

Abstract
The cell microenvironment, which is critical for stem cell maintenance, contains both cellular and non-cellular components, including secreted growth factors and the extracellular matrix1,2,3. Although Notch and other signalling pathways have previously been reported to regulate quiescence of stem cells4,5,6,7,8,9, the composition and source of molecules that maintain the stem cell niche remain largely unknown. Here we show that adult muscle satellite (stem) cells in mice produce extracellular matrix collagens to maintain quiescence in a cell-autonomous manner. Using chromatin immunoprecipitation followed by sequencing, we identified NOTCH1/RBPJ-bound regulatory elements adjacent to specific collagen genes, the expression of which is deregulated in Notch-mutant mice. Moreover, we show that Collagen V (COLV) produced by satellite cells is a critical component of the quiescent niche, as depletion of COLV by conditional deletion of the Col5a1 gene leads to anomalous cell cycle entry and gradual diminution of the stem cell pool. Notably, the interaction of COLV with satellite cells is mediated by the Calcitonin receptor, for which COLV acts as a surrogate local ligand. Systemic administration of a calcitonin derivative is sufficient to rescue the quiescence and self-renewal defects found in COLV-null satellite cells. This study reveals a Notch–COLV–Calcitonin receptor signalling cascade that maintains satellite cells in a quiescent state in a cell-autonomous fashion, and raises the possibility that similar reciprocal mechanisms act in diverse stem cell populations.
Similar content being viewed by others


Main
Notch activation antagonizes myogenesis by induction of transcriptional repressors (members of the HES/HEY family) and sequestration of the co-activator Mastermind-like 1 from the muscle differentiation factor MEF2C10,11. However, Notch signalling has broader functions in muscle cells, including the maintenance of quiescence4,5. To explore these functions, we carried out chromatin immunoprecipitation following by sequencing (ChIP–seq) screening12 and observed that intracellular Notch (NICD) and its downstream effector RBPJ occupied and regulated enhancers proximal to the collagen genes Col5a1, Col5a3, Col6a1 and Col6a2, which code for collagens that are amongst the most abundant of those produced by satellite cells (Fig. 1a, b and Extended Data Fig. 1a–e). By analysing mouse genetic models with altered Notch activity, we showed that the expression of these collagens tightly correlated with Notch activity in vivo (Extended Data Fig. 2a–e). Moreover, transcriptional induction of Col5a1 and Col5a3 by NICD translated to elevated COLV protein levels, specifically the a1(V)a2(V)a3(V) isoform (α3-COLV), in fetal forelimb (Fig. 1c) and adult hindlimb (tibialis anterior muscle) myogenic cells (Fig. 1d and Extended Data Fig. 2f for α3-COLV antibody specificity). Furthermore, we isolated collagen-depleted myofibres after treatment with collagenase, to monitor de novo α3-COLV production. As Col5a1 and Col5a3 transcripts are downregulated upon exit from quiescence (Extended Data Figs. 1a, 2g), no α3-COLV was detected in freshly isolated or activated satellite cells. Instead, genetic overexpression of NICD resulted in abundant, newly synthesized α3-COLV (Fig. 1e, f).

a, NICD and RBPJ ChIP–seq tracks from C2C12 cells indicating enhancers associated with the Col5a1, Col5a3, Col6a1 and Col6a2 loci. Orange rectangle, NICD and RBPJ enhancers; asterisk, enhancers used for luciferase assays (Extended Data Fig. 1c). b, Top, RBPJ ChIP in proliferating primary myogenic cells on Delta-like 1 (n = 4 ChIPs). Bottom, RBPJ ChIP in quiescent satellite cells, fixed before isolation25 (n = 3 ChIPs). Data are mean ± s.d.; two-sided unpaired t-test. c, Forelimb muscles of embryonic day (E)17.5 Myf5Cre-NICD mouse fetuses show upregulation of COLV. Inset shows low α3-COLV expression (higher exposure time). Note the membrane GFP-marked (mGFP) fibres in control and mononucleated NICD+PAX7+ cells in Myf5Cre-NICD mice26. d, Anti-GFP (satellite cells) and anti-α3-COLV immunostaining on transverse sections of quiescent adult tibialis anterior muscles expressing NICD–IRES–GFP (Pax7CT2-NICD). All GFP+ cells overexpressed COL5A3 (50 cells per mouse, n = 3 mice). e, Freshly fixed single myofibres from Pax7CT2-NICD extensor digitorum longus muscles at 0 h (left) or after 24 h in culture (right), stained for GFP and α3-COLV. f, Vertical and horizontal optical sections of myofibre presented in e from Pax7CT2-NICD mice (24 h in culture) showing COLV surrounding NICD–GFP+ satellite cells. Scale bars, 50 μm (c) and 10 μm (d–f). In c, d, insets are shown at 2× magnification of main panels. NS, not significant.
To assess the functional role of COLV, isolated satellite cells were incubated with COLI, COLV or COLVI in the presence of 5-ethynyl-2′-deoxyuridine (EdU) to assess proliferation and stained for PAX7, which marks muscle stem and/or progenitor cells, and the muscle commitment (MYOD) and differentiation (Myogenin) proteins. Only the COLV-complemented medium delayed entry of quiescent cells into the cell cycle (32 h, Fig. 2a), and consequently delayed their amplification and differentiation (72 h, Fig. 2b; 10 days, Extended Data Fig. 3a–c). As previously shown4,13, Rbpj−/− cells underwent precocious differentiation and this was partially antagonized by COLV, consistent with the finding that Col5a1 and Col5a3 genes are targets of NICD and RBPJ (Fig. 2c, d and Extended Data Fig. 3d–g). Taken together, these results show that COLV, specifically, sustains primary muscle cells in a more stem-like PAX7+ state, indicating that COLV could potentially have a role in maintaining the quiescent niche.

a, EdU pulse (for 2 h) of freshly isolated satellite cells cultured for 32 h: COLI (35%), COLVI (34%) and COLV (18%) (n = 4 mice, ≥250 cells, 2 wells per condition). Tg:Pax7-nGFP mice express nuclear (n)GFP driven by Pax7 regulatory elements. b, Immunostaining of freshly isolated satellite cells cultured for 72 h. PAX7: 58%, 55% and 81%; Myogenin: 56%, 57% and 24% for COLI, COLVI and COLV, respectively (n = 4 mice, ≥250 cells, 2 wells per condition). c, Experimental scheme for satellite cells plated overnight before collagen treatment. cKO, conditional knockout. d, Immunostainings of freshly isolated satellite cells incubated with collagens for 60 h (n = 3 mice, ≥200 cells, 2 wells per condition). Percentage (%) is presented over total GFP+ cells. Data are mean ± s.d.; two-sided paired t-test; *, P value calculated by two-sided unpaired t-test. Scale bars, 50 μm.
To determine whether COLV produced by satellite cells is a functional component of the niche, we generated compound Tg:Pax7-CreERT2;Col5a1flox/flox;R26mTmG (hereafter referred to as ‘Col5a1 cKO’) mice, in which COLV was depleted and simultaneously lineage-traced in GFP+ satellite cells4,14 (Fig. 3a and Extended Data Fig. 4a). Because the α1-chain of COLV is present in all COLV isoforms (which are trimeric), Col5a1 deletion produces cells completely lacking COLV protein14. Unexpectedly—given the general stability of collagens—targeted deletion of Col5a1 resulted in upregulation of the differentiation marker genes Myod (also known as Myod1) and Myog, and a concomitant reduction of the quiescence marker Calcr, as well as Pax7, only 18 days after tamoxifen treatment (Fig. 3b). Mutant cells also showed ectopic expression of Myogenin (Fig. 3c), increased 5-bromo-2′-deoxyuridine (BrdU) incorporation (Fig. 3d) and showed a significant decline in PAX7+ satellite cells (Fig. 3e). The Col5a1 cKO cells did not undergo apoptosis (data not shown), but fused to give rise to GFP-marked myofibres (Fig. 3f). Therefore, blocking de novo synthesis of COLV resulted in the spontaneous exit of satellite cells from quiescence, and differentiation, a phenotype reminiscent of Notch loss-of-function4,5.

a, Experimental schemes for control (Tg:Pax7-CT2;Col5a1+/+;R26mTmG), heterozygous (Tg:Pax7-CT2;Col5a1flox/+;R26mTmG) and conditional knockout (Tg:Pax7-CT2;Col5a1flox/flox;R26mTmG) mice. TA, tibialis anterior muscle. b, RT–qPCR of satellite cell (Pax7, Calcr) and differentiation (Myod, Myog) markers on Col5a1−/− and control satellite cells isolated by fluorescence-activated cell sorting from resting muscle (n = 3 mice per genotype). Ctr, control; Het, heterozygous; cKO, conditional knockout. c, Representative images of membrane-bound GFP+ (mGFP) satellite cells from total muscle preparations from control and Col5a1-null mice plated for 12 h. Arrow, mGFP+Myogenin+ cell (n = 3 mice per genotype, ≥200 cells). d, mGFP+ satellite cells from total muscle preparations plated for 12 h. Asterisk, non-recombined BrdU+ cell; arrows, mGFP+BrdU+ cells (n = 3 mice per genotype, ≥250 cells). e, Satellite cell quantification in quiescent tibialis anterior muscles (seven weeks after tamoxifen treatment) in control, heterozygous and Col5a1 cKO mice (n = 3 (control) and 4 (heterozygous and cKO) tibialis anterior muscles per genotype). f, Immunostaining of sections from control and Col5a1 cKO tibialis anterior muscles seven weeks after tamoxifen treatment (n = 3 mice per genotype). g, Immunostaining of sections from control and Col5a1 cKO tibialis anterior muscles 21 days after injury (n = 3 mice per genotype). h, Muscle cross-sectional area distribution 21 days after injury (shown as violin plots) was significantly different in control versus Col5a1 cKO tibialis anterior muscles, based on Kruskal–Wallis test (n = 3 mice per genotype, 1,000 fibres analysed per mouse). i, Immunostaining of sections 18 days after cardiotoxin injury of control and Col5a1 cKO tibialis anterior muscles (n = 3 mice per genotype). Percentage (%) is presented over total GFP+ cells. Data are mean ± s.d.; two-sided unpaired t-test. Scale bars, 50 μm (c, d) and 100 μm (f, g, i).
To investigate the role of Col5a1 in regeneration, we examined the morphology of tibialis anterior muscles of Col5a1 cKO mice, 18 days after cardiotoxin-mediated injury (Fig. 3a). Notably, mutant myogenic cells produced smaller nascent myofibres compared to control cells (Fig. 3g, h). Unexpectedly, fewer self-renewing PAX7+ cells were observed in the Col5a1 cKO mice (Fig. 3i) in spite of abundant COLV in regenerating muscle (data not shown), probably produced by the resident fibroblasts, suggesting a cell-autonomous role for Col5a1. To investigate self-renewal in a more tractable system, we targeted COLV using short interfering RNA (siRNA) on isolated myofibres in culture in which satellite cells proliferate and self-renew on the myofibre. Consistent with our in vivo observations, Col5a1 knockdown by siRNAs resulted in a marked decrease in the number of the self-renewing PAX7+MYOD− cells, compared to scramble control cells (Extended Data Fig. 4b, c). Of note, Col5a3 siRNA phenocopied Col5a1 siRNA, which demonstrates that the active triple helix contains α3-COLV (Extended Data Fig. 4c).
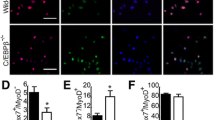
Substrate rigidity and geometry have previously been demonstrated to control stem cell properties, including differentiation and self-renewal15,16. However, we observed that COLV interacted with myogenic cells only when added in the medium, and not when present as a coating substrate (data not shown), which led us to speculate that it acted as a signalling molecule rather than a biomechanical modulator. To identify the cell surface receptor of COLV on satellite cells, we used a myotube-formation assay (see Extended Data Fig. 3b), coupled to inhibitors against known collagen receptors, including Integrins and the RTK receptor DDR117,18, but these did not obstruct the anti-myogenic activity of COLV (Extended Data Fig. 5a). Because collagens have also previously been shown to bind G-protein coupled receptors (GPCRs)19,20, we focused on Calcitonin receptor (CALCR), which is a GPCR critical for the maintenance of satellite cells21. Only cells that expressed CALCR showed decreased proliferation in the presence of COLV (Extended Data Fig. 5b), and Calcr−/− satellite cells isolated from conditional knockout Pax7CreERT2;Calcrflox/flox mice failed to respond to COLV treatment (Fig. 4a and Extended Data Fig. 5c–e), demonstrating that CALCR constitutes an essential mediator of the COLV signal (Extended Data Fig. 4e). Accordingly, as CALCR is rapidly cleared after satellite cell activation21, COLV had no effect on cultured myogenic cells that had been activated in vivo (three days after injury; Extended Data Fig. 5f). However, we note that addition of COLV on freshly isolated satellite cells appeared to stabilize residual CALCR and retain Calcr gene expression, thus allowing their prolonged interaction (Extended Data Fig. 5g–i). In summary, we show that CALCR is a critical mediator of the effect of COLV on maintaining quiescence and on the stemness properties of satellite cells.

a, Control (Ctr; Pax7CT2/+;Calcr+/+;R26stop-YFP) and Calcr-deficient (Calcr cKO; Pax7CT2/+;Calcrflox/flox;R26stop-YFP) satellite cells incubated for 10 days with COLI or COLV and immunostained for differentiation (n = 3 mice, ≥250 cells). b, Binding assay of COLV and CALCR by colorimetric on-cell enzyme-linked immunosorbent assay based on the measurements of horseradish peroxide absorbance. Runs test P value < 0.0001. Results presented as ratio of absorbance over non-treated cells (NT, orange line = 1) at 20 min of horseradish peroxide development. c, cAMP measurements of Calcr-transduced C2C12 cells after three hours of treatment with acetic acid (HOAc), COLI, COLV or elcatonin. Graph represents fold cAMP induction over average of mock cells treated with HOAc (n = 4 assays). d, Dose–response curve of fold cAMP concentration in Calcr-transduced C2C12 cells treated for 3 h with increasing concentrations of COLV. Half-maximal effective concentration (EC50) = 25.05 μg ml−1 (n = 4 independent assays). e, Experimental scheme of tamoxifen and elcatonin administration to Col5a1 cKO mice and their corresponding control mice. f, RT–qPCR of satellite cells (Pax7, Calcr) and differentiation (Myog) markers on Col5a1 cKO mutant mice and control mice (n = 3 mice per condition) treated with elcatonin or saline. g, Representative images of mGFP+ satellite cells from total muscle preparations from Col5a1-null mice injected with saline or elcatonin, plated for 12 h. Arrows, mGFP+MYOG+ cells (n = 3 mice per condition, ≥200 cells). h, EdU (for 2 h) and mGFP staining of satellite cells from total muscle preparations from control mice treated with saline or elcatonin, plated for 36 h. Asterisk, mGFP−EdU+ cell (n = 3 mice per genotype, ≥400 cells). i, Experimental scheme of tamoxifen and elcatonin administration to control and Col5a1 cKO mice. j, PAX7+ cells on tibialis anterior sections 21 days after injury in mice treated with saline or elcatonin (n = 6 mice for control and 8 mice for cKO, per treatment). Percentage (%) is presented over total GFP+ cells. Data are mean ± s.d.; a–c, two-sided paired t-test; f–j, two-sided unpaired t-test. Scale bars, 25 μm.
To date, it has been assumed that CALCR in satellite cells is activated by circulating calcitonin peptide hormones, which are principally expressed by parafollicular thyroid cells; this points to systemic regulation of stem cell quiescence. Based on our findings, we reasoned that COLV serves as a local ligand for the CALCR receptor. Indeed, on-cell enzyme-linked immunosorbent assay experiments showed that COLV—but not COLI—selectively bound to cells expressing CALCR (Fig. 4b). Notably, this binding was functional as COLV—but not COLI—displayed rapid activation kinetics and upregulation of levels of intracellular cAMP, which is a downstream reporter of CALCR activation22 (Fig. 4c, d and Extended Data Fig. 6a). In vitro binding assays using the extracellular domain of CALCR did not result in robust interaction with COLV (data not shown). Therefore, we propose that binding of COLV to CALCR requires a specific configuration of the receptor, possibly involving the extracellular loops or co-factors. Taken together, these data demonstrate that COLV physically and functionally interacts with CALCR.
In this study, we showed that blocking COLV production from satellite cells resulted in rupture of quiescence and impaired self-renewal in vivo. Combined with our ex vivo results, the similarity of these phenotypes to Notch and CALCR signalling abrogation points to a cell-autonomous Notch–COLV–CALCR axis that sustains muscle stem cells in their niche. Consistent with this notion, administration of the CALCR ligand elcatonin to control and Col5a1-null mice resulted in upregulation of the stem cell markers Pax7 and Calcr, indicating that the injected ligand was readily delivered to the quiescent satellite cells (Fig. 4e, f). Notably, elcatonin mitigated the precocious Myog transcription and protein expression levels in Col5a1 mutant cells (Fig. 4f, g). Elcatonin also prolonged the G0-to-S transition of control satellite cells exiting quiescence (Fig. 4h), which suggests that hyperactivation of CALCR could drive cells into a deeper, more dormant-like quiescent state marked by higher Pax7 expression23. Therefore, CALCR activity appears to control quiescence quantitatively, shown by the loss of satellite cells in the absence of ligand COLV, and qualitatively, shown by the presence of dormant-like satellite cells upon hyperactivation. Elcatonin restored the number of PAX7+ satellite cells in regenerating Col5a1 cKO muscles to wild-type levels (Fig. 4i, j), and in an ex vivo self-renewal reserve-cell model (Extended Data Fig. 6b, c). Therefore, we show that endogenous calcitonin levels are not sufficient to maintain Col5a1-null satellite cells, and that exogenous administration of a calcitonin derivative rescued the defects, probably via the activation of CALCR.
Here we describe a self-sustained signalling cascade orchestrated by the Notch pathway and propagated by the extracellular matrix of the immediate skeletal muscle stem cell niche (Extended Data Fig. 7). We propose that Notch acts as a sensor of the homeostatic environment by reinforcing the niche with active COLV that provides cell-autonomous signals and maintains stem cell quiescence. Upon disruption of the niche and physical separation of the ligands, Notch signalling is sharply downregulated and stem cells exit quiescence4,24. This halts further production of COLV and thus favours satellite cell activation, as shown in our model (Extended Data Fig. 7). It would be of interest to investigate whether the Notch–COLV–CALCR signalling cascade described here applies to stem cells in other tissues and organisms, in which an extracellular matrix protein produced by the stem cell can act as a local ligand for cell-autonomous stability of the niche through a GPCR. The regulatory mechanism that we identify provides a framework to construct a more complete view of the stem cell niche, and to manipulate stem cell behaviour in a therapeutic context.
Methods
Mouse strains
Mouse lines used in this study have been described and provided by the corresponding laboratories: Myf5Cr mice27, Pax7CreERT2 mice28 (used to recombine R26stop-NICD allele), R26stop-NICD-nGFP mice29, R26mTmG mice30 (ROSA 26 gene trap with membrane-Tomato floxed/membrane-GFP), Rbpjflox/flox mice31, Pax7CT2/+;Calcrflox/flox;R26stop-YFP/stop-YFP mice21 (triple mutant mice provided by S.F.) and Col5a1flox/flox mice32. Tg:Pax7-CreERT2 (used to recombine Rbpj and Col5a1) and Tg:Pax7-nGFP lines have previously been described4,33. All adult mice analysed were between 8 and 12 weeks old. Animals were handled according to national and European community guidelines, and protocols were approved by the ethics committee at Institut Pasteur and the French Ministry.
Muscle injury, tamoxifen, BrdU and elcatonin administration
For muscle injury, Tg:Pax7-CreERT2;Col5a1flox/flox;R26mTmG mice and their corresponding controls were anaesthetized with 0.5% Imalgene/2% Rompun and the tibialis anterior muscle was injected with 50 μl of cardiotoxin (10 μM; Latoxan). Tg:Pax7-CreERT2;Rbpjflox/flox;R26mTmG mice and their corresponding controls were injected intraperitoneally with tamoxifen three times (250 to 300 μl, 20mg/ml; Sigma T5648; diluted in sunflower seed oil/5% ethanol). Pax7CreERT2;Calcrflox/flox;R26stop-YFP mice and their corresponding controls were injected intraperitoneally with tamoxifen twice (1 mg per 5 g of body weight) and euthanized two weeks later. Pax7CreERT2;R26stop-NICD-ires-nGFP and Tg:Pax7-CreERT2;Col5a1flox/flox;R26mTmG mice and their corresponding controls were fed a diet containing tamoxifen for one and two weeks, respectively (Envigo, TD55125). Six days before being euthanized, Tg:Pax7-CreERT2;Col5a1flox/flox;R26mTmG mice and their corresponding controls were given the thymidine analogue BrdU (0.5 mg/ml, #B5002; Sigma) in the drinking water supplemented with sucrose (25 mg/ml). Elcatonin (2.5 ng per g of mouse, final concentration in 0.9% NaCl; Mybiosource, MBS143228) was injected subcutaneously eight times, every other day. Comparisons were done between age-matched littermates using 8–12-week-old mice.
Muscle enzymatic dissociation and stem cell isolation
Adult and fetal limb muscles were dissected, minced and incubated with a mix of Dispase II (Roche, 04942078001) 3 U/ml, collagenase A (Roche, 11088793001) 100 μg/ml and DNase I (Roche, 11284932001) 10 mg/ml in Hank’s Balanced Salt Solution (Gibco) supplemented with 1% penicillin–streptomycin (PS; Gibco) at 37 °C at 60 r.p.m. in a shaking water bath for 2 h. The muscle suspension was successively filtered through 100-μm and 70-μm cell strainers (Miltenyi, 130-098-463 and 130-098-462) and then spun at 50g for 10 min at 4 °C to remove large tissue fragments. The supernatant was collected and washed twice by centrifugation at 600g for 15 min at 4 °C. Before fluorescence-activated cell sorting (FACS), the final pellet was resuspended in cold Dulbecco’s modified Eagle’s medium (DMEM) and 1% PS supplemented with 2% fetal bovine serum (FBS), and the cell suspension was filtered through a 40-μm strainer. Satellite cells were sorted with Aria III (BD Biosciences) using either the GFP (Tg:Pax7-nGFP or Tg:Pax-CreERT2;Rbpjflox/flox;R26mTmG or Tg:Pax7-CreERT2;Col5a1flox/flox;R26mTmG) or the YFP (Pax7CT2;Calcrflox/flox;R26stop-YFP) cell markers. Isolated, mononuclear cells were collected in DMEM/1% PS/2% FBS. Enzymatically dissociated muscle was also plated directly without FACS on Matrigel-coated dishes (Corning, 354248; 30 min at 37 °C), and fixed 12 h later with 4% paraformaldehyde (PFA)/PBS. Cells were immunostained following the protocol described above.
Chromatin immunoprecipitation
Cultured myoblasts
Satellite cells were isolated from adult Tg:Pax7-nGFP mice and plated on dishes, coated with Delta-like 1, for 72 h to maintain active Notch signalling, as previously described4,34. Cells were then processed for ChIP using a dual cross-linking protocol35, with slight modifications. In brief, cells were fixed on the dish with 2 mM di(N-succinimidyl) glutarate (Sigma, 80424) in PBS for 45 min at room temperature. After two washes with PBS, cells were re-fixed with 1% formaldehyde/PBS for 10 min at room temperature, before quenching the reaction with 1/20 volume of 2.5 M glycine for 5 min at room temperature. The cells were then collected with a cell scraper in PBS supplemented with 1% BSA and protease inhibitors (Roche, 11697498001), and collected by spinning. Cell lysis and chromatin isolation were done using the Ideal ChIP–seq kit for histones (Diagenode, C01010051). Chromatin was sheared using a Bioruptor Pico (Diagenode B01060001) with 10 cycles of 30 s on/off sonication. The samples were prepared in triplicates from different plates. Primary myogenic cells (2 × 106) were used per ChIP and 2 × 104 cells were used per input. The immunoprecipitations were performed following the manufacturer’s guidelines using 6 μl of anti-RBPJ antibody (Cell Signalling, #5313) or 1.5 μl of rabbit control IgG antibody (Diagenode, C15410206) in a final volume of 300 μl per ChIP. The purification of the immunoprecipitated DNA was performed using DiaPure columns (Diagenode, C03040001). RT–qPCR was performed using FastStart Universal SYBR Green Master mix (Roche, 04913914001) and analysis was performed using the 2−ΔΔCt method36 normalized to the Neg16 region.
Quiescent satellite cells
Satellite cells were isolated from adult Tg:Pax7-nGFP mice using in situ fixation to preserve Notch signalling from dissociation-induced downregulation25. Cells were fixed as above in 2 mM di(N-succinimidyl) glutarate for 45 min, followed by 10 min with 1% formaldehyde at room temperature. Cell lysis and chromatin isolation were performed using Auto-TrueMicrochip kit (Diagenode, C01010140). Chromatin was sheared as above with 10 cycles of 30 s on/off sonication using a Bioruptor Pico. Two hundred thousand cells were used per ChIP and 2 × 103 per input and IPs were performed using 2 μl of anti-RBPJ antibody (Cell Signalling, 5313) or 0.5 μl of rabbit control IgG antibody following the manufacturer’s guidelines. Immunoprecipitated chromatin preparations and input were purified using the Auto IPure kit v2 (Diagenode). RT–qPCR was performed using FastStart Universal SYBR Green Master mix (Roche, 04913914001) and analysis was performed using the 2−ΔΔCt method36 normalized to the Neg16 region. Primers used for ChIP–qPCR are listed in Supplementary Table 1.
Cell culture and collagen incubation
Satellite cells isolated by FACS were plated at 3 × 103 cells per cm2 on ibi-treated μ-slides (Ibidi, 80826) pre-coated with 0.1% gelatin for 2 h at 37 °C. Cells were cultured in satellite cell growth medium containing DMEM (Gibco) supplemented with F12 (50:50; Gibco), 1% PS, 20% FBS (Gibco) and 2% Ultroser (Pall; 15950-017) at 37 °C, 3% O2, 5% CO2 for the indicated time. Twelve hours after plating, collagens (COLI rat tail, BD Biosciences, 354236; COLV human placenta, Sigma, C3657; COLVI human placenta, AbD Serotec 2150-0230) resuspended in HOAc acid at 1 mg/ml, were added to the culture medium at a final concentration of 50 μg/ml and cells were fixed with 4% PFA for 10 min at room temperature. To assess proliferation, cells were pulsed with the thymidine analogue EdU, 1 × 10−6 M at 2 h before fixation (ThermoFisher Click-iT Plus EdU kit, C10640). Inhibitors used: Obtustatin (Integrin α1β1, Tocris, 4664, 100 nM), TC-I 15 (Integrin α2β1 Tocris, 4527, 100 μM), RGDS peptide (all Integrins, Tocris, 3498, 100 μM), 7rh37 (DDR1, a gift from K. Ding, 20 nM).
Muscle fixation and histological analysis
Embryo forelimbs were fixed in 4% PFA/0.1% Triton for 2 h, washed overnight with 1× PBS, immersed in 20% sucrose/PBS overnight, embedded in OCT, frozen in liquid nitrogen and sectioned transversