

Abstract
The spliceosome catalyses the excision of introns from pre-mRNA in two steps, branching and exon ligation, and is assembled from five small nuclear ribonucleoprotein particles (snRNPs; U1, U2, U4, U5, U6) and numerous non-snRNP factors1. For branching, the intron 5′ splice site and the branch point sequence are selected and brought by the U1 and U2 snRNPs into the prespliceosome1, which is a focal point for regulation by alternative splicing factors2. The U4/U6.U5 tri-snRNP subsequently joins the prespliceosome to form the complete pre-catalytic spliceosome. Recent studies have revealed the structural basis of the branching and exon-ligation reactions3, however, the structural basis of the early events in spliceosome assembly remains poorly understood4. Here we report the cryo-electron microscopy structure of the yeast Saccharomyces cerevisiae prespliceosome at near-atomic resolution. The structure reveals an induced stabilization of the 5′ splice site in the U1 snRNP, and provides structural insights into the functions of the human alternative splicing factors LUC7-like (yeast Luc7) and TIA-1 (yeast Nam8), both of which have been linked to human disease5,6. In the prespliceosome, the U1 snRNP associates with the U2 snRNP through a stable contact with the U2 3′ domain and a transient yeast-specific contact with the U2 SF3b-containing 5′ region, leaving its tri-snRNP-binding interface fully exposed. The results suggest mechanisms for 5′ splice site transfer to the U6 ACAGAGA region within the assembled spliceosome and for its subsequent conversion to the activation-competent B-complex spliceosome7,8. Taken together, the data provide a working model to investigate the early steps of spliceosome assembly.
Similar content being viewed by others


Main
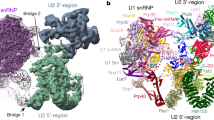
To gain structural insights into early spliceosome assembly, we prepared the yeast prespliceosome A-complex on the UBC4 pre-mRNA that carries a mutation in the pre-mRNA branch point sequence, which was previously used to stall the A-complex9 (UACUAAC to UACAAAC, in which A is the branch point adenosine and A is the mutated nucleotide) (Extended Data Fig. 1a, b). The purified A-complex contained stoichiometric amounts of the U1 and U2 snRNP proteins (Extended Data Fig. 1b), and was used to determine cryo-electron microscopy (cryo-EM) densities of the A-complex at 4.0 Å (U1 snRNP, map A2) and 4.9–10.4 Å (U2 snRNP, maps A1 and A3) resolution, respectively (Extended Data Figs. 1c–e, 2). From these densities we built a near-complete atomic model of the A-complex (Fig. 1, Supplementary Videos 1, 2, Supplementary Data, Extended Data Fig. 1f), comprising 34 proteins, U1 and U2 snRNAs, and 34 nucleotides of pre-mRNA. The final model lacks the mobile cap-binding complex, Prp5 or the U1 subunit Prp40 (Extended Data Fig. 1b, d, e; Extended Data Table 1). The elongated U1 and U2 snRNPs bind the pre-mRNA 5′ splice site (5′SS) and branch-point sequences, respectively, and associate in a parallel manner to form the A-complex (Fig. 2a). The U1 snRNP structure contains all the essential regions of the U1 snRNA and 16 proteins (Fig. 1). The U1 snRNP ‘core’ is highly similar to its human counterpart10 (Extended Data Figs. 3, 4), comprising the seven-membered Sm ring and orthologues of the human U1 snRNP proteins (Snp1, human U1-70k; Mud1, human U1A; Yhc1, human U1C), and is bound to the peripheral yeast U1 proteins Luc7, Nam8, Prp39, Prp42, Snu56 and Snu7111 (Extended Data Figs. 3, 4). The U2 snRNP has a bipartite structure as observed in B-complex8, comprising the SF3b subcomplex (‘5′ region’) and the U2 3′ domain and SF3a subcomplex (‘3′ region’) that are organized around the 5′ and 3′ regions of the U2 snRNA, respectively (Figs. 1, 2a, Extended Data Fig. 5). At the current resolution, the conformation of the U2 5′ region appears unchanged from the B-complex8, in which the pre-mRNA branch-point sequence is base-paired with the U2 snRNA and the branch point adenosine is bulged out and accommodated in a pocket formed by the U2 SF3b subunits Hsh155 and Rds3. After we completed the A-complex structure, the cryo-EM structure of the free yeast U1 snRNP was reported12. This model is in good agreement with the U1 snRNP in our A-complex structure, but there are important differences12.

Two orthogonal views of the yeast A-complex structure. Subunits are coloured according to snRNP identity (U1, shades of purple, U2, shades of green), and the pre-mRNA intron (black) and its 5′ exon (orange) are highlighted. The orthologous human protein name is shown after the solidus. The location of the cap-binding complex (CBC) is indicated by a brown oval (see Extended Data Fig. 1e).

a, The A-complex U1–U2 snRNP interfaces (A and B) and the RNA network are shown as cartoons, and are superimposed on the transparent surfaces of the prespliceosome proteins. The U2 subunit Hsh155 surface (grey oval), which interacts with the tri-snRNP in the B-complex, is freely accessible in the A-complex. The U1 snRNP proteins Nam8 (orange, human TIA-1), Luc7 (purple, human LUC7L), Prp39 (magenta, human PRPF39) and Yhc1 (dark magenta, human U1C) and the U2 snRNP proteins Lea1 (light green, human U2-A′), Rse1 (dark green, human SF3B3), and Prp9 (teal, human SFA3) are shown as ribbons. BP, branch point. b, The pre-mRNA 5′SS is recognized by the U1 snRNA 5′ end, and is stabilized by Luc7 and Yhc1. Notably, the Yhc1 ZnF and Luc7 ZnF2 domains are arranged with pseudo-C2 symmetry around the U1–5′SS helix. c, Nam8 binds the U1 snRNP through its linker (yellow), RNA recognition motif 3 (RRM3, light orange) and C-terminal regions (orange), whereas its RRM1 and RRM2 domains are mobile and project towards the intron to bind uridine-rich sequences downstream of the pre-mRNA 5′SS (dashed black line), as with its human counterpart TIA-118. Nam8 contacts the Yhc1 (human U1C) C terminus, and human TIA-1 biochemically also interacts with human U1C18. Snu56 (blue), Prp39 (magenta), Prp42 (violet), and Hsh49 (light green) are shown as transparent ribbon models and other protein and U1 snRNA elements were removed for clarity.
