubilized and purified (Extended Data Fig. 2a). Vitrified complexes were imaged by single-particle cryo-electron microscopy (cryo-EM) on a Titan Krios. Following 2D and 3D classification, the most abundant class was resolved to 3.0 Å (Extended Data Fig. 2c–f, Supplementary Table 1). The cryo-EM density map allowed unambiguous assignment of the TT-OAD2-binding site and pose, and clear rotamer placement for most amino acids within the receptor core and G protein (Fig. 2, Extended Data Figs. 3, 4a, b). The GLP-1R extracellular domain (ECD) and the Gαs α-helical domain were not resolved at high resolution, consistent with their greater mobility. Rigid body fitting of an available X-ray structure of the GLP-1R ECD domain (PDB code 3C5T)20 was performed into the density to generate a full-length model.

Top, orthogonal views of the TT-OAD2–GLP-1R–Gs complex cryo-EM map (left) and the structure after refinement in the cryo-EM map (right), colour-coded to protein chains; GLP-1R (blue), TT-OAD2 (red), heterotrimeric Gs (α: gold, β: dark cyan, γ: purple, Nb35: salmon). Middle, TT-OAD2 interacts with the top of the GLP-1R bundle. Interacting residues of GLP-1R (blue) with TT-OAD2 (red). Bottom, TT-OAD2-mediated cAMP production by receptors containing alanine mutants of key residues assessed in ChoFlpIn cells. Data are mean + s.e.m. of four independent experiments performed in duplicate. WT, wild type.
TT-OAD2 bound high up in the helical bundle interacting with residues within TM1, TM2, TM3, ECL1 and ECL2 (Fig. 2, Extended Data Fig. 4a). Most interactions are hydrophobic in nature (Fig. 2), including numerous π–π stacking interactions between receptor aromatic residues and phenolic regions within the ligand. Unexpectedly, TT-OAD2 adopts a ‘boomerang-like’ orientation within the binding site with the 3,4-dichloro-benzyl ring of TT-OAD2 protruding beyond the receptor core through transmembrane helices 2 and 3, interacting with W2032.73, and embedding in the detergent micelle, consistent with probable interactions with the lipid bilayer in a native system. F2303.33 and W297ECL2 interact with the 2,3-dimethyl-pyridin-4-yl-phenol region, Y220ECL1 forms a hydrogen bond with the 2,3-dimethyl-pyridine ring and K1972.67 forms a polar interaction with the propionic acid part of the ligand. Additional hydrophobic contacts are formed with TT-OAD2 by Y1451.40, L2012.71, I1962.69, A2002.70, L217ECL1, V2293.32 and M2043.36 (Fig. 2, Extended Data Fig. 4a). Molecular dynamics simulations of the TT-OAD2–GLP-1R–Gs complex predicted further transient interactions with TM1, TM2, TM3, ECL1, ECL2 and the ECD of GLP-1R (Extended Data Table 1). Assessment of TT-OAD2-induced cAMP production at alanine mutants of key receptor residues within the binding site revealed reduced potency (negative logarithm of the half-maximal effective concentration, pEC50), reduced maximal responses (Emax) or both relative to the wild-type receptor (Fig. 2, Supplementary Table 2). Application of the operational model of agonism revealed these mutations directly alter TT-OAD2 functional affinity (KA) and/or efficacy (τ) (Supplementary Table 2), which highlights the importance of these residues in TT-OAD2 function.
Peptide versus non-peptide binding sites
The TT-OAD2-binding pose has very limited overlap with full-length peptides, GLP-1 and exendin-P5 (ExP5)3,6 (Fig. 3, Extended Data Fig. 5). Structural comparisons, combined with associated molecular dynamics simulations performed on models generated from the cryo-EM data, identified only 10 out of 29 residues that interact with both TT-OAD2 and GLP-1. Moreover, the persistence and nature of ligand interactions formed by common residues differed (Fig. 3c, Extended Data Table 1). In contrast to TT-OAD2, peptide ligands engage transmembrane helices 5–7 in addition to extensive interactions deep within the bundle in transmembrane helices 1–3 (Fig. 3, Extended Data Fig. 5, Extended Data Table 1).

a, b, Superimposition of the GLP-1R from PDB 5VAI (GLP-1R or G protein: orange, GLP-1: green) and the TT-OAD2 structure (GLP-1R or G protein: blue, TT-OAD2: red) reveals partial overlap of peptide- and TT-OAD2-binding sites and conformational differences in the receptor. a, Left, full complex; middle, close up of ECD and the top of the seven-transmembrane (7-TM) bundle; right, close up of the transmembrane bundle. b, Left, 16 Å, 7 Å and 6 Å differences occur in the location of TM6/ECL3, TM7 and TM1, respectively. Middle, a 4 Å shift in the location of the top of TM2 result in distinct conformations of ECL1. Right, the intracellular region of the GLP-1R helical bundles have similar overall backbone conformations. c, Comparison of the GLP-1R–TT-OAD2 and GLP-1R–GLP-1 contacts during molecular dynamics simulations performed on the GLP-1R–TT-OAD2–Gs and GLP-1R–GLP-1–Gs complexes. Top (left) and side (right) views of the GLP-1R transmembrane domain (ribbon representation, TT-OAD2 in red sticks, GLP-1 not shown). TT-OAD2 made contacts (red coloured ribbon) with ECL1 and residues located at the top of TM2 and TM3. GLP-1 was able to engage TM5, TM6 and TM7 of the receptor and side chains located deep in the bundle (blue coloured ribbon). Residues that are involved both in the GLP-1R–TT-OAD2–Gs and GLP-1R–GLP-1–Gs complexes are indicated by asterisks, and coloured according to the algebraic difference in occupancy (contact differences in percentage frames) between GLP-1R–TT-OAD2–Gs and GLP-1R–GLP-1–Gs. Red indicates regions more engaged by TT-OAD2 and blue more engaged by GLP-1. The ECD is not shown. Plotted data are summarized in Extended Data Table 1.
The relatively limited overlap between the peptide- and TT-OAD2-binding sites suggests that this compound series may modulate peptide function in a physiological setting. To address this, we assessed the effect of TT-OAD2 on the signalling of two physiological ligands (Extended Data Fig. 6). TT-OAD2 inhibited GLP-1- and oxyntomodulin-mediated cAMP, calcium, pERK1/2 and β-arrestin responses in a concentration-dependent manner (Extended Data Fig. 6). This suggests that the profile of signalling observed from the GLP-1R when using TT-OAD2-like compounds as drugs may depend on the dose administered; at high concentrations, their presence would probably inhibit all endogenous peptide effects, biasing receptor responses primarily to cAMP formation mediated by the compound itself. However, at lower concentrations, some endogenous peptide signalling may still occur. Notably, TTP273 was reported to exhibit greater clinical efficacy at lower concentrations, indicating that maintenance of some aspects of physiological signalling may be important for clinical efficacy19.
GLP-1R conformational changes and activation
At a gross level, the TT-OAD2-complexed GLP-1R helical bundle displays the key hallmarks of activated, peptide-occupied, class B GPCRs2,3,4,5,6. At the extracellular face, this includes the large outward movement of TM6, ECL3 and TM7, inward movements of TM1, helical extensions within TM2 and TM3, a reordering of ECL1, and conformational transitions within ECL2 that increases upward towards the extracellular side (Extended Data Fig. 5). At the intracellular side, there is an equivalent large outward movement of TM6 away from the centre of the helical bundle, and the smaller outward movement of TM5. It is important to note that the fully active state is driven in part by allosteric conformational changes, including those in the extracellular face, linked to G protein binding21. Nonetheless, all the GLP-1R structures are solved with the same G protein yet reveal conformational differences at their extracellular face, including within the extent of movement of TM6, ECL3, ECL7 and the conformation of the ECD, TM2–ECL1 and ECL2 that are linked to the bound agonists (Fig. 3a, b, Extended Data Fig. 5b, c). This suggests that distinct receptor activation triggers converge to common changes at the intracellular face that allow coupling to transducers.
