

Abstract
Interleukin (IL)-2 is a pleiotropic cytokine that is necessary to prevent chronic inflammation in the gastrointestinal tract1,2,3,4. The protective effects of IL-2 involve the generation, maintenance and function of regulatory T (Treg) cells4,5,6,7,8, and the use of low doses of IL-2 has emerged as a potential therapeutic strategy for patients with inflammatory bowel disease9. However, the cellular and molecular pathways that control the production of IL-2 in the context of intestinal health are undefined. Here we show, in a mouse model, that IL-2 is acutely required to maintain Treg cells and immunological homeostasis throughout the gastrointestinal tract. Notably, lineage-specific deletion of IL-2 in T cells did not reduce Treg cells in the small intestine. Unbiased analyses revealed that, in the small intestine, group-3 innate lymphoid cells (ILC3s) are the dominant cellular source of IL-2, which is induced selectively by IL-1β. Macrophages in the small intestine produce IL-1β, and activation of this pathway involves MYD88- and NOD2-dependent sensing of the microbiota. Our loss-of-function studies show that ILC3-derived IL-2 is essential for maintaining Treg cells, immunological homeostasis and oral tolerance to dietary antigens in the small intestine. Furthermore, production of IL-2 by ILC3s was significantly reduced in the small intestine of patients with Crohn’s disease, and this correlated with lower frequencies of Treg cells. Our results reveal a previously unappreciated pathway in which a microbiota- and IL-1β-dependent axis promotes the production of IL-2 by ILC3s to orchestrate immune regulation in the intestine.
Similar content being viewed by others


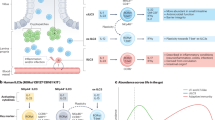
Main
To determine whether IL-2 is constitutively required for the maintenance of Treg cells and immunological homeostasis in the intestine, we administered isotype-control or anti-IL-2 neutralizing antibodies every other day to adult mice for two weeks. Within this short time period, the neutralization of IL-2 promoted an enlargement of the spleen and mesenteric lymph nodes, and caused significant reductions of Treg cells and significant increases in the proliferation of CD4+ T cells throughout the gastrointestinal tract and associated lymphoid tissues, including the mesenteric lymph nodes, large intestine and small intestine (Extended Data Fig. 1a–g). Blockade of IL-2 resulted in significantly enhanced IFNγ production by CD4+ T cells in both the small and large intestine, as well as increased IL-17A production in the large intestine (Extended Data Fig. 1h–k). Previous studies have suggested that CD4+ T cells are the dominant cellular source of IL-21,2. Therefore, we generated mice with a lineage-specific deletion of IL-2 in T cells by crossing IL-2-floxed mice10 with Lckcre mice. LckcreIl2f/f mice exhibited a complete loss of IL-2 protein staining in T cells, and we observed a significant reduction in the number of Treg cells, and an increase in CD4+ T cell proliferation and effector function in the mesenteric lymph nodes and large intestine (Extended Data Fig. 2a–g). By contrast, deletion of T-cell-derived IL-2 did not alter Treg cells, CD4+ T cell proliferation or effector cytokine production within the small intestine (Extended Data Fig. 2h–k). Collectively, these data demonstrate that T-cell-derived IL-2 is required for maintaining immunological homeostasis in lymphoid tissues and the large intestine but is dispensable in the small intestine, which indicates the existence of other critical cellular sources of IL-2.
We next examined the relevant cellular sources of IL-2 in the healthy mammalian small intestine by unbiased flow cytometry analyses. Notably, we observed that the major population of IL-2+ cells lack lineage markers for T cells, B cells, macrophages and dendritic cells, but are CD127+, CD90.2+ and express retinoic-acid-related orphan receptor γt (RORγt) (Fig. 1a), which represents a phenotype that is consistent with ILC3s11,12,13. To directly compare Il2 transcript levels between CD4+ T cells and ILC3s in the healthy small intestine, we performed RNA sequencing on sorted cell populations. In comparison to differentially expressed genes found in ILC3s (Rorc, Il22, Il1r (also known as Il1r1) and Il23r) or CD4+ T cells (Cd3d), expression of Il2 was more highly enriched in ILC3s (Fig. 1b). Significantly higher expression of Il2 was confirmed in ILC3s relative to CD4+ T cells, dendritic cells or B cells after quantitative PCR analysis of populations that were purified from the healthy mouse small intestine (Fig. 1c). ILC3s were the most-abundant IL-2+ cell type in terms of frequency and total cell number among other subsets of innate lymphoid cells and total CD4+ T cells from the small intestine (Fig. 1d–f, Extended Data Fig. 3), and were present at higher cell numbers than effector and memory CD4+ T cells (Extended Data Fig. 4a). This is in contrast to the large intestine, in which the majority of IL-2 was produced by CD4+ T cells and there was a limited presence of innate lymphoid cells that produce IL-2 (Extended Data Fig. 4b–d). ILC3s are a heterogeneous population that includes both CCR6+ lymphoid-tissue-inducer-like ILC3s and T-bet+ ILC3s11,12,13. IL-2 in the small intestine was produced by both subsets of ILC3s, with a significantly higher frequency of T-bet+ ILC3s that produce IL-2 (Extended Data Fig. 4e). Production of IL-2 by ILC3s was confirmed by flow cytometry analyses of the small intestine of Rag1−/− mice, which revealed that the major population of IL-2+ cells comprises CD127+CD90.2+RORγt+ ILC3s (Extended Data Fig. 4f–h), which include T-bet+ and CCR6+ ILC3s (Extended Data Fig. 4i, j). Unbiased analyses of the large intestine of Rag1−/− mice indicated that the major population of IL-2+ cells is comprised of innate lymphoid cells (Extended Data Fig. 4k). Furthermore, the frequency of IL-2+ cells that we observed in the small intestine of Rag2−/− mice was significantly reduced in Rag2−/−Il2rg−/− mice deficient in innate lymphoid cells, or Rag2−/− mice depleted of innate lymphoid cells with anti-CD90.2 antibody (Fig. 1g). Collectively, these findings demonstrate that IL-2 is dominantly produced by ILC3s in the healthy small intestine.

a, Flow cytometry plots show IL-2 staining in small-intestinal lamina propria cells of C57BL/6 mice. Lineage 1, CD11b, CD11c and B220; lineage 2, CD3ε, CD5 and CD8α. b, Heat map showing expression Z-scores of the indicated genes in CD4+ T cells (CD45+CD3+CD4+) and ILC3s (CD45+CD3−RORγtGFP+CD127+) from small-intestinal lamina propria cells of Rorc(gammat)-GfpTG (also known as Tg(Rorc-EGFP)1Ebe)) mice, as measured by RNA sequencing. c, Transcription of Il2 in the sort-purified CD4+ T cells, ILC3s, dendritic cells (DCs; CD45+CD11chighMHCII+CD64−) and B cells (CD45+CD19+) from small-intestinal lamina propria cells of Rorc(gammat)-GfpTG mice, as determined by quantitative PCR analysis (n = 6). Il2 expression was normalized to Hprt1, and then further normalized to the CD4+ T cells within each mouse. ND, not detectable. d–f, Flow cytometry plots (d), graph of frequency (e) and quantification of cell number (f) of IL-2+ cells in small-intestinal lamina propria cells of C57BL/6 mice (n = 8). ILC1s, group-1 innate lymphoid cells; ILC2s, group-2 innate lymphoid cells. g, Flow cytometry plots show IL-2 staining in small-intestinal lamina propria cells (genotypes of mice, and antibodies, listed above plots). Bar graph shows percentages in each group of mice (n = 4). Data in c, e and f are pooled from two independent experiments. Data in a and g are representative of four and two independent experiments with similar results, respectively; at least three mice per group. Results are shown as mean ± s.e.m. Statistics are calculated by paired or unpaired two-tailed Student’s t-test.
