

Abstract
Autophagy increases the lifespan of model organisms; however, its role in promoting mammalian longevity is less well-established1,2. Here we report lifespan and healthspan extension in a mouse model with increased basal autophagy. To determine the effects of constitutively increased autophagy on mammalian health, we generated targeted mutant mice with a Phe121Ala mutation in beclin 1 (Becn1F121A/F121A) that decreases its interaction with the negative regulator BCL2. We demonstrate that the interaction between beclin 1 and BCL2 is disrupted in several tissues in Becn1F121A/F121A knock-in mice in association with higher levels of basal autophagic flux. Compared to wild-type littermates, the lifespan of both male and female knock-in mice is significantly increased. The healthspan of the knock-in mice also improves, as phenotypes such as age-related renal and cardiac pathological changes and spontaneous tumorigenesis are diminished. Moreover, mice deficient in the anti-ageing protein klotho3 have increased beclin 1 and BCL2 interaction and decreased autophagy. These phenotypes, along with premature lethality and infertility, are rescued by the beclin 1(F121A) mutation. Together, our data demonstrate that disruption of the beclin 1–BCL2 complex is an effective mechanism to increase autophagy, prevent premature ageing, improve healthspan and promote longevity in mammals.
Similar content being viewed by others

Main
Autophagy, an evolutionarily conserved lysosomal degradation pathway, has a key role in tissue homeostasis, health and disease4. In 2003, we showed that the Caenorhabditis elegans autophagy gene bec-1 (orthologue of yeast ATG6, mammalian beclin 1) was required for lifespan extension in nematodes with a loss of function in the insulin signalling pathway5. Subsequently, numerous loss-of-function studies in C. elegans and Drosophila have confirmed an essential role for the autophagy machinery in longevity1,2, and tissue-specific deletion of core autophagy genes have shown that autophagy delays age-related changes in mouse tissues, including kidney and heart6,7. Moreover, physiological inducers (such as caloric restriction) as well as pharmacological inducers (such as spermidine) of autophagy increase lifespan in mice1,8,9. Despite these clues that autophagy may be a longevity pathway in mammals, definitive evidence that increased basal autophagy extends mammalian healthspan and lifespan is lacking.
An earlier study10 demonstrated an increase in lifespan of mice that transgenically overexpress ATG5. However, it is unclear how overexpression of ATG5, a protein necessary for autophagy but not directly involved in the regulation of autophagy levels, results in increased autophagy. Moreover, ATG5 has other key functions, such as the regulation of inflammation11, and these roles are not shared by other genes in the autophagy pathway. Therefore, it is imperative to use a more direct and specific genetic approach to assess the effects of enhanced basal autophagy on mammalian lifespan and healthspan. To do so, we focused on the mammalian autophagy protein, beclin 1 (encoded by Becn1)12, which is part of an autophagy-specific class III phosphatidylinositol-3-OH kinase (PI3K) complex13 that has a key role in the regulation of the initiation of autophagosome formation14.
We recently reported the construction of mice with a Phe-to-Ala knock-in substitution mutation in the BH3 domain of beclin 1 (F121A; corresponding to F123A in human beclin 1)15 that decreases the binding of two negative regulators of autophagy (BCL2 and BCL-XL) to beclin 1 in vitro16,17. Using these mice, we performed co-immunoprecipitation of endogenous beclin 1 with BCL2 in muscle, heart, kidney and liver of two-month-old wild-type and homozygous knock-in mice. We observed a marked reduction in beclin 1 co-immunoprecipitation with BCL2 in the tissues of the knock-in mice (Fig. 1a, b). In parallel, we analysed autophagic flux by crossing wild-type or knock-in mice with animals that transgenically express green fluorescent protein (GFP)-tagged LC318, a fluorescent marker of autophagosomes. In skeletal muscle, heart, renal glomeruli, proximal convoluted tubules and liver, knock-in mice had significantly increased numbers of GFP–LC3 puncta compared to wild-type control littermates (Fig. 1c, d; Extended Data Fig. 1). In all tissues except for the liver, there was a further increase in GFP–LC3 puncta after treatment with chloroquine, an inhibitor of lysosomal acidification and autophagosome–lysosomal fusion, indicating that the increased numbers of GFP–LC3 puncta in knock-in mice represents a true increase in basal autophagic flux, rather than a block in autophagosomal maturation. We further confirmed that knock-in mice had increased autophagic flux by western blot analyses. Both hearts and kidneys had increased conversion of LC3-I to LC3-II (the lipidated, autophagosome-associated form of LC3), decreased levels of total LC3 and decreased levels of the autophagy substrate p6219 (Fig. 1e, f). Similar findings were also observed in the hearts and kidneys of six- to eight-month-old mice (Extended Data Fig. 2), indicating that the effects of the knock-in mutation are sustained over time in adulthood.