Although the low resolution of the receptor ECD for the TT-OAD2 complex indicates extensive mobility, it occupied a distinct orientation relative to the transmembrane core in comparison to peptide-bound complexes, whereas both GLP-1- and ExP5-bound receptors stabilized a similar conformation3,6 (Extended Data Fig. 5a). Similarly, the short 11-mer peptide HepP5 forms few interactions with the ECD18 and occupies a distinct orientation relative to GLP-1 and ExP5, but this conformation also differs from that stabilized by TT-OAD2 (Extended Data Fig. 5c). The cryo-EM map of the TT-OAD2-bound receptor complex supports extended interactions of the ECD with ECL1 and ECL2 (Extended Data Fig. 4c) and this is supported by molecular dynamics simulations that predicts interactions of R40ECD with D215ECL1 and E34ECD with R299ECL2 (Extended Data Table 2). This later interaction is particularly important as R299ECL2 directly, and stably interacts with peptide ligands, but in the TT-OAD2-bound receptor, stabilizes the N terminus of the ECD in a position that may have an analogous role to the peptide in stabilizing ECL2. Indeed, in our models, the position of the far N-terminal ECD helix overlapped with the location of the C-terminal region of GLP-1 and ExP5 when comparing the TT-OAD2- and peptide-bound structures (Fig. 3a). Thus, the ECD is likely to be important for both stabilizing the TT-OAD2-binding site and facilitating receptor activation, as previously proposed for different classes of peptide ligands22,23.
Distinctions from peptide-bound receptors observed within TM2/ECL1 and ECL2 (Fig. 3b) are probably driven by direct ligand interactions by TT-OAD2 (Fig. 2), whereas those within TM6 and TM7 by direct interactions formed by peptide agonists. Molecular dynamics simulations also support a role of membrane lipid interactions in directly stabilizing both these regions within the TT-OAD2-bound structure (Extended Data Fig. 7). Notably, the helical bundle of the TT-OAD2-complexed receptor is in a more open conformation than the peptide-occupied receptors, largely owing to the top of TM6/ECL3, TM7 and TM1 residing 16 Å, 6 Å and 7 Å further outwards relative to the GLP-1-bound structure (measured from the Cα atoms of D3726.62/ECL3, F3817.37 and P1371.32, respectively (Fig. 3b). The orientation of TM6, ECL3 and TM7 also differs between ExP5- and GLP-1-bound structures, with ExP5 adopting a more open conformation3; however, the outward positioning of ECL3 induced by TT-OAD2 is much larger (Extended Data Fig. 5b). Peptide-bound structures of all solved class B GPCRs revealed direct interactions of the engaged peptide with residues within TM5, TM6, TM7 and ECL3 with the peptide volume (minimally) presumed to actively contribute to the outward conformational change in this region2,3,4,8,9,24. In the apo-state of the glucagon receptor, interactions occur between ECL3 and the ECD that contribute to maintenance of receptor quiescence7,8,25,26. Molecular dynamics simulations on the GLP-1R structures, performed after the removal of either TT-OAD2 or GLP-1, predict that the GLP-1R ECD also adopts both open and closed conformations in the apo-state, in which it can form transient interactions with both ECL2 and ECL325 (Extended Data Fig. 8). Combining this information with the GLP-1R active structures suggests that interactions, with either peptide or non-peptide agonists, can release ECL3-ECD constraints, lowering the energy barrier for receptor activation. However, the degree of ligand interaction with TM6–ECL3–TM7 determines the extent to which the transmembrane bundle opens, and this in turn directly contributes to G-protein efficacy and biased agonism, as these regions (TM6–ECL3–TM7 and TM1) have been identified as key drivers for these phenomena, particularly for the GLP-1R3,27,28,29.
Despite the different binding modes, commonalities observed in interactions with TT-OAD2 and peptide with transmembrane helices 1–3 and stabilization of ECL2 are sufficient to initiate conformational transitions that propagate to a similar reorganization of the class B GPCR conserved central polar network that is linked to activation, albeit the mechanism for this differs for peptide agonists versus TT-OAD2 (Fig. 4a, Supplementary Video 1, Extended Data Fig. 9). Molecular dynamics simulations of the GLP-1-bound GLP-1R predicted persistent interactions between Y1521.47, R1902.60, Y2413.44 and E3646.53 and the N terminus of GLP-1 that directly engage the central polar network (Fig. 4a, Extended Data Tables 1, 2, Supplementary Video 1). By contrast, TT-OAD influences the central polar network allosterically via interactions with K1972.67, Y1451.40 and Y1481.43. TT-OAD2 also promotes unique hydrogen bond networks with crucial residues in TM2 (Fig. 4a, Extended Data Table 2) that result in different interaction patterns at the top of TM1 and TM2 relative to peptide-occupied receptors. These effects propagate to the polar network through transient contacts between TT-OAD2 with Y1481.43 and Y1521.47 that in turn interact with R1902.60 of the central polar network (Supplementary Video 2). When bound by GLP-1, the polar network is stabilized by ligand and a network of water molecules, whereas for TT-OAD2, this occurs via a distinct network of structural waters rather than by the ligand (Fig. 4b, Supplementary Video 1). These differences in the mechanism of conformational transitions and stabilization of conserved polar networks (summarized in Extended Data Fig. 9) may contribute to the different kinetic profiles of G-protein activation, as well as the full versus partial agonism for cAMP production.

Summaries of interactions observed in molecular dynamics simulations (Supplementary Video 2) on TT-OAD2- and GLP-1-bound GLP-1R that predict interactions stabilizing the active conformation of the central polar network. a, Left, GLP-1 (brown ribbon) residue D9 (brown stick) forms an ionic interaction (red dotted lines) with R1902.60, which is involved in key hydrogen bonds with N2403.43 (in turn interacting with S1862.56). At the top of TM2, K1972.67, D1982.68 and Y1451.40 are stabilized in polar interactions (red dotted lines). Right, TT-OAD2 (brown stick and transparent surface) forms ionic interaction (red dotted lines) with K1972.67 and hydrophobic contacts with Y1451.40 and Y1481.43 (cyan transparent surfaces) modifying the interaction network at the top of TM1. Y1481.43 transiently interacts with R1902.60 and partially reorients N2403.43 and S1862.56. TM6 and TM7 were removed for clarity. b, GLP-1R transmembrane helix sites are occupied by structural water molecules; blue spheres indicate receptor volumes occupied by low-mobility water molecules (occupancy more than 75% frames). Left, the GLP-1R–GLP-1–Gs complex stabilizes the central transmembrane polar residues by waters interacting with Y1521.47, T3917.46, R1902.60 and E3645.53 (Supplementary Video 1). Right, the GLP-1R–TT-OAD2–Gs complex is characterized by structural water molecules interacting with N3205.50 and E3646.53 (Supplementary Video 1).
Collectively, our work provides key advances in understanding the activation of class B GPCRs and Gs protein efficacy, identifying a non-peptide binding site within the GLP-1R that can promote distinct efficacy and biased signalling relative to peptide ligands, and this may extend to other class B GPCRs. The demonstration that non-peptide agonists of the GLP-1R are not required to mimic the extensive receptor contacts formed by peptides within the transmembrane cavity to promote receptor activation will advance the pursuit of non-peptide agonists for therapeutically important class B receptors.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation.
