

Abstract
The translocase of the outer mitochondrial membrane (TOM) is the main entry gate for proteins1,2,3,4. Here we use cryo-electron microscopy to report the structure of the yeast TOM core complex5,6,7,8,9 at 3.8-Å resolution. The structure reveals the high-resolution architecture of the translocator consisting of two Tom40 β-barrel channels and α-helical transmembrane subunits, providing insight into critical features that are conserved in all eukaryotes1,2,3. Each Tom40 β-barrel is surrounded by small TOM subunits, and tethered by two Tom22 subunits and one phospholipid. The N-terminal extension of Tom40 forms a helix inside the channel; mutational analysis reveals its dual role in early and late steps in the biogenesis of intermembrane-space proteins in cooperation with Tom5. Each Tom40 channel possesses two precursor exit sites. Tom22, Tom40 and Tom7 guide presequence-containing preproteins to the exit in the middle of the dimer, whereas Tom5 and the Tom40 N extension guide preproteins lacking a presequence to the exit at the periphery of the dimer.
Similar content being viewed by others


Main
Mitochondria have central roles in cellular energy production, metabolic pathways, lipid biogenesis, signalling and programmed cell death. Mitochondrial functions rely on the assembly of around 1,000 resident proteins, 99% of which are synthesized as preproteins in the cytosol and imported into mitochondria by translocator complexes1,2,3. The TOM complex functions as the main entry gate for more than 90% of mitochondrial proteins. The import pathway then branches into different sorting pathways to the outer membrane, intermembrane space (IMS), inner membrane and matrix, depending on the class of preproteins. In yeast, as in humans, the TOM complex consists of the channel-forming β-barrel protein Tom40 and six α-helical membrane-integrated subunits: the receptors Tom20, Tom22 and Tom70, and regulators Tom5, Tom6 and Tom7. The TOM complex has been observed in three-channel (trimer) and two-channel (dimer) forms4,5,6,7,8,9 (Extended Data Fig. 1a).
Preprotein translocation across membranes requires high fidelity for both substrate recognition and substrate movement through a proteinaceous channel. Protein secretion in the bacterial inner membrane and eukaryotic endoplasmic reticulum is driven by a translocon (the SecYEG or Sec61 complex) composed primarily of α-helical membrane-spanning segments10,11,12,13,14. Through coordinated movement of the helical segments relative to each other, substrate proteins can be translocated across the membrane or laterally gated into the membrane, depending on their intrinsically determined route through the translocon10,11,12,13,14. In the bacterial outer membrane, three protein translocases comprising a β-barrel channel are used either for protein translocation across the membrane (FhaC15) or lateral transfer into the membrane (BamA16 and TamA17). The TOM complex represents a fundamentally distinct form of translocator (α/β translocator), composed of a β-barrel channel together with α-helical subunits that coordinate its activity through interactions in the plane of the membrane. To date, the structural information available regarding this complex comes from in situ crosslinking of the trimer4 and a 6.8-Å medium-resolution cryo-electron microscopy (cryo-EM) structure of the dimer9. To understand the architecture and function of the TOM complex, we combined high-resolution cryo-EM and functional analyses. The results reveal how the TOM complex facilitates transport of around 1,000 different preproteins with divergent structural and chemical properties into mitochondria.
Structure of the TOM core complex
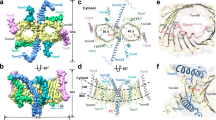
We purified the TOM complex from the yeast Saccharomyces cerevisiae (Extended Data Fig. 1b–e). The purified TOM complex is a mixture of trimers and dimers8 (Extended Data Fig. 1a). High-speed atomic force microscopy (HS-AFM) analysis showed that the trimer could be converted to the dimer (Extended Data Fig. 1f, Supplementary Videos 1, 2). The cryo-EM structure was determined at an effective overall resolution of 3.8 Å as determined by Fourier shell correlation (FSC) (local resolution varying from 3.6 to 5.7 Å) (Fig. 1a, b, Extended Data Fig. 2, Extended Data Table 1). Main chains and many bulky side chains of each subunit were built from the electron microscopy density map or Coulomb potential map and, with the exception of disordered N- or C-terminal segments and short loops, the main chains were visualized as continuous density. The structure showed two molecules each of Tom40, Tom22, Tom5, Tom6 and Tom7, with a two-fold symmetry and dimensions of 66 Å × 48 Å × 65 Å for each protomer, corresponding to the previously observed TOM core complex7,8,9 (Extended Data Fig. 1a). Initially, the purified TOM complex fraction contained the receptor Tom20 (Extended Data Fig. 1d), but the association of Tom20 with the TOM core complex is labile, and thus the stable particles used for determination of the dimeric structure lacked Tom20, consistent with the reported increased stability of the dimeric form in the absence of Tom207,8,18. The channel subunit Tom40 forms a 19-stranded transmembrane β-barrel, with β-strand lengths varying from 5 to 12 residues (Extended Data Fig. 3a). The Tom22, Tom5, Tom6 and Tom7 transmembrane α-helices are closely apposed to the β-barrel (Extended Data Fig. 3b).

a, b, Cryo-EM density of the TOM complex dimer (a) and its cartoon representation (b). Each subunit is shown in a different colour. The TOM-bound phospholipid (PL), modelled as phosphatidylethanolamine, is shown as a space-filling model. The broken lines (b, left panel) indicate the membrane–water interface, which was estimated from the positions of the transmembrane segments for Tom2234, Tom5, Tom6 and Tom7 calculated by the TMpred server (https://embnet.vital-it.ch/software/TMPRED_form.html). c, Close-up view of the two Tom22 molecules and bound phospholipid. Tom22 (cartoon representation) and PL (space-filling model) are superimposed according to the EM density. d, The cavity formed by Tom40 and Tom22 for binding of the phospholipid. Tom40 is shown by cutaway representation. e, The association of the first and last β-strands of Tom40 (blue, the EM density is superimposed) is stabilized by Tom22 (red) and phospholipid (green). The head group of the phospholipid was not visible, except for the phosphate, but phosphatidylethanolamine was modelled on the basis of previously reported biochemical results46. f, Association of Tom7 (green, the EM density is superimposed) with the outer wall of the Tom40 β-barrel.
α–β contacts and lipid in the dimer
Comparative sequence analysis suggested the evolution of the TOM complex from an ancestral complex consisting of Tom40, Tom22 and Tom719,20. Bioinformatics assessment of Tom7 homologues from the broad range of eukaryotic lineages revealed a universal code of aromatic and hydrophilic residues stamped in its predicted transmembrane segment21. The universally conserved glycine and proline residues (P34, G40 and P48 in the yeast protein) of Tom7 were a particularly distinct feature, given that proline is rarely found in transmembrane segments22. Proline residues are also found universally in the transmembrane segment of Tom22 at a conserved position with respect to the cis membrane surface, corresponding to P112 in the yeast protein21. Consistent with these conserved, helix-breaking residues, a distinct feature of the TOM complex is the arrangement of noticeably kinked transmembrane α-helices of Tom7 and Tom22 around the two Tom40 β-barrels (Fig. 1a, b).
The two Tom40 β-barrels are tethered by the two molecules of Tom22 inserted between them. Tom22 forms a continuously apposed transmembrane α-helix, kinked by 30° through G110 and P112 (Fig. 1c) to follow the contour of the Tom40 β-barrel. The α–β contacts between Tom40 and Tom22 are largely mediated through hydrophobic residues (Fig. 1c). The IMS or C-terminal halves of the kinked Tom22 helices run in parallel between the Tom40 β-barrel dimer, keeping the β-barrels apart. The IMS halves of the Tom22 helices and the Tom40 β-barrels form an intramembrane cavity at the dimer interface, in which a phospholipid molecule was observed, with its head group facing the IMS (Fig. 1c, d). The acidic phosphate group was located close to the conserved R330 of Tom40. The cytosolic halves of the Tom22 helices run outward from the β-barrel dimer interface, and the acyl chains of the phospholipid are held in the resultant space between the Tom40 β-barrels in the vicinity of hydrophobic residues (Fig. 1b). The Tom40 β-barrels directly contact each other near the membrane–cytosol interface. The insertion of the IMS halves of the kinked Tom22 helices together with the phospholipid head group between the IMS side of the Tom40 protomers provides a structural explanation for the observed tilt9 of the Tom40 β-barrels relative to each other (Fig. 1a, b, side views), which probably distorts the lipid bilayer.
The first (β1) and last (β19) β-strands of Tom40 associate to stably close the β-barrel with seven hydrogen bonds (Extended Data Fig. 3a). The structure of the Tom40 β-barrel is further stabilized by association of the two Tom22 transmembrane helices and the acyl chains of the bound phospholipid (Fig. 1e, Extended Data Fig. 3b). The Tom40 β-barrel could potentially undergo a lateral opening for insertion of outer-membrane proteins23 analogous to the bacterial system of BamA17 and TamA16, and Sam50 (also known as Tob55) of the mitochondrial sorting and assembly machinery24. This lateral gating of Tom40 would require Tom22 to shift sideways, a dynamic movement previously envisaged in the TOM complex dimer–trimer transition in intact mitochondria4,5 (Extended Data Fig. 1a).
Tom7 has a highly unusual membrane topology in which a short hydrophobic helix, α2, spans the membrane core, presents a subsequent hydrophilic segment to the IMS side of the membrane and then diverts back into the lipid core by forming a short C-terminal hydrophobic helix α3. This topology traverses six Tom40 β-strands (Fig. 1e, Extended Data Fig. 3b). At the position of contact with helix α2 of Tom7, hydrogen bonds among β-strands 4, 5 and 6 of Tom40 are partly broken (Fig. 1e, Extended Data Fig. 3a). The contacts of Tom5 and Tom6 with the Tom40 β-barrel are described below.