a, Co-immunoprecipitation of beclin 1 and BCL2 in indicated tissues from two-month-old Becn1+/+ (wild-type, WT) and Becn1F121A/F121A (knock-in, KI) animals. b, Quantification of beclin 1 co-immunoprecipitated with BCL2 in a. H, heart; K, kidney; L, liver; M, skeletal muscle. c, Representative images of GFP–LC3 puncta (autophagosomes) in indicated tissues from wild-type and knock-in mice that had been crossed with mice that transgenically express GFP–LC3, with or without 50 mg kg−1 chloroquine (CQ) for 6 h. Scale bars, 10 μm. (See Extended Data Fig. 1 for enlarged images.) White arrows denote representative GFP–LC3 puncta. d, Quantification of GFP–LC3 puncta with or without chloroquine in indicated tissues. e, Western blot analysis of autophagy markers in the hearts and kidneys of two-month-old wild-type and knock-in mice. f, Quantification of p62 and total LC3 levels (normalized to β-actin) and LC3-II/LC3-I ratio in e. Data are mean ± s.e.m. for three mice per genotype. In a and e, each lane represents a different mouse. P values were determined by a one-sided unpaired t-test. PCT, renal proximal convoluted tubules; WCL, whole cell lysate. For uncropped gels, see Supplementary Fig. 1.
We further evaluated the effect of the knock-in mutation on autophagy using mouse embryonic fibroblasts (MEFs) derived from knock-in or wild-type littermate controls. In knock-in MEFs, there was decreased co-immunoprecipitation of beclin 1 with BCL2, increased numbers of GFP–LC3 puncta, decreased levels of p62 and total LC3, and increased numbers of autophagic structures (autophagosomes and autolysosomes) observed by quantitative electron microscopy (Extended Data Fig. 3a–d). Treatment with the lysosomal inhibitor bafilomycin A1 indicated a bona fide increase in autophagic flux in knock-in MEFs. To evaluate possible effects of the beclin 1 knock-in mutation other than autophagy, we examined endocytosis, a process thought to involve the beclin 1–VPS34 complex20. We did not observe any significant differences in endocytosis between knock-in and wild-type MEFs, as measured by the kinetics of endocytic uptake of fluorescent transferrin (Extended Data Fig. 3e). This is expected, as BCL2 binding to beclin 1 has not been shown to regulate beclin 1-dependent functions other than autophagy.
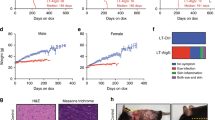
Taken together, these findings demonstrate that the Becn1F121A/F121A knock-in mouse model is useful for studying the effects of constitutively increased basal autophagy on mammalian lifespan and healthspan. Therefore, we evaluated the lifespan of a large cohort of beclin 1 knock-in and wild-type littermates (Fig. 2) on an inbred C57BL/6J background. The combined data for males and females showed a significant lifespan extension (Fig. 2a) of knock-in mice compared to wild-type littermate controls (median survival: 26 months, wild type; 29 months, knock-in). This extension in longevity was observed both in females (median survival: 27 months, wild type; 30 months, knock-in) and males (median survival: 25 months, wild type; 28 months, knock-in) (Fig. 2b, c; Extended Data Table 1), showing a sex-independent effect of the beclin 1(F121A) mutation on lifespan. Knock-in mice also had an increase in maximal lifespan (Extended Data Table 1). Thus, this gain-of-function mutation in a core autophagy gene, Becn1, extends mammalian lifespan.

a–c, Kaplan–Meier survival curves for Becn1+/+ wild-type and Becn1F121A/F121A knock-in mice, showing the lifespan of all mice in the cohort (a), females alone (b) or males alone (c). n denotes the number of mice per group. P values were determined by log-rank (Mantel–Cox) test.
Next, we evaluated age-related phenotypes in two vital organs, kidney and heart, where we showed that the beclin 1(F121A) mutation disrupted beclin 1–BCL2 binding and increased basal autophagy. During ageing, the kidney develops pathological changes, including fibrosis and nuclear damage, and proximal convoluted tubule-specific deletion of the autophagy gene, Atg5, exacerbates these phenotypes6,21. In 20-month-old knock-in mice and wild-type control littermates, the proximal convoluted tubules had increased vacuolar changes in the wild-type mice as compared to the knock-in mice (Fig. 3a). In this region, there was increased nuclear DNA damage, as measured by increased TUNEL staining of non-pyknotic/apoptotic-appearing nuclei (Fig. 3b) that did not stain positive for the apoptotic marker, active caspase 3 (Extended Data Fig. 4a). Consistent with this evidence of decreased cellular damage in the proximal convoluted tubules of knock-in mice, there was also decreased fibrosis (Fig. 3c). Similarly, we observed increased non-pyknotic/apoptotic-appearing, active caspase 3-negative, TUNEL-positive nuclei in the hearts of 20-month-old wild-type as compared to knock-in mice (Fig. 3e; Extended Data Fig. 4b). This phenotype was most pronounced in the subendocardial region of the left ventricle (the part of the myocardium subjected to greatest haemodynamic stress), and was accompanied by an increase in the cardiomyocyte cross-sectional area in the wild-type mice (Fig. 3f). Moreover, there was a significant increase in fibrosis, a hallmark of cardiac ageing, in wild-type versus knock-in mice (Fig. 3g). In young (two-month-old) mice, no genotype-specific differences were observed in any of the renal and cardiac parameters assessed, indicating that the decreased pathology in older knock-in mice genuinely reflects a reduction in age-related changes (rather than developmental differences) in these organs. Thus, the beclin 1(F121A) knock-in mutation decreases age-related renal and cardiac changes, including fibrosis.