TT-OAD2 synthesis
Several azoanthracene-based derivatives are reported as potent agonists of the GLP-1R (WO10114824), and a compound from this series known as OAD2 was selected for our studies (WO14113357). OAD2, (S)-2-{[(3S,8S)-3-[4-(3,4-dichloro-benzyloxy)-phenyl]-7-((S)-1-phenyl-propyl)-2,3,6,7,8,9-hexahydro-[1,4]dioxino[2,3-g]isoquinoline-8-carbonyl]-amino}-3-[4-(2,3-dimethyl-pyridin-4-yl)-phenyl]-propionic acid, was synthesized using procedures previously described (see example 179 in WO10114824), and a dihydrochloride salt form (OAD2.2HCl) was prepared by standard methods from the free base. Therefore, TT-OAD2 is the dihydrochloride salt of OAD2 in patent WO14113357. The purity of TT-OAD2 was determined by liquid chromatography–mass spectrometry (LC–MS) to be 98.62%.
Constructs
GLP-1R was modified to contain either a 2xcMyc-N-terminal epitope tag (for signalling and radioligand-binding assays) or a Nanoluc tag (with a 12xGly linker; for NanoBRET binding studies) after the native signal peptide. For β-arrestin recruitment assays, a C-terminal Rluc8 was fused to the C terminus of the receptor. For G-protein conformational assays, a Nanoluc flanked by SGGGGS linkers was inserted into Gαs and Gαi2 after G(h1ha10) in Gαs or E(HA.03) in Gαi2 as previously described30,31. These were used in conjunction with an N-terminally Nluc-labelled Gγ2. For G-protein steady-state assays, G-protein NanoBit-split luciferase constructs were generated by fusing the LgBIT after G(h1ha10) in Gαs or E(HA.29) in Gαi2 and the SmBIT to Gγ2. For structural studies, human GLP-1R in the pFastBac vector was modified to include an N-terminal Flag tag epitope and a C-terminal 8×histidine tag; both tags are removable by 3C protease cleavage. These modifications did not alter the pharmacology of the receptor3. A dominant-negative Gαs construct was generated previously by site directed mutagenesis to incorporate mutations that alter nucleotide handling, stabilize the G0 state and interactions with the βγ subunits30.
Insect cell expression
GLP-1R, human dominant-negative Gαs, His6-tagged human Gβ1 and Gγ2 were expressed in Tni insect cells (Expression systems) using baculovirus. Cell cultures were grown in ESF 921 serum-free media (Expression Systems) to a density of 4 million cells per ml and then infected with three separate baculoviruses at a ratio of 2:2:1 for GLP-1R, dominant-negative Gαs and Gβ1γ2. Cells were obtained by centrifugation 60 h after infection and the cell pellet was stored at −80 °C.
Purification of the TT-OAD2–GLP-1R–Gs complex
Cell pellet was thawed in 20 mM HEPES, pH 7.4, 50 mM NaCl, 2 mM MgCl2 supplemented with cOmplete Protease Inhibitor Cocktail tablets (Roche). Complex formation was initiated by addition of 50 μM TT-OAD2, Nb35–His (10 μg ml−1) and apyrase (25 mU ml−1, NEB) to catalyse hydrolysis of unbound GDP and allow for stabilization of the G0 state; the suspension was incubated for 1 h at room temperature. Membrane was solubilized by 0.5% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace) supplemented with 0.3% (w/v) cholesteryl hemisuccinate (CHS, Anatrace) for 2 h at 4 °C. Insoluble material was removed by centrifugation at 30,000g for 30 min and the solubilized complex was immobilized by batch binding to M1 anti-Flag affinity resin in the presence of 3 mM CaCl2. The resin was packed into a glass column and washed with 20 column volumes of 20 mM HEPES pH 7.4, 100 mM NaCl, 2 mM MgCl2, 3 mM CaCl2, 1 μM OAD, 0.01% (w/v) MNG and 0.006% (w/v) CHS before bound material was eluted in buffer containing 5 mM EGTA and 0.1 mg ml−1 Flag peptide. The complex was then concentrated using an Amicon Ultra Centrifugal Filter (molecular mass cut off 100 kDa) and subjected to size-exclusion chromatography on a Superdex 200 Increase 10/300 column (GE Healthcare) that was pre-equilibrated with 20 mM HEPES pH 7.4, 100 mM NaCl, 2 mM MgCl2, 1 μM OAD, 0.01% (w/v) MNG and 0.006% (w/v) CHS to separate complex from contaminants. Eluted fractions consisting of receptor and G-protein complex were pooled and concentrated. Final yield of purified complex was approximately 0.15 mg per litre of insect cell culture.
Electron microscopy
Samples (3 µl) were applied to a glow-discharged Quantifoil R1.2/1.3 CuRh 200 mesh holey carbon grid (Quantifoil GmbH) and were flash frozen in liquid ethane using the Vitrobot mark IV (Thermo Fisher Scientific) set at 100% humidity and 4 °C for the prep chamber. Data were collected on a Titan Krios microscope (Thermo Fisher Scientific) operated at an accelerating voltage of 300 kV with a 50 μm C2 aperture at an indicated magnification of 105 K in nanoprobe EFTEM mode. Gatan K3 direct electron detector positioned post a Gatan Quantum energy filter, operated in a zero-energy-loss mode with a slit width of 25 eV was used to acquire dose fractionated images of the GLP-1R TT-OAD2-bound sample without an objective aperture. Movies were recorded in hardware-binned mode (previously called counted mode on the K2 camera) yielding a physical pixel size of 0.826 Å pixel−1 with an exposure time of 3.715 s amounting to a total dose of 65.6 e− Å−2 at a dose rate of 12.2 e− pixel−1 s−1, which was fractionated into 62 subframes. A second dataset of 1,568 micrographs was also recorded using the same microscope but in ‘super-resolution’ mode on the K3 detector, the physical pixel size was 0.413 Å with an exposure time of 4.015 s amounting to a total dose of 63.5 e− Å−2, which was fractionated into 67 subframes. Defocus range was set between −0.7 and −1.5 μm. A total of 3,158 plus 1,568 movies were collected in two data collection sessions.
Electron microscopy data processing
Movies were motion-corrected with UCSF MotionCor232 (movies collected in super-resolution mode were Fourier scaled by a factor of ×2 to match the pixel size of the larger data set). This was followed by CTF estimation sing the GCTF software packag33. Particles were picked from the micrographs using the automated reference-free procedure in RELION34,35. Reference free 2D and 3D classification (by generating multiple ab initio models with no structural identity enforced) was carried out in CryoSPARC (v.2.5.0)36. A homogeneous subset of particles was then subjected to movie refinement and Bayesian particle polishing as implemented in RELION (v.3.0). This homogeneous subset of polished particles was used in a 3D refinement in RELION and then further classified into 3D classes with alignment of Euler angles not taken into account. Particles belonging to the 3D class that yielded the best resolved map were then subjected to signal subtraction to subtract density due to the detergent micelle and the alpha domain of the G protein. Final 3D refinement was performed in RELION (3.0) yielded a map of resolutions 3.01 Å. Local resolution estimations were performed using the ResMAP software packag37.
Atomic model refinement
Fitting the model to the cryoEM electron density map was achieved using the MDFF routine in namd38. The fitted model was further refined by rounds of manual model building in coot39 and real space refinement as implemented in the Phenix software package40, the model restraints for the TT ligand were prepared by using the coordinates generated from Chem3D and the ELBOW software package41. The ligands were fitted after the first round of real-space refinements, manually first in coot39, then refined using Phenix real-space refinement42. Ramachandran, rotamer and secondary structure restraints were applied for the first round of real-space refinement, and after manual inspection and adjustment of the model in coot further real-space refinements were carried out with only Ramachandran and rotamer restraints applied and the model/data weight was allowed to freely refine. The density around the extracellular domain was poorly resolved (local resolution estimated at >8 Å) and was not modelled.