Dimer–trimer conversion
A comparison of the cryo-EM structure of the purified dimeric TOM complex with the trimer model derived from site-specific photocrosslink mapping in intact mitochondria4 (Extended Data Figs. 1a, 3c) showed that the relative positions of Tom5 and Tom6 to Tom40 are similar between the trimer and the dimer (Extended Data Fig. 4a). Conversely, the orientation of both Tom22 molecules and perhaps the positions of the two Tom7 molecules partially differ between the dimer and trimer (Extended Data Fig. 4a). Examination of the crosslinked residues that do not fit to the cryo-EM structure—residues 90, 309, 335 and 350 of Tom40 and residues 112 and 114 of Tom22 (Supplementary Table 1, Extended Data Fig. 4b, c)—suggests a possible geometrical rearrangement of the TOM subunits, including rotation of the Tom22 transmembrane helix and shift of Tom7 from the current position on dimer–trimer conversion (Extended Data Fig. 4d, e). Kinks in the membrane-embedded helices of Tom22 and Tom7 may enable twisting around the axis of the helix, potentially helping to transmit changes in positioning of the cytosolic and IMS domains of Tom22 with respect to other TOM subunits. The transmembrane helix of Tom6 also interacts with the outer wall of the Tom40 β-barrel through hydrophobic interactions with strands β13–β15 (Fig. 2a, Extended Data Fig. 3b). The initial N-terminal segment (residues 1–24) is disordered, with residues 25–31 oriented towards the neighbouring Tom40 molecule (Fig. 1b). This, together with previous crosslinking results4,5, suggests that the transmembrane segment (residues 32–49) locks onto the β-barrel and the kinked N-terminal helix may bridge the two Tom40 molecules, providing a structural insight into how Tom6 prevents dissociation of Tom22 from the TOM complex.

a, Association of Tom6 (yellow, the EM density is superimposed) with the outer wall of the Tom40 β-barrel. b, Crosslinked products of Tom40 containing BPA at the indicated positions in the C-terminal segment were purified via His-tagged Tom40 and detected using the indicated antibodies. Crosslinked products between Tom40 and Tim50 (40–50) or Tom22 (40–22) are indicated. Blots are representative of two independent experiments. Asterisks, non-specific bands. c, Association of Tom5 (red, the EM density is superimposed) with the outer wall of the Tom40 β-barrel.
The distribution of the conserved residues on the Tom40 β-barrel outer wall (Supplementary Fig. 1, Extended Data Fig. 5) shows that some key residues are distributed differently in each docking interface, suggesting specific docking interfaces for different α-helical TOM subunits. Indeed, conserved residues of the outer wall of the Tom40 β-barrel are enriched in the interface regions for Tom22 and Tom7 (Extended Data Fig. 5, Supplementary Table 2), which is consistent with the proposed ancestral TOM complex consisting of Tom40, Tom22 and Tom719,20. Eight residues at conserved positions on the β-barrel surface of Tom40 were found to be in proximity to ten residues at conserved positions on the Tom22 transmembrane helices (Extended Data Fig. 5a). Two conserved residues on the Tom40 β-barrel outer wall were found to be close to two conserved residues of Tom7. In particular, G129 of Tom40, which is also conserved in another Tom7-binding protein Mdm1025,26, may minimize possible steric hindrance in binding of the bulky side chain of conserved I33 of Tom7 to the groove of Tom40 (Extended Data Fig. 5b); W32, I33 and V36 of Tom7 are inserted into the groove formed mainly by G129, G139 and A118 of Tom40. Three residues at conserved positions of Tom40 were found to be in proximity to three residues at conserved positions of Tom6, which may be sufficient to fix the position of the transmembrane helix of Tom6 (Extended Data Fig. 5c). There was no conserved proximity pair between the Tom40 β-barrel and the Tom5 transmembrane segment (Extended Data Fig. 5d), yet the IMS segment of Tom5 interacts with the N-terminal segment of Tom40, as described below.
Exit site for the presequence pathway
Functional studies suggested that Tom727 and the IMS regions of Tom2228,29,30 and Tom4030,31 are involved in forming a trans presequence-binding site on the IMS side of the outer membrane. The proximity of the hydrophilic loop between the Tom7 helices α2 and α3, the C-terminal acidic and disordered IMS domain of Tom22 and the hydrophilic and disordered C-terminal segment and loop connecting β-strands β1 and β2 of Tom40 (Extended Data Fig. 6a, b) provides structural insight into this mechanism. In addition to the observations that the presequence of a TOM-arrested preprotein is crosslinked to Tim5032,33 and Tim50 is in the direct vicinity of the C-terminal IMS domain of Tom2234,35 in organello, proximity relationships of the IMS domain of Tom22, the C-terminal IMS-facing segment of Tom40 and the IMS-exposed domain of Tim50 were shown by in vivo crosslinking using photoreactive p-benzoylphenylalanine (BPA) (Fig. 2b, Extended Data Fig. 6c; Mia40 (also known as Tim40) was not crosslinked to the Tom22 or Tom40 C-segment, but to the Tom40 N-terminal segment, as described below). Deletion of the C-terminal segment of Tom40 impaired the accumulation of the presequence-containing Oxa1 precursor at the TOM complex trans site (Extended Data Fig. 6d–f), similar to the reported roles of the Tom22 IMS and Tom7 in formation of the trans site27,36. The biogenesis of the presequence-lacking precursor of Tim9 was not inhibited by the C-terminal truncation of Tom40 (Extended Data Fig. 6g) (as described below, the import of Tim9 did not involve Tom22 or Tom7). We conclude that presequences are guided to the trans site composed of the IMS-facing regions of Tom40, Tom22 and Tom7 and leave the Tom40 channel in the middle of the dimeric TOM structure to be transferred from the IMS domains of Tom40 and Tom22 to Tim50 of the TIM23 complex (Extended Data Fig. 6h).
Tom5 and the N extension of Tom40
Tom5 forms a transmembrane α-helix located in the distal position of the TOM core dimer, with a kink at P39–W42 (Fig. 2c). The N-terminal hydrophilic part of the helix is oriented towards the cytosol. The central part of the Tom5 helix is hydrophobic and binds to β-strands 9–11 of Tom40. A long N-terminal segment of Tom40 (residues 1–82, termed N extension) preceding β-strand β1 passes through the β-barrel pore to the IMS side; the α1-helix (residues 48–53 of Tom40) is situated in the IMS, and the α2-helix (residues 63–74 with a short helix break at residues 66–67) is located inside the β-barrel pore (Fig. 3a–c). The α1-helix interacts with the IMS part of the Tom5 helix (Fig. 3a), whereas the α2-helix interacts with the concave inner wall of the Tom40 β-barrel (β-strands 11–17) mainly through hydrophilic residues (Fig. 3b, c).

a, Association of the α2-helix of the Tom40 N-terminal segment (dark blue, the EM density is superimposed) with the pore interior of the Tom40 β-barrel. b, c, Close-up views of the interactions between the α2-helix of the Tom40 N-terminal segment (dark blue, the EM density is superimposed) with the pore interior of the Tom40 β-barrel. d, Close-up view of the interactions between the α1-helix of the Tom40 N-terminal segment (dark blue, the EM density is superimposed) and the IMS region of Tom5.
Residues up to position 59 of the N extension are located in the proximity of the IMS part of Tom5 (Fig. 3d). We screened the growth of N-terminal and C-terminal truncation mutants of Tom40 and found that a truncation including residue 59 was critical for respiratory growth (Fig. 4a). Residue 59 is located at the beginning of the first conserved region of Tom40 (Supplementary Fig. 1). Under fermenting growth conditions, a double mutant with the N-terminal deletion Tom40(Δ59) and a deletion of TOM5 showed a synthetic growth defect (Fig. 4b), supporting the view that Tom5 and the N extension of Tom40 are functionally related.

a, Growth analysis of yeast strains expressing Tom40 variants with the indicated N- or C-terminal truncations on non-fermentable medium. Data are representative of three independent experiments. WT, wild-type. b, Growth analysis of wild-type and tom5∆ strains expressing the indicated Tom40 N-terminal truncations on fermentable medium. Data are representative of two independent experiments. c, Crosslinked products of Tom40 containing BPA at the indicated positions were purified via His-tagged Tom40 and detected by the indicated antibodies. Crosslinked products between Tom40 and Mia40 (40–40) are indicated. Blots are representative of three (top left) or two (other blots) independent experiments. d, e, Import of radiolabelled Tim9 and Tim12 into isolated wild-type, Tom40(Δ58) and Tom40(Δ59) mitochondria, followed by analysis via native gel electrophoresis and autoradiography. Data are representative of three (d) or two (e) independent experiments. TIM22, TIM22 complex; Tim9–Mia40 and Tim12–Mia40, mixed disulfide intermediates of Tim precursors with Mia40. f, Import of radiolabelled pSu9–DHFR into isolated mitochondria, followed by treatment with proteinase K and SDS–PAGE analysis. Data are representative of two independent experiments. Δψ, membrane potential; p, precursor form; m, mature form. g, Wild-type, Tom40(Δ58) and Tom40(Δ59) mitochondria were treated with 4,4′-dipyridyl disulfide (4-DPS) or dithiothreitol (DTT) and the redox state of Mia40 was analysed by non-reducing SDS–PAGE and western blotting. Data are representative of two independent experiments. Red., reduced form; oxid., oxidized form.
Exit site for proteins lacking presequences
Site-specific crosslinking revealed a close contact of small TIM chaperones to the IMS-exposed portion of the Tom40 N extension4. These chaperones bind the precursors of inner-membrane carrier proteins and outer-membrane β-barrel proteins upon their translocation through Tom401,2,3,4. Deletion mutagenesis of the N extension of Tom40 demonstrated that removal of residues 1–57 inhibited the import of carrier precursors and β-barrel precursors, but not of presequence-containing precursors (which were inhibited by a larger truncation including residue 62)4. As the N extension leaves the Tom40 channel on the opposite site of the Tom22–Tom7–Tom40 trans site for presequence-containing precursors (Fig. 1a, b), the structure of the TOM complex reveals the existence of two different IMS exit sites of Tom40: a site for the presequence pathway located in the middle of the dimeric TOM structure and a peripheral site for the carrier and β-barrel precursors lacking a presequence (Extended Data Fig. 6h).
The fourth essential protein import pathway of mitochondria transports cysteine-rich precursors through the TOM complex to the mitochondrial IMS import and assembly (MIA) system37,38,39,40. The core component Mia40 was crosslinked to the disordered Tom40 N extension via BPA introduced at residues 34 and 44 of the extension (Fig. 4c), suggesting that the N extension recruits Mia40 to the IMS outlet of the Tom40 channel (Tom22 and Tim50 were crosslinked not to the N extension, but to the C-segment of Tom40; Fig. 2b). The import of MIA substrates such as Tim9 was highly dependent on the presence of Tom5, but not on other small TOM proteins or TOM receptors38,41 (Extended Data Fig. 7a–e). In Tom40(Δ59) mitochondria, the steady-state level of Tom5 decreased at elevated temperature (37 °C) (Extended Data Fig. 7f); the isolated mitochondria were competent in inserting the Tom5 precursor into the outer membrane (Extended Data Fig. 7g), indicating that the N extension of Tom40 stabilizes the assembly of Tom5 into the TOM complex. To analyse whether the N extension itself has a role in the MIA import pathway, we isolated mitochondria from cells grown at 30 °C that contained wild-type levels of Tom5 (Extended Data Fig. 7f). Under these conditions, the TOM complex analysed by native gel electrophoresis became smaller owing to the truncation of Tom40, but formed a stable complex; only a small fraction was destabilized in Tom40(Δ59) mitochondria (Extended Data Fig. 7h). The biogenesis of MIA substrates (Tim9 and Tim12) can be dissected in an early targeting step to the Mia40-bound intermediate and a late folding/assembly step into the oxidized Tim9–Tim10 and TIM22 complexes39,42. Tom40(Δ59) mitochondria were impaired in the early targeting to Mia40, and therefore the late folding/assembly step was diminished (Fig. 4d, e). The presequence import pathway, analysed with the Atp2 precursor and model preproteins Su9–mouse dihydrofolate reductase (Su9–DHFR) and b2(167)ΔDHFR, was not affected in these Tom40 mutants (Fig. 4f, Extended Data Fig. 7i). The redox state and native electrophoretic mobility of Mia40 and the intactness of the outer membrane (Fig. 4g, Extended Data Fig. 7h, j) were not altered in the Tom40 truncation mutants, excluding the possibility that the import of MIA substrates was indirectly affected by an altered Mia40 activity or the loss of IMS components.
The results so far suggest that the N extension of Tom40 interacts with Tom5. Tom5 and the IMS-exposed portion of the N extension including residue 59 are required for the efficient targeting of precursors to Mia40, indicating that, like carrier and β-barrel precursors, MIA substrates leave the Tom40 channel near the N extension–Tom5 (Extended Data Fig. 6h).
Point mutations in the conserved Tom40 residues L57 and T62 showed a synthetic growth defect (Fig. 5a). T62 of the N extension interacts with D170 in strand β7 of the β-barrel wall, whereas L57 is in the vicinity of S174 in the loop connecting the β7 and β8 strands (Fig. 5b); Tom5 and Mia40 interacting regions are retained in these point mutants. In Tom40(L57A/T62A) double-mutant mitochondria, the early targeting of Tim9 precursor to the Mia40-bound intermediate was possible, but the late biogenesis step towards formation of oxidized Tim9–Tim10 and TIM22 complexes was impaired (Fig. 5c). The presequence and carrier import pathways (Su9–DHFR and ADP–ATP carrier (AAC), respectively) were unaffected or only mildly affected in Tom40(L57A/T62A) mitochondria (Extended Data Fig. 7k). Addition of reduced glutathione (GSH) promotes the formation of oxidized MIA pathway substrates, probably by preventing the formation of non-productive long-lived Mia40-substrate intermediates43. GSH stimulated Tim9 assembly in wild-type mitochondria, but not in Tom40(L57A/T62A) mitochondria (Fig. 5d). Tom5 levels, TOM native electrophoretic mobility and Mia40 redox state were similar to the wild type in Tom40(L57A/T62A) mitochondria (Extended Data Figs. 7l, 8a, b). We conclude that these conserved Tom40 residues are involved in promoting late steps of the MIA pathway, including oxidative folding and subsequent assembly into the final complexes. This is consistent with the report that deletion of the conserved NPGT sequence in Neurospora crassa Tom40 (corresponding to yeast residues 59–62; Supplementary Fig. 1) caused severe growth defects44.

a, Growth analysis of yeast strains expressing Tom40(L57A), Tom40(T62A) and Tom40(L57A/T62A) on non-fermentable medium. Data are representative of five independent experiments (three for T62A). b, Close-up view of the N-terminal segment around L57 and T62 of Tom40. c, Import of radiolabelled Tim9 precursor into isolated wild-type, Tom40(L57A) and Tom40(L57A/T62A) mitochondria, analysed by blue native electrophoresis. Data are representative of seven (L57A/T62A) or two (L57A) independent experiments. d, Import of radiolabelled Tim9 into Tom40(L57A/T62A) mitochondria in the presence of the indicated concentrations of GSH (at higher concentrations, GSH also inhibited the assembly in wild-type mitochondria). Data are representative of two independent experiments. e, Close-up view of L202 of β-strand β9 and the N-terminal segment of Tom40. f, Radiolabelled Tim9 was imported into wild-type and Tom40(L202A) mitochondria for 5 min. The mitochondria were reisolated and incubated for the indicated chase periods, followed by analysis by blue native electrophoresis. Data are representative of three independent experiments. g, Schematic model of the two distinct precursor exit sites of the TOM complex. The views for a channel unit from the cytosol (left) and a unit from the IMS (rotated by 180°; right) are shown. Tom22, which links the channel units, indicates the middle of the dimeric TOM complex.
Replacement of the conserved residue L202 in the Tom40 β9-strand by alanine (Supplementary Fig. 1) caused respiratory growth defects at elevated temperature (Extended Data Fig. 8c). L202 appears to be responsible for the positioning of helix α2 of the N extension by forming a hydrophobic linkage to type-conserved V63 and L66 in the N extension together with type-conserved A210 and L233 in the inner wall of the Tom40 β-barrel (Fig. 5e, Supplementary Fig. 1). The precursor of Tim9 was targeted to Mia40 in Tom40(L202A) mitochondria, but the subsequent chase into the oxidized form, which involves the formation of intramolecular disulfide bonds42,45, was impaired (Fig. 5f), similar to the effect of iodoacetamide addition to prevent formation of further disulfides in the precursor (Extended Data Fig. 8d). Tom40(L202A) mitochondria accumulated the Tim9(C55S) variant precursor42 as the Mia40 mixed disulfide intermediate with an efficiency close to that of wild-type mitochondria (Extended Data Fig. 8e), offering further evidence that targeting to Mia40 was possible in Tom40(L202A) mitochondria. The Tim9 precursor was lost from Tom40(L202A) mitochondria when the chase to the oxidized forms was attempted (Fig. 5f), but not when the oxidation was blocked by iodoacetamide; that is, when the precursor remained bound to Mia40 (Extended Data Fig. 8d). The Mia40 redox state, TOM native electrophoretic mobility, intactness of the outer membrane, presequence and carrier import pathways were not affected in Tom40(L202A) mitochondria (Extended Data Fig. 8b, g, h).
We conclude that the N extension of Tom40 plays a dual role in the MIA pathway. First, the IMS-exposed portion of the extension (up to residue 59) interacts with Mia40 and Tom5 and is required for the early import step—that is, translocation of precursors to Mia40 and formation of a Mia40-precursor mixed intermediate. Second, Tom40 residues involved in the positioning of the N extension inside the β-barrel (including residues 57, 62 and 202) are required for late biogenesis steps that involve the complete import of precursors into the IMS and the formation of intramolecular disulfide bonds. Mutants in which this late biogenesis step is disturbed exhibited a loss of precursor from mitochondria, indicating that the full translocation of the precursor through Tom40 occurs only at this late stage.
Conclusions
Four protein import pathways into mitochondria are essential for cell viability. In each of these pathways, the precursor proteins are translocated through the TOM complex. The cryo-EM structure of the core TOM complex reveals an α/β-membrane-integrated translocator. Given that the core machinery is conserved in all eukaryotes19,20, the structure of the TOM complex from yeast provides a fundamental model for the function of this exquisite nanomachine. Conserved proline residues provide kinks in the α-helical transmembrane segments that cope with embracing the rounded outer face of the Tom40 β-barrel and provide intricate structural features such as the Tom7 crook into the presequence-binding domain and a potential Tom22 rotational manoeuvre for opening of the putative lateral gate in Tom40.
The β-barrel structure of Tom40 discloses surfaces that are proposed to guide protein substrates through the Tom40 channel4 (Extended Data Fig. 9). Site-specific crosslinking indicated that presequences and hydrophobic precursors interact with different residues of the channel interior4. The cryo-EM structure demonstrates the inferred alignment4 of acidic patches and hydrophobic patches, respectively, and thus outlines different translocation paths for presequence-containing precursors and hydrophobic precursors. The N extension of Tom40 passes through the β-barrel into the IMS. Full activity of the MIA import pathway depends on proper positioning of the N extension inside the barrel lumen.
The TOM structure reveals two preprotein exit sites towards the IMS (Fig. 5g). Presequence-containing preproteins that constitute around 60% of mitochondrial proteins leave the Tom40 pore at the trans presequence-binding site, which is formed by Tom22–Tom7–Tom40 near the middle of the Tom40 dimer, and are then transferred to Tim50 of the TIM23 complex. Presequence-lacking preproteins, which include three different classes (MIA substrates, β-barrel and carrier precursors), leave the Tom40 channel in the proximity of Tom5 and the N extension of Tom40 at the periphery of the Tom40 dimer. The IMS-exposed portion of the N extension of Tom40 recruits small TIM chaperones and Mia40 close to the exit site to ensure an efficient transfer of precursor proteins. The TOM complex thus functions as a hub for the intramitochondrial sorting of precursor proteins.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigations were not blinded to allocation during experiments and outcome assessment.