The first ten nucleotides of U1 snRNA are disordered in the free U1 snRNP12, but become ordered in our A-complex structure by pairing with the pre-mRNA 5′SS (Fig. 2a, b). Additional density appeared adjacent to the U1–5′SS helix, into which we could build a newly ordered Yhc1 peptide (human U1C) that contacts the 5′SS phosphate backbone (+5 and +6 positions, the ‘Yhc1–5′SS loop’) and a near-complete model of Luc7 (in the previous study Luc7 was attributed to what is now assigned as Snu7112) (Extended Data Figs. 3a, c, 4a). Although Luc7 is disordered in the free U1 snRNP, it associates stably with the U1–5′SS helix in the A-complex (Extended Data Fig. 4a), suggesting a mechanism for the selection of weak 5′SS sequences13. In our structure Luc7 is anchored by its N-terminal α-helix 1 to the Sm ring subunit SmE, and its C3H-type zinc finger 1 (ZnF1) domain binds where the 5′ exon emerges from the U1–5′SS helix, in excellent agreement with RNA–protein crosslinks13 (Fig. 2b). The adjacent Luc7 C2H2-type ZnF2 contacts the U1–5′SS helix minor groove and the U1 snRNA phosphate backbone (nucleotides U5–C8). This interaction mirrors that between the Yhc1 ZnF domain and the 5′SS nucleotides +1 to +4 downstream of the 5′SS junction10 (Fig. 2b). Thus, Yhc1 and Luc7 make no base-specific interactions with the U1–5′SS helix, and instead cradle the U1–5′SS helix phosphate backbone to stabilize 5′SS binding. Consistent with the structure, weakening of any of these interactions can impair splicing and bypass the requirement for Prp28 helicase activity13,14,15,16.
The A-complex structure reveals structural insights into the functions of the human alternative splicing factors LUC7-like (LUC7L, yeast Luc7) and TIA-1 (yeast Nam8) (Extended Data Fig. 4c, d). Luc7 and its human homologues LUC7L1–3 are highly conserved, suggesting that the LUC7L N-terminal α-helix also anchors it to the SmE protein and that the invariant ZnF2 helix α8 similarly stabilizes the U1–5′SS helix to promote the inclusion of weak alternative splice sites13 (Fig. 2b, Extended Data Figs. 3c, 6a). The yeast U1 snRNP subunit Nam8 and its human homologue TIA-1 contain three RNA recognition motif (RRM) domains and a C-terminal Gln-rich extension (Extended Data Fig. 6b). Human TIA-1 binds to uridine-rich sequences downstream of the 5′SS predominantly through the RRM2 domain17,18 to allow the use of weak 5′SSs. The Nam8 RRM2 shows high sequence similarity to the TIA-1 RRM2, including the nearly identical RNP1 and RNP2 motifs, indicating that Nam8 also binds uridine-rich sequences through its RRM2 also (Extended Data Fig. 6b). In the A-complex structure the Nam8 RRM3 and its C-terminal region bind in a cavity of the Prp39–Prp42 heterodimer and contact the Yhc1 C-terminal region near the U1–5′SS helix (Fig. 2c). From this location, Nam8 could project its mobile RRM2 domain to bind uridine-rich intron sequences downstream of the 5′SS, consistent with crosslinking experiments17, and thereby promote meiotic pre-mRNA splicing19 (Fig. 2a, c).
In the A-complex, the U1 snRNP binds to the U2 snRNP through two interfaces, A and B (Fig. 2a). In interface A, the N-terminal helices α1–2 of the U1 protein Prp39 stably bind the U2 3′ domain subunit Lea1 (human U2A′) (Fig. 2a, Extended Data Fig. 5). The Prp39–Prp42 heterodimer binds Yhc1 to anchor the U2 snRNP 3′ domain to the U1 snRNP. Similar interactions were observed biochemically between the human alternative-splicing factor PRPF39 homodimer and U1C12 (yeast Yhc1), suggesting that PRPF39 may contact the human U2 3′ domain in a similar manner, although it is not an obligate component of the human A-complex20 (Fig. 2a). Different, non-overlapping Lea1 surfaces are used to interact with the NTC protein Syf1 in the yeast C- and C*/P-complex conformations of the spliceosome21 (Extended Data Fig. 5c), suggesting that Lea1 aids in the repositioning of the U2 3′ domain in multiple stages of splicing. Interface B is transient and found only in a subset of cryo-EM images (Extended Data Figs. 2a, 5a, b). It involves weak interactions between the yeast-specific U1 snRNA stem loop 3–3 and the U2 SF3b Rse1 subunit β-propellers B and C (BPB and BPC) and the C terminus of U2 SF3a Prp9. The pre-mRNA 5′SS and branch point branching reactants are positioned approximately 150 Å apart in the A-complex, with 40 nucleotides of the UBC4 intron looped out in between (Fig. 2a, Extended Data Fig. 1e, f). The small interfaces between the U1 and U2 snRNPs orient the snRNPs relative to each other, and this may facilitate 5′SS transfer in the assembled spliceosome and the subsequent dissociation of the U1 snRNP, consistent with the structural and biochemical data7,8. Although the precise U1–U2 snRNP interfaces may differ in the human A-complex, a key function of U1–U2 (alternative) splicing factors could be to ensure that U1 and the U1–5′SS helix are oriented correctly relative to the U2 snRNP.
Before A-complex formation, the yeast Msl5–Mud2 heterodimer recognizes the branch point sequence through Msl5 and binds the U1 snRNP subunit Prp40 (human PRPF40) in the E complex, looping out the intron between the 5′SS and branch point sequences22 (Extended Data Fig. 4e). Although Prp40 was not identified in the free U1 snRNP12 or in our A-complex structure, Prp40 crosslinks to Luc7 and Snu7112 and unassigned cryo-EM density in the A-complex may indicate its peripheral location near Luc7 (Extended Data Figs. 1e, 4a, e). Msl5–Mud2 may then be destabilized by the Sub2 helicase, allowing the Prp5 helicase to remodel U2 snRNA for the stable association of the U2 snRNP with the branch point sequence in the A-complex9. Prp5 was shown to physically interact with the U2 SF3b subunit Hsh155 HEAT repeats 1–6 and 9–1223 and with U2 snRNA at and surrounding the branch point-interacting stem loop9. Thus, after Prp5 activity, Prp5 needs to dissociate to fully expose the Hsh155 HEAT repeats 11–13 together with the U2 snRNA 5′ end in the A-complex, to allow for the subsequent U4/U6.U5 tri-snRNP association to assemble the spliceosome7,8,9 (Fig. 2a).
The A-complex structure also provides new insights into formation of the fully assembled pre-B-complex spliceosome, which requires integration of the tri-snRNP with the A-complex. The subsequent Prp28 helicase-mediated transfer of the 5′SS from U1 to U6 snRNA and destabilization of the U1 snRNP produces the B-complex spliceosome24. We first modelled a fully assembled yeast spliceosome, by superimposing the U2 snRNP SF3b-containing domains of the yeast A-complex (from this study) and the yeast B-complex structure8. As in the B-complex structure8, the U2 snRNP would associate with tri-snRNP via the U2/U6 helix II and Prp3 (Extended Data Fig. 7). The modelling shows that the U1 snRNP would clash with large parts of the Brr2-containing ‘helicase’ domain (‘U1-B-complex’; Extended Data Figs. 7b, 8b), which may be relieved owing to their known flexibilities8 (Extended Data Fig. 5a). However the known binding site for Prp28 at the U5 Prp8 N-terminal domain (Prp8N) observed in human tri-snRNP25 would be sterically occluded by the pre-bound B-complex proteins7,8,26. We therefore considered an alternative model for the assembled yeast ‘pre-B-complex’ spliceosome, by combining the available data from yeast and human systems8,25,27,28 (Fig. 3a, Extended Data Figs. 7a, 8a). First, the isolated human25 and yeast tri-snRNP26,29 structures differ in their protein composition and conformation, indicating that different complexes accumulate at steady-state. In the human tri-snRNP structure25 the BRR2 helicase is held near SNU114 by the SAD1 protein and PRP28 is bound to the PRP8 N-terminal domain. In the yeast tri-snRNP26,29 and the yeast and human B-complex structures7,8 Brr2 is repositioned and loaded onto its U4 snRNA substrate and the B-complex proteins replace Prp28 at the Prp8N domain, ready for spliceosome activation. Second, in humans, an ATPase-deficient PRP28 helicase stalls spliceosome assembly at the pre-B-complex stage, before disruption of the U1–5′SS interaction28 and this complex comprises the U1 and U2 snRNPs, a loosely associated tri-snRNP, and SAD128. Third, in yeast, Sad1 is essential for splicing and is very transiently associated with the tri-snRNP27. Given the high conservation of the major spliceosome components in yeast and humans, the yeast spliceosome may likewise assemble with a human-like tri-snRNP that contains Prp28, Sad1 and a repositioned Brr2 helicase25,28. On the basis of these assumptions, we modelled a yeast pre-B-complex spliceosome that comprises all five snRNPs with a combined molecular mass of approximately 3.1 megadalton and with only minor clashes (Fig. 3a, Extended Data Fig. 7a, b). Notably, this model indicates that the U2 snRNP positions the U1 snRNP to deliver the U1–5′SS helix to the exposed U6 ACAGAGA stem in tri-snRNP, only approximately 20 Å away from where Prp28 is likely to mediate 5′SS transfer, consistent with protein–RNA crosslinks30 (Fig. 3b). This suggests that the subsequent repositioning of the Brr2 helicase onto the U4 snRNA, observed in the B complex structure7,8,would coincide with the release of the U1 snRNP owing to a steric clash, rendering Brr2 competent for spliceosome activation only after successful 5′SS transfer (Extended Data Figs. 7b, 8a). The model thus indicates a new molecular checkpoint to couple 5′SS transfer with U1 snRNP release and formation of the B-complex (Extended Data Figs. 7b, 8a).

a, One of the two alternative pre-B-complex models, suggesting that the U2 snRNP orients the U1 snRNP to deliver the pre-mRNA 5′SS to the U6 ACAGAGA stem. The model was obtained by superposing the yeast A- (from this study) and B-complex structures (RCSB Protein Data Bank code (PDB ID) 5NRL) and by modifying the locations of Brr2, U4 Sm ring, Sad1, and Prp28 to resemble a human-like pre-B-complex conformation on the basis of the biochemical data and the human U4/U6.U5 tri-snRNP structure (PDB ID 3JCR) (see ‘Structural modelling’ in Methods). Colouring as in Fig. 1 and a previously published work8. b, The pre-B-complex RNA network and the Prp28 helicase are shown as cartoons and are superimposed on transparent surfaces of the spliceosome proteins. Prp28 is positioned at the Prp8 N-terminal domain as in human tri-snRNP25 and may clamp onto the pre-mRNA near the U1–5′SS helix to destabilize it and transfer the 5′SS from U1 snRNA to the U6 snRNA ACAGAGA stem (red arrow), which are separated by approximately 20 Å in the pre-B model. The positions of proteins marked with asterisks are based on the human tri-snRNP structure (PDB ID 3JCR).
In summary, the prespliceosome structure reveals how the U1 and U2 snRNPs recognize the two reactants of the branching reaction and associate together with the tri-snRNP into the fully assembled spliceosome. The results further suggest how the human alternative-splicing factors LUC7L and TIA-1 may influence splice-site selection.
Methods
Prespliceosome preparation and purification
To obtain the prespliceosome A-complex for structural studies, we prepared yeast S. cerevisiae containing a genomic TAPS affinity tag on the U2 snRNP subunit Hsh155, essentially as described31. Yeast cells were grown in a 120-l fermenter, and splicing extract was prepared using the liquid-nitrogen method, essentially as described32. Capped UBC4 pre-mRNA containing a point mutation (U > A) two nucleotides upstream of the branch point adenosine and three MS2 stem loops at the 3′ end was produced by in vitro transcription9,33. The RNA product was labelled with Cy5 at its 3′ end to monitor complex purification34. The pre-mRNA substrate was bound to the MS2–MBP fusion protein and added to an in vitro splicing reaction carried out for 90 min at 23 °C, essentially as described33. The reaction mixture was then centrifuged through a 40% glycerol cushion in buffer A (20 mM HEPES (pH 7.9), 50 mM KCl, 0.2 mM EDTA, 1 mM dithiothreitol (DTT), 0.04% NP-40). The cushion was diluted with buffer A containing 1% glycerol, and applied to amylose resin (NEB) pre-washed with buffer B (20 mM HEPES (pH 7.9), 75 mM KCl, 5% glycerol, 0.2 mM EDTA, 1 mM DTT, 0.03% NP-40). After 12 h incubation at 4 °C, the resin was washed with buffer B and eluted in buffer B containing 50 mM KCl and 12 mM maltose. Fractions containing A-complex were pooled and applied to Strep-Tactin resin (GE Healthcare), pre-washed with buffer B, and incubated for 4 h at 4 °C. The resin was washed with buffer B containing 2 mM MgCl2, and eluted with buffer B containing 50 mM KCl, 2.5 mM desthiobiotin, and 2 mM MgCl2. The A-complex fractions were pooled and crosslinked using 1.1 mM BS3 (Sigma) on ice for 1 h, and subsequently quenched with 50 mM ammonium bicarbonate. The sample was concentrated to ~0.4 mg ml−1 and immediately used for EM sample preparation. Mass spectrometry (data not shown), indicated that homogenous A-complex was purified, containing sub-stoichiometric amounts of Prp5 (Extended Data Fig. 1b). The splicing assay in Extended Data Fig. 1a was carried out as for A-complex purification, but in a volume of 25 μl and in the absence of MS2–MBP fusion protein, and was visualized after 30 min of splicing at 23 °C on a denaturing 14% polyacrylamide TBE gel with a Typhoon scanner (GE Healthcare).
Electron microscopy
For cryo-EM analysis the A-complex sample was applied to R2/2 holey carbon grids (Quantifoil), precoated with a 5–7-nm homemade carbon film. Grids were glow-discharged for 20 s before deposition of 2.5 μl sample (~0.4 mg ml−1), and subsequently blotted for 2–3.5 s and vitrified by plunging into liquid ethane with a Vitrobot Mark III (FEI) operated at 4 °C and 100% humidity. Cryo-EM data were acquired on three separate FEI Titan Krios microscopes (datasets one to three) operated in EFTEM mode at 300 keV, each equipped with a K2 Summit direct detector (Gatan) and a GIF Quantum energy filter (slit width of 20 eV, Gatan). Datasets one and three were recorded using ‘Krios 1’ and ‘Krios 2’ at the MRC-LMB, respectively, and dataset three using ‘Krios 2’ at the Astbury Biostructure Laboratory (University of Leeds). For dataset one, 5,935 movies were acquired using EPU (FEI) with a defocus range of –0.4 μm to –4.4 μm at a nominal magnification of 105,000× (1.13 Å pixel–1). The camera was operated in ‘counting’ mode with a total exposure time of 13 s fractionated into 20 frames, a dose rate of 4.25 e− pixel−1 s−1, and a total dose of 43 e− Å−2 per movie. Dataset two was collected in the same manner, except that 727 movies were recorded using SerialEM35, at a nominal magnification of 105,000× (1.14 Å pixel−1), a total exposure time of 8 s fractionated into 20 frames, a dose rate of 4.33 e− pixel−1 s−1 and a total dose of 27 e− Å−2 per movie. Dataset three was collected with EPU (FEI) similar to dataset one, except that 2,745 movies were collected at a nominal magnification of 130,000× (1.07 Å pixel−1), a total exposure time of 8 s fractionated into 20 frames, a dose rate of 7.94 e− pixel−1 s−1 and a total dose of 56 e− Å−2 per movie.
Image processing
Movies were aligned using MOTIONCOR236 with 5 × 5 patches and applying a theoretical dose-weighting model to individual frames. Contrast transfer function (CTF) parameters were estimated using Gctf37. Resolution is reported on the basis of the gold-standard Fourier shell correlation (FSC) (0.143 criterion) as described38 and B-factors were determined and applied automatically in RELION 2.139,40. Particles from dataset one were automatically picked using Gautomatch (K. Zhang) and screened manually, and were then extracted in RELION with a 5,602 pixel box size and pre-processed. Particles from datasets two and three were picked and pre-processed in the same way, and were then rescaled to the pixel size of dataset one (1.13 Å pixel−1) in RELION 2.1 by Fourier cropping during particle extraction with a 5,602 pixel box. For rescaling, we first calculated 3D refinements in RELION 2.1 for each dataset (one to three) and performed real space correlation fits in UCSF Chimera to identify scaling factors for datasets two and three relative to dataset one. Because the absolute magnification values differed slightly for the different microscopes, we re-determined the CTF values for datasets two and three using the new pixel sizes with Gctf37, and then re-extracted and rescaled the particles to the 5,602 pixel box. Combining datasets one to three yielded a total dataset of 406,272 particles that were used for subsequent processing.
The first 22,319 particles from dataset one were used to generate an ab initio 3D reference for the A-complex using default parameters and three classes in cryoSPARC41 (Extended Data Fig. 2a). The complete dataset (one to three) was subjected to a ‘heterogeneous’ (multi-reference) refinement in cryoSPARC using default parameters and four classes: the ab initio A-complex reference and three ‘junk’ references (Extended Data Fig. 2a; round 1). Class one contained 153,570 particles (37.8%, percentage of particles form the full dataset) and was used for a 3D refinement in RELION 2.1 with a soft mask in the shape of the A-complex. This yielded a density (map A1) with an overall resolution of 4.9 Å and a B-factor of −188 Å2, comprising U1 snRNP and the U2 snRNP 3′ region (Extended Data Figs. 1e, d, 2, 9). To improve the U1 snRNP density, we prepared a soft mask enveloping the U1 snRNP with the volume eraser in UCSF Chimera42 and RELION 2.139,40. This enabled the focused refinement of the U1 snRNP (map A2) from the same 153,570 particles to an overall resolution of 4.0 Å and a B-factor of −146 Å2 (Extended Data Figs. 1e, d, 2, 9). In the A-complex the U2 snRNP 5′ region is flexible relative to the U1 and the U2 3′ region (Extended Data Fig. 2). To position the U2 snRNP 5′ region in the A-complex, we used a soft mask surrounding the U2 5′ region and carried out 3D classification without image alignment with six classes (Extended Data Fig. 2a; round 2). This revealed a class with defined U2 5′ region from 19,937 particles (4.9%) that could be refined to an overall resolution of 10.4 Å (Extended Data Figs. 2, 9). Local resolution was estimated using ResMap43 (Extended Data Fig. 2d, e).
Structural modelling
We prepared a composite model of the A-complex by combining the A1-3 densities (Extended Data Fig. 1e, f). Model building was carried out in COOT44. The U1 snRNP coordinates were refined into the sharpened A2 density in PHENIX45 using the phenix.real_space_refine routine, and applying secondary structure, rotamer, nucleic acid and metal ion restraints. Homology models for yeast Yhc1, Snp1, and Mud1 were generated using MODELLER46 from the human U1 snRNP crystal structures10 (PDB ID 4PJO, 4PKD) and were fitted and manually adjusted in the A2 map. The yeast B-complex U5 Sm ring model was used as the initial model for the U1 Sm ring, and was manually adjusted in the A2 density. Initial models for Prp39 and Prp42 were generated by I-TASSER47 and were subsequently adjusted and extended manually. The Prp39 N-terminal residues 47–339 were modelled as poly-alanine owing to a lower local resolution of ~5–6 Å (Extended Data Figs. 2d, e, 3c). Snu56, the Yhc1 C terminus, the Snu71 N terminus were modelled de novo; Yhc1 residues 48–82 and 135–142 were modelled as poly-alanine. To build the Luc7 model a C3H-type ZnF (from PDB ID 1RGO) for ZnF1 and a C2H2-type ZnF (from Yhc1) for ZnF2 were used to guide modelling in the A2 density, with a local resolution of 4–5 Å (Extended Data Fig. 3c). The helices connecting Luc7 ZnF1 and ZnF2 (α5–7) were modelled as poly-alanine, and were assigned on the basis of density connectivity. The U1 snRNP protein model is in excellent agreement with biochemical and protein crosslinking results12. The U1 snRNA model was generated on the basis of similarity to the U1 snRNA in the human U1 snRNP crystal structures (PDB ID 3CW1, 4PJO, 4PKD) and according to the yeast U1 snRNA secondary structure prediction48. All base-pairing U1 snRNA regions (helix H; SL1; SL2-1 and -2; SL3-1, -2, -3, -4, -5 and -6), except for the SL3-7 and the tip of SL3-3, were modelled (Extended Data Fig. 3f, g). The human SL1 loop (PDB ID 4PKD) was rigid-body-fitted together with the homology model of the yeast Snp1 (described above), and the human U1 snRNA sequence was replaced with that of yeast. The loops connecting SL2-1 to SL2-2 as well as SL3-3 to SL3-4 and SL3-4 to SL3-5 and the tips of SL2-2, SL3-3, -4 and -5 were not built, owing to a lower local resolution (~4.5 Å). The location of a region of U1 snRNA SL3-7 was modelled as a phosphate backbone only and may correspond to the sequence surrounding residues 378–391 and 428–440. The U1–5′SS was modelled de novo, and the UBC4 pre-mRNA contained 12 nucleotides, ten from the intron (+1 to +10) and two from the 5′ exon (−1 to −2).
The U2 snRNP 3′ region (U2 3′ domain and SF3a subcomplexes) from the yeast B-complex structure (PDB ID 5NRL) were fitted into the A1 density using UCSF Chimera42, and the positions of Lea1, Msl1 and U2 snRNA residues 139–1169 were adjusted as a rigid body in COOT44. The U2 snRNP 5′ region from the yeast B-complex structure (PDB ID 5NRL) was fitted into the A3 density in UCSF Chimera. This provided an excellent fit, suggesting that the U2 5′ region structure is not changed substantially from that observed in the yeast B-complex8. To generate the complete A-complex model, the refined U1 snRNP model and the U2 snRNP 3′ region were fitted into the A3 density in UCSF Chimera, together with the fitted U2 snRNP 5′ region. The final model comprises 34 proteins, U1 and U2 snRNAs, and the pre-mRNA substrate.
To generate the alternative pre-B-complex model shown in Fig. 3, we modified and combined structural models using COOT44, on the basis of structural and biochemical data from yeast and human systems8,25,28. We first superimposed our A-complex structure on the yeast B-complex structure8 using the U2 SF3b-containing domain. The free human tri-snRNP structure (PDB ID 3JCR), which may resemble the pre-B conformation7,25, was used to model the yeast tri-snRNP in the pre-B-complex conformation. We first removed the B-complex proteins from the yeast B-complex structure, because these are absent in the purified human pre-B-complex28. Human pre-B instead contained the PRP28 helicase and SAD1, and we therefore placed crystal structures of the yeast Prp28 helicase49 (PDB ID 4W7S) and yeast Sad150 (PDB ID 4MSX) in their human tri-snRNP locations25. We then positioned the U4 Sm ring and Brr2 as in the human tri-snRNP structure, in which the Brr2 PWI domain makes a conserved contact with Sad151. We removed a Snu66 peptide bound to Brr2 from the model, because its binding at this site is uncertain in the pre-B-complex conformation. Several minor differences remain between the free human tri-snRNP structure25 and the pre-B-complex model, and these were not modelled. The final pre-B model contained only minor clashes, and one observed clash between the highly flexible Prp28 RecA-2 lobe25 and the flexible U6 snRNA 5′ stem loop8,26 could be resolved by a minor repositioning of either domain. The final pre-B model comprises 66 proteins, five snRNAs, the pre-mRNA substrate, and has a combined molecular mass of ~3.1 MDa.
Figures were generated with PyMol (https://www.pymol.org) and UCSF Chimera.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Three-dimensional cryo-EM density maps A1, A2 and A3 have been deposited in the Electron Microscopy Data Bank under the accession numbers EMD-4363, EMD-4364 and EMD-4365, respectively. The coordinate file of the A-complex has been deposited in the Protein Data Bank under the accession number 6G90.
References
Will, C. L. & Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 3, a003707 (2011).
Papasaikas, P. & Valcárcel, J. The spliceosome: the ultimate RNA chaperone and sculptor. Trends Biochem. Sci. 41, 33–45 (2016).
Wilkinson, M. E., Lin, P. C., Plaschka, C. & Nagai, K. Cryo-EM studies of pre-mRNA splicing: from sample preparation to model visualization. Annu. Rev. Biophys. 47, 175–199 (2018).
Behzadnia, N. et al. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J. 26, 1737–1748 (2007).
Gao, G. & Dudley, S. C. Jr. RBM25/LUC7L3 function in cardiac sodium channel splicing regulation of human heart failure. Trends Cardiovasc. Med. 23, 5–8 (2013).
Hackman, P. et al. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann. Neurol. 73, 500–509 (2013).
Bertram, K. et al. Cryo-EM structure of a pre-catalytic human spliceosome primed for activation. Cell 170, 701–713.e711 (2017).
Plaschka, C., Lin, P. C. & Nagai, K. Structure of a pre-catalytic spliceosome. Nature 546, 617–621 (2017).
Liang, W. W. & Cheng, S. C. A novel mechanism for Prp5 function in prespliceosome formation and proofreading the branch site sequence. Genes Dev. 29, 81–93 (2015).
Kondo, Y., Oubridge, C., van Roon, A. M. & Nagai, K. Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5′ splice site recognition. eLife 4, e04986 (2015).
Neubauer, G. et al. Identification of the proteins of the yeast U1 small nuclear ribonucleoprotein complex by mass spectrometry. Proc. Natl Acad. Sci. USA 94, 385–390 (1997).
Li, X. et al. CryoEM structure of Saccharomyces cerevisiae U1 snRNP offers insight into alternative splicing. Nat. Commun. 8, 1035 (2017).
Puig, O., Bragado-Nilsson, E., Koski, T. & Séraphin, B. The U1 snRNP-associated factor Luc7p affects 5′ splice site selection in yeast and human. Nucleic Acids Res. 35, 5874–5885 (2007).
Agarwal, R., Schwer, B. & Shuman, S. Structure-function analysis and genetic interactions of the Luc7 subunit of the Saccharomyces cerevisiae U1 snRNP. RNA 22, 1302–1310 (2016).
Schwer, B. & Shuman, S. Structure–function analysis of the Yhc1 subunit of yeast U1 snRNP and genetic interactions of Yhc1 with Mud2, Nam8, Mud1, Tgs1, U1 snRNA, SmD3 and Prp28. Nucleic Acids Res. 42, 4697–4711 (2014).
Chen, J. Y. et al. Specific alterations of U1-C protein or U1 small nuclear RNA can eliminate the requirement of Prp28p, an essential DEAD box splicing factor. Mol. Cell 7, 227–232 (2001).
Puig, O., Gottschalk, A., Fabrizio, P. & Séraphin, B. Interaction of the U1 snRNP with nonconserved intronic sequences affects 5′ splice site selection. Genes Dev. 13, 569–580 (1999).
Förch, P., Puig, O., Martínez, C., Séraphin, B. & Valcárcel, J. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5′ splice sites. EMBO J. 21, 6882–6892 (2002).
Spingola, M. & Ares, M. Jr. A yeast intronic splicing enhancer and Nam8p are required for Mer1p-activated splicing. Mol. Cell 6, 329–338 (2000).
Agafonov, D. E. et al. ATPγS stalls splicing after B complex formation but prior to spliceosome activation. RNA 22, 1329–1337 (2016).
Fica, S. M. & Nagai, K. Cryo-electron microscopy snapshots of the spliceosome: structural insights into a dynamic ribonucleoprotein machine. Nat. Struct. Mol. Biol. 24, 791–799 (2017).
Abovich, N. & Rosbash, M. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell 89, 403–412 (1997).
Tang, Q. et al. SF3B1/Hsh155 HEAT motif mutations affect interaction with the spliceosomal ATPase Prp5, resulting in altered branch site selectivity in pre-mRNA splicing. Genes Dev. 30, 2710–2723 (2016).
Staley, J. P. & Guthrie, C. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein Prp28p. Mol. Cell 3, 55–64 (1999).
Agafonov, D. E. et al. Molecular architecture of the human U4/U6.U5 tri-snRNP. Science 351, 1416–1420 (2016).
Nguyen, T. H. D. et al. Cryo-EM structure of the yeast U4/U6.U5 tri-snRNP at 3.7 Å resolution. Nature 530, 298–302 (2016).
Huang, Y. H., Chung, C. S., Kao, D. I., Kao, T. C. & Cheng, S. C. Sad1 counteracts Brr2-mediated dissociation of U4/U6.U5 in tri-snRNP homeostasis. Mol. Cell. Biol. 34, 210–220 (2014).
Boesler, C. et al. A spliceosome intermediate with loosely associated tri-snRNP accumulates in the absence of Prp28 ATPase activity. Nat. Commun. 7, 11997 (2016).
Wan, R. et al. The 3.8 Å structure of the U4/U6.U5 tri-snRNP: Insights into spliceosome assembly and catalysis. Science 351, 466–475 (2016).
Ismaïli, N., Sha, M., Gustafson, E. H. & Konarska, M. M. The 100-kda U5 snRNP protein (hPrp28p) contacts the 5′ splice site through its ATPase site. RNA 7, 182–193 (2001).
Nguyen, T. H. D. et al. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature 523, 47–52 (2015).
Umen, J. G. & Guthrie, C. A novel role for a U5 snRNP protein in 3′ splice site selection. Genes Dev. 9, 855–868 (1995).
Galej, W. P. et al. Cryo-EM structure of the spliceosome immediately after branching. Nature 537, 197–201 (2016).
Wang, H., Ach, R. A. & Curry, B. Direct and sensitive miRNA profiling from low-input total RNA. RNA 13, 151–159 (2007).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Scheres, S. H. W. & Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012).
Kimanius, D., Forsberg, B. O., Scheres, S. H. & Lindahl, E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 5, e18722 (2016).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Webb, B. & Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 5.6.1–5.6.32 (2014).
Yang, J. et al. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015).
Kretzner, L., Krol, A. & Rosbash, M. Saccharomyces cerevisiae U1 small nuclear RNA secondary structure contains both universal and yeast-specific domains. Proc. Natl Acad. Sci. USA 87, 851–855 (1990).
Jacewicz, A., Schwer, B., Smith, P. & Shuman, S. Crystal structure, mutational analysis and RNA-dependent ATPase activity of the yeast DEAD-box pre-mRNA splicing factor Prp28. Nucleic Acids Res. 42, 12885–12898 (2014).
Hadjivassiliou, H., Rosenberg, O. S. & Guthrie, C. The crystal structure of S. cerevisiae Sad1, a catalytically inactive deubiquitinase that is broadly required for pre-mRNA splicing. RNA 20, 656–669 (2014).
Absmeier, E., Santos, K. F. & Wahl, M. C. Functions and regulation of the Brr2 RNA helicase during splicing. Cell Cycle 15, 3362–3377 (2016).
Ester, C. & Uetz, P. The FF domains of yeast U1 snRNP protein Prp40 mediate interactions with Luc7 and Snu71. BMC Biochem. 9, 29 (2008).
Kimura, E. et al. Serine-arginine-rich nuclear protein Luc7l regulates myogenesis in mice. Gene 341, 41–47 (2004).
Yan, C., Wan, R., Bai, R., Huang, G. & Shi, Y. Structure of a yeast activated spliceosome at 3.5 Å resolution. Science 353, 904–911 (2016).
Wilkinson, M. E. et al. Postcatalytic spliceosome structure reveals mechanism of 3′-splice site selection. Science 358, 1283–1288 (2017).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Buchan, D. W., Minneci, F., Nugent, T. C., Bryson, K. & Jones, D. T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357 (2013).
Acknowledgements
We thank C. Savva, S. Chen, G. Cannone, G. McMullan, J. Grimmett and T. Darling for maintaining electron microscopy and computing facilities; the mass spectrometry facility for protein identification; A. Murzin for discussions; E. Hesketh and R. Thompson for assistance with cryo-EM data collection of dataset three and S.-C. Cheng, A. Newman, L. Strittmatter, M. E. Wilkinson for critical reading of the manuscript. We thank J. Löwe, V. Ramakrishnan, D. Barford and R. Henderson for their continuing support. The project was supported by the Medical Research Council (MC_U105184330) and European Research Council Advanced Grant (693087-SPLICE3D). C.P. was supported by an EMBO Long-Term Fellowship (984-2015).
Author information
Authors and Affiliations
Contributions
C.P. and P.-C.L. established complex preparation, performed cryo-EM data analysis, model building, and refinement. C.C. assisted with cryo-EM analysis. C.P., P.-C.L. and K.N. analysed the structure and wrote the manuscript. C.P. and P.-C.L. designed the project and K.N. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Biochemical characterization and cryo-EM of the prespliceosome A-complex.
a, Mutation of the UBC4 pre-mRNA branch point sequence (UACUAAC to UACAAAC, in which A is the branch-point adenosine and A is the mutated nucleotide) stalls splicing before the first step, as described9. Splicing reactions were carried out for 30 min at 23 °C in yeast extract using wild-type (lane one) or mutant (U > A, lane two) pre-mRNA. This experiment was performed three times. The asterisk indicates a degradation product. For gel source data see Supplementary Fig. 1a. b, Protein analysis of purified A-complex (SDS–PAGE stained with Coomassie blue). The U2-associated Prp5 protein is sub-stoichiometric and not observed in the A-complex structure. The purification and analysis of protein compositions were performed at least five times with similar results. For gel source data see Supplementary Fig. 1b. c, Cryo-EM micrograph of the A-complex. Scale bar, 100 nm. d, 2D class averages of the A-complex were determined in RELION 2.139,40, and reveal a bipartite architecture, comprising the U1 snRNP and the U2 snRNP 3′ and 5′ regions, respectively. e, Composite cryo-EM density of the A-complex shown in two orthogonal views (compare to Fig. 1). The respective densities used for modelling the U1 snRNP (A2, grey), the U2 3′ region (A1, cyan), and the U2 5′ region (A3, green) are coloured and superimposed on a transparent outline of the full A3 map (Methods). The overall resolution of each map as well as the percentage from the cleaned dataset of 153,556 particles are shown in parentheses. Non-modelled regions are indicated and putatively assigned. f. Composite cryo-EM density with the final A-complex model superimposed in a cartoon representation. The path of 40 nucleotides of the disordered UBC4 pre-mRNA intron are indicated. A-complex components are coloured as in Fig. 1. Views as in e.
Extended Data Fig. 2 Cryo-EM image classification and refinement.
a, Image processing workflow for analysis of the A-complex cryo-EM dataset (see ‘Image processing’ in Methods). To visualize differences between the reconstructions, the U1 snRNP (grey), U2 3′ (cyan) and U2 5′ regions (green) are coloured. For each round of three-dimensional classification, the percentage of the data and the type of soft-edged mask are indicated. The type of mask and overall resolution are indicated for each 3D refinement (blue box). b, Orientation distribution plots for all particles that contribute to the respective A1, A2, and A3 cryo-EM reconstructions. c, Gold-standard Fourier shell correlation (FSC = 0.143) of the respective A1, A2 and A3 cryo-EM reconstructions. d, Two views of the composite A-complex cryo-EM density (maps A1, A2 and A3) coloured by local resolution as determined by ResMap43. e, As panel d, but for a central slice through the composite A-complex cryo-EM map.
Extended Data Fig. 3 Details of the U1 snRNP.
a, U1 snRNP structure with subunits coloured as in Fig. 1, except for Nam8 (orange), Snu56 (light blue), Snu71 (blue), Luc7 (dark purple), Mud1 (red) and the U1 snRNA (various). The pre-mRNA nucleotides are labelled relative to the first nucleotide (+1) of the intron. The Nam8 RRM1 and RRM2 domains are flexible and project downstream of the 5′SS. The protein attributed to Luc7 in the free U1 snRNP structure12 was re-assigned to Snu71. C-term, C terminus; N-term, N terminus; SL, stem loop. In the structure we do not observe any evidence that the C-terminal tails of SmB, SmD1, and SmD3 interact with the 5′SS, consistent with their absence in the human 5′SS–minimal U1 snRNP crystal structure10. b, Representative regions of the sharpened U1 snRNP density determined at 4 Å resolution (map A2) are superimposed on the refined coordinate model. The density reveals side-chain details, and here segments from the Prp42 N terminus (TPR repeat 1), the Sm ring subunit SmB, and the Snu56 α-helical domain are shown. c, The A2 cryo-EM density is shown superimposed on the coordinate models of a selection of U1 snRNP proteins: Luc7, Snu71, Yhc1 and Prp39. In the structure most of Snu71 is disordered, except for a small N-terminal domain (residues 2–43) that binds between the Prp42 N terminus and the Snu56 KH-like fold, consistent with protein crosslinking12. Functional regions and disordered domains are indicated. d, The U1 snRNA–pre-mRNA 5′ splice site (U1–5′SS) model is superimposed on its cryo-EM density (map A2). A secondary structure diagram of the U1–5′SS interaction is shown underneath the model. The register of the U1–5′SS is shifted by one nucleotide with respect to U1C (Yhc1) compared to the minimal human 5′SS–U1 snRNP crystal structure, owing to an additional nucleotide in the yeast U1 snRNA10 (U11). Lines indicate Watson–Crick base pairs and dots indicate pseudouridine (ψ)-containing base pairs. e, The Prp39–Prp42 heterodimer is coloured to indicate each of their respective TPR repeats. f, Cryo-EM density of U1 snRNA from maps A2 (dark grey) and A3 (light grey) without (top) and with the superimposed coordinate model of yeast U1 snRNA (bottom). The model is labelled and coloured according to functional regions of U1 snRNA (5′ end, pink; H helix, cyan; SL1, dark blue; SL2-1, green; SL3-1, light blue; SL2-2 and SL3-2 to -7, grey; 3′end and Sm site, yellow). g, Secondary-structure diagram of U1 snRNA. Bold letters indicate residues included in the model, lines indicate Watson–Crick base pairs, and dots G–U wobble and pseudouridine-containing base pairs. Compare to e. The conserved U1 snRNA ‘core’ is outlined with a grey box. The region of the putative phosphate backbone model of part of the U1 SL3-7 region is indicated with a grey box.
Extended Data Fig. 4 Comparisons of yeast and human U1 snRNPs and implications for alternative splicing.
a, Formation of the U1–5′SS helix induces stable binding of Luc7. In the absence of a pre-mRNA 5′SS in the free U1 snRNP density (left, EMD-8622), Luc7 and the U1 5′ end are disordered. Upon 5′SS recognition at the U1 5′ end (centre, map A2), Luc7 becomes ordered and stabilizes the U1–5′SS interaction, suggesting a mechanism for the selection of weak 5′SS sequences. The free U1 snRNP and the 5′SS-bound (map A2) cryo-EM densities are superimposed on the right. Although the long α-helical density next to Luc7 cannot be assigned with confidence, protein–protein crosslinking data12 and protein secondary structure prediction are consistent with the presence of either Prp40 or Snu71. On the basis of additional biochemical data on the interaction between the α-helical Prp40 FF1 domain and Luc7 ZnF252, we would speculate that the Prp40 FF1 domain is the most likely candidate for this density. b, Comparison of the yeast U1 snRNP ‘core’ with the human U1 snRNP crystal structure (PDB ID 3CW1). Protein and RNA (top) and RNA only (bottom) are shown side by side (left and centre) and superimposed by a global alignment in PyMOL (right). Coloured as in Extended Data Fig. 3a. c, The yeast U1 snRNP model suggests regulatory mechanisms for human alternative splicing factors. The human homologues of the peripheral yeast U1 proteins may function through stabilization of the U1–5′SS interaction (region 1), of the U1–U2 3′ region interface (region 2), or the U1–U2 5′ interface (region 3). The yeast U1 snRNP ‘core’ is shown superimposed on a surface representation of the U1 snRNP model (top), compared with the similarly coloured human U1 snRNP (below). Interaction sites with the U2 snRNP are labelled (top). d, The location of yeast U1 snRNP components with homology to human splicing factors are indicated in the U1 snRNP structure. The Prp39–Prp42 heterodimer (human PRPF39 homodimer), Nam818 (human TIA-1 and TIA-R), Luc753 (human LUC7L1–3), and the Yhc1 C terminus (human U1C) have clear counterparts in the human system. The yeast-specific U1 snRNA insertions may be replaced in the human system by alternative splicing factors that modulate interactions with the U2 5′ region. e, Model of the yeast E complex on the basis of the U1 snRNP structure and biochemical data22. Luc7, Snu71 and Prp40 form a heterotrimer in vitro52, and their interacting regions may be located near unassigned density (compare to Extended Data Fig. 1e) at the tip of an unassigned 40-residue α-helix next to Luc7 ZnF2. This helix is likely to belong to the U1 subunit Snu71 or Prp40, consistent with protein crosslinking12 and protein secondary-structure prediction. Prp40 could then bind the yeast branch point-binding protein (BBP, human SF1), which in turn interacts with Mud2 (human U2AF65) to tether the pre-mRNA branch-point sequence in the E complex22.
Extended Data Fig. 5 Conformational flexibility of the U2 snRNP.
a, Two defined positions of the U1 snRNP-U2 3′ region could be identified relative to the U2 5′ region. A-complex models were fitted into class two and four from round two of the 3D image classification (compare Extended Data Fig. 2a). The classes are aligned via their U2 5′ region, illustrating their relative flexibility. b, Cartoon schematic of observed positions of the U2 3′ region relative to the U2 5′ region in the A-complex (left), B-complex8 (centre), and activated B-complex (Bact) (right, modelled from previously published work54). Although in the B-complex the U2 3′ region is free, in the A- and Bact-complexes the position of the U2 3′ region is influenced by interactions with Prp39 as well as Syf1 and Clf1, respectively. c, The U2 snRNP subunit Lea1 (human U2A′) aids to position the U2 snRNP 3′ domain in different spliceosome states. In our A-complex structure, the Prp39 TPR repeat T1 contacts the helical C terminus of Lea1. In the yeast C-complex structure, the non-modelled density for the Syf1 N terminus binds a neighbouring but non-overlapping surface of Lea1 (PDB ID 5LJ5). In the C*/P-complex55 (PDB ID 6EXN), the Syf1 N terminus binds yet another Lea1 surface and the U2 3′ domain is repositioned relative to its C-complex location. Together, this suggests that the Lea1 provides multiple interfaces that can be used to position the U2 3′ domain in different spliceosomal complexes. d, Fit of the U2 3′ region coordinate model to the A1 cryo-EM density. The dashed black lineseparates the U2 3′ domain (Sm ring, Msl1 and Lea1 subunits and U2 snRNA, left) and the SF3a subcomplex (Prp9, Prp11 and Prp21, right). Two orthogonal views are shown (Supplementary Video 2). e, Fit of the U2 5′ region coordinate model to the A3 cryo-EM density. A density consistent with the U2 snRNA stem IIa/b and the branch helix is observed. Two density thresholds are shown side by side (left, 0.0163; right, 0.0121), and orthogonal views are shown underneath (Supplementary Video 2).
Extended Data Fig. 6 Luc7 and Nam8 sequence alignments.
a, The Luc7 (human LUC7-like) amino-acid sequence alignment comparing S. cerevisiae, Kluyveromyces lactis, Schizosaccharomyces pombe, Danio rerio, Xenopus tropicalis, Mus musculus, Bos taurus and Homo sapiens was generated with Clustal Omega and visualized with ESPript 356,57. For the human sequence, LUC7L1 was used. Secondary structure elements are indicated above the sequence and derive from the A-complex structure (purple) or PSIPRED58 secondary structure prediction (grey). Modelled regions (dashed line) and the Zn-coordinating residues of ZnF1 and ZnF2 (asterisks) are indicated. Invariant or conserved residues are highlighted with a red box or red letter font, respectively. b. As in panel a but for Nam8 (human TIA-1) comparing S. cerevisiae, K. lactis, S. pombe, Drosophila melanogaster, D. rerio, X. tropicalis, M. musculus, B. taurus, and H. sapiens amino acid sequences.
Extended Data Fig. 7 Details of the pre-B-complex model.
a, Multiple views of the pre-B-complex model, generated by combining functional and structural data from yeast and human systems8,25. The mobility of the U1 snRNP relative to the U2 snRNP in the A-complex (this study) as well as of the U2 snRNP relative to tri-snRNP in the B-complex structure8 are indicated (left). The pre-B model contained only minor clashes, and a clash between the highly flexible Prp28 C-terminal RecA-2 lobe (from the human tri-snRNP25) and the highly flexible U6 snRNA 5′ stem loop (from the yeast B-complex8) may be resolved by small movements of either domain. b, Structural comparisons of the yeast pre-B model (from this study) and the yeast B-complex structure (PDB ID 5NRL8) suggest the existence of a molecular checkpoint to couple 5′SS transfer to U1 snRNP release and formation of the activation-competent B-complex. In the pre-B model (left) Sad1 tethers Brr2 through its interaction with the conserved Brr2 PWI domain51, and the U1 snRNP and its U1–5′SS helix are positioned near the U6 ACAGAGA region and the helicase Prp28. Subsequent to Prp28-mediated 5′SS transfer, Brr2 is repositioned onto its U4 snRNA substrate, guided by the B-complex-specific proteins (right). In this conformation the Brr2 helicase and its associated factors would clash with the U1 snRNP, consistent with U1 snRNP destabilization and release yeast and human B-complexes7,8. Brr2 is now ready to initiate spliceosome activation and formation of the active site in the Bact-complex. Regions that are changed between pre-B- and B-complex models (black outline) and the clash between the Brr2-containing ‘helicase’ domain and the U1 snRNP in B-complex (red X) are indicated. The lower right panel would conform to the alternative ‘U1-B-complex’ model.
Extended Data Fig. 8 Model for early splicing events.
a, Cartoon schematic of proposed early splicing events, detailing (i) assembly of the pre-B-complex spliceosome from the A-complex and the U4/U6.U5 tri-snRNP and (ii) the subsequent conversion to the pre-catalytic B-complex spliceosome. In the pre-B model the mobile U1 snRNP is next to Prp28, which is bound at the Prp8N domain. To initiate 5′SS transfer, Prp28 could clamp the pre-mRNA at, or next to, the U1–5′SS helix to destabilize it and to hand over the 5′SS to the U6 ACAGAGA region of tri-snRNP, consistent with protein–RNA crosslinks30. Transfer of the 5′SS may induce the binding of the B-complex proteins to replace Prp28 at the Prp8N domain and induce the large movement of Brr2 to its B-complex location on U4 snRNA. The U1 snRNP, now loosely tethered to U2, may dissociate from the B-complex owing to the steric clash with the Brr2-containing ‘helicase’ domain8 (Extended Data Fig. 7b). Consistent with this, the human pre-B-complex converts to a B-complex-like state in the presence of a 5′SS oligonucleotide, which coincides with U1 snRNP release28. This model can explain how Brr2 is kept inactive to prevent premature U4/U6 duplex unwinding26. The model thereby implies the existence of a molecular checkpoint, coupling 5′SS transfer from U1 to U6 snRNA with Brr2 helicase repositioning and U1 snRNP release to generate the activation-competent B-complex spliceosome. b, Cartoon schematic of an alternative model for spliceosome assembly and 5′SS transfer that relies only on the yeast A-complex (from this work), tri-snRNP26,29 and B-complex structures8. In this model the tri-snRNP that associates with the A-complex already contains the Brr2 helicase bound to the U4 snRNA substrate and the yeast B-complex proteins at the Prp8 N-terminal domain. The tri-snRNP then binds the A-complex (transition I, ‘Assembly’), requiring a substantial readjustment to avoid a steric clash of the Brr2-containing ‘helicase’ domain and the U1 snRNP (‘U1-B-complex’). The Prp28 helicase is then recruited to the U1 snRNP directly as the Prp28-binding site on the Prp8 N-terminal domain in human tri-snRNP is occupied by B-complex proteins25. Prp28 then disrupts the U1–5′SS helix, leading to 5′SS transfer (transition II, ‘Transfer’). Similar to the ‘pre-B-complex’ assembly model in a, the U1 snRNP, now freed from the 5′SS, may then be released owing to a steric clash with the Brr2-containing ‘helicase’ domain. This model does not require Sad1. Compare to a.
Extended Data Fig. 9 Data collection, refinement statistics and validation.
a, Cryo-EM data collection and refinement statistics of the A-complex structure. Maps A1 and A3 were used to position the U2 snRNP 3′ and 5′ regions, respectively. b. FSC between the A2 cryo-EM density and the refined A-complex U1 snRNP coordinate model.
Supplementary information
Supplementary Figure 1
Uncropped gels shown in Extended Data Figure 1a (a) and 1b (b).
Supplementary Data
PyMol session of the A complex structure (PDB 6G90). Subunits are coloured as in Figure 1.
Video 1: 360° rotation of the A complex coordinate model (PDB 6G90).
Coloured as in Figure 1.
Video 2: 360° rotation of the composite A complex cryo-EM density.
A1, grey, EMD-4363; A2, cyan, EMD-4364; A3, green, EMD-4365. Coloured as in Extended Data Figure 1e.
Rights and permissions
About this article

Cite this article
Plaschka, C., Lin, PC., Charenton, C. et al. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature 559, 419–422 (2018). https://doi.org/10.1038/s41586-018-0323-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-018-0323-8
- Springer Nature Limited
This article is cited by
-
Structural insights into the cross-exon to cross-intron spliceosome switch
Nature (2024)
-
Structural basis of human U5 snRNP late biogenesis and recycling
Nature Structural & Molecular Biology (2024)
-
Intramolecular autoinhibition regulates the selectivity of PRPF40A tandem WW domains for proline-rich motifs
Nature Communications (2024)
-
Variation of C-terminal domain governs RNA polymerase II genomic locations and alternative splicing in eukaryotic transcription
Nature Communications (2024)
-
Co-transcriptional gene regulation in eukaryotes and prokaryotes
Nature Reviews Molecular Cell Biology (2024)