Modelling methods for preparation of molecular dynamic simulations
The two missing receptor loops, namely the stalk region and ICL3, were generated using PLOP43; ICL3 was also minimized in the presence of Gα to eliminate steric clashes. On the basis of the electron density of our structures, TM1 for the GLP-1-bound 5VAI structure6 was replaced by TM1 from the P5-bound structure (PDB code 6B3J)3 by the method of molecular superposition. The missing residues in the stalk region were reconstructed using Modeller44 subject to the constraint that the high variability positions45 in the GLP-1R multiple sequence alignment (E133–R134) faced outwards. The missing loops in the G protein were generated by molecular superposition, using VMD46, of the corresponding loops in the β2-adrenergic receptor–G protein complex47, PDB code 3SN6 to the flank either side of the gap, since this particular X-ray structure (with 99% identity to the G protein used in this study) generally gave a lower root mean squared deviation value on molecular superposition than plausible alternative G-protein structures (for example, PDB 5VAI). The joining point was taken as the closest atom pairs (usually separated by approximately 0.2 Å) that maintained an appropriate Cα–Cα distance (3.7–3.9 Å) across the join; selected residues spanning the join were minimized using PLOP where additional refinement was deemed necessary. The exception to this was the loop between A249–N264, which was completed using the shorter loop from the adenosine A2A receptor–G-protein complex, PDB code 5G5348. The helical domain, between residues G47 and G207, which is not visible in the cryo-EM structure, was omitted as in earlier work.
Molecular dynamics methods
Four GLP-1R complexes (GLP-1R–TT-OAD2–Gs; GLP-1R–TT-OAD2; GLP-1R–GLP-1–Gs; and GLP-1R–GLP-1; Supplementary Table 3) and two apo GLP-1R structures (obtained by removing both the Gs protein and the ligands; Supplementary Table 3) were prepared for simulation with the CHARMM36 force field49, through use of in-house python htmd50 and TCL (Tool Command Language) scripts. The pdb2pqr51 and propka52 software were used to add hydrogen atoms appropriate for a pH of 7.0; the protonation of titratable side chains was checked by visual inspection. The coordinates were superimposed on the corresponding GLP-1R coordinates from the OPM database53 so as to orient the receptor before insertion54 in a rectangular pre-built 125 Å × 116 Å 1-palmitoyl-2-oleyl-sn-glycerol-3-phosphocholine (POPC) bilayer; lipid molecules overlapping the receptor were removed. TIP3P water molecules were added to the 125 Å × 116 Å × 195 Å simulation box using the VMD Solvate plugin 1.5 (Solvate Plugin, v.1.5; http://www.ks.uiuc.edu/Research/vmd/plugins/solvate/). Overall charge neutrality was maintained by adding Na+ and Cl− counter ions to a final ionic concentration of 150 mM using the VMD Autoionize plugin 1.3 (Autoionize Plugin, v.1.3; http://www.ks.uiuc.edu/Research/vmd/plugins/autoionize/). CGenFF force field parameters55,56,57 and topology files for TT-OAD2 were retrieved from the Paramch56 webserver. No further optimization was performed because the obtained parameters were associated to low penalty scores.
Systems equilibration and molecular dynamics simulation settings
ACEMD58 was used for both equilibration and molecular dynamics productive simulations. Isothermal-isobaric conditions (Langevin thermostat59 with a target temperature of 300 K and damping of 1 ps−1 and Berendsen barostat60 with a target pressure 1 atm) were used to equilibrates the systems through a multi-stage procedure (integration time step of 2 fs). Initial steric clashes between lipid atoms were reduced through 3,000 conjugate-gradient minimization steps, then a 2 ns molecular dynamics simulation was run with a positional constraint of 1 kcal mol−1 Å−2 on protein atoms and lipid phosphorus atoms. Subsequently, 20 ns of molecular dynamics simulations were performed constraining only the protein atoms. In the final equilibration stage, protein backbone alpha carbons constraints were applied for a further 60 ns.
Productive trajectories in the canonical ensemble (NVT) at 300 K (four 500-ns-long replicas for each GLP-1R complex; Supplementary Table 3) were computed using a thermostat damping of 0.1 ps−1 with an integration time step of 4 fs and the M-SHAKE algorithm61 to constrain the bond lengths involving hydrogen atoms. The cut-off distance for electrostatic interactions was set at 9 Å, with a switching function applied beyond 7.5 Å. Long-range Coulomb interactions were handled using the particle mesh Ewald summation method (PME)62 by setting the mesh spacing to 1.0 Å. Trajectory frames were written every 100 ps of simulations.
Molecular dynamics analysis
The first half (500 ns) of the molecular dynamics replicas involving GLP-1R–TT-OAD2, GLP-1R–GLP-1 complexes as well as the apo-GLP-1R (TT-OAD2), and apo-GLP-1R (GLP-1) systems (Supplementary Table 3) were considered as part of the equilibration stage and therefore not considered for analysis. Atomic contacts (atom distance less than 3.5 Å) were computed using VMD46. Hydrogen bonds were identified using the GetContacts analysis tool (https://getcontacts.github.io/), with the donor-acceptor distance set to 3.3 Å and the angle set to 150°. Videos were generated using VMD46 and avconv (https://libav.org/avconv.html). Root mean square fluctuation (RMSF) values were computed using VM46 after superposition of the molecular dynamic trajectories frames on the alpha carbon of the transmembrane domain (residues E1381.33–V4047.60). The orientation of the N-terminal helix of the ECD of GLP-1R was drawn in VMD considering a representative frame every 10 ns. To detect volumes within the transmembrane domain of GLP-1R occupied by water molecules with low mobility (structural water molecules), the AquaMMapS63 analysis was performed on 10-ns-long molecular dynamics simulations of the GLP-1R–TT-OAD2–Gs and GLP-1R–GLP-1–Gs complexes (coordinates were written every 10 ps of simulation); all the alpha carbons were restrained in analogy with the approach proposed previously64.
Whole-cell radioligand binding assays
HEK293 cells (confirmed mycoplasma negative) were seeded at 30,000 cells per well in 96-well culture plates and incubated overnight in DMEM containing 5% FBS at 37 °C, 5% CO2. Media was replaced with HBSS containing 25 mM HEPES and 0.1% (w/v) BSA with 0.1 nM 125I-exendin(9–39) and increasing concentrations of unlabelled agonist. Cells were incubated overnight at 4 °C, washed three times in ice-cold buffer and then solubilized in 0.1 M NaOH. Radioactivity was determined by gamma counting. Non-specific activity was defined using 1 μM exendin(9–39).
cAMP accumulation assays
HEK293 cells (confirmed mycoplasma negative) were seeded at a density of 30,000 cells per well into 96-well culture plates and incubated overnight in DMEM containing 5% FBS at 37 °C in 5% CO2. cAMP detection was performed as previously described in the presence of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthin65. All values were converted to cAMP concentration using a cAMP standard curve performed in parallel and data were subsequently normalized to the response of 100 μM forskolin in each cell line. In one series of experiments, vehicle or increasing concentrations of TT-OAD2 was added 30 min before assay of peptide response.
cAMP kinetics studies
HEK293A cells (confirmed mycoplasma negative) were transfected with an Epac-cAMP sensor (CAMYEL) and human GLP-1R at an optimized ratio. Ligand-mediated cAMP production was measured 48 h after transfection. In brief, culture media was replaced with assay buffer (1× HBSS, 10 mM HEPES, 0.1% BSA, pH 7.4). BRET signals were measured at 1 min intervals using a PHERAstar plate reader (BMG LabTech) in the absent or present of increasing concentration of ligands. Forskolin (100 μM) was used as a positive control, and data were normalized to the forskolin response.