ILC3 development and function can be markedly influenced by the intestinal microbiota11,12,13. To investigate whether the intestinal microbiota also regulates ILC3-intrinsic IL-2 production, we examined the small intestine of conventionally housed specific-pathogen-free mice, specific-pathogen-free mice exposed to broad-spectrum antibiotics and germ-free mice. Notably, germ-free mice or specific-pathogen-free mice that were exposed to broad-spectrum antibiotics displayed significantly decreased IL-2 production by ILC3s, relative to specific-pathogen-free mice (Fig. 2a). To determine the signals that directly induce ILC3-intrinsic IL-2 production, we stimulated sort-purified ILC3s from the small intestine with IL-1β, IL-6, IL-23 or the aryl hydrocarbon receptor agonist FICZ, which are all known to directly stimulate ILC3s11,12,13. Among these, we found that IL-1β selectively induced Il2 transcription and protein production in ILC3s (Fig. 2b, c). We observed comparable results with stimulation using IL-1α (Fig. 2d). Furthermore, IL-1β was sufficient to significantly boost IL-2 production in ILC3s from germ-free mice and specific-pathogen-free mice exposed to broad-spectrum antibiotics (Fig. 2e, f).

a, Flow cytometry plots with graph of frequency of IL-2+ ILC3s in small-intestinal lamina propria cells of conventional specific-pathogen-free (SPF), antibiotics-treated specific-pathogen-free (ABX) and germ-free (GF) mice (n = 4). b, Quantitative PCR examination of Il2 transcript in sort-purified ILC3s in the presence of various stimuli (n = 6). Il2 expression was normalized to Hprt1, and then further normalized to the medium control within each mouse. c, Flow cytometry plots with graph of frequency of IL-2+ ILC3s from the small-intestinal lamina propria cells of specific-pathogen-free mice after ex vivo culture (n = 4). d, Small-intestinal lamina propria cells from specific-pathogen-free mice were cultured ex vivo for 4 h with or without IL-1α, and examined for IL-2+ ILC3s by flow cytometry (n = 7). e, f, Small-intestinal lamina propria cells from specific-pathogen-free mice treated with antibiotics (e), or germ-free mice (f), were cultured ex vivo with or without IL-1β, and examined for IL-2 in ILC3s by flow cytometry (n = 4). g, Quantitative PCR analysis of Il1b transcript in sort-purified macrophages (CD45+CD11b+CD64+F4/80+), conventional dendritic cell 1 (cDC1) (CD45+CD11c+MHCII+CD64−XCR1+CD172a−) and conventional dendritic cell 2 (cDC2) (CD45+CD11c+MHCII+CD64−XCR1−CD172a+) in small-intestinal lamina propria cells (n = 6). Il1b expression was normalized to Gapdh. h, i, Quantitative PCR analysis of Il1b transcript in sort-purified small-intestinal macrophages (h) or flow cytometry analysis of ILC3-derived IL-2 expression (i) in specific-pathogen-free, germ-free and segmented filamentous bacteria (SFB) mono-colonized mice (n = 6). j, Quantitative PCR analysis of Il1b transcript in sort-purified small-intestinal macrophages in wild-type (WT), Myd88−/− and Nod2−/− mice (n = 3). k, Flow cytometry analysis of small-intestinal IL-2+ ILC3s in wild-type mice, Nod2−/− mice and Nod2−/− mice after ex vivo culture (n = 8). Data in a, c, e, f and j are representative of two or three independent experiments with similar results (at least three mice per group). Data in b, d, g–i and k are pooled from two independent experiments. Results are shown as mean ± s.e.m. Statistics are calculated by paired or unpaired two-tailed Student’s t-test.
Gene-expression analyses of intestinal phagocytes revealed that the majority of IL-1β in the small intestine is produced by macrophages (Fig. 2g). Consistent with this, macrophages isolated from the small intestine of germ-free mice exhibited significantly reduced IL-1β expression and could be partially restored by mono-colonization with segmented filamentous bacteria (Fig. 2h), a component of the mouse microbiota that is known to colonize the distal small intestine, induce IL-1β, activate ILC3s and promote expansion of Treg cells14,15,16. Mono-colonization with segmented filamentous bacteria was sufficient to significantly induce ILC3-derived IL-2 production (Fig. 2i). To explore the sensing modules that are required to promote the IL-1β–ILC3–IL-2 axis, we examined Myd88−/− mice and observed a significant reduction in expression of IL-1β by macrophages (Fig. 2j). The intracellular pattern recognition receptor NOD2 has also previously been linked to the processing and secretion of IL-1β17. Consistent with this, we observed comparable transcript levels of IL-1β in macrophages that were sorted from the small intestine of Nod2−/− mice, but ILC3-derived IL-2 was significantly reduced and could be completely restored by stimulation with recombinant IL-1β (Fig. 2j, k). These data reveal that MYD88- and NOD2-dependent sensing of the intestinal microbiota is essential to promote macrophage production of IL-1β and activate ILC3s to produce IL-2.
To directly investigate whether IL-1R signalling is essential for IL-2 production by ILC3s in vivo, we crossed Ncr1cre and IL-1R-floxed mice to selectively delete IL-1R in the NKp46+ subset of ILC3s18,19. In comparison to littermate controls, Ncr1creIl1rf/f mice exhibit reduced IL-2 production by ILC3s, and display a significant decrease of Treg cells in the small intestine (Fig. 3a, b). To directly investigate the role of ILC3-derived IL-2, we generated mice with a deletion of IL-2 in ILC3s by using Ncr1creIl2f/f mice. This approach resulted in an efficient loss of IL-2 in T-bet+ ILC3s in the small intestine, but did not alter the frequency of IL-2 production in CD4+ T cells or dendritic cells (Extended Data Fig. 5a). We observed minimal production of IL-2 in natural killer cells and group-1 innate lymphoid cells in the small intestine; these populations do not contribute to Treg cell homeostasis, as no changes were observed after depletion with anti-NK1.1 antibody (Extended Data Fig. 5b–d). By contrast, we observed a significant reduction in the frequency and overall number of Treg cells in the small intestine of Ncr1creIl2f/f mice, compared to littermate controls (Fig. 3c, Extended Data Fig. 6a). This reduction in Treg cells was associated with a significant increase in the numbers of T helper type 1 (TH1) cells, whereas TH17 cells and the proliferation of CD4+ T cells were unchanged (Fig. 3d, Extended Data Fig. 6b–d). These results were tissue-specific, as no changes in Treg cells were observed in the large intestine of Ncr1creIl2f/f mice and no changes were observed in ILC3 homeostasis or cytokine production in the small intestine of Ncr1creIl2f/f mice (Extended Data Fig. 6e–g). This was surprising, given that ILC3s can respond to IL-2; however, we observed that Treg cells exhibit higher levels of the high-affinity IL-2R (also known as CD25) and more-efficiently bind IL-2, relative to ILC3s from the small intestine (Extended Data Fig. 6h, i).