Protein expression and purification
The TOM complex was purified from the S. cerevisiae strain expressing Tom22 with the C-terminally attached His10 tag from the chromosome (the Tom22-His10-pep4∆ strain). After cultivation in 24 l YPD medium for 24 h at 30 °C, cells were collected and resuspended in 10–20 ml of breaking buffer (0.6 M sorbitol, 10 mM Tris-HCl, pH 7.4, 1 mM EDTA). The cells were frozen by dropwise addition to liquid nitrogen in a 1000 ml stainless beaker to form small particles, which were subsequently grounded by Multi-Beads Shocker (Yasui Kikai). The resultant frozen cell powder was slowly thawed at room temperature and centrifuged (1,500g, 20 min, 4 °C) to remove debris. Mitochondria were isolated by centrifugation (13,000g, 20 min, 4 °C) and washed by SEM buffer (250 mM sucrose, 10 mM MOPS, pH 7.2, 1 mM EDTA). After lysis of mitochondria with solubilization buffer (50 mM Hepes-KOH, pH 7.4, 150 mM NaCl, 5% glycerol and 1.1% digitonin (Merck)) for 16 h at 4 °C, insoluble materials were removed by centrifugation (23,000g, 30 min, 4 °C). The supernatant was mixed with 2 ml of TALON metal affinity resin (Clontech) and incubated for 30 min. The TOM complex was eluted by step gradient of 10–160 mM imidazole in TALON buffer (20 mM Hepes-KOH, pH 7.4, 150 mM NaCl, 0.1% GDN (Anatrace)). The eluted fraction was subjected to gel filtration with a Superose 6 Increase 10/300 GL column (GE Healthcare) equilibrated with gel-filtration buffer (20 mM Hepes-KOH, pH 7.4, 150 mM NaCl, 0.01% GDN). The purified TOM complex was concentrated to 5-10 mg ml−1 with an Amicon Ultra-0.5 filter (Merck Millipore, 100 kDa MW cut-off).
Cryo-EM grid preparation
The concentrated TOM complex was loaded onto a glow-discharged grid (Quantifoil R1.2/1.3, Cu/Rh, 200 mesh), blotted by Vitrobot Mark IV (FEI) at 6 °C, and plunge-frozen in liquid ethane.
Cryo-EM data acquisition and processing
Grids were screened for ice quality and cryo-EM data were acquired on a Titan Krios transmission EM (Thermo Fisher Scientific) at 300 kV under low-dose conditions (49 frames at 50 e− Å−2). A total of 2,057 micrographs were collected on a Falcon III direct electron detector (Thermo Fisher Scientific). The movie frames were aligned using MotionCor247. CTF parameters were estimated by CTFFIND 448. Then, 1,866 micrographs were submitted to auto-picking. Subsequently, 699,268 particles were extracted and 2D classified by RELION-3.049. For building an initial model, an additional two rounds of 2D classification were performed. Selected 573,344 particles from the first 2D classification were submitted to 3D classification, with the initial model as 3D reference. Two rounds of 3D classification were performed, the first one with no symmetry and the subsequent second one with C2 symmetry imposed. Finally, 124,653 particles were selected for several cycles of 3D auto-refinement followed by CTF refinement and Bayesian polishing (Extended Data Table. 1). The overall gold-standard resolution was 3.81 Å, with the local resolution in the N-terminal helix of the Tom40 channel extending to 3.66 Å.
Model building and refinement
The atomic models of all five different subunits of the TOM complex were built de novo into the EM density map in COOT50. As guide structures, we used a homology model of S. cerevisiae (Sc) Tom40 based on the N. crassa (Nc) Tom40 model (PDB: 5O8O) and the cryo-EM structure of NcTOM complex (EMDB: 3761). The atomic model of the ScTOM complex was refined using PHENIX51. Molecular graphics were illustrated with UCSF Chimera52, Molecular Operating Environment (MOE) software (2016.08; Chemical Computing Group) and CueMol 2.0 (http://www.cuemol.org).
Yeast strains, plasmids and primers
The S. cerevisiae strains used in this study are listed in Supplementary Table 353. For the preparation of the yeast TOM complex, the DNA fragments for Tom22−His1034 and the kanMX4 marker were combined by PCR using the primers Tom22His10-KanMX-f and Tom22His10-KanMX-r (Supplementary Table 4) and integrated into the TOM22 locus of the D273-10B strain, then G418+ transformants were selected to yield the Tom22-His10 strain. The DNA fragment for YAP1 was amplified by PCR using primers of PEP4-YAP1-f and PEP4-YAP1-r (Supplementary Table 4), and was integrated into the PEP4 locus in the Tom22-His10 strain, then cycloheximide resistant transformants were selected to yield the Tom22-His10-pep4∆ strain.
The strains tom5∆, tom6∆, tom7∆, tom20∆, tom22∆, tom70∆ and their corresponding wild-type strains YPH499 and YPH500 have been described54,55. Yeast strains expressing wild-type or mutant Tom40 proteins except for those in Fig. 4b were generated by transforming pFL39-TOM40 or the related plasmids encoding Tom40 mutant versions into the yeast strain YPH499-tom40Δ + Yep-TOM40 followed by selection on 5-fluoroorotic acid (5-FOA) plates (9.33 mM 5-FOA (MELFORD), 1.68% (w/v) yeast nitrogen base without amino acids (Difco, BD), 5% (w/v) glucose and 0.19% (w/v) CSM-URA (MP Biomedicals), 0.013% (w/v) uracil (Sigma)). Yeast cells were grown in YPD (1% (w/v) yeast extract, 2% (w/v) bacto peptone, 2% (w/v) glucose), YPS (1% (w/v) yeast extract, 2% (w/v) bacto peptone, 2% (w/v) sucrose) or YPG (1% (w/v) yeast extract, 2% (w/v) bacto peptone, 3% (w/v) glycerol, pH 5.0) at 24–30 °C. For growth analysis, yeast cells grown in YPD until OD600 1.0 were diluted to OD600 0.01 in sterilized water56. The cells were plated at fivefold serial dilution on YPD or YPG plates.
To construct the strain W303-1A-tom40Δtom5Δ + pRS316-TOM40, the DNA fragment for the hygMX marker was amplified by PCR using the primers tom5-del-fw and tom51-del-rv from the pBS-hygMX plasmid. The amplified DNA fragment was integrated into the 3′-half region of the TOM5 gene, and hygromycin B-resistant transformants were selected. Yeast strains used in Fig. 4b were further generated by transforming pRS314-TOM40 or the related plasmids encoding Tom40 mutants into the strains W303-1A-tom40Δ + pRS316-TOM40 or W303-1A-tom40Δtom5Δ + pRS316-TOM40 followed by cultivation in SCD medium (0.67% (w/v) yeast nitrogen base without amino acids (Difco, BD), 2% (w/v) glucose (Nacalai Tesque), 0.5% (w/v) casamino acid (Difco, BD), 0.002% (w/v) uracil (Fuji-film Wako)) containing 5.2 mM 5-FOA. For growth analysis, yeast cells were plated at tenfold dilution on SCD (with 5.2 mM 5-FOA) plates at 30 °C.
Cloning
The DNA constructs for the N-terminally truncated Tom40 mutants except for those used for Fig. 4b were made by two-step PCR based on pFL39-TOM40. The first round of PCR was performed using primer sets Tom40up/Tom40R and Tom40Δ2-x/Tom40down (x represents the C-terminal amino-acid position of the truncated region), respectively (Supplementary Table 4). The resulting PCR products were mixed in equimolar amounts as template and amplified using the primer set Tom40up/Tom40down to generate the final PCR product, which was subsequently digested with BamHI and HincII to replace the corresponding fragment of pFL39-TOM40. The DNA constructs for the C-terminally truncated Tom40 mutants were generated using the same strategy. The first round of PCR was performed using primer sets Tom40Cup/Tom40Δy-387 and Tom40CF/Tom40Cdown (y represents the N-terminal amino-acid position of the truncated region), respectively. The resulting PCR products were mixed in equimolar amounts as template and amplified using the primer set Tom40Cup/Tom40Cdown to generate the final PCR product, which was subsequently digested with HincII and HindIII to replace the corresponding fragment of pFL39-TOM40. All point mutations were introduced by the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies) using the corresponding primer pairs (Supplementary Table 4) according to the manufacturer’s instructions based on pFL39-TOM40. The plasmid pFL39-Tom40-L57A/T62A was generated by introducing L57A via the primer set L57Afw/L57Arv into the plasmid pFL39-Tom40-T62A. All plasmids were sequenced. Besides the expected mutations, the plasmids encoding Tom40 truncation mutants (except pFL39-Tom40-Δ364-387) and Tom40-T62A contained an undesired mutation, which led to an E360G substitution in the translated protein. For these mutants, pFL39-Tom40-E360G, which did not lead to any growth phenotype of yeast cells compared to pFL39-TOM40, served as the control plasmid.
The DNA constructs for wild-type Tom40 and the N-terminally truncated Tom40 mutants used for Fig. 4b were made as follows. The pRS314 plasmid containing the TOM40 promoter and terminator containing the BamHI/XhoI site in between them were used to integrate the DNA fragments for Tom40 or its N-terminally truncated mutants that were PCR-amplified with the primer pairs of Tom40-fw or tom40Δz-fw (z represents the N-terminal amino-acid position of the truncated region) and Tom40-rv (Supplementary Table 4) at the BamHI/XhoI site.
HS-AFM observation
HS-AFM observation was performed in the tapping mode using a laboratory-built apparatus57. The short cantilevers used (BL-AC10DS-A2) were purchased from Olympus; resonant frequency, ~600 KHz in water; quality factor, ~2 in water; and spring constant, ~0.1 N/m. The general procedure for HS-AFM observation of protein molecules was described previously58. In brief, a glass sample stage (diameter, ~2 mm; height, ~2 mm) with a thin mica disc (1 mm in diameter and 0.05 mm thick) glued to the top by epoxy was attached onto the top of the Z-scanner by a drop of nail polish. A freshly cleaved mica surface was prepared by removing the top layers of mica using Scotch tape. Then, a drop (~2 μl) of the solubilized TOM complex diluted with a buffer containing 20 mM Hepes-KOH (pH7.4), 150 mM KCl and 0.01% GDN (final concentration of the TOM complex 0.25 nM) was deposited onto the mica surface. After 5 min of incubation, the sample stage was immersed in a liquid cell containing ~60 μl of the same buffer. The cantilever’s free oscillation amplitude A0 was set at 1−2 nm and the set point amplitude was set at 0.9 × A0, such that the loss of cantilever’s oscillation energy per tap was adjusted to be 2−3 kBT on average. The imaging rate, scan size and pixel size were a combination of 5 frames s−1, 50 × 50 nm2 and 50 × 50 or of 3.3 frames s−1, 200 × 200 nm2 and 120 × 120, respectively.