β-arrestin recruitment assays
HEK293 cells (confirmed mycoplasma negative) were transiently transfected with GLP-1R-Rluc8 and β-arrestin1-Venus at a 1:4 ratio and seeded at a density of 30,000 cells per well into 96-well culture plates and incubated for 48 h in DMEM containing 5% FBS at 37 °C in 5% CO2. β-arrestin recruitment was performed as previously described66. In one series of experiments, vehicle or increasing concentrations of TT-OAD2 was added 30 min before assay of peptide response.
ERK1/2 phosphorylation assays
HEK293 cells (confirmed mycoplasma negative) expressing stably expressing the GLP-1R were seeded at a density of 30,000 cells per well into 96-well culture plates and incubated overnight at 37 °C in 5% CO2. Receptor-mediated pERK1/2 was determined using the AlphaScreen ERK1/2 SureFire protocol as previously described14. Data were normalized to the maximal response elicited by 10% FBS determined at 6 min. In one series of experiments, vehicle or increasing concentrations of TT-OAD2 was added 30 min before assay of peptide response.
Ca2+ mobilization assays
HEK293 cells (confirmed mycoplasma negative) stably expressing the GLP-1R were seeded at a density of 30,000 cells per well into 96-well culture plates and incubated overnight at 37 °C in 5% CO2, and receptor- mediated intracellular calcium mobilisation determined as previously described65. Fluorescence was determined immediately after ligand addition, with an excitation wavelength set to 485 nm and an emission wavelength set to 520 nm, and readings taken every 1.36 s for 120 s. The peak value was used to create concentration-response curves. Data were normalized to the maximal response elicited by 100 μM ATP. In one series of experiments, vehicle or increasing concentrations of TT-OAD2 was added 30 min before assay of peptide response.
Generation of stable cell lines containing wild-type and mutant GLP-1R
Mutant receptors were generated in a 2xc-Myc epitope-tagged receptor using QuikChange site-directed mutagenesis (Invitrogen) and sequences confirmed. Wild-type and mutant receptors were stably expressed in CHOFlpIn cells (confirmed mycoplasma negative) using the FlpIn Gateway technology system and selected using 600 μg ml−1 hygromyocin B.
NanoBRET ligand binding
HEK293A cells were transiently transfected with Nluc-hGLP-1R. Forty-eight hours after transfection, cells were collected and plasma membrane was extracted as described previously31. Cell membrane (1 μg per well) was incubated with furimazine (1:1,000 dilution from stock) in assay buffer (1× HBSS, 10 mM HEPES, 0.1% (w/v) BSA, 1× P8340 protease inhibitor cocktail, 1 mM DTT and 0.1 mM PMSF, pH 7.4). RhodamineX-Ex4 (Rox-Ex4) was used as fluorescent ligand in the NanoBRET binding assay. BRET signal between Nluc-hGLP-1R and Rox-Ex4 was measured using PHERAstar (BMG LabTech) at 10 s interval (25 °C), a 2 min baseline was taken before addition of Rox-Ex4 (Kd concentration 3.16nM, determined previously), the measurement continued for 15 min followed by adding increasing concentration of TT-OAD2, or unlabelled Ex4 as a control. Data were corrected for baseline and vehicle treated samples.
G-protein conformation assays
HEK293AΔS/Q/12/13 cells stably expressing GLP-1R (tested and confirmed to be free from mycoplasma) were transfected with a 1:1:1 ratio of Nanoluc-Gαs (Nanoluc inserted at position 72):Gβ1:Venus-Gγ2 24 h before collection and preparation of cell plasma membranes. Cell membrane (5 μg per well) was incubated with furimazine (1:1,000 dilution from stock) in assay buffer (1× HBSS, 10 mM HEPES, 0.1% (w/v) BSA, 1× P8340 protease inhibitor cocktail, 1 mM DTT and 0.1 mM PMSF, pH 7.4). The GLP-1R-induced BRET signal between Gαs and Gγ was measured at 30 °C using a PHERAstar (BMG LabTech). Baseline BRET measurements were taken for 2 min before addition of vehicle or ligand. BRET was measured at 15-s intervals for a further 7 min. All assays were performed in a final volume of 100 μl.
G-protein NanoBIT assays
HEK293A wild-type cells stably express human GLP-1R were transiently transfected with Gα-LgBIT, Gβ1, Gγ2-SmBIT (1:5:5) 48 h before the assays. Cells were then incubated with coelenterazine H (5 μM) for 1 h at room temperature. Luminescence signals were measured using a Clariostar plate reader (BMG LabTech) at 30 s intervals before and after ligand addition (25 °C). Data were corrected to baseline and vehicle treated samples.
In vivo IVGTT assays
Intravenous glucose tolerance tests were performed in male human GLP-1R knock-in and knockout mice (all on C57/BL6 background67). Catheters were placed in the right carotid artery and left jugular vein of mice 6–11 months of age. Approximately one week later, mice (n = 4–5 per group) were fasted overnight and the catheters were exteriorized as mice acclimated to test cages. Vehicle (5% DMSO, 20% Captisol in NaHPO4, pH 2, 1 ml kg−1), GLP-1(7-36)NH2 at 10 μg kg−1, GIP(1-42) at 25 μg kg−1, or OAD2 at 3 mg kg−1 was administered intravenously one minute before glucose load (0.5 g kg−1). Blood samples were collected at −10, 0, 2, 4, 6, 10, 20 and 30 min to determine blood glucose concentrations via glucometer (Roche, Aviva) and plasma insulin measurement (Alpco, 80-INSMSU-E10). All mouse experiments were performed in accordance with the Institutional Animal Care and Use Committee of Eli Lilly and Company and the NIH Guide for the Use and Care of Laboratory Animals.
Data analysis
Pharmacological data were analysed using Prism 7 (GraphPad). Concentration response signalling data were analysed using a three-parameter logistic equation, or via operational analysis. Changes in the rate of change in BRET kinetic data were fitted to one-phase association curve. Statistical analysis was performed with either one-way analysis of variance and a Dunnetts post-test or a paired t-test, and significance accepted at P < 0.05.
Graphics
Molecular graphics images were produced using the UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco (supported by NIH P41 RR-01081).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All relevant data are available from the authors and/or included in the manuscript or Supplementary Information. Atomic coordinates and the cryo-EM density map have been deposited in the Protein Data Bank (PDB) under accession number 6ORV and Electron Microscopy Data Bank (EMDB) accession EMD-20179.
References
Bortolato, A. et al. Structure of class B GPCRs: new horizons for drug discovery. Br. J. Pharmacol. 171, 3132–3145 (2014).
Liang, Y. L. et al. Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497 (2018).
Liang, Y. L. et al. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555, 121–125 (2018).
Liang, Y. L. et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123 (2017).
Zhao, L. H. et al. Structure and dynamics of the active human parathyroid hormone receptor-1. Science 364, 148–153 (2019).
Zhang, Y. et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253 (2017).
Siu, F. Y. et al. Structure of the human glucagon class B G-protein-coupled receptor. Nature 499, 444–449 (2013).
Zhang, H. et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 546, 259–264 (2017).
Zhang, H. et al. Structure of the glucagon receptor in complex with a glucagon analogue. Nature 553, 106–110 (2018).
Song, G. et al. Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators. Nature 546, 312–315 (2017).
Htike, Z. Z. et al. Efficacy and safety of glucagon-like peptide-1 receptor agonists in type 2 diabetes: A systematic review and mixed-treatment comparison analysis. Diabetes Obes. Metab. 19, 524–536 (2017).
Aroda, V. R. et al. PIONEER 1: Randomized clinical trial comparing the efficacy and safety of oral semaglutide monotherapy with placebo in patients with type 2 diabetes. Diabetes Care 42, dc190749 (2019).
Pratley, R. et al. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet 394, 39–50 (2019).
Wootten, D. et al. Differential activation and modulation of the glucagon-like peptide-1 receptor by small molecule ligands. Mol. Pharmacol. 83, 822–834 (2013).
Nolte, W. M. et al. A potentiator of orthosteric ligand activity at GLP-1R acts via covalent modification. Nat. Chem. Biol. 10, 629–631 (2014).
Freeman, J. W. S. et al. TTP273: Oral, G protein pathway selective clinical-stage GLP-1 receptor (GLP-1R) agonist. Keystone Symposium, Keystone, CO February 22 http://vtvtherapeutics.com/wp-content/uploads/pdf/GLP1_poster_animation_Keystone_CO_FINAL.pdf (2016).
Chen, D. et al. A nonpeptidic agonist of glucagon-like peptide 1 receptors with efficacy in diabetic db/db mice. Proc. Natl Acad. Sci. USA 104, 943–948 (2007).
Jazayeri, A. et al. Crystal structure of the GLP-1 receptor bound to a peptide agonist. Nature 546, 254–258 (2017).
Freeman, J. D. C., Dunn, I. & Valcarce, C. TTP273, Oral (nonpeptide) GLP-1R agonist: improved glycemic control without nausea and vomiting in phase 2. American Diabetes Association 77th Scientific Sessions, San Diego, CA, June 9–13, 2017 http://vtvtherapeutics.com/wp-content/uploads/pdf/ADA_Logra_study_results_poster_1220-P%20Final.pdf (2017).
Runge, S., Thøgersen, H., Madsen, K., Lau, J. & Rudolph, R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J. Biol. Chem. 283, 11340–11347 (2008).
DeVree, B. T. et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 535, 182–186 (2016).
Yin, Y. et al. An intrinsic agonist mechanism for activation of glucagon-like peptide-1 receptor by its extracellular domain. Cell Discov. 2, 16042 (2016).
Zhao, L. H. et al. Differential requirement of the extracellular domain in activation of class B G protein-coupled receptors. J. Biol. Chem. 291, 15119–15130 (2016).
Ehrenmann, J. et al. High-resolution crystal structure of parathyroid hormone 1 receptor in complex with a peptide agonist. Nat. Struct. Mol. Biol. 25, 1086–1092 (2018).
Yang, L. et al. Conformational states of the full-length glucagon receptor. Nat. Commun. 6, 7859 (2015).
Wootten, D., Simms, J., Miller, L. J., Christopoulos, A. & Sexton, P. M. Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc. Natl Acad. Sci. USA 110, 5211–5216 (2013).
dal Maso, E. et al. The molecular control of calcitonin receptor signaling. ACS Pharmacol. Transl. Sci. 2, 31–51 (2019).
Wootten, D. et al. The extracellular surface of the GLP-1 receptor is a molecular trigger for biased agonism. Cell 165, 1632–1643 (2016).
Lei, S. et al. Two distinct domains of the glucagon-like peptide-1 receptor control peptide-mediated biased agonism. J. Biol. Chem. 293, 9370–9387 (2018).
Liang, Y.-L. et al. Dominant negative G proteins enhance formation and purification of agonist-GPCR-G protein complexes for structure determination. ACS Pharmacol. Transl. Sci. 1, 9 (2018).
Furness, S. G. B. et al. Ligand-dependent modulation of G protein conformation alters drug efficacy. Cell 167, 739–749.e711 (2016).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Nakane, T., Kimanius, D., Lindahl, E. & Scheres, S. H. Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. eLife 7, e36861 (2018).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Chan, K. Y., Trabuco, L. G., Schreiner, E. & Schulten, K. Cryo-electron microscopy modeling by the molecular dynamics flexible fitting method. Biopolymers 97, 678–686 (2012).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D 65, 1074–1080 (2009).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018).
Jacobson, M. P. et al. A hierarchical approach to all-atom protein loop prediction. Proteins 55, 351–367 (2004).
Eswar, N. et al. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter 5, Unit 5.6 (2006).
Vohra, S. et al. Similarity between class A and class B G-protein-coupled receptors exemplified through calcitonin gene-related peptide receptor modelling and mutagenesis studies. J. R. Soc. Interface 10, 20120846 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph 14, 33–38 (1996).
Rasmussen, S. G. et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 477, 549–555 (2011).
Carpenter, B., Nehmé, R., Warne, T., Leslie, A. G. & Tate, C. G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 536, 104–107 (2016).
Huang, J. & MacKerell, A. D. Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 (2013).
Doerr, S., Harvey, M. J., Noé, F. & De Fabritiis, G. HTMD: high-throughput molecular dynamics for molecular discovery. J. Chem. Theory Comput. 12, 1845–1852 (2016).
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 (2004).
Olsson, M. H., Søndergaard, C. R., Rostkowski, M. & Jensen, J. H. PROPKA3: consistent treatment of internal and surface residues in empirical pka predictions. J. Chem. Theory Comput. 7, 525–537 (2011).
Lomize, M. A., Lomize, A. L., Pogozheva, I. D. & Mosberg, H. I. OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625 (2006).
Sommer, B. Membrane packing problems: a short review on computational membrane modeling methods and tools. Comput. Struct. Biotechnol. J. 5, e201302014 (2013).
Vanommeslaeghe, K. et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010).
Vanommeslaeghe, K. & MacKerell, A. D. Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 52, 3144–3154 (2012).
Vanommeslaeghe, K., Raman, E. P. & MacKerell, A. D. Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 52, 3155–3168 (2012).
Harvey, M. J., Giupponi, G. & Fabritiis, G. D. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 5, 1632–1639 (2009).
Loncharich, R. J., Brooks, B. R. & Pastor, R. W. Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N'-methylamide. Biopolymers 32, 523–535 (1992).
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684 (1984).
Kräutler, V. G., van Gunsteren, W. F. & Hünenberger, P. H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 22, 501–508 (2001).
Essmann, U. P. & Berkowitz, L. M.L. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577 (1995).
Cuzzolin, A., Deganutti, G., Salmaso, V., Sturlese, M. & Moro, S. AquaMMapS: an alternative tool to monitor the role of water molecules during protein-ligand association. ChemMedChem 13, 522–531 (2018).
Wall, M. E., Calabró, G., Bayly, C. I., Mobley, D. L. & Warren, G. L. Biomolecular solvation structure revealed by molecular dynamics simulations. J. Am. Chem. Soc. 141, 4711–4720 (2019).
Koole, C. et al. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Mol. Pharmacol. 78, 456–465 (2010).
Savage, E. E., Wootten, D., Christopoulos, A., Sexton, P. M. & Furness, S. G. A simple method to generate stable cell lines for the analysis of transient protein-protein interactions. Biotechniques 54, 217–221 (2013).
Jun, L. S. et al. A novel humanized GLP-1 receptor model enables both affinity purification and Cre-LoxP deletion of the receptor. PLoS One 9, e93746 (2014).
Acknowledgements
The work was supported by the Monash University Ramaciotti Centre for Cryo-Electron Microscopy, the Monash MASSIVE high-performance computing facility, the National Health and Medical Research Council of Australia (NHMRC) project grants (1061044, 1065410, 1120919 and 1126857) and NHMRC program grants (1055134 and 1150083), the Japan Society for the Promotion of Science (JSPS) KAKENHI no. 18H06043 and Japan Science and Technology Agency (JST) PRESTO no. 18069571 (to R.D.). P.M.S. and A.C. are NHMRC Senior Principal Research Fellows and D.W. is an NHMRC Senior Research Fellow. S.G.B.F. is an ARC Future Fellow. A.I. was funded by the PRIME JP17gm5910013 and the LEAP JP17gm0010004 from the Japan Agency for Medical Research and Development, and JSPS KAKENHI 17K08264. We are grateful to G. Christopoulos, V. Julita, T. Fields, C. Lafuente, J. M. Minguez, G. C. Sanz and F. Qu for assay and technical support.
Author information
Authors and Affiliations
Contributions
P.Z. designed and performed most of the pharmacological studies with assistance from T.T.T.; Y.-L.L. expressed and purified the complex; R.D. performed cryo-sample preparation and imaging to acquire electron microscopy data; M.J.B. and R.D. processed the electron microscopy data and performed electron microscopy map calculations; M.J.B. built the model and performed refinement; M.M.F. performed the mutagenesis studies, L.C. performed studies in the HEK293 CRISPR-knockout cells; G.D. and C.A.R. designed, performed and analysed the molecular dynamics simulations; F.S.W. and M.G.B. provided TT-OAD2. M.E.C., M.G.B. and K.W.S. designed and oversaw the in vivo studies; P.Z., Y.-L.L., M.J.B., G.D., C.A.R., F.S.W., K.W.S., R.D., P.M.S. and D.W. performed data analysis; P.Z., Y.-L.L., M.J.B., G.D., C.A.R., F.S.W., K.W.S., A.C., L.J.M., M.-W.W. and R.D. assisted with data interpretation, figure and manuscript preparation; P.M.S. and D.W. designed and supervised the project, interpreted the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
F.W.S., M.E.C. and K.W.S. are employees of Eli Lilly and Company.
Additional information
Peer review information Nature thanks Doryen Bubeck, Dave D’Alessio, Nita R. Shah and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Binding, transducer coupling and signalling mediated by TT-OAD2.
a, Kinetic ligand-binding assay using ROX-exendin-4 as the fluorescent probe. TT-OAD2 is only able to partially displace the probe and with slower kinetics relative to exendin-4 that shows complete displacement of the probe with rapid kinetics. b, cAMP accumulation studies using GLP-1 and TT-OAD2 as the agonist in wild-type HEK293 cells and HEK293 cells in which Gs/olf (ΔGs) or all Gi/o/z (ΔGi/o/z) have been depleted using CRISPR–Cas9. c, HEK293A cells transiently transfected with the GLP-1R and the NanoBit constructs for Gαs and Gαi2 (Gα-LgBIT, Gγ2-SmBIT). Luminescence signal was assessed over time (0–20 min) in the presence of increasing concentrations of GLP-1 and TT-OAD2. Concentration response curves are expressed as AUC (0–20 min) for each concentration and normalized to the negative response observed by GLP-1 at 1 μM. d, Agonist-induced changes in trimeric Gs protein conformation. Ligand-induced changes in BRET were measured in plasma membrane preparations performed in kinetic mode until kinetic equilibrium was reached for vehicle or increasing concentrations of GLP-1 (left) and TT-OAD2 (right). The addition of GTP dissociated the trimeric G protein complex stabilized by GLP-1-occupied and TT-OAD2-occupied GLP-1R. e, Agonist-induced changes in trimeric Gi2 protein conformation. Left, ligand-induced changes in BRET were measured in plasma membrane preparations performed in kinetic mode until kinetic equilibrium with a saturating concentration of GLP-1 and TT-OAD2. The BRET signal decreased in the presence of GTP, which suggests that GTP dissociated the Gi2 protein complex stabilized by GLP-1-occupied and TT-OAD2-occupied GLP-1R. Quantification of the plateau (middle) and the rate of ligand-induced conformational change (right) for each agonist (1 μM GLP-1 and 10 μM TT-OAD2) was calculated by applying a one-phase association curve to the kinetic data with values from each individual experiment show in black circles. f, Concentration–response curves of production in live HEK293 cells expressing the GLP-1R and an EPAC BRET biosensor in the presence of different concentrations of GLP-1 and TT-OAD2. Left, cAMP response taken 25 min after ligand addition. Right, area under the curve (AUC) analysis of the response calculated as AUC across the full kinetic trace for each ligand concentration (from data in Fig. 2d). Data are mean + s.e.m. of 4–6 independent experiments performed in duplicate or triplicate.
Extended Data Fig. 2 Purification, cryo-EM data imaging and processing of the TT-OAD2–GLP-1R–Gs complex.
a, Representative elution profile of Flag-purified complex on Superdex 200 Increase 10/30 SEC. b, Representative micrograph of the TT-OAD2–GLP-1R–Gs complex. Red circles highlight examples of individual particles. c, Two-dimensional class averages of the complex in maltose-neopentyl glycol (MNG) micelle. d, Cryo-EM data processing workflow. e, Gold-standard Fourier shell correlation (FSC) curves, showing the overall nominal resolution at 3.0 Å. f, 3D histogram representation of the Euler angle distribution of all the particles used for the in the reconstruction overlaid on the density map drawn on the same coordinate axis (map is coloured according to local resolution as in g). g, Cryo-EM density map coloured according to resolution. Left, map with the GLP-1R ECD masked; right, map including the ECD of GLP-1R.
Extended Data Fig. 3 The atomic resolution model of the TT-OAD2–GLP-1R–Gαs heterotrimer in the cryo-EM density map.
Electron microscopy density map and the model are shown for all seven transmembrane helices and helix 8 (H8) of the receptor, the α5 helix of the GαS Ras-like domain and TT-OAD2. All transmembrane helices exhibit good density, with TM6 that displays flexibility being the least well resolved region.
Extended Data Fig. 4 Cryo-EM density supports ligand interactions in the TT-OAD2-binding site.
a, Interacting residues predicted by LigPlot using the full-length model with ECD. b, The pose of TT-OAD2 and interactions with residues within TM1, TM2, TM3, ECL1 and ECL2 are supported by well-resolved density in the cryo-EM map. c, Density for the ECD was visible in the cryo-EM and supports extended interactions of the ECD with ECL1 and ECL2, as well as with the ligand TT-OAD2.
Extended Data Fig. 5 Comparison of the TT-OAD2–GLP-1R–Gs complex with peptide agonist-bound GLP-1R structures and the inactive class B GPCR glucagon receptor transmembrane helices.
a, Structures of agonist-bound GLP-1R; from left to right: GLP-1R (orange) bound to GLP-1 peptide (green) in an active conformation, GLP-1R (pink) bound to ExP5 peptide (cyan) in an active conformation, GLP-1R (blue) bound to non-peptide TT-OAD2 (red) in an active conformation, GLP-1R (pale green) bound to 11-mer peptide HepP5 (purple) in a partially active conformation. Far right, overlay of GLP-1R agonist-bound structures highlighting variations within the ECD position in the different structures. Inset, differences in the location of the ECD are supported by density in the cryo-EM maps; shown are the GLP-1-bound (orange) and TT-OAD2 bound (blue) GLP-1R. b, c, Various overlays of these structures (using the same colours) to compare conformational differences between the different structures. b, Overlay of TT-OAD2-bound GLP-1R Gs structure with the full-length peptide bound Gs structures and the inactive glucagon receptor (GCGR; grey) bundle reveals common conformational transitions occur in all agonist-bound structures relative to the inactive GCGR, but the extent of these movements differ. A more open helical bundle is observed for the TT-OAD2-bound GLP-1R than either GLP-1- or ExP5-bound owing to a distinction in the conformations of TM1, TM6, TM7 and ECL3 at the extracellular side of the receptor induced by the binding of the different ligands (left and middle). Middle, differences in the conformation of TM2 between the inactive and peptide-agonist-bound structures is also evident. Right, at the intracellular face all active structures display a similar large outward movement of TM6 and a smaller movement within TM5. c, Comparison of TT-OAD2-bound GLP-1R with the small peptide HepP5-bound GLP-1R structure. Left, TT-OAD2 and Hep-P5 occupy a partially overlapping binding site but promote distinct conformations of the ECD and transmembrane bundle of the GLP-1R. Middle, HepP5 engages deeper in the helical bundle than TT-OAD2 and promotes a more closed helical bundle owing to differences induced in the conformation of TM1, TM6, TM7 and ECL3. Right, overlay of the TT-OAD2-, Hep-P5- and GLP-1-bound GLP-1R transmembrane bundles reveals HepP5 induces a similar conformation of the helical bundle to GLP-1 whereas TT-OAD2 induces a distinct conformation.