a, b, Flow cytometry plots with graph of the frequencies of IL-2+ ILC3s (a) or Treg cells (b) in small-intestinal lamina propria cells of Il1rf/f and Ncr1creIl1rf/f mice (n = 8). c, d, Flow cytometry plots and quantification of cell numbers of Treg cells (c) and TH1 cells (d) in small-intestinal lamina propria cells of Il2f/f and Ncr1creIl2f/f mice at steady state (n = 8). e, Naive OT-II CD4+ T cells were transferred into Il2f/f and Ncr1creIl2f/f mice, and recipient mice were fed with OVA for 12 days. Donor T cells were analysed for FOXP3 staining in the small-intestinal lamina propria cells (n = 5). f, g, Delayed-type hypersensitivity response was assessed in designated mice by ear swelling (f) and serum concentration of anti-OVA IgG1 was tested by enzyme-linked immunosorbent assay (ELISA) (g) (n = 5 mice, non-tolerized group; n = 8 mice, tolerized group). Data in a–d, f and g are pooled from two independent experiments. Data in e are representative of two independent experiments with similar results (at least four mice per group). Results are shown as mean ± s.e.m. Statistics are calculated by unpaired two-tailed Student’s t-test (a–e) or two-way analysis of variance (ANOVA) (f, g).
Loss of ILC3-specific IL-1R or IL-2 significantly reduced the frequencies of peripherally induced Treg cells in the small intestine (marked by low staining of neuropilin-1) in addition to significant reductions of both RORγt+ and GATA3+ subsets of Treg cells (Extended Data Fig. 7a–d). ILC3-derived IL-2 did not affect expression of Lag3, Tgfb1, Ctla4 and Ebi3 in Treg cells, but did result in a modest decrease in expression of Il10 (Extended Data Fig. 7e). Consistent with this, Treg cells from the small intestine of control and Ncr1creIl2f/f mice exhibited a comparable potential to suppress effector T cell proliferation when ratios were normalized (Extended Data Fig. 7f, g). Furthermore, previous studies have identified a role for ILC3s in regulating adaptive immunity in the large intestine through granulocyte–macrophage colony-stimulating factor (GM-CSF) and MHCII20,21,22. However, loss of GM-CSF or ILC3-specific MHCII did not affect Treg cells in the small intestine or IL-2 production by ILC3s, and—similarly—ILC3-derived IL-2 did not affect ILC3 expression of GM-CSF or MHCII20 (Extended Data Fig. 8). These data collectively demonstrate that ILC3-derived IL-2 is a newly identified and non-redundant pathway that supports the population size and homeostasis of peripherally induced Treg cells in the small intestine.
Peripheral Treg cells in the small intestine are induced by dietary antigens and maintain oral tolerance23. Therefore, we next asked whether deficiency of ILC3-intrinsic IL-2 affects the de novo maintenance of small intestinal Treg cells upon oral administration of ovalbumin (OVA). Notably, the induction of OVA-specific inducible Treg cells (NRP-1low) in the small intestine was significantly impaired in Ncr1creIl2f/f mice, compared to littermate controls (Fig. 3e). We assessed whether oral tolerance was intact in these mice using a delayed-type hypersensitivity model24. In contrast to littermate controls, feeding OVA to Ncr1cre-Il2f/f mice did not induce oral tolerance, and did not protect mice from a delayed-type hypersensitivity reaction characterized by ear swelling and higher titres of anti-OVA immunoglobulin G1 (IgG1) in the serum upon OVA challenge in the periphery (Fig. 3f, g, Extended Data Fig. 6j). These data are consistent with previous literature that describes maintenance and expansion of antigen-specific Treg cells in the lamina propria of the intestine as a critical step in oral tolerance25. These data critically demonstrate that NKp46+ ILC3s are an essential and non-redundant source of IL-2 for maintaining Treg cell homeostasis and promoting oral tolerance uniquely in the small intestine.
We next targeted IL-2 in all subsets of ILC3s by generating RorccreIl2f/fRag1−/− mice and associated controls, and reconstituted these mice with an adoptive transfer of unfractionated wild-type CD4+ T cells. In comparison to Il2f/fRag1−/− recipient mice, RorccreIl2f/fRag1−/− recipient mice exhibited rapid and substantial weight loss, shorter colon length, spontaneous colonic inflammation characterized by increased inflammatory cell infiltration and significantly increased levels of faecal lipocalin-2 (Extended Data Fig. 9a–d). Notably, we observed significantly decreased numbers of colonic Treg cells, associated with significantly increased numbers of IL-17A+IFNγ− and IL-17A+IFNγ+ T cells in the large intestine, as well as increased numbers of TH1 cells in the small intestine of RorccreIl2f/fRag1−/− recipient mice relative to controls, even though both groups exhibited comparable small-intestinal Treg cells (Extended Data Fig. 9e–h). These findings suggest a model in which ILC3-derived IL-2 can also modulate immunological homeostasis and prevent inflammation throughout the entire gastrointestinal tract in the context of lymphopenia.
Finally, to investigate whether ILC3s support Treg cells in the human intestine, we examined the frequency of these populations in intestinal biopsies from healthy controls and patients with Crohn’s disease. As previously reported26,27, we observed significantly reduced frequencies of Treg cells and ILC3s in the small intestine of patients with Crohn’s disease relative to healthy controls, and—notably—we also identified a significant positive correlation between the frequencies of these two cell types within the same biopsy (Fig. 4a–c). To determine whether the impaired responses of Treg cells are associated with reduced ILC3-derived IL-2, we sort-purified ILC3s from resected small intestinal tissues of patients with Crohn’s disease and identified significantly reduced levels of IL2 transcript isolated from inflamed versus non-inflamed regions (Fig. 4d). In addition, IL-2+ ILC3s were significantly reduced in terminal-ileum biopsies from patients with Crohn’s disease relative to healthy controls, and no significant change was observed in IL-2+CD4+ T cells (Fig. 4e, f). These data indicate that—similar to their role in mice—ILC3s support Treg cells within the human small intestine, and suggest that this pathway becomes dysregulated in the context of Crohn’s disease.

a–f, Lamina propria cells were isolated from terminal-ileum biopsies from healthy controls or patients with Crohn’s disease (CD), and the frequency of Treg cells (CD45+CD3+CD4+FOXP3+ of CD4+ T cells) (a) and ILC3s (CD45+LIN−CD127+CD117+NKp44+ of total innate lymphoid cells) (b) were quantified. c, The frequency of ILC3s was correlated with the frequency of Treg cells (n = 16 healthy controls; n = 20 patients with Crohn’s disease). d. Human ILC3s were sort-purified from resected tissues of patients with Crohn’s disease and IL2 transcript was examined by quantitative PCR. e, f, The frequency of IL-2+ ILC3s (e) or IL-2+CD4+ T cells (f) were analysed in healthy controls (n = 11) and patients with Crohn’s disease (n = 11) by flow cytometry. Data in a–c, e and f, statistical analyses performed using a Mann–Whitney U-test (unpaired). Correlative analyses were compared by parametric Pearson’s rank correlation coefficient (R2). Data in d are performed using Wilcoxon matched-pairs test (paired). Results are shown as mean ± s.e.m. Statistics are calculated by two-tailed test.