a–d, Representative images and quantification of pathological score (a), TUNEL-positive nuclei (b), interstitial fibrosis (c), and endogenous LC3 puncta (autophagosomes) (see enlarged images in Extended Data Fig. 4c) (d) in the cortical region of the kidney. e–h, Representative images and quantification of TUNEL-positive nuclei (e), cardiomyocyte cross-sectional fibre size (f), cardiac interstitial fibrosis (g), and endogenous LC3 puncta (see enlarged images in Extended Data Fig. 4d) (h) in the heart. Two-month-old wild-type and knock-in mouse kidneys and hearts (n = 6 per genotype) were analysed. For kidney analyses, 20-month-old wild-type (n = 20 and n = 16) and knock-in (n = 26 and n = 19) mice were used for histopathological and autophagy analyses, respectively. For heart analyses, 20-month-old wild-type (n = 19 and n = 15) and knock-in (n = 22 and n = 19) mice were used for histopathological and autophagy analyses, respectively. Scatter plot bars represent median ± interquartile ranges. P values were determined by Mann–Whitney test. Autophagy analyses were one-sided and all other analyses were two-sided. i, j, Percentage of wild-type and knock-in mice (aged 20 months) with spontaneous tumours, including lymphoproliferative disease (LPD), lymphomas, and non-lymphoid malignancies (i) and representative images of the most frequently observed neoplastic lesions from wild-type mice (j). See text for statistical analyses of data. Scale bars, 50 µm (a–c, e–g, j) and 10 µm (d, h). In d and h, red arrows denote representative endogenous LC3 puncta.
In this cohort of 20-month-old mice, autophagosome numbers, as measured by endogenous staining of LC3, were markedly decreased in the kidneys and hearts of both wild-type and knock-in mice (Fig. 3d, h; Extended Data Fig. 4c, d), consistent with a predicted age-related decline in autophagic capacity2,22. However, the knock-in mouse kidneys and hearts still had more autophagosomes than their wild-type counterparts. Taken together, these data demonstrate that the beclin 1(F121A) knock-in mutation delays, but does not prevent, age-related decline in mouse autophagic function in parallel with observed decreases in age-related organ pathology. This finding is predicted, as downstream autophagy gene expression decreases with ageing2,22; thus, decreased negative regulation of beclin 1 per se is not sufficient to reverse age-related decline in autophagosome formation.
In mice and humans, ageing results in increased susceptibility to a variety of cancers. Specifically, mice with a C57BL/6J genetic background display age-related increases in lymphoproliferative disease (LPD) and lymphomas, histiocytic sarcomas, lung cancers and liver cancers23. In a cohort of 20-month-old knock-in mice and wild-type littermates, there was a significant decrease in age-related spontaneous tumorigenesis in the knock-in mice (Fig. 3i, j), either when comparing all malignancies (P = 0.034, chi-square) or non-lymphoid malignancies alone (P = 0.024, chi-square). Thus, beclin 1(F121A) knock-in mice with increased basal autophagy have a decreased incidence of age-related spontaneous cancer. The similar prevalence of LPD in both genotypes, coupled with increased prevalence of lymphomas in wild-type mice, is consistent with delayed onset of LPD and/or progression of LPDs to frank lymphoma in knock-in animals.
The central role of beclin 1–BCL2 disruption in promoting increased basal autophagy, lifespan extension and improved healthspan raised the question of whether known anti-ageing factors might exert their longevity-promoting activity by disrupting beclin 1–BCL2 binding and autophagy. Klotho is a single-pass membrane-bound protein that can be cleaved and released into the circulation; it is highly expressed in the kidney, and it regulates ageing24. Klotho expression decreases with ageing in mice and humans24. Klotho deficiency in mice, either via hypomorphic expression or gene knockout, results in a syndrome resembling (but not completely recapitulating) ageing3,25, including premature lethality (death within a few months of birth), infertility and multisystem defects. Klotho overexpression or administration of soluble klotho protein extends lifespan and rescues most phenotypes in klotho hypomorphic mice26,27. Moreover, soluble klotho promotes autophagic flux in the kidney and, in the setting of renal ischaemic injury, mitigates renal fibrosis and retards progression to end-stage renal disease28.
Therefore, we crossed the Becn1F121A/F121A mice with a well-characterized premature ageing model in which animals harbour a hypomorphic mutation in the klotho gene3 (kl/kl mice; termed here KlHM/HM). Klotho deficiency resulted in a marked increase in beclin 1 co-immunoprecipitation with BCL2 in the kidney (Fig. 4a, b), in support of the concept that disruption of beclin 1 and BCL2 binding may have a mechanistic role in klotho-induced autophagy. Consistent with this, recombinant soluble klotho decreased beclin 1 and BCL2 binding in cultured HeLa cells (Extended Data Fig. 5). The beclin 1(F121A) mutation decreased beclin 1 and BCL2 binding in KlHM/HM kidney to levels similar to those observed in wild-type mice without discernible effects on klotho protein expression (Fig. 4a, b). In parallel, the beclin 1(F121A) mutation restored levels of autophagy in KlHM/HM mice to those observed in wild-type mice, as measured by LC3-II conversion and p62 degradation (Fig. 4c, d). Notably, this reversal of the enhanced beclin 1–BCL2 binding and decreased autophagy in the beclin 1 knock-in/KlHM/HM double-mutant mice nearly completely rescued the premature lethality phenotype of both female and male mice (Fig. 4e–g); 100% of the KlHM/HM mice were dead by approximately 12 weeks, whereas most of the double-mutant beclin 1 knock-in and kl/kl mice (Becn1F121A/F121AKlHM/HM) survived a 45-week observation period. Furthermore, both female and male infertility was rescued in the double-mutant (Becn1F121A/F121AKlHM/HM) mice. Finally, the severe growth retardation of KlHM/HM mice was almost completely reversed by the beclin 1(F121A) mutation (Fig. 4h, i). Taken together, these data indicate that a gain-of-function mutation in beclin 1, which disrupts beclin 1–BCL2 binding and increases autophagy, reverses the premature ageing phenotypes resulting from klotho deficiency.

a, Co-immunoprecipitation of beclin 1 and BCL2 in the kidneys from mice of indicated genotype (n = 3 animals per genotype). b, Quantification of beclin 1 co-immunoprecipitated with BCL2 in a. c, Western blot analysis of autophagy markers in kidney from mice of indicated genotype. d, Quantification of western blot analysis of p62 levels and LC3-II/LC3-I ratios (combined results are for six mice per genotype; each independent experiment had one mouse per genotype). Data are mean ± s.e.m. e–g, Kaplan–Meier survival curves for mice of indicated genotype, showing the lifespan of all the mice in the cohort (e), females alone (f) or males alone (g). n denotes the number of mice per group. h, Representative images of ten-week-old mice of indicated genotype for body size comparison. Scale bars, 1 cm. i, Serial body weights in female and male mice of indicated genotype. Data are mean ± s.e.m. for 12 mice per genotype. P values were determined by one-sided unpaired t-test (b, d) or log-rank Mantel–Cox test (e–g). HM, KlHM/HM (hypomorphic mutation) mice; KI/HM, Becn1F121A/F121AKlHM/HM mice. For uncropped gels, see Supplementary Fig. 1.
Our findings demonstrate that a knock-in gain-of-function point mutation in a key autophagy regulatory gene, Becn1, in mice results in increased basal levels of autophagy, extends lifespan in both males and females, and improves healthspan, including a decrease in age-related renal and cardiac changes and age-related spontaneous malignancies. Importantly, the decreased beclin 1–BCL2 binding in vivo is not only associated with longevity and improved ageing-related phenotypes, but it also has a marked effect on rescuing the early lethality and infertility of mice deficient in a well-established anti-ageing protein, klotho. Taken together, our results suggest that activation of the beclin 1 class III PI3K autophagy-initiating complex may be an effective and safe way to bypass upstream ageing signals to promote mammalian healthspan and lifespan. Specifically, we propose that disruption of beclin 1–BCL2 binding may be one such strategy, as our findings provide genetic proof that chronic in vivo disruption of this complex exerts substantial beneficial effects on mammalian lifespan and healthspan. More speculatively, it is possible that disruption of the beclin 1–BCL2 complex, and ensuant autophagy induction, underlies the anti-ageing mechanism of klotho and perhaps other longevity signalling pathways.
Methods
Cell culture
HeLa cells were obtained from the ATCC (American Type Culture Collection), tested negative for mycoplasma by PCR and authenticated by ATCC Cell Line Authentication Service. Primary MEFs were isolated from mouse embryos at embryonic day (E) 13.5 and cultured as described29.
Mice
Becn1F121A/F121A knock-in mice were generated as described15 and backcrossed for more than 12 generations to C57BL/6J mice (Jackson Laboratories). Becn1+/+ (wild-type) and Becn1F121A/F121A (knock-in) littermate mice were crossed with GFP–LC3 transgenic animals18 in a pure C57BL/6J background and tissues of offspring were used for autophagic flux analyses. Klotho hypomorphic mice (best known as kl/kl; referred to as KlHM/HM in this manuscript) have been previously described3 and were crossed with Becn1+/+ (wild-type) and Becn1F121A/F121A (knock-in) mice to obtain double mutants. Mice were genotyped for the Kl and Becn1 alleles as described3,15. All mice were housed on a 12-h light/dark cycle and both males and females were used for all analyses. For sample size and randomization information, please see the Life Sciences Reporting Summary. For survival analysis, mice were monitored weekly for the duration of the observation period. For western blot autophagy analyses, animals were starved overnight and re-fed for 3 h before sample collection to minimize variability due to differences in food intake. All animal procedures were performed in accordance with institutional guidelines and with approval from the UT Southwestern Medical Center Institutional Animal Care and Use Committee.
Immunoprecipitations
For beclin 1–BCL2 co-immunoprecipitation, frozen tissues were weighed and homogenized in ice-cold lysis buffer (25 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100; 1 ml per 100 mg tissue) containing cOmplete, mini protease (Roche) and Halt phosphatase (Thermo Scientific) inhibitor cocktails for 30 min at 4 °C. Lysates were centrifuged (16,000g at 4 °C for 30 min) and the supernatants were pre-cleared with 60 μl protein-G agarose beads for 2 h and incubated overnight with BCL2-agarose (or IgG) antibody. Immunoprecipitates were washed five times, resuspended in 2× SDS–PAGE loading buffer, boiled for 5 min and analysed by western blot using anti-beclin 1 (sc-7382, Santa Cruz; 1:500 dilution), anti-BCL2 (sc-7382, Santa Cruz; 1:100 dilution), anti-klotho (KO603, Trans Genic Inc. Japan; 1:1,000 dilution) and anti-actin (sc-47778, Santa Cruz, 1:5,000 dilution) antibodies.
For in vitro analyses of the effects of klotho on beclin 1 and BCL2 binding, soluble full-length mouse klotho protein was purified as previously described27, and HeLa (ATCC) cells were cells were treated either with PBS or klotho protein (0.4 nM or 2.0 nM) for 24 h before co-immunoprecipitation using the same protocol described above for mouse tissues.
Autophagy analyses
To assess in vivo autophagy levels in different tissues, two- and six-month-old homozygous Becn1F121A/F121A;GFP–LC3 or Becn1+/+;GFP–LC3 mice (three mice per group) were treated with either PBS or chloroquine (50 mg kg−1) for 6 h. Mice were then perfused with 4% paraformaldehyde (PFA) in PBS and tissues were collected and processed for frozen sectioning as described18. The total number of GFP–LC3 puncta was counted per 2,500 μm2 area (more than 15 randomly chosen fields were used per mouse) and the average value for each tissue for each mouse was determined by an observer blinded to genotype. The mouse muscle, heart, and liver tissue sections were imaged using a 63× objective and for renal glomeruli and proximal convoluted tubules, tissue sections were imaged using a 40× objective on a Zeiss AxioPlan 2 microscope.
For western blot analysis, frozen tissues were lysed in ice-cold lysis buffer (Tris-HCl, pH 7.5, 50 mM, NaCl 150 mM, 1 mM EDTA, 1% Triton X-100) with cOmplete, mini protease (Roche) and Halt phosphatase (Thermo Scientific) inhibitor cocktails for 30 min at 4 °C. Lysates were centrifuged at 16,000g for 30 min. Cleared lysates were diluted in 2× SDS–PAGE loading buffer and analysed using anti-p62 (GP62-C, Progen, 1:1,000 dilution), anti-LC3B (L7543, Sigma, 1:10,000 dilution) and anti-actin (sc-47778, Santa Cruz, 1:5,000 dilution) antibodies. Endogenous LC3 immunofluorescence staining of paraffin-embedded tissues was performed as previously described30.
Electron microscopic analyses of MEFs derived from wild-type and knock-in littermate mice was performed and analysed as described16.
Measurement of endocytosis
Analysis of transferrin uptake by wild-type and knock-in MEFs as an indicator of endocytosis activity was performed as previously described31,32.