Extended Data Fig. 6 Pharmacological responses exhibited by endogenous ligands GLP-1 and oxyntomodulin in the presence of TT-OAD2.
Signalling profiles of GLP-1 and oxyntomodulin, after 30 min preincubation of vehicle (0) or increasing concentrations of TT-OAD2. Data were performed in HEK293A cells stably expressing the GLP-1R, and are mean + s.e.m. of 3–4 independent experiments performed in duplicate.
Extended Data Fig. 7 GLP-1R domains are stabilized by either ligand contacts or lipid interactions.
a, Top, RMSF values of alpha carbons computed during MD simulations of the GLP-1R–GLP-1–Gs complex (black line) and the GLP-1R–TT-OAD2–Gs complex (red line); transmembrane helices, intracellular loops (ICLs), and ECLs positions are indicated. Bottom left, RMSF values plotted on the GLP-1R structure bound to GLP-1 (transparent ribbon). Bottom right, RMSF values plotted on the GLP-1R structure bound to TT-OAD2 (transparent stick representation). ECL1 and ECL3 were more dynamic in the GLP-1-bound receptor than the TT-OAD2-bound structure. By contrast, ECL2 and the top end of TM5 were more mobile in the GLP-1R–TT-OAD2–Gs complex. b, GLP-1R contacts formed with membrane lipids during molecular dynamic simulations of the GLP-1R–TT-OAD2–Gs and the GLP-1R–GLP-1–Gs systems. Two sides views of the receptor are shown (ribbon and transparent surface). When bound to TT-OAD2, ECL1, TM3, the distal end of TM6, and ECL3 are more in contact with the membrane lipids (magenta). By contrast, TM1 and TM7 are more prone to interact with the membrane when GLP-1 is bound (green). The outward movement of ECL3 in the GLP-1R–TT-OAD2–Gs complex (stabilized by a hydrogen bond network different than GLP-1R–GLP-1–Gs; Extended Data Table 2) produces more interactions with the lipids, possibly further stabilizing the open conformation of TM6, ECL3 and TM7.
Extended Data Fig. 8 Dynamics of the ECD of GLP-1R.
a, The vector (shown here as a green arrow) connecting S49ECD and E34ECD alpha carbons (ECD N-terminal helix) are shown in the box. b, Left, ECD N-terminal helix orientations observed during the molecular dynamics simulation of the GLP-1R–GLP-1–Gs (black arrows), the GLP-1R–GLP-1 complex (obtained by removing G protein; blue arrows), and the apo-GLP-1R (obtained by removing both the Gs protein and GLP-1; cyan arrows) are shown on the left viewed from the top and side of the bundle. The receptor is shown as a dark grey ribbon. During molecular dynamic simulations with GLP-1 bound, the N-terminal helix was oriented vertically (black and blue arrows), whereas in the apo-form the ECD N-terminal helix was more dynamic and experienced both open and closed conformations (this is analogous to the suggested ECD dynamics for the glucagon receptor). Right, ECD N-terminal helix orientations of the GLP-1R–TT-OAD2–Gs (red arrows), the GLP-1R–TT-OAD2 complex (obtained by removing G protein; orange arrows), and the apo-GLP-1R (obtained by removing both the Gs protein and TT-OAD2; yellow arrows) are shown. The receptor is shown as a red ribbon. The distal end (S49ECD) of the helix was more mobile than the proximal one (E34ECD), which had an overall tendency to remain in the proximity of the TT-OAD2-binding site, driven by transient interactions with the ligand (Extended Data Table 1) and hydrogen bonds with the R299ECL2 side chain (Extended Data Table 2). Molecular dynamics simulations therefore suggest a different behaviour for residue R299ECL2, which is stably involved in interactions with the peptide in the GLP-1-R–GLP-1–Gs complex (Extended Data Table 1), and instead interacts with E34ECD and other residues located at the ECL2 (E294ECD, D293ECD and N300ECD) in the GLP-1R–TT-OAD2–Gs complex (Extended Data Table 2).
Extended Data Fig. 9 Proposed activation mechanism of class B GPCRs.
Left, in the inactive conformation, the top of the transmembrane domain is stabilized by interactions of the ECD with the TM6–ECL3–TM7 region. Top, activation of class B GPCRs by peptides occurs via a two domain mechanism. Top left, engagement of the peptide with the receptor ECD releases ECD constraints on the transmembrane domain promoting outward movements of TM1, TM6 and TM7 by peptide. Middle, interaction of the peptide N terminus in the bundle within TM1, TM2, TM3, TM5, TM6 and TM7 promotes TM1, TM6 and TM7 to close in around the peptide. Direct engagement of peptides with the central polar network facilitates conformational transitions required for G protein coupling and activation. Top right, the active conformation of the central polar network is stabilized by a series of structural waters. Bottom, interaction of the non-peptide TT-OAD2 at the top of the GLP-1R transmembrane bundle releases ECD constraints on the transmembrane bundle resulting in movements of TM1, TM6 and TM7 outwards. TT-OAD2 does not engage TM5–TM7 and the bundle remains open. TT-OAD allosterically promotes conformational rearrangement of the central polar network to stabilize the fully active receptor conformation that allows coupling to G protein. Bottom right, the central polar network is stabilized by a distinct network of structural waters relative to peptide-mediated activation.
Supplementary information
Supplementary Tables
This file contains Supplementary Tables 1-3.
Video 1
Different hydration of polar networks within the GLP-1R transmembrane (TM) domain. In the GLP-1R:TT-OAD2 Gs complex (left), structural water molecules form stable hydrogen bonds with the N3205.50 side chain and the Y2413.44 backbone atoms as well with E3646.53. The bound GLP-1 (right) stabilizes a water molecule network in the proximity of the peptide N-terminal residues H7 and A8 as well of Y1521.47, T3917.46, R1902.60, E3645.53.
Video 2
TT-OAD2 interactions lead to reorganisation and stabilisation of the central polar network via a distinct mechanism to GLP-1. The GLP-1R:TT-OAD2 interactions modify the hydrogen bond network between TM1 and TM2. Left; GLP-1 (brown ribbon representation) residue 3 (D9) (white stick) forms an ionic interaction (red dotted lines) with R1902.60, which is involved in key hydrogen bonds with N2403.43 (in turn interacting with S1862.56). At the top of TM2, K1972.67, D1982.68, and Y1451.40 are stabilized in polar interactions (red dotted lines). Right; The TT-OAD2 (brown stick and transparent van der Waals surface) binding triggers the reorganization of the interaction network at the top of TM1 and leads the Y1481.43 and Y1521.47 to make contact and form hydrogen bond interactions with the R1902.60. The ECL3 of the GLP-1R:TT-OAD complex was removed for clarity.
Rights and permissions
About this article

Cite this article
Zhao, P., Liang, YL., Belousoff, M.J. et al. Activation of the GLP-1 receptor by a non-peptidic agonist. Nature 577, 432–436 (2020). https://doi.org/10.1038/s41586-019-1902-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1902-z
- Springer Nature Limited
This article is cited by
-
G protein-coupled receptors (GPCRs): advances in structures, mechanisms and drug discovery
Signal Transduction and Targeted Therapy (2024)
-
Cryo-electron microscopy for GPCR research and drug discovery in endocrinology and metabolism
Nature Reviews Endocrinology (2024)
-
GLP-1R signaling neighborhoods associate with the susceptibility to adverse drug reactions of incretin mimetics
Nature Communications (2023)
-
Conserved class B GPCR activation by a biased intracellular agonist
Nature (2023)
-
GWAS of random glucose in 476,326 individuals provide insights into diabetes pathophysiology, complications and treatment stratification
Nature Genetics (2023)