Isolation of mitochondria
Yeast cells were grown in YPG, YPS or YPD at 24–37 °C until an early logarithmic growth phase. Mitochondria were isolated by differential centrifugation55. Yeast cells were collected, washed in water and incubated in prewarmed DTT softening buffer (100 mM Tris-H2SO4, pH 9.4, 10 mM DTT) for 20 min at growth temperature. Cells were washed and incubated in zymolyase buffer (20 mM K2HPO4, pH 7.4, 1.2 M sorbitol) supplemented with 4 mg per g cells zymolyase (Seikagaku Biobusiness) at growth temperature for 30 min to open the cell wall. The obtained spheroplasts were washed and homogenized in cold homogenizing buffer (600 mM sorbitol, 10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.2% (w/v) fatty-acid free bovine serum albumin, 2 mM PMSF) using a teflon glass homogenizer. Subsequently, cell debris was removed (2,700g, 6 min, 4 °C) and mitochondria were isolated from the supernatant (17,000g, 15 min, 4 °C). Mitochondria were resuspended in SEM buffer (250 mM sucrose, 1 mM EDTA, 10 mM MOPS, pH 7.2) at a protein concentration of 10 mg ml−1 and were shock-frozen in liquid nitrogen. Mitochondria were stored in aliquots at −80 °C until use. For western blotting, mitochondrial proteins were separated by blue native electrophoresis or SDS–PAGE. To compare the levels of various proteins in parallel, the identical batches of isolated mitochondria were loaded onto several gels in order to obtain the optimal separation range for decoration with specific antisera for each protein of interest.
Synthesis of radiolabelled precursor proteins
To generate radiolabelled mitochondrial precursor proteins, pGEM4Z plasmids or PCR-generated templates were used for in vitro translation in the presence of [35S]methionine by the Flexi Rabbit Reticulocyte Lysate System (Promega) or the TNT SP6 Coupled Reticulocyte Lysate System (Promega) according to the manufacturer’s instructions. To prepare Tim9 precursors for import, lysates were either treated with 10 mM DTT and used for import experiments or precipitated with two volumes of saturated (NH4)2SO4 supplemented with 20 mM EDTA and 50 mM DTT for 30 min on ice. After centrifugation (20,000g, 15 min, 4 °C), the precipitated proteins were resuspended in urea denaturation buffer (8 M urea, 30 mM MOPS, pH 7.2, 20 mM DTT) and shaken at 25 °C for 15 min. After a clarifying spin (20,000g, 5 min, room temperature), the supernatant containing denatured Tim9 precursors was used for import experiments.
Mitochondrial protein import
The import of small Tim precursors was performed according to the previously reported method59. In brief, Tim9 precursors were incubated at 30 °C for the indicated periods with isolated mitochondria in import buffer lacking BSA (250 mM sucrose, 80 mM KCl, 5 mM MgCl2, 10 mM KH2PO4, 5 mM l-methionine, 10 mM MOPS-KOH, pH 7.2) supplemented with 2–10 mM GSH when indicated. The import reaction was stopped by addition of iodoacetamide to 50 mM final concentration. To chase the precursor, mitochondria were reisolated after 5 min import and subsequently resuspended in the import buffer without BSA for the chasing period. To remove the small Tim precursors exposed on the mitochondrial surface after import, the samples were treated with 10 μg ml−1 proteinase K for 15 min on ice. To study the arrest of Tim9 precursors at the Mia40 stage after 5 min import, samples were treated with 50 mM (final concentration) iodoacetamide and were further incubated at 30 °C for the indicated periods. Other radiolabelled precursor proteins were incubated with isolated mitochondria in import buffer (3% (w/v) fatty-acid free BSA, 250 mM sucrose, 80 mM KCl, 5 mM MgCl2, 2 mM KH2PO4, 5 mM l-methionine, 10 mM MOPS-KOH pH 7.2) supplemented with 4 mM ATP, 4 mM NADH, 5 mM creatine phosphate and 0.1 mg ml−1 creatine kinase at 20 °C or 25 °C for the indicated periods. To dissipate the membrane potential (Δψ), 8 μM antimycin A, 1 μM valinomycin and 20 μM oligomycin were used. After import, the samples were treated with proteinase K (25–50 μg ml−1) for 15 min on ice where indicated. To study the insertion of Tom5 into the outer membrane, radiolabelled Tom5 precursor was incubated in import buffer for 10 min at 25 °C. After a clarifying spin to remove aggregated precursor, isolated mitochondria were added, followed by an import incubation at 25 °C. The mitochondria were reisolated immediately after the import incubation, washed and incubated in 0.1 M sodium carbonate. After 30 min on ice, carbonate-resistant membrane sheets were pelleted (125,000g, 30 min, 4 °C) and analysed by SDS–PAGE60.
Proteinase K treatment, reduction and oxidation of mitochondrial proteins
Isolated mitochondria were incubated in SEM buffer or EM buffer (1 mM EDTA, 10 mM MOPS, pH 7.2) for 15 min on ice. Subsequently, mitochondria were treated with 10–50 μg ml−1 proteinase K for 15 min on ice. To inhibit the proteinase K activity, mitochondria were incubated with 2 mM PMSF for 10 min on ice. To reduce or oxidize mitochondrial proteins, isolated mitochondria were treated with 50 mM DTT or 4-DPS (180–650 μM) in SEM buffer for 5 min on ice, respectively. To terminate the redox reaction, samples were supplemented with 50 mM iodoacetamide and incubated on ice for 10 min. The proteins were separated by reducing or non-reducing SDS–PAGE followed by western blotting.
In vivo photocrosslinking
In vivo BPA crosslinking was performed as described previously4. For BPA crosslinking of Tom22, TSY1/pTS1-X (X: 132, 136 and 140) cells24 were grown in 5 ml of SD (−Trp, −Leu) for 36 h at 30 °C. The cells were collected by centrifugation (1,500g, 5 min, 25 °C), resuspended in 50 ml of SGal (−Trp, −Leu, + 1 mM BPA) and cultured for 18 h at 30 °C. Half of the culture was irradiated by UV (365 nm) for 7 min, and the other half kept in the dark. Then, the cells were collected by centrifugation (1,500g, 5 min, 4 °C), and Tom22–His10 was purified with Ni-NTA (see below). For BPA crosslinking of Tom40, TSY5/pTS6-X (X: 34, 44, 372, 375, 379, 383 and 387) cells4 were grown in SCGal (−Ura, −Trp) for 48 h at 30 °C, then diluted tenfold in SCD (−Ura, −Trp, + 1 mM BPA), and cultured for 22 h at 30 °C. Then the culture was subjected to crosslinking by UV irradiation as in the case of Tom22.
For Ni-NTA purification of the crosslinked products, cells with and without UV irradiation were resuspended in TE buffer (10 mM Tris-HCl pH 8.0 and 1 mM EDTA), treated with 300 mM NaOH, 7% (v/v) 2-mercaptoethanol for 5 min on ice, and then received 10% trichloroacetic acid. The precipitated proteins were washed with ice-cold acetone after centrifugation (20,000g, 10 min, 4 °C), and then solubilized with 1% SDS buffer (40 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% (w/v) SDS, 1 mM PMSF) for 5 min at 60 °C. Insoluble materials were removed by centrifugation (20,000g, 10 min, 25 °C), and the supernatant was diluted fivefold with 0.5% TX-100 buffer (0.5% (v/v) Triton X-100, 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 40 mM imidazole) and incubated for 2 h at 4 °C with Ni-NTA agarose resin. Ni-NTA resin was washed with 0.5% TX-100 buffer 4 times, and bound proteins were eluted by elution buffer (0.5% (v/v) Triton X-100, 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 400 mM imidazole). Eluted proteins were mixed with 0.03% (w/v) sodium deoxycholate and 10% trichloroacetic acid, and kept on ice for 20 min. The precipitated proteins were washed with ice-cold acetone after centrifugation (20,000g, 10 min, 4 °C), then solubilized with 1% SDS buffer for 5 min at 60 °C and subjected to SDS–PAGE followed by western blotting.
Analysis of gel-separated proteins
Radiolabelled proteins were separated by blue native electrophoresis or SDS–PAGE and analysed by autoradiography using Typhoon FLA 7000 phosphoimager and Fuji FLA 9000 phosphoimager. Non-radiolabelled proteins were detected by western blotting. Antibodies were used that had been generated in rabbits against proteins of S. cerevisiae by injection of peptides (Cit1, Cyc1, Por1, Rip1, Tim13, Tim23, Tim50, Tom5, Tom40, Tom70) and recombinant full-length or truncated proteins (Mia40, Tom22, Tom40, Sam50, Tim50). The sera were used for western blotting with 1:250 to 1:5,000 dilution. Detection was performed using chemiluminescence, X-ray films or LAS 3000/4000 (FujiFilm). The figure legends indicate how many times the experiments were performed (independent experiments with similar set-ups and results). Data were processed using ImageJ, MultiGauge, Adobe Photoshop and Illustrator CS5, CS6 and CC 2017. Adjustments of brightness and contrast were applied to whole images in linear settings. Uncropped gels and source data are shown in Supplementary Fig. 2.
Mass spectrometry analysis
For matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI–TOF-MS) analysis, the purified TOM complex solution was desalted by ZipTip C18 (Merck) and mixed 1:10 with matrix solution (10 mg ml−1 α-cyano-4-hydroxycinnamic acid in 0.1% trifluoroacetic acid and 80% acetonitrile). The mixture was then applied onto a target plate (MTP 384 target plate ground steel BC, Bruker), and MS spectra were measured with an autoflex speed MS spectrometer (Bruker) in a positive ion mode protocol. The acquired spectra were analysed with flexAnalysis 3.4 (Bruker). For nano liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis, the purified TOM complex was separated by SDS–PAGE, and small TOM proteins (~10 kDa) were extracted from a gel stained with Coomassie brilliant blue and were in-gel digested by trypsin. The peptides were subjected to nanoLC-MS/MS analysis and proteins were identified by Mascot search (Japan Proteomics).
Patch analysis, multiple alignment and conservation analysis
Analyses of basic, acidic and hydrophobic patches were performed using Patch Analyzer in MOE 2015.10 software. The multiple alignments in this paper were generated by MAFFT61 and displayed using Jalview62. The TOM complex evolved independently into various complex forms in eukaryotic lineages20. In Opisthokonta (fungi and metazoan) the TOM complex probably maintains the same forms as the yeast TOM complex20. For conservation analysis, homologous sequences of TOM complex subunits were obtained from reference proteomes belonging to Opisthokonta using HMMER 3.2.1 (http://hmmer.org); 260, 281, 162, 117 and 100 sequences for Tom40, Tom22, Tom7, Tom6 and Tom5, respectively. The multiple alignments were generated by MAFFT61 and conservation score are analysed using ConSurf63.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The cryo-EM density maps have been deposited in the Electron Microscopy Data Bank under accession code EMD-9851. The atomic coordinates have been deposited in the Protein Data Bank under accession code 6JNF. All the data from this study are available in the Article, its Extended Data and Supplementary Information. The uncropped blots, gels and plates are provided in Supplementary Fig. 2.
References
Endo, T. & Yamano, K. Multiple pathways for mitochondrial protein traffic. Biol. Chem. 390, 723–730 (2009).
Neupert, W. A perspective on transport of proteins into mitochondria: a myriad of open questions. J. Mol. Biol. 427, 1135–1158 (2015).
Pfanner, N., Warscheid, B. & Wiedemann, N. Mitochondrial proteins: from biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 20, 267–284 (2019).
Shiota, T. et al. Molecular architecture of the active mitochondrial protein gate. Science 349, 1544–1548 (2015).
Sakaue, H. et al. Porin associates with Tom22 to regulate the mitochondrial protein gate assembly. Mol. Cell 73, 1044–1055 (2019).
Künkele, K.-P. et al. The preprotein translocation channel of the outer membrane of mitochondria. Cell 93, 1009–1019 (1998).
Ahting, U. et al. The TOM core complex: the general protein import pore of the outer membrane of mitochondria. J. Cell Biol. 147, 959–968 (1999).
Model, K., Meisinger, C. & Kühlbrandt, W. Cryo-electron microscopy structure of a yeast mitochondrial preprotein translocase. J. Mol. Biol. 383, 1049–1057 (2008).
Bausewein, T. et al. Cryo-EM structure of the TOM core complex from Neurospora crassa. Cell 170, 693–700 (2017).
Van den Berg, B. et al. X-ray structure of a protein-conducting channel. Nature 427, 36–44 (2004).
Voorhees, R. M., Fernández, I. S., Scheres, S. H. W. & Hegde, R. S. Structure of the mammalian ribosome–Sec61 complex to 3.4 Å resolution. Cell 157, 1632–1643 (2014).
Gogala, M. et al. Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 506, 107–110 (2014).
Itskanov, S. & Park, E. Structure of the posttranslational Sec protein–translocation channel complex from yeast. Science 363, 84–87 (2019).
Wu, X., Cabanos, C. & Rapoport, T. A. Structure of the post-translational protein translocation machinery of the ER membrane. Nature 566, 136–139 (2019).
Baud, C. et al. Translocation path of a substrate protein through its Omp85 transporter. Nat. Commun. 5, 5271 (2014).
Lundquist, K., Bakelar, J., Noinaj, N. & Gumbart, J. C. C-terminal kink formation is required for lateral gating in BamA. Proc. Natl Acad. Sci. USA 115, E7942–E7949 (2018).
Bamert, R. S. et al. Structural basis for substrate selection by the translocation and assembly module of the β-barrel assembly machinery. Mol. Microbiol. 106, 142–156 (2017).
Model, K. et al. Protein translocase of the outer mitochondrial membrane: role of import receptors in the structural organization of the TOM complex. J. Mol. Biol. 316, 657–666 (2002).
Dolezal, P., Likic, V., Tachezy, J. & Lithgow, T. Evolution of the molecular machines for protein import into mitochondria. Science 313, 314–318 (2006).
Fukasawa, Y., Oda, T., Tomii, K. & Imai, K. Origin and evolutionary alteration of the mitochondrial import system in eukaryotic lineages. Mol. Biol. Evol. 34, 1574–1586 (2017).
Maćasev, D. et al. Tom22′, an 8-kDa trans-site receptor in plants and protozoans, is a conserved feature of the TOM complex that appeared early in the evolution of eukaryotes. Mol. Biol. Evol. 21, 1557–1564 (2004).
Senes, A., Gerstein, M. & Engelman, D. M. Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with β-branched residues at neighboring positions. J. Mol. Biol. 296, 921–936 (2000).
Harner, M., Neupert, W. & Deponte, M. Lateral release of proteins from the TOM complex into the outer membrane of mitochondria. EMBO J. 30, 3232–3241 (2011).
Höhr, A. I. C. et al. Membrane protein insertion through a mitochondrial β-barrel gate. Science 359, eaah6834 (2018).
Yamano, K., Tanaka-Yamano, S. & Endo, T. Tom7 regulates Mdm10-mediated assembly of the mitochondrial import channel protein Tom40. J. Biol. Chem. 285, 41222–41231 (2010).
Ellenrieder, L. et al. Separating mitochondrial protein assembly and endoplasmic reticulum tethering by selective coupling of Mdm10. Nat. Commun. 7, 13021 (2016).
Esaki, M. et al. Mitochondrial protein import. Requirement of presequence elements and TOM components for precursor binding to the TOM complex. J. Biol. Chem. 279, 45701–45707 (2004).
Court, D. A. et al. Role of the intermembrane-space domain of the preprotein receptor Tom22 in protein import into mitochondria. Mol. Cell. Biol. 16, 4035–4042 (1996).
Moczko, M. et al. The intermembrane space domain of mitochondrial Tom22 functions as a trans binding site for preproteins with N-terminal targeting sequences. Mol. Cell. Biol. 17, 6574–6584 (1997).
Kanamori, T. et al. Uncoupling of transfer of the presequence and unfolding of the mature domain in precursor translocation across the mitochondrial outer membrane. Proc. Natl Acad. Sci. USA 96, 3634–3639 (1999).
Rapaport, D., Neupert, W. & Lill, R. Mitochondrial protein import. Tom40 plays a major role in targeting and translocation of preproteins by forming a specific binding site for the presequence. J. Biol. Chem. 272, 18725–18731 (1997).
Yamamoto, H. et al. Tim50 is a subunit of the TIM23 complex that links protein translocation across the outer and inner mitochondrial membranes. Cell 111, 519–528 (2002).
Mokranjac, D. et al. Tim50, a novel component of the TIM23 preprotein translocase of mitochondria. EMBO J. 22, 816–825 (2003).
Shiota, T., Mabuchi, H., Tanaka-Yamano, S., Yamano, K. & Endo, T. In vivo protein-interaction mapping of a mitochondrial translocator protein Tom22 at work. Proc. Natl Acad. Sci. USA 108, 15179–15183 (2011).
Tamura, Y. et al. Tim23–Tim50 pair coordinates functions of translocators and motor proteins in mitochondrial protein import. J. Cell Biol. 184, 129–141 (2009).
Chacinska, A. et al. Mitochondrial presequence translocase: switching between TOM tethering and motor recruitment involves Tim21 and Tim17. Cell 120, 817–829 (2005).
Naoé, M. et al. Identification of Tim40 that mediates protein sorting to the mitochondrial intermembrane space. J. Biol. Chem. 279, 47815–47821 (2004).
Gornicka, A. et al. A discrete pathway for the transfer of intermembrane space proteins across the outer membrane of mitochondria. Mol. Biol. Cell 25, 3999–4009 (2014).
Chacinska, A. et al. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 23, 3735–3746 (2004).
Mesecke, N. et al. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell 121, 1059–1069 (2005).
Kurz, M. Martin, H. Rassow, J., Pfanner, N. & Ryan, M. T. Biogenesis of Tim proteins of the mitochondrial carrier pathway: differential targeting mechanisms and crossing over with the main import pathway. Mol. Biol. Cell 10, 2461–2474 (1999).
Stojanovski, D. et al. Mitochondrial protein import: precursor oxidation in a ternary complex with disulfide carrier and sulfhydryl oxidase. J. Cell Biol. 183, 195–202 (2008).
Bien, M. et al. Mitochondrial disulfide bond formation is driven by intersubunit electron transfer in Erv1 and proofread by glutathione. Mol. Cell 37, 516–528 (2010).
Rapaport, D. et al. Structural requirements of Tom40 for assembly into preexisting TOM complexes of mitochondria. Mol. Biol. Cell 12, 1189–1198 (2001).
Habich, M. et al. Vectorial import via a metastable disulfide-linked complex allows for a quality control step and import by the mitochondrial disulfide relay. Cell Rep. 26, 759–774 (2019).
Becker, T. et al. Role of phosphatidylethanolamine in the biogenesis of mitochondrial outer membrane proteins. J. Biol. Chem. 288, 16451–16459 (2013).
Zheng, W. et al. Adaptive optics improves multiphoton super-resolution imaging. Nat. Methods 14, 869–872 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Sikorski, R. S. & Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 (1989).
Model, K. et al. Multistep assembly of the protein import channel of the mitochondrial outer membrane. Nat. Struct. Biol. 8, 361–370 (2001).
Opaliński, Ł. et al. Recruitment of cytosolic J-proteins by TOM receptors promotes mitochondrial protein biogenesis. Cell Rep. 25, 2036–2043 (2018).
Qiu, J. et al. Coupling of mitochondrial import and export translocases by receptor-mediated supercomplex formation. Cell 154, 596–608 (2013).
Ando, T., Uchihashi, T. & Fukuma, T. High-speed atomic force microscopy for nano-visualization of dynamic biomolecular processes. Prog. Surf. Sci. 83, 337–437 (2008).
Uchihashi, T., Kodera, N. & Ando, T. Guide to video recording of structure dynamics and dynamic processes of proteins by high-speed atomic force microscopy. Nat. Protoc. 7, 1193–1206 (2012).
Milenkovic, D. et al. Identification of the signal directing Tim9 and Tim10 into the intermembrane space of mitochondria. Mol. Biol. Cell 20, 2530–2539 (2009).
Thornton, N. et al. Two modular forms of the mitochondrial sorting and assembly machinery are involved in biogenesis of α-helical outer membrane proteins. J. Mol. Biol. 396, 540–549 (2010).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M. & Barton, G. J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Ashkenazy, H. et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016).
Acknowledgements
We thank the members of the Endo and Pfanner labs for discussion and critical comments on the manuscript. This work was supported by JSPS KAKENHI to T.E. (15H05705 and 2222703), K.I. (16K21680 and 18K11543), T.S. (19K16077 and 18KK0197) and T.A. (26119003) and JST CREST grants to T.E. (JPMJCR12M1) and to M.K. (JPMJCR14M1); a European Research Council (ERC) Consolidator grant to N.W. (no. 648235); Deutsche Forschungsgemeinschaft grants to T.B. (BE 4679/2-2) and N.P. (PF 202/9-1); and Germany’s Excellence Strategy to T.B., N.W. and N.P. (CIBSS – EXC-2189—Project ID 390939984; GSC-4 Spemann Graduate School). The following grants are also acknowledged: a grant from Takeda Science Foundation (to T.E.); Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under grant numbers JP19am01011115 (to M.K.) and JP19am0101114 (to K.I.); grants from the Ichiro Kanehara Foundation for the Promotion of Medical Sciences and Medical Care, Waksman Foundation of Japan, Tokyo Biochemical Research Foundation, Sumitomo Foundation, Naito Foundation, and Uehara Memorial Foundation (to T.S.); and the Australian Research Council Discovery Project DP160100227 (to T.L.). We acknowledge the Cryo-EM Facility of the University of Tokyo. Y.A. was supported by a Research Fellowship for Young Scientists from the Japan Society of the Promotion of Science (15J07687). Work included in this study has also been performed in partial fulfillment of the requirements for the doctoral thesis of C.L. at the University of Freiburg. We thank T. Nishizawa for discussion and advice on cryo-EM data processing, M. H. Schuler for analysis of yeast mutants, R. Beckmann and A. Heuer for advice on cryo-EM measurements and H. Nishitoh and Y. Inaba for instruments and reagents for T.S.
Author information
Authors and Affiliations
Contributions
Y.A. performed most of the experiments and wrote the paper. A.T. performed and M.K. supervised cryo-EM measurements and data processing. J.Q., J. Song, C.L., L.-S.W., M.W. and C.S. performed functional studies on the role of TOM proteins in preprotein import into mitochondria, characterized yeast mutants, studied the import of precursor proteins into isolated mitochondria and analysed data. K.I. performed bioinformatics analyses and wrote a part of the paper. T.S. performed crosslinking and T.L. designed and supervised crosslinking and wrote a part of the paper. H.S. analysed yeast cell growth of double mutants of tom5Δ and Tom40 N-terminal truncation. K.Y., S.K. and J. Suzuki assisted with designing preparation of the materials including the yeast TOM complex. H.A. and T.A. performed HS-AFM measurements. N.W., T.B. and N.P. designed and supervised functional studies on the role of TOM proteins in preprotein import into mitochondria, analysed data and wrote a part of the paper. T.E. designed the research and wrote the paper. All authors discussed the results of the experiments and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Purification of the TOM complex.
a, Forms of the TOM complex. Newly imported Tom40 takes a β-barrel structure and forms an assembly ‘dimer’ lacking Tom22 and Tom20. The dimer is matured into the ‘trimer’ with the receptors Tom22 and Tom204. Even after the maturation, the trimer dynamically exchanges with a minor population of the assembly dimer4. After solubilization of mitochondria, the TOM complex can be found as a trimer, containing Tom20 at least partly. Our previous in vivo crosslinking analyses revealed the subunit arrangement of the trimer4,32, and the present study revealed the high-resolution cryo-EM structure of the dimeric TOM core complex that contains Tom22, but lacks Tom20. b, Elution profile of gel filtration of the purified TOM complex. c, Blue native electrophoresis gel of the TOM complex stained with Coomassie brilliant blue. The apparent molecular weight of the TOM complex is ~440 kDa on blue native electrophoresis, which corresponds to the trimer. d, SDS–PAGE gel of the TOM complex stained with Coomassie brilliant blue. The bands for Tom40, Tom22–His10, Tom20 and Tom5 were assigned by western blotting with antibodies against each subunits. The presence of Tom5, Tom6 and Tom7 were confirmed by mass spectrometry (MS) analyses. The bands for Tom5 and Tom7 were assigned by MS/MS analyses of the tryptic fragments of the proteins contained in each band. e, MALDI–TOF MS of purified TOM complex in the m/z range below 10,000. Calculated molecular weights of yeast small TOM subunits are 5986.0 (Tom5), 6407.6 (Tom6), and 6871.0 (Tom7). Tom7 appears to lack the N-terminal Met, and Tom6 may be acetylated. The data are representative of six (b, c), four (d) and two (e) independent experiments. f, Typically observed particles of HS-AFM images of the TOM complex trimer and dimer on mica.
Extended Data Fig. 2 Cryo-EM analysis of the TOM complex.
a, A micrograph of the TOM complex. b, Selected 2D classes showing various orientations. c, FSC curve after the application of soft mask in RELION, showing FSC of 0.143. d, The schematic workflow of the steps followed in data processing leading to the final structure of the TOM core complex. e, Angular particle distribution in the final reconstruction. f, Local map resolution of the final TOM complex structure with colours according to the scale on the right side. g, FSC curve between the EM map and the atomic model of the TOM complex. h, An example of the fit of the model into the density map; the region around β-strands 11–18 of Tom40 is shown.
Extended Data Fig. 3 β-barrel structure of Tom40.
a, Hydrogen bonds between the 19 β-strands on the basis of the EM structure. Circles, pentagons and squares indicate residues in loops, α-helices and β-strands, respectively. Disordered regions are coloured in grey. Hydrogen bond, CO–NH distance < 4.0 Å; weak hydrogen bond, 4.0 Å ≤ CO–NH distance ≤ 4.5 Å; no hydrogen bond, 4.5 Å < CO–NH distance. b, Relative positions of the other TOM subunits and phospholipid in contact with the Tom40 β-barrel. c, Summary of previous crosslinking results of Tom404. BPA-incorporated residues are labelled in bold. Crosslinked partners are indicated by different colours. Residues labelled with a red asterisk are not within a distance of 5 Å from their crosslinked partners in the EM structure.
Extended Data Fig. 4 Model for transition between the Tom22-containing dimer and trimer.
a, Comparison of the cryo-EM structure of Tom22-containing dimer (D) and crosslinked-based structure model of Tom22-containing trimer (T). b, The residues whose proximity relationships are inconsistent between the trimer (crosslinking) and the dimer (EM structure) are mapped on the EM structure. Top view from the cytosolic side (left) and side view (right) of the EM dimer structure (cartoon model) with the side chains (space-filling model) of the residues that were crosslinked to other subunits4 but were not within a distance of 4.5 Å from the crosslinked partners in the EM structure (Supplementary Table 1), and therefore judged to be important for formation of the mature trimer4. c, The residues suggesting the possible difference in the subunit interfaces between the trimer and dimer are mapped on the EM structure. Although Tom22A P112 and Tom40B Y309 face each other in the EM structure, the Tom22 transmembrane helix was crosslinked to two Tom40 molecules through Tom22 P112 and Tom40 Y3094, suggesting that these two residues should not face each other in the trimer. Tom22 E120 and Tom40 R310 are evolutionary conserved (Extended Data Fig. 7 and Supplementary Table 2) and electrostatic interaction between the two residues would contribute to stabilization of the dimer. d, e, Upon formation of the trimer from the dimer, the Tom22 transmembrane helix in the EM structure may rotate (with a possible conformational change), so that Tom22A P112 faces the Tom40A molecule and Tom22A S114 interacts with the Tom40B molecule. This rotation would lead to a switch of the electrostatic-interaction pairs from Tom22 E120 and Tom40 R310 to Tom22 E120 and Tom40 K90 with possible concomitant movement of the Tom7 C-terminal segment; the second short C-terminal helix of Tom7 appears to be stabilized by the hydrogen bond between the carbonyl O of L51 in Tom7 and the side-chain NH of K90 in Tom40 in the dimer, which is probably lost in the trimer. The rotation of the Tom22 transmembrane helix would also allow K94 and R89 of Tom22 to interact with D350 of the neighbouring Tom40 molecule. Basic and acidic residues are coloured in blue and red, respectively.
Extended Data Fig. 5 Conserved interactions between the Tom40 β-barrel and the α-helical subunits.
a–d, Interactions are shown between the Tom40 β-barrel and the α-helical subunits Tom22 (a), Tom7 (b), Tom6 (c) and Tom5 (d). Tom40 residues of the β-barrel outer wall and loops are shown in space-filling form and coloured by evolutionary conservation (darker blue represents more conserved residues). Tom40 residues at ‘conserved amino acid’ (conserved) positions within 4.5 Å distance of the α-helical subunits are labelled. The main chains of the α-helical subunits are shown in ribbon form, and residues at conserved positions within 4.5 Å distance of Tom40 are shown in stick form, labelled, and coloured by evolutionary conservation (deeper colour represents more highly conserved residues). Residues in parentheses are the most frequently observed residue at each conserved position. Pairs of residues at conserved positions of Tom40 β-barrel and α-helical subunits in proximity (≤4.5 Å) are underlined and bolded: Tom40B R310 (R) and Tom22 E120 (E), Tom40B S312 (S) and Tom22 S116 (A), Tom40B Y314 (F) and Tom22 L109 (L), Tom40B V324 (V) and Tom22 L109 (L), Tom40B V324 (V) and Tom22 L108 (L), Tom40B V324 (V) and Tom22 T105 (S), Tom40B L328 (L) and Tom22 P112 (P), Tom40B R330 (K) and Tom22 A119 (E), Tom40A L336 (F) and Tom22 L115 (L), Tom40B H346 (H) and Tom22 T105 (S), Tom40B H346 (H) and Tom22 T104 (T), and Tom40B H346 (H) and Tom22 W100 (W) (a); Tom40 F98 (F) and Tom7 G40 (G) and Tom40 G129 (G) and Tom7 I33 (I) (b); Tom40 L259 (Y) and Tom6 V46 (V), Tom40 L259 (Y) and Tom6 Q50 (Q), Tom40 G299 (G) and Tom6 N38 (S) and Tom40 S320 (S) and Tom6 N38(S) (c). No conserved proximate residue pair was found between Tom40 and Tom5 (d). Phospholipid (modelled as phosphatidylethanolamine) is shown in space-filling form; carbon, nitrogen, oxygen and phosphorus are coloured light green, blue, red and magenta, respectively (a).
Extended Data Fig. 6 Formation of the two exit sites of the TOM channel for different classes of precursor proteins.
a, Disordered regions of Tom22 and Tom40 facing the IMS. b, The TOM complex viewed from the IMS side shows that the trans presequence binding site (orange) is formed by the hydrophilic loop of Tom7 (residues 43–49), the hydrophilic β1–β2 loop (residues 92–96, M94 is disordered) and C-terminal domain (residues 363–387, residues after L374 are disordered, G363–E366 form a loop and E367–G373 form a helix) of Tom40 and the acidic IMS domain of Tom22 (residues 120–152, residues after 131 are disordered, the transmembrane helix is extended up to F130). Disordered parts are shown with beads. c, Crosslinked products of Tom22 containing BPA at the indicated positions were detected by the indicated antibodies. Crosslinked products between Tom22 and Tim50 (22–50) are indicated. Representative data of two independent experiments. d, Radiolabelled Oxa1 precursor was imported into wild-type and Tom40(Δ364–387) mitochondria for the indicated periods in the absence or presence of a membrane potential (Δψ) and analysed by blue native electrophoresis and autoradiography. Representative data of three independent experiments. Quantification of Oxa1 bound to the TOM complex; mean values ± s.e.m. (n = 3 biologically independent experiments); the amount of bound Oxa1 in wild-type mitochondria after 20 min (−Δψ) was set as 100% (control). e, Mitochondria isolated from wild-type and Tom40(Δ364–387) yeast grown at 30 °C were analysed by SDS–PAGE and western blotting using the indicated antisera. Representative data of four independent experiments. Amount of total mitochondrial protein is indicated. f, Mitochondria isolated from wild-type and Tom40(Δ364–387) yeast grown at 30 °C were analysed by blue native electrophoresis and western blotting with Tom40 antiserum. Representative data of four independent experiments. g, Radiolabelled Tim9 was imported and assembled into the TIM22 complex in wild-type and Tom40(Δ364–387) mitochondria; data are mean ± s.e.m. (n = 3 biologically independent experiments); the amount in wild-type mitochondria after 40 min import was set as 100% (control). h, The trans presequence binding sites for presequence-containing proteins (b) and the N extension of Tom40 recruiting small TIM chaperones (for presequence-lacking β-barrel and carrier proteins) and Mia40 (for MIA substrates) are spatially separated to form distinct precursor exit sites at the outlet of the TOM channel. The two exit sites are connected to the different translocation paths (yellow broken lines) that include either the aligned acidic patches (red) or the aligned hydrophobic patches (green) inside the Tom40 β-barrel pore (Extended Data Fig. 9). Disordered parts are shown with beads.
Extended Data Fig. 7 Roles of Tom5 and the N extension of Tom40 in MIA import and assembly.
a–e, Import of radiolabelled Tim9 precursor into isolated wild-type, tom5∆, tom6∆, tom7∆, tom22∆, tom20∆ and tom70∆ mitochondria, analysed by blue native electrophoresis (top) and SDS–PAGE (bottom). TIM22, TIM22 complex; Tim9–Mia40, mixed disulfide intermediate of Tim9 with Mia40; TIM9–10, Tim9–Tim10 complex. Representative data of three (a, b, top), four (d, top) or two (other panels) independent experiments. f, Mitochondria were isolated from wild-type, Tom40(∆58) and Tom40(∆59) yeast cells, which were grown at 30 °C and, where indicated, additionally shifted to 37 °C for 12 h, and analysed by SDS–PAGE and western blotting using the indicated antisera. Representative data of two independent experiments. g, Radiolabelled Tom5 precursor was imported into mitochondria isolated from wild-type and Tom40(∆59) yeast cells, which were grown at 30 °C and shifted to 37 °C for 12 h, followed by incubation with sodium carbonate. The carbonate-resistant pellets were analysed by SDS–PAGE and autoradiography. Representative data of three independent experiments. h, Mitochondria of wild-type, Tom40(∆58) and Tom40(∆59) cells grown at 30 °C were analysed by blue native electrophoresis and western blotting using Tom40- and Mia40-specific antisera. Representative data of two independent experiments. i, Imports of radiolabelled Atp2 and b2(167)∆DHFR into wild-type, Tom40(Δ58) and Tom40(Δ59) mitochondria were analysed by SDS–PAGE. Representative data of three independent experiments. ∆ψ, membrane potential; p, precursor form; m, mature form; i, intermediate form. j, Mitochondria of wild-type, Tom40(∆58) and Tom40(∆59) cells grown at 30 °C were subjected to hypo-osmotic swelling and treatment with proteinase K (Prot. K) as indicated. Proteins were analysed by SDS–PAGE and western blotting using the indicated antisera. Representative data of two independent experiments. The IMS protein Tim13 shows that the outer membranes of (non-swollen) Tom40(∆58) and Tom40(∆59) mitochondria were intact like those of wild-type mitochondria. k, Import of radiolabelled Su9–DHFR precursor (top) or AAC (bottom) into wild-type, Tom40(L57A) and Tom40(L57A/T62A) mitochondria was analysed by SDS–PAGE (top) or blue native gel (bottom). Representative data of three (Su9–DHFR) or two (AAC) independent experiments. l, Mitochondria of wild-type, Tom40(L57A) and Tom40(L57A/T62A) strains grown at 30 °C and, where indicated, additionally shifted to 37 °C for 29.5 h, were analysed by SDS–PAGE and western blotting using the indicated antisera. Representative data of two independent experiments.
Extended Data Fig. 8 MIA import and assembly are disturbed in point mutants of the Tom40 N extension.
a, Wild-type, Tom40(L57A) and Tom40(L57A/T62A) mitochondria from cells grown at 30 °C were analysed by blue native electrophoresis and western blotting using Tom40- and Mia40-specific antisera. Representative data of two independent experiments. b, Wild-type, tom5∆, Tom40(L202A) and Tom40(L57A/T62A) mitochondria were treated with DTT or 4-DPS and the redox state of Mia40 was analysed by non-reducing SDS–PAGE and western blotting. Representative data of three independent experiments. Red, reduced form; oxid., oxidized form. c, Growth analysis of yeast strains expressing the indicated Tom40 variants on YPG. Representative data of two independent experiments. d, Radiolabelled Tim9 was imported into isolated wild-type and Tom40(L202A) mitochondria for 5 min, followed by addition of iodoacetamide (IA), and incubated further as indicated. The import was analysed by blue native electrophoresis. Representative data of two independent experiments. TIM22, TIM22 complex; Tim9–Mia40, mixed disulfide intermediate of Tim9 with Mia40; TIM9–10, Tim9–Tim10 complex. e, Import of radiolabelled Tim9(C55S) into wild-type and Tom40(L202A) mitochondria followed by treatment with proteinase K as indicated. The import was analysed by blue native electrophoresis (top) and non-reducing SDS–PAGE (bottom). Representative data of two independent experiments. Tim9–Mia40, mixed disulfide intermediate of Tim9(C55S) with Mia40. f, Wild-type and Tom40(L202A) mitochondria from cells grown at 30 °C were analysed by blue native electrophoresis and western blotting using Tom40 and Mia40 specific antisera. Representative data of two independent experiments. g, Wild-type and Tom40(L202A) mitochondria from cells grown at 30 °C were treated with proteinase K where indicated. Representative data of two independent experiments. The stability of IMS-exposed proteins indicates that the outer membranes of Tom40(L202A) mitochondria were intact like those of wild-type mitochondria. h, Import of radiolabelled Su9–DHFR (top) and AAC (bottom) into wild-type and Tom40(L202A) mitochondria was analysed by SDS–PAGE (top) or blue native electrophoresis (bottom). Representative data of three independent experiments.
Extended Data Figure 9 The pore interior of Tom40 and precursor import paths.
a, b, The views from the cytosol (top) and cut-away views (bottom) for Tom40. Section planes for the cut-away views are indicated with broken lines in the top panels. The side chains of the Tom40 residues crosslinked to the translocation intermediates of a presequence-containing precursor pSu9–DHFR (a) and of a hydrophobic presequence-lacking precursor AAC–DHFR (b) are shown on the EM structure with colour reflecting the amount of crosslinked products detected4. The acidic patches (red) and hydrophobic patches (green) are shown in the cut-away views for the β-barrel side of the pore interior of Tom40. Possible precursor translocation paths are indicated with arrows by broken lines (bottom). c, Section curved plane for the cut-away views (centre, bottom) is indicated with a broken line in the view from the cytosolic side (top). The acidic patches (red), hydrophobic patches (green) and possible precursor translocation paths (broken lines) are shown (bottom).
Supplementary information
Supplementary Figures
This file contains Supplementary Figures 1 (alignment of the N-terminus to β-strand 14 of Tom40 from various organisms) and 2 (the uncropped version of all the blots, gels and plates in this study).
Supplementary Tables
This file contains Supplementary Tables 1-4.
Video 1
HS-AFM observation of the purified TOM complex on a mica plate (200 nm × 200 nm).
Video 2
HS-AFM observation of the purified TOM complex scattered on a mica plate showing conversion of the trimer to the dimer (50 nm × 50 nm).
Source data
Rights and permissions
About this article

Cite this article
Araiso, Y., Tsutsumi, A., Qiu, J. et al. Structure of the mitochondrial import gate reveals distinct preprotein paths. Nature 575, 395–401 (2019). https://doi.org/10.1038/s41586-019-1680-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1680-7
- Springer Nature Limited
This article is cited by
-
A hybrid TIM complex mediates protein import into hydrogenosomes of Trichomonas vaginalis
BMC Biology (2024)
-
Mitochondrial stress: a key role of neuroinflammation in stroke
Journal of Neuroinflammation (2024)
-
The ER-SURF pathway uses ER-mitochondria contact sites for protein targeting to mitochondria
EMBO Reports (2024)
-
The architecture of substrate-engaged TOM–TIM23 supercomplex reveals preprotein proximity sites for mitochondrial protein translocation
Cell Discovery (2024)
-
NLG1, encoding a mitochondrial membrane protein, controls leaf and grain development in rice
BMC Plant Biology (2023)