Histopathological analyses
Wild-type and knock-in control littermates aged two months and 20 months were perfused with 4% PFA in PBS before tissue collection, fixation, and preparation of paraffin-embedded sections for histopathological analyses. Standard haematoxylin and eosin (H&E) staining was performed for analyses of age-related neoplasia and renal histopathological score. H&E-stained tissues sections of several organs were evaluated per mouse in a genotype-blinded manner by a pathologist with expertise in the diagnosis of human and murine neoplasms and all cases of lymphoproliferative disease and non-lymphoid malignancies were recorded. TUNEL staining was performed according to manufacturer’s instructions (ApopTag Peroxidase In situ Apoptosis Detection Kit, Millipore) and active caspase 3 staining was performed using anti-active caspase 3 (ab3202, Abcam, 1:20 dilution) antibody overnight at 4 °C and an ABC kit according to manufacturer’s instructions. The total numbers of TUNEL-positive nuclei and active caspase 3-positive cells were counted in sections of the entire renal cortex of each mouse to calculate the number of TUNEL-positive nuclei and active caspase 3-positive cells per unit area, and the total numbers of TUNEL-positive nuclei and active caspase 3-positive cells were counted in each longitudinal section of the heart to calculate the number of TUNEL-positive nuclei and active caspase 3-positive cells per unit area. To determine the renal pathological score, ten random fields of the renal cortex sections were evaluated per mouse. Each field was given a pathological score using the following scale: 0, absence of damage; 1, <15% tissue area with damage; 2, 15–50% tissue area with damage; 3, >50% tissue area with damage. The scores of each field were summed to give a final histolopathological score for each mouse, ranging from 0 to 30. Wheat germ agglutinin (WGA) staining was performed by pre-incubating slides in citrate buffer at 50 °C for 13 min, blocking with 1% bovine serum albumin (BSA), 5% goat serum in PBS for 1 h, and staining with Alexa Fluor 594-WGA (W11262, ThermoFisher Scientific, 1:100 dilution) for 1 h at room temperature. The average cross-sectional size of cardiomyocytes was determined by ImageJ; 100 cells were analysed per mouse (using 20 cells per field and acquiring 5 fields per heart). Masson’s trichrome staining was performed according to the manufacturer’s instructions (ab150686, Abcam) and used for analyses of renal cortex and cardiac fibrosis. The percentage area of tissue with fibrosis was measured. The fibrotic areas and total field size were manually outlined and quantified using ImageJ for 10 randomly chosen fields for the renal cortex per mouse and 5 randomly chosen fields for the heart per mouse. Quantification of all histopathological analyses was performed by an observer blinded to experimental age and genotype.
Statistical analyses
Data were analysed using the GraphPad Prism 7 and OASIS 2 software. log-rank (Mantel–Cox) tests were used to analyse Kaplan–Meier curves, and a Booshloo’s test for maximum lifespan analysis (at 90% survival) was performed as described33. Chi-square test was used to compare the percentage of mice with spontaneous malignancies. One-tailed unpaired Student’s t-tests were used for analyses of beclin 1–BCL2 binding and autophagy in wild-type versus knock-in mice. For data with a non-normal distribution, the ROUT (robust regression and outlier removal) method34 was used to eliminate outliers with Q coefficient set at 1% and data were analysed by two-tailed Mann–Whitney tests.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Full scans for all western blots are provided in Supplementary Fig. 1. Source data for all graphs in this manuscript have been provided. All other data are available from the corresponding authors on reasonable request.
Change history
19 June 2018
In this Letter, the graphs in Fig. 2a and c were inadvertently the same owing to a copy and paste error from the original graphs in Prism. The Source Data files containing the raw data were correct. Fig. 2c has been corrected online.
References
Madeo, F., Zimmermann, A., Maiuri, M. C. & Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Invest. 125, 85–93 (2015).
Rubinsztein, D. C., Mariño, G. & Kroemer, G. Autophagy and aging. Cell 146, 682–695 (2011).
Kuro-o, M. et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51 (1997).
Levine, B. & Kroemer, G. Autophagy in the pathogenesis of disease. Cell 132, 27–42 (2008).
Meléndez, A. et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387–1391 (2003).
Yamamoto, T. et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 12, 801–813 (2016).
Eisenberg, T. et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 22, 1428–1438 (2016).
Eisenberg, T. et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314 (2009).
Mercken, E. M. et al. SIRT1 but not its increased expression is essential for lifespan extension in caloric-restricted mice. Aging Cell 13, 193–196 (2014).
Pyo, J. O. et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 4, 2300 (2013).
Kimmey, J. M. et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528, 565–569 (2015).
Liang, X. H. et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676 (1999).
Kihara, A., Kabeya, Y., Ohsumi, Y. & Yoshimori, T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2, 330–335 (2001).
Levine, B., Liu, R., Dong, X. & Zhong, Q. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 25, 533–544 (2015).
Rocchi, A. et al. A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease. PLoS Genet. 13, e1006962 (2017).
Pattingre, S. et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939 (2005).
Sinha, S., Colbert, C. L., Becker, N., Wei, Y. & Levine, B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the γ-herpesvirus 68 Bcl-2 homolog M11. Autophagy 4, 989–997 (2008).
Mizushima, N., Yamamoto, A., Matsui, M., Yoshimori, T. & Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111 (2004).
Mizushima, N., Yoshimori, T. & Levine, B. Methods in mammalian autophagy research. Cell 140, 313–326 (2010).
McKnight, N. C. et al. Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS Genet. 10, e1004626 (2014).
Lim, J. H. et al. Age-associated molecular changes in the kidney in aged mice. Oxid. Med. Cell. Longev. 2012, 171383 (2012).
Lapierre, L. R., Kumsta, C., Sandri, M., Ballabio, A. & Hansen, M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 11, 867–880 (2015).
Brayton, C. in The Mouse in Biomedical Research Vol. 2 (eds Fox, J. et al.), 623–717 (Elsevier, Amsterdam, 2007).
Kuro-o, M. Klotho and aging. Biochim. Biophys. Acta 1790, 1049–1058 (2009).
Tsujikawa, H., Kurotaki, Y., Fujimori, T., Fukuda, K. & Nabeshima, Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol. Endocrinol. 17, 2393–2403 (2003).
Kurosu, H. et al. Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833 (2005).
Chen, T. H. et al. The secreted Klotho protein restores phosphate retention and suppresses accelerated aging in Klotho mutant mice. Eur. J. Pharmacol. 698, 67–73 (2013).
Shi, M. et al. αKlotho mitigates progression of AKI to CKD through activation of autophagy. J. Am. Soc. Nephrol. 27, 2331–2345 (2016).
Su, T. et al. Deletion of histidine triad nucleotide-binding protein 1/PKC-interacting protein in mice enhances cell growth and carcinogenesis. Proc. Natl Acad. Sci. USA 100, 7824–7829 (2003).
Sebti, S. et al. BAT3 modulates p300-dependent acetylation of p53 and autophagy-related protein 7 (ATG7) during autophagy. Proc. Natl Acad. Sci. USA 111, 4115–4120 (2014).
Fielding, A. B., Willox, A. K., Okeke, E. & Royle, S. J. Clathrin-mediated endocytosis is inhibited during mitosis. Proc. Natl Acad. Sci. USA 109, 6572–6577 (2012).
Tacheva-Grigorova, S. K., Santos, A. J., Boucrot, E. & Kirchhausen, T. Clathrin-mediated endocytosis persists during unperturbed mitosis. Cell Reports 4, 659–668 (2013).
Wang, C., Li, Q., Redden, D. T., Weindruch, R. & Allison, D. B. Statistical methods for testing effects on “maximum lifespan”. Mech. Ageing Dev. 125, 629–632 (2004).
Motulsky, H. J. & Brown, R. E. Detecting outliers when fitting data with nonlinear regression — a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 7, 123 (2006).
Acknowledgements
We thank S. Sciarretta for discussions; N. Mizushima for reagents; L. Nguyen for technical assistance and H. Smith for assistance with manuscript preparation. This work was supported by NIH grants RO1-CA109618 (B.L.), U19-AI199725 (B.L.), RO1-DK091392 and RO1-DK092461 (M.C.H. and O.W.M.), P30-DK07938 (O.W.M.), K99R00-DK094980 (C.H.), Cancer Prevention Research Institute of Texas grant RP120718 (B.L.) and a Fondation Leducq grant 15CBD04 (B.L., A.F.F. and S.S.).
Reviewer information
Nature thanks D. Harrison and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
A.F.F., S.S., Y.W., C.H., T.T., Y.L., M.C.H. and B.L. designed the study. A.F.F., S.S., Y.W., Z.Z., M.S., K.L.M. and W.-C.C. performed biochemical analyses. A.F.F., S.S. and Z.Z. performed autophagy microscopic analyses. A.F.F. and S.S. performed renal and cardiac histopathological analyses. G.B. characterized malignancies. A.F.F., S.S., D.K.M., G.G.S, G.B., O.W.M., M.C.H. and B.L. discussed and analysed data. A.F.F., S.S., M.C.H. and B.L. wrote the manuscript. A.F.F. and S.S. contributed equally and the order of these authors was determined arbitrarily.
Corresponding authors
Ethics declarations
Competing interests
B.L. is a Scientific Founder of Casma Therapeutics, Inc.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Increased basal autophagy in tissues of beclin 1(F121A) knock-in mice.
a, Representative images and quantification of GFP–LC3 puncta (autophagosomes) in glomeruli from Becn1+/+;GFP-LC3 (wild-type, WT) and Becn1F121A/F121A;GFP-LC3 (knock-in, KI) mice with or without 50 mg kg−1 chloroquine for 6 h. Scale bars, 10 μm. Data are mean ± s.e.m. for three mice per genotype. b, Enlarged versions of the images shown in Fig. 1c. P values determined by one-sided unpaired t-test. Arrows denote representative GFP–LC3 puncta.
Extended Data Fig. 2 Sustained increase in basal autophagy during adulthood in beclin 1(F121A) knock-in mice.
a, Co-immunoprecipitation of beclin 1 and BCL2 in representative samples of hearts and kidneys from eight-month-old Becn1+/+ (wild-type) and Becn1F121A/F121A (knock-in) animals. b, Quantification of beclin 1 co-immunoprecipitated with BCL2 in indicated tissues of eight-month-old wild-type and knock-in mice (n = 3 mice per genotype). c, Quantification of GFP–LC3 puncta in hearts and tissues from six-month-old Becn1+/+;GFP-LC3 (wild-type) and Becn1F121A/F121A;GFP-LC3 (knock-in) mice with or without chloroquine (50 mg kg−1, 6 h). d, Western blot analysis of autophagy markers in the hearts and kidneys from eight-month-old wild-type and knock-in mice. Each lane represents a different mouse. e, Quantification of p62 and total LC3 levels (normalized to β-actin), as well as LC3-II/LC3-I ratios from samples in d. Data are mean ± s.e.m. for three mice per genotype. P values were determined by one-sided unpaired t-test. For uncropped gels, see Supplementary Fig. 1.
Extended Data Fig. 3 Increased autophagy, but not endocytosis, in beclin 1(F121A) MEFs.
a, Co-immunoprecipitation of beclin 1 and BCL2 in MEFs derived from Becn1+/+ (wild-type) and Becn1F121A/F121A (knock-in) animals. b, Representative images (top) and quantification (bottom) of GFP–LC3 puncta in wild-type and knock-in cells with or without 10 nM bafilomycin A1 (BafA1) for 3 h. Scale bars, 10 µm. c, Western blot analysis of autophagy markers in wild-type and knock-in MEFs with or without BafA1 (100 nM, 2 h). d, Representative images and quantitative electron microscopic analysis of autophagic structures in wild-type and knock-in MEFs with or without BafA1 (100 nM, 3 h). Insets show representative autophagosome (arrowhead) and autolysosome (arrow). Scale bars, 1 µm. e, Representative images and quantification of transferrin uptake kinetics in wild-type and knock-in cells. Scale bars, 20 µm. Results shown are representative of two and four independent experiments respectively for a and c. Data are mean ± s.e.m. for three replicates in b and for 50 cells per genotype and condition in d. Data points on line graph in e denote mean ± s.e.m. for cells at 7 min (n = 63, WT; n = 54, KI), 15 min (n = 58, WT; n = 52 KI), and 30 min (n = 76, WT; n = 83, KI). P values determined by unpaired one-sided (b) and two-sided (d, e) t-test. For uncropped gels, see Supplementary Fig. 1.
Extended Data Fig. 4 Apoptosis and autophagy analyses in kidneys and hearts of aged mice.
a, b, Representative images and quantification of active caspase 3-positive cells in kidneys (a) and hearts (b) from Becn1+/+ (wild-type) and Becn1F121A/F121A (knock-in) animals. Two month-old wild-type and knock-in mouse kidneys and hearts (n = 6 per genotype) were analysed. For kidney analyses, aged (20-month-old) wild-type (n = 20) and knock-in (n = 26) mice were used. For heart analyses, aged (20-month-old) wild-type (n = 19) and knock-in (n = 26) mice were used. Scatter plot bars represent median ± interquartile ranges. P values were determined by two-sided Mann–Whitney test. c, d, Enlarged versions of the endogenous LC3 puncta (autophagosome) images shown in Fig. 3d (c) and Fig. 3h (d). Arrows denote representative LC3 puncta.
Extended Data Fig. 5 In vitro klotho treatment disrupts the beclin 1–BCL2 interaction.
Co-immunoprecipitation of beclin 1 and BCL2 in HeLa cells treated with PBS or the indicated concentrations of recombinant full-length mouse klotho protein for 24 h. Result shown is representative of two independent experiments. For uncropped gels, see Supplementary Fig. 1.
Supplementary information
Supplementary Figures
This file contains Supplementary Figure 1 which shows the uncropped gels for all the main and Extended Data Figures
Rights and permissions
About this article

Cite this article
Fernández, Á.F., Sebti, S., Wei, Y. et al. Disruption of the beclin 1–BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140 (2018). https://doi.org/10.1038/s41586-018-0162-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-018-0162-7
- Springer Nature Limited
This article is cited by
-
Biological role and expression of translationally controlled tumor protein (TCTP) in tumorigenesis and development and its potential for targeted tumor therapy
Cancer Cell International (2024)
-
Autophagy dysfunction contributes to NLRP1 inflammasome-linked depressive-like behaviors in mice
Journal of Neuroinflammation (2024)
-
Focusing on mitochondria in the brain: from biology to therapeutics
Translational Neurodegeneration (2024)
-
Molecular mechanisms of aging and anti-aging strategies
Cell Communication and Signaling (2024)
-
Glial senescence enhances α-synuclein pathology owing to its insufficient clearance caused by autophagy dysfunction
Cell Death Discovery (2024)