Collectively, our studies reveal a pathway of immune regulation that uniquely occurs in the healthy small intestine (Extended Data Fig. 10). This pathway is continuously required, involves MYD88- and NOD2-dependent microbial sensing by macrophages, production of IL-1β and induction of ILC3-derived IL-2. Subsequently, this pathway supports intestinal Treg cells, immunological homeostasis and oral tolerance. Our findings in mouse models translated into human samples, and indicate that impaired ILC3-derived IL-2 is linked to a reduction of Treg cells and impaired immune regulation within the small intestine of patients with Crohn’s disease. Our results will inform ongoing strategies of therapeutically administering low doses of IL-2 to patients with inflammatory bowel disease, or to individuals with lymphopenia, immune deficiencies or who have been treated with chemotoxic agents. ILC3-derived IL-2 does not exhibit functional redundancy or hierarchies with previously known pathways by which ILC3s regulate adaptive immunity in the large intestine. Rather, our data define tissue-specific differences in the mechanisms by which ILC3s orchestrate immune tolerance in the small and large intestines. This is probably driven by substantial differences in microbial burdens, mucus layers, tissue permeability and metabolite access between these two organs28,29,30. Our data provide insights into the pathways that support Treg cells in the small intestine, and thus add an important complement to the well-known mechanisms that support Treg cells in the large intestine4,5,6,7,8,30. Finally, our results substantially advance our understanding of the role and regulation of IL-2 throughout the gastrointestinal tract, and identify a previously unappreciated direct communication between ILC3s and Treg cells.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
Mice
Wild-type, Rag1−/−, THY1.1, OT-II, Lckcre, Il1rf/f, H2-Ab1f/f, Csf2−/−, Myd88−/− and Nod2−/− mice on a C57BL/6 background were purchased from the Jackson Laboratory. Rag2−/− and Rag2−/−Il2rg−/− on a C57BL/6 background were purchased from Taconic Farms. C57BL/6 Rorccre mice and Rorc(gammat)-GfpTG mice were provided by G.E. C57BL/6 Il2f/f mice were provided by K.A.S. C57BL/6 Ncr1cre were provided by E.V., and only heterozygous Ncr1cre mice were used in this study. All mice were bred and maintained in specific-pathogen-free facilities at Weill Cornell Medicine and littermates were used as controls in all experiments. C57BL/6 germ-free mice were maintained at the gnotobiotic facility at Weill Cornell Medicine. Sex- and age-matched mice between 8 and 14 weeks of age were used for experiments if not otherwise indicated. Sex was not found to influence results of any experiments. No mice were excluded from the analysis unless clearly indicated. All mouse experiments were approved by, and performed in accordance with, the Institutional Animal Care and Use Committee guidelines at Weill Cornell Medicine.
In vivo administration of antibodies and antibiotics
Anti-IL-2 monoclonal antibodies (JES6-1A12 and S4B6-1, BioXCell) were administered intraperitoneally every other day at a dose of 300 μg (150 μg JES6-1A12 plus 150 μg S4B6-1) per mouse starting on day 0 and ending on day 14. Anti-CD90.2 monoclonal antibody (30H12, BioXCell) was administered intraperitoneally every 3 days at a dose of 250 μg per mouse starting on day 0 and ending on day 14, as previously described21. Anti-NK1.1 monoclonal antibody (PK136, BioXCell) was administered intraperitoneally every 3 days at a dose of 250 μg per mouse starting on day 0 and ending on day 14. A cocktail of antibiotics (0.25 mg/ml of vancomycin, 0.5 mg/ml of ampicillin, neomycin, gentamicin and metronidazole, and 4 mg/ml sucralose) was continuously administered via drinking water for two weeks.
Isolation of cells from the intestinal lamina propria of mice and humans
Intestines of mice were removed, cleaned from remaining fat tissue and washed in ice-cold PBS (Corning). Peyer’s patches on the small intestine were identified and removed. Intestines were opened longitudinally and washed in ice-cold PBS. Afterwards, mucus was gently removed by forceps and intestines were cut into approximately 0.5-cm sections. Dissociation of epithelial cells was performed by incubation on a shaker in HBSS (Sigma Aldrich) containing 5 mM EDTA (Thermo Fisher Scientific), 1 mM DTT (Sigma Aldrich) and 2% FBS twice for 20 min at 37 °C. After each step, samples were vortexed and the epithelial fraction discarded. Afterwards, samples were washed by PBS and enzymatic digestion was performed in RPMI containing 10% FBS and 0.4 U/ml dispase (Thermo Fisher Scientific), 1 mg/ml collagenase III (Worthington) and 20 μg/ml DNase I (Sigma Aldrich) on a shaker for 45 min at 37 °C. Leukocytes were further enriched by a 40/80% Percoll gradient centrifugation (GE Healthcare).
For human samples, de-identified intestinal biopsies from the terminal ileum of paediatric individuals with Crohn’s disease or age-matched controls who did not have inflammatory bowel disease were obtained following Institutional-Review-Board-approved protocols from either the Children’s Hospital of Philadelphia or the JRI IBD Live Cell Bank Consortium at Weill Cornell Medicine. Informed consent was obtained from all subjects. Tissues were processed by first incubating in 1 mM EDTA, 1 mM DTT and 5% FBS (all from Thermo Fisher Scientific) for 30 min at 37 °C with shaking to remove intestinal epithelial cells. Supernatants were then discarded and the remaining tissues were incubated in 0.5 mg/ml collagenase and 20 μg/ml DNase I for 30 min at 37 °C with shaking to obtain the lamina propria fraction. Any remaining tissues were also included following mechanical dissociation and filtering through a 70-μm cell strainer. All cells were then viably cryopreserved at −150 °C in 90% FBS and 10% DMSO for future side-by-side analyses.
Surgical-resection samples from the small intestine of patients with Crohn’s disease were obtained through Institutional-Review-Board-approved protocols from the Center for Advanced Digestive Care at Weill Cornell Medicine. Informed consent was obtained from all subjects. Inflamed and non-inflamed regions were isolated by a trained pathologist. Single-cell suspensions from intestinal tissues were obtained by incubating tissues for 30 min at 37 °C with shaking in stripping buffer (1 mM EDTA, 1 mM DTT and 5% FCS) to remove the epithelial layer. Supernatants were then discarded. Tissues were then mechanically dissociated with a sterile scalpel. The lamina propria fraction was obtained by incubating the dissociated tissues for 1 h at 37 °C with shaking in 2 mg/ml collagenase D (Roche), 0.1 mg/ml DNase I (Sigma Aldrich) and 1 mg/ml of trypsin inhibitor (Gibco) digestion solution. Remaining tissues were then filtered through a 70-μm cell strainer. All cells were then viably cryopreserved in 90% FBS and 10% DMSO for side-by-side analysis at a later time point. Following thawing and filtering through a 70-μm cell strainer, cells were stained with antibodies for flow cytometry acquisition.
Flow cytometry and cell sorting
Single-cell suspensions were incubated on ice with conjugated antibodies in PBS containing 2% FBS and 1 mM EDTA. Dead cells were excluded with Fixable Aqua Dead Cell Stain (Thermo Fisher Scientific). The staining antibodies for flow cytometry were purchased from Thermo Fisher Scientific, Biolegend or BD Biosciences. For mouse cell-surface staining: B220 (RA3-6B2), CCR6 (29-2L17), CD3ε (145-2C11), CD4 (GK1.5 or RM4-5), CD5 (53-7.3), CD8α (53-6.7), CD11b (M1/70), CD11c (N418), CD19 (eBio1D3), CD25 (PC61.5), CD44 (IM7), CD45 (30-F11), CD45RB (C363-16A), CD62L (MEL-14), CD64 (X54-5/7.1), CD90.2 (30-H12), CD127 (A7R34), CD172a (P84), F4/80 (BM8), MHCII (M5/114.15.2), NK1.1 (PK136), NRP-1 (3E12), TCRβ (H57-597), THY1.1 (OX-7) and XCR1 (ZET). For mouse intracellular staining: Eomes (Dan11mag), FOXP3 (FJK-16S), GATA3 (L50-823), GM-CSF (MP1-22E9), IL-2 (JES6-5H4), IL-6 (MP5-20F3), IL-17A (eBio 17B7), IL-22 (IL22JOP), IFNγ (XMG1.2), Ki-67 (SolA15), RORγt (B2D or Q31-378), T-bet (eBio4B10) and TNF (MP6-XT22). Human samples were stained for CD3 (UCHT1), CD4 (SK3), CD5 (UCHT2), CD11b (CBRM1/5), CD11c (3.9), CD14 (TuK4), CD19 (HIB19), CD34 (581), CD45 (HI30), CD94 (DX22), CD117 (104D2), CD123 (6H6), CD127 (A019D5), FcεR1 (AER-37), FOXP3 (PCH101), IL-2 (MQ1-17H12) and NKp44 (44.189).
For intracellular transcription factor or cytokine staining, cells were stained for surface markers, followed by fixation and permeabilization according to the manufacturer’s protocol (FOXP3 staining buffer set from Thermo Fisher Scientific). For intracellular cytokine staining, cells were first incubated for 4 h in RPMI with 10% FBS, 50 ng/ml phorbol 12-myristate 13-acetate (PMA), 750 ng/ml ionomycin and 10 μg/ml brefeldin A, all obtained from Sigma Aldrich. IL-2 binding capacity was assessed using a biotinylated IL-2 fluorokine assay kit (R&D Systems), following the manufacturer’s instructions. All flow cytometry experiments were performed using a Fortessa II flow cytometer and the FACS Diva software (BD Biosciences), and analysed with FlowJo v.10 software (TreeStar) or sort-purified by using FACSAria II cell sorter (BD Biosciences).
T cell adoptive transfer
Naive OT-II T cells (CD4+CD25−CD44lowCD62Lhigh) (1 × 106) were sort-purified from the spleen and lymph nodes of THY1.1+OT-II mice and adoptively transferred by retro-orbital injection into recipient mice (Il2f/f and Ncr1creIl2f/f mice). One day later, recipient mice were gavaged with OVA (grade V, Sigma Aldrich) 20 mg per mouse every other day, and simultaneously fed ad libitum with OVA dissolved in drinking water (10 mg/ml) for 12 days. Treg cell conversion of naive OT-II cells was analysed in the small intestine on day 13.
CD3+CD4+ T cells were sorted from the spleen and lymph nodes of THY1.1+ mice, and 3 × 106 cells were transferred intravenously to each recipient mouse (Il2f/fRag1−/− and RorccreIl2f/fRag1−/− mice). The weights of recipient mice were monitored through the progression of the experiment.
In vitro stimulation
Sort-purified ILC3s (CD45+CD3−RORγtGFP+CD127+) from the small intestine of Rorc(gammat)-GfpTG mice were plated in a 96-well plate (1 × 104 cells per well for quantitative PCR) and incubated in DMEM with high glucose, supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, non-essential amino acids, 80 μM 2-mercaptoethanol, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (all from Gibco) in the presence of recombinant mouse IL-1β (20 ng/ml, Thermo Fisher Scientific), IL-6 (20 ng/ml, Thermo Fisher Scientific), IL-23 (20 ng/ml, Thermo Fisher Scientific) or FICZ (500 nM, Sigma Aldrich) at 37 °C. For quantitative PCR, cells were treated for 4 h.
Induction and measurement of delayed-type hypersensitivity responses
Mice were tolerized by gavage with of 50 mg OVA (Grade III, Sigma Aldrich) on 2 consecutive days. One week later, mice were immunized by subcutaneous injection between the shoulder blades of 300 μg OVA (Grade VI, Sigma Aldrich) in 200 μl PBS/Complete Freud’s Adjuvant emulsion (Sigma Aldrich). Two weeks after immunization, mice were challenged by subcutaneous injection of 50 μg OVA (grade VI, Sigma Aldrich) in 20 μl PBS into the right ear pinna; 20 μl PBS without OVA were injected into the left ear pinna for control purposes. Ear thickness was measured in a blinded fashion before and 48 h after injection with a digital precision caliper. OVA-specific ear swelling was calculated as (right ear thickness − left ear thickness) at 48 h − (right ear thickness − left ear thickness) at 0 h.
Quantitative PCR
Sort-purified cells were lysed in RLT buffer (Qiagen). RNA was extracted via RNeasy mini kits (Qiagen), as per the manufacturer’s instructions. Reverse transcription of RNA was performed using Superscript reverse transcription according to the protocol provided by the manufacturer (Thermo Fisher Scientific). Real-time PCR was performed on cDNA using SYBR green chemistry (Applied Biosystems). Reactions were run on a real-time PCR system (ABI 7500; Applied Biosystems). Samples were normalized to Hprt1 or Gapdh, and displayed as a fold change compared to controls.
ELISA
For OVA-specific IgG1 quantification, 40 μg/ml OVA (grade VI) was coated on an ELISA plate, mouse serum was diluted from 1:104 to 1:106, and an anti-OVA IgG1 (Cayman Chemical) standard curve ranging from 0.781 ng/ml to 100 ng/ml was applied. OVA-specific IgG1 was detected using an anti-mouse IgG1-HRP antibody (BD Biosciences). For faecal lipocalin-2 detection, faecal samples were collected and weighted for data normalization, then were homogenized in PBS and centrifuged at 12,000 r.p.m. to remove aggregates, and the resulting supernatant was collected. Afterwards, a sandwich ELISA was performed using mouse lipocalin-2/NGAL DuoSet ELISA (R&D Systems), according to the manufacturer’s instructions.
Regulatory T cell suppression assay
Dendritic cells (CD11c+MHCII+) or naive T cells (CD3+CD4+CD25−CD45RBhigh) were sort-purified from the spleen of wild-type or THY1.1+ mice, respectively. Small-intestinal Treg cells (CD45+CD3+CD4+CD25+) were sort-purified from either Ncr1creIl2f/f mice or littermate controls. and were subsequently found to be at least 95% FOXP3+ T cells. Dendritic cells were plated at 4 × 103 per well in the presence of 1 μg/ml soluble purified anti-CD3 antibody (clone 145-2C11, BD Biosciences). CFSE-labelled T cells (2 × 104 per well) were mixed with Treg cells at defined ratios and co-cultured with dendritic cells in the presence of anti-CD3. After a three-day culture, T cell proliferation was measured by CFSE dilution via flow cytometry. Treg cell suppression was calculated by gating on T effector cells, and quantifying the percentage of CFSE dilution in comparison to cells cultured in the absence of Treg cells.
Histological staining
Tissue samples from the intestines of mice were fixed with 4% paraformaldehyde, embedded in paraffin and 5-μm sections were stained with haematoxylin and eosin.
RNA sequencing
ILC3s (CD45+CD3−RORγtGFP+CD127+) and CD4+ T cells (CD45+CD3+CD4+) were sort-purified from small intestine of Rorc(gammat)-GfpTG mice. Sorted cells were used to prepare RNA sequencing libraries by the Epigenomics Core at Weill Cornell Medicine, using the Clontech SMARTer Ultra Low Input RNA Kit V4 (Clontech Laboratories). Sequencing was performed on an Illumina HiSeq 2500, yielding 50-bp single-end reads. Raw sequencing reads were demultiplexed with Illumina CASAVA (v.1.8.2). Adapters were trimmed from reads using FLEXBAR (v.2.4) and reads were aligned to the NCBI GRCm38/mm10 mouse genome using the STAR aligner (v.2.3.0) with default settings. Reads per gene were counted using Rsubread. One sample of the CD4+ T cell group was removed, as its library size was anomalously small compared to those of the other samples. Genes with at least ten counts in each sample were considered for further analysis. Differential expression was assessed using DESeq2 version 1.14.0 with default parameters and with a false discovery rate of 0.1.
Statistical analysis
P values of mouse datasets were determined by paired or unpaired two-tailed Student’s t-test with a 95% confidence interval. Variance was analysed using F-test. Welch’s correction was performed in case of unequal variance. Where appropriate, Mann–Whitney U-test, Wilcoxon matched-pairs test or two-way ANOVA followed by Bonferroni post-tests were performed. All statistical tests were performed with Graph Pad Prism v.6 software. P values of less than 0.05 were considered to be significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
RNA sequencing data are available at Gene Expression Omnibus under accession number GSE126580. All datasets generated and/or analysed during the current study are presented in this published article, the accompanying Source Data or Supplementary Information, or are available from the corresponding author upon reasonable request.
Change history
23 April 2019
The Reporting Summary for this article was originally missing and an unrelated document was substituted in place. This has been corrected online.
References
Boyman, O. & Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 12, 180–190 (2012).
Malek, T. R. The biology of interleukin-2. Annu. Rev. Immunol. 26, 453–479 (2008).
Sadlack, B. et al. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 75, 253–261 (1993).
Josefowicz, S. Z., Lu, L. F. & Rudensky, A. Y. Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 30, 531–564 (2012).
Belkaid, Y. & Tarbell, K. Regulatory T cells in the control of host–microorganism interactions. Annu. Rev. Immunol. 27, 551–589 (2009).
Izcue, A., Coombes, J. L. & Powrie, F. Regulatory lymphocytes and intestinal inflammation. Annu. Rev. Immunol. 27, 313–338 (2009).
Tanoue, T., Atarashi, K. & Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 16, 295–309 (2016).
Zhou, L. & Sonnenberg, G. F. Essential immunologic orchestrators of intestinal homeostasis. Sci. Immunol. 3, eaao1605 (2018).
Klatzmann, D. & Abbas, A. K. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat. Rev. Immunol. 15, 283–294 (2015).
Popmihajlov, Z., Xu, D., Morgan, H., Milligan, Z. & Smith, K. A. Conditional IL-2 gene deletion: consequences for T cell proliferation. Front. Immunol. 3, 102 (2012).
Artis, D. & Spits, H. The biology of innate lymphoid cells. Nature 517, 293–301 (2015).
Spits, H. et al. Innate lymphoid cells—a proposal for uniform nomenclature. Nat. Rev. Immunol. 13, 145–149 (2013).
Sonnenberg, G. F. & Artis, D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat. Med. 21, 698–708 (2015).
Atarashi, K. et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 163, 367–380 (2015).
Gaboriau-Routhiau, V. et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31, 677–689 (2009).
Sano, T. et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 163, 381–393 (2015).
Philpott, D. J., Sorbara, M. T., Robertson, S. J., Croitoru, K. & Girardin, S. E. NOD proteins: regulators of inflammation in health and disease. Nat. Rev. Immunol. 14, 9–23 (2014).
Narni-Mancinelli, E. et al. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc. Natl Acad. Sci. USA 108, 18324–18329 (2011).
Robson, M. J. et al. Generation and characterization of mice expressing a conditional allele of the interleukin-1 receptor type 1. PLoS ONE 11, e0150068 (2016).
Mortha, A. et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 343, 1249288 (2014).
Hepworth, M. R. et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 498, 113–117 (2013).
Hepworth, M. R. et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science 348, 1031–1035 (2015).
Kim, K. S. et al. Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 351, 858–863 (2016).
Hadis, U. et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34, 237–246 (2011).
Pabst, O. & Mowat, A. M. Oral tolerance to food protein. Mucosal Immunol. 5, 232–239 (2012).
Boschetti, G. et al. Therapy with anti-TNFα antibody enhances number and function of Foxp3+ regulatory T cells in inflammatory bowel diseases. Inflamm. Bowel Dis. 17, 160–170 (2011).
Bernink, J. H. et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 14, 221–229 (2013).
Johansson, M. E. & Hansson, G. C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 16, 639–649 (2016).
Mowat, A. M. & Agace, W. W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 14, 667–685 (2014).
Maloy, K. J. & Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306 (2011).
Acknowledgements
We thank members of the Sonnenberg Laboratory for discussions and critical reading of the manuscript, and T. Shima and Y. Umesaki from Yakult Central Institute for providing segmented filamentous bacteria and advice. Research in the Sonnenberg Laboratory is supported by the National Institutes of Health (R01AI143842, R01AI123368, R01AI145989, R21DK110262 and U01AI095608), the NIAID Mucosal Immunology Studies Team (MIST), the Crohn’s and Colitis Foundation, the Searle Scholars Program, the American Asthma Foundation Scholar Award, Pilot Project Funding from the Center for Advanced Digestive Care (CADC), an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund, a Wade F. B. Thompson/Cancer Research Institute CLIP Investigator grant, the Meyer Cancer Center Collaborative Research Initiative and the Jill Roberts Institute (JRI) for Research in IBD. L.Z. and J.G. are supported by fellowships from the Crohn’s and Colitis Foundation (608975 and 519428). N.J.B. is supported by a fellowship from the NIH (F32AI124517). We thank the Epigenomics Core of Weill Cornell Medicine, and all contributing members of the JRI IBD Live Cell Bank, which is supported by the JRI, Jill Roberts Center for IBD, Cure for IBD, the Rosanne H. Silbermann Foundation and Weill Cornell Medicine Division of Pediatric Gastroenterology and Nutrition. Research in the Vivier laboratory is supported by funding form the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (TILC, grant agreement no. 694502); the Agence Nationale de la Recherche; Equipe Labellisée ‘La Ligue’, Ligue Nationale contre le Cancer, MSDAvenir, Innate Pharma and institutional grants to the CIML (INSERM, CNRS and Aix-Marseille University) and to Marseille Immunopôle. Research reported in this publication was supported by the National Center for Advancing Translational Science of the National Institute of Health, under award number UL1TR002384.
Reviewer information
Nature thanks Koji Hase, Scott Snapper and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
L.Z. and G.F.S. conceived the project. L.Z. performed most experiments and analysed the data. F.T., C.C., N.J.B., J.G., H.K. and E.K.S. helped with experiments. G.G.P. performed bioinformatics analyses. J.R.K., R.N.B., M.A.S. and R.E.S. provided human samples, scientific advice and expertise. E.V., G.E. and K.A.S. provided essential mouse models, scientific advice and expertise. L.Z. and G.F.S. wrote the manuscript, with input from all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 IL-2 blockade results in disrupted T cell homeostasis throughout the intestinal tract and associated lymphoid tissues.
a–c, Sex- and age- matched C57BL/6 mice were treated with anti-IL-2 monoclonal antibodies every other day for two weeks, and examined for the size of the spleen and mesenteric lymph nodes (mLN) (a), the frequency of Treg cells (b) and Ki-67+CD4+ T cells (c) of mesenteric lymph nodes by flow cytometry (n = 10). d–g, Mice from a–c were also analysed for the frequencies of Treg cells (d) and Ki-67+CD4+ T cells (e) in large-intestinal lamina propria cells, and for the frequencies and numbers of Treg cells (f) and Ki-67+CD4+ T cells (g) in small-intestinal lamina propria cells by flow cytometry (n = 10). h–k, Mice from a–c were analysed for the frequency of TH1 cells (h) and TH17 cells (i) in large-intestinal lamina propria cells, and the frequencies and numbers of TH1 cells (j) and TH17 cells (k) in small-intestinal lamina propria cells by flow cytometry (n = 10). Data in a are representative of two independent experiments with similar results. Data in b–k are pooled from two independent experiments. Results are shown as mean ± s.e.m. All statistics are calculated by unpaired two-tailed Student’s t-test. LI, large intestine; SI, small intestine.
Extended Data Fig. 2 T-cell-derived IL-2 is essential for maintaining immunological homeostasis in the mesenteric lymph node and large intestine.
a, Sex- and age- matched Il2f/f and LckcreIl2f/f mice were examined for the deletion efficiency of IL-2 in CD4+ T cells in the large and small intestines. b, c, Mice in a were examined for the frequency of Treg cells (b) and Ki-67+ CD4+ T cells (c) from the mesenteric lymph nodes by flow cytometry (n = 6). d–g, Mice in a were analysed for the frequencies and numbers of Treg cells (d), Ki-67+CD4+ T cells (e), TH1 cells (f) and TH17 cells (g) of large-intestinal lamina propria cells by flow cytometry (n = 6). h–k, Mice in a were analysed for the frequencies and numbers of Treg cells (h), Ki-67+CD4+ T cells (i), TH1 cells (j) and TH17 cells (k) of small-intestinal lamina propria cells by flow cytometry (n = 6). Data in a are representative of two independent experiments with similar results. Data in b–k are pooled from two independent experiments. Results are shown as mean ± s.e.m. All statistics are calculated by unpaired two-tailed Student’s t-test.
Extended Data Fig. 3 Gating strategy to analyse subsets of innate lymphoid cells and CD4+ T cells in the small intestine.
Gating strategy for flow cytometry analysis of innate lymphoid cells and CD4+ T cells in small-intestinal lamina propria cells. Lineage 1, CD11b, CD11c and B220; lineage 2, CD3ε, CD5 and CD8α. CD4+ T cells were identified as CD45+Lineage 2+CD4+; group-1 innate lymphoid cells were identified as CD45+Lineage−CD127+CD90.2+T-bet+RORγt−; group-2 innate lymphoid cells were identified as CD45+Lineage−CD127+CD90.2+GATA3+; ILC3s were identified as CD45+Lineage−CD127+CD90.2+RORγt+; and subsets of ILC3s further identified as CCR6+T-bet− ILC3s or CCR6−T-bet+ ILC3s.
Extended Data Fig 4 IL-2+ cells in the large intestine of wild-type mice and in the small and large intestines of Rag1−/− mice.
a, Flow cytometry plots with graph of frequency and numbers of IL-2 in ILC3s and effector and memory (E/M) CD4+ T cells (CD3+CD4+FOXP3−CD44highCD62Llow) in small-intestinal lamina propria cells of wild-type mice (n = 8). b, Flow cytometry plots show IL-2+ cells in large-intestinal lamina propria cells of C57BL/6 mice. Lineage 1, CD11b, CD11c and B220; lineage 2, CD3ε, CD5 and CD8α. c, d, Flow cytometry plots with graph of frequency (c) and absolute numbers (d) of IL-2+ cells in large-intestinal lamina propria cells of C57BL/6 mice (n = 6). e. The frequency and number of IL-2+ subsets of ILC3s in small-intestinal lamina propria cells of C57BL/6 mice (n = 8). f, Flow cytometry plots show IL-2+ cells in small-intestinal lamina propria cells of Rag1−/− mice. g, h, Flow cytometry plots with graph of frequency (g) and absolute numbers (h) of IL-2+ cells in small-intestinal lamina propria cells of Rag1−/− mice (n = 5). i, j, Flow cytometry plots with graph of frequency (i) and absolute numbers (j) of IL-2+ subsets of ILC3s in small-intestinal lamina propria cells of Rag1−/− mice (n = 5). k, Flow cytometry plots show IL-2+ cells in large-intestinal lamina propria cells of Rag1−/− mice. Lineage 1, CD11b, CD11c and B220; lineage 2, CD3ε, CD5 and CD8α. Data in b and f–k are representative of two independent experiments with similar results. Data in a and c–e are pooled from two independent experiments. Results are shown as mean ± s.e.m. Statistics are calculated by paired or unpaired two-tailed Student’s t-test.
Extended Data Fig. 5 Natural killer cells and group-1 innate lymphoid cells are dispensable for maintenance of Treg cells in the small intestine.
a, IL-2 was assessed in T-bet+ ILC3s, total ILC3s, CD4+ T cells, natural killer cells, group-1 innate lymphoid cells and dendritic cells in small-intestinal lamina propria cells of Il2f/f and Ncr1creIl2f/f mice. b, The number of IL-2+ cells was quantified in small-intestinal lamina propria cells of Il2f/f and Ncr1creIl2f/f mice (n = 4). c, d, Sex- and age- matched C57BL/6 mice were treated with anti-NK1.1 monoclonal antibody every three days for two weeks and examined for efficiency of depletion of natural killer cells (c), and for the frequency and number of Treg cells in small-intestinal lamina propria cells (d) (n = 7). Data in a–c are representative of two independent experiments with similar results. Data in d are pooled from two independent experiments. Results are shown as mean ± s.e.m. Statistics are calculated by unpaired two-tailed Student’s t-test.
Extended Data Fig. 6 ILC3-derived IL-2 is dispensable for the maintenance of small-intestinal TH17 cells, ILC3 homeostasis and large-intestinal Treg cells.
a–d, Il2f/f and Ncr1creIl2f/f mice were analysed for the percentage of Treg cells (a), TH1 cells (b), Ki-67+CD4+ T cells (c) and the frequency and cell number of TH17 cells (d), in small-intestinal lamina propria cells at steady state by flow cytometry (n = 8). e, Il2f/f and Ncr1creIl2f/f mice were examined for the frequency and number of Treg cells in large-intestinal lamina propria cells by flow cytometry (n = 8). f, Il2f/f and Ncr1creIl2f/f mice were examined for the frequency and number of ILC3s in small-intestinal lamina propria cells by flow cytometry (n = 8). g, IL-22 was assessed in ILC3s from small-intestinal lamina propria cells of Il2f/f or Ncr1creIl2f/f mice. h, Representative histograms and bar graph examination of CD25 staining on Treg cells and IL-2+ ILC3s. i, Representative histograms that demonstrate IL-2 binding capacity and quantification of bound IL-2 mean fluorescence intensity in Treg cells and ILC3s. j, Experimental design of the delayed-type hypersensitivity model. Data in f–h are representative of two independent experiments with similar results (at least three mice per group). Data in a–e and i are pooled from two independent experiments. Results are shown as mean ± s.e.m. Statistics are calculated by paired or unpaired two-tailed Student’s t-test.
Extended Data Fig. 7 Deletion of ILC3-intrinsic IL-2 affects the population size of peripherally induced Treg cells but not their suppressive capacity.
a, b, The frequency of peripheral Treg cells (labelled ‘Nrp-1lo pTregs’) and thymic Treg cells (labelled ‘Nrp-1hi tTregs’) were characterized in small-intestinal lamina propria cells of Il1rf/f and Ncr1creIl1rf/f mice (a) or Il2f/f and Ncr1creIl2f/f mice (b) (n = 5). c, d, The frequency of subsets of Treg cells were analysed in small-intestinal lamina propria cells of Il2f/f and Ncr1creIl2f/f mice (n = 5). e, Small-intestinal Treg cells were examined for expression of Lag3, Tgfb1, Ctla4, Ebi3 and Il10 in Il2f/f and Ncr1creIl2f/f mice (n = 7). f, g, Sort-purified small-intestinal CD45+CD3+CD4+CD25+ Treg cells were co-cultured with sort-purified CFSE-labelled splenic effector T cells (CD3+CD4+CD25−CD45RBhigh) in the presence of purified splenic dendritic cells and soluble anti-CD3 for three days. CFSE dilution was analysed and quantified (n = 6). Data in a–d and f are representative of two independent experiments with similar results. Data in e and g are pooled from two independent experiments. Results are shown as mean ± s.e.m. Statistics are calculated by unpaired two-tailed Student’s t-test.
Extended Data Fig. 8 ILC3-derived IL-2 does not exhibit functional redundancy or hierarchies with ILC3-specific GM-CSF or MHCII.
a, Flow cytometry plots with graph of frequency and quantification of cell numbers of IL-2+ ILC3s in small-intestinal lamina propria cells of wild-type and Csf2−/− mice (n = 8). b, c, Flow cytometry plots with graph of frequency and quantification of cell numbers of Treg cells (b) and IL-2+ ILC3s (c) in small-intestinal lamina propria cells of H2-Ab1f/f littermate controls and mice lacking ILC3-specific MHCII (MHCIIΔILC3 mice) (n = 7). d, e, Flow cytometry plots with graph of frequency and quantification of cell numbers of MHCII+ ILC3s (d) and GM-CSF+ ILC3s (e) in small-intestinal lamina propria cells of Il2f/f and Ncr1creIl2f/f mice (n = 7). Data are pooled from two independent experiments. Results are shown as mean ± s.e.m. Statistics are calculated by unpaired two-tailed Student’s t-test.
Extended Data Fig. 9 ILC3-derived IL-2 promotes essential immune regulation in the intestine.
a–h, CD4+ T cells were adoptively transferred into Il2f/fRag1−/− or RorccreIl2f/fRag1−/− recipient mice. a–d, Recipient mice were examined for changes in weight (a), colon length (b), histological haematoxylin and eosin staining in the terminal colon (c) and lipocalin-2 presence in faecal samples (d) (n = 8). e, Flow cytometry plots with graph of percentage and absolute cell number of Treg cells in large-intestinal lamina propria cells in defined recipient mice. f, Absolute cell number of Treg cells in small-intestinal lamina propria cells in defined recipient mice. g, Flow cytometry plots and graph of frequency and absolute number of IFNγ−IL-17A+ and IFNγ+IL-17A+ cells in large-intestinal lamina propria cells in defined recipient mice. h, Cell number of TH1 and TH17 cells in small-intestinal lamina propria cells in defined recipient mice. n = 7 mice, Il2f/fRag1−/− group; n = 8 mice, RorccreIl2f/fRag1−/− (e–h). Data in a–h are pooled from two independent experiments. Results are shown as mean ± s.e.m. Statistics are calculated by unpaired two-tailed Student’s t-test.
Extended Data Fig. 10 A IL-1β–ILC3–IL-2 circuit is essential for the maintenance of Treg cells and immunological homeostasis uniquely within the small intestine.
Here we define a pathway of immune regulation in the small intestine. This pathway is continuously required, and involves MYD88- and NOD2-dependent microbial sensing by macrophages, production of IL-1β and induction of ILC3-derived IL-2 to support the maintenance of peripherally induced intestinal Treg cells. Consequently, this is essential to maintain immunological homeostasis and oral tolerance, and becomes dysregulated in inflammatory bowel disease in humans.
Supplementary information
Source data
Rights and permissions
About this article

Cite this article
Zhou, L., Chu, C., Teng, F. et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 568, 405–409 (2019). https://doi.org/10.1038/s41586-019-1082-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1082-x
- Springer Nature Limited
This article is cited by
-
Increased regulatory activity of intestinal innate lymphoid cells type 3 (ILC3) prevents experimental autoimmune encephalomyelitis severity
Journal of Neuroinflammation (2024)
-
Donor regulatory T cells rapidly adapt to recipient tissues to control murine acute graft-versus-host disease
Nature Communications (2024)
-
Group 3 innate lymphoid cells in intestinal health and disease
Nature Reviews Gastroenterology & Hepatology (2024)
-
Dysregulation of Wnt/β-catenin signaling contributes to intestinal inflammation through regulation of group 3 innate lymphoid cells
Nature Communications (2024)
-
The renaissance of oral tolerance: merging tradition and new insights
Nature Reviews Immunology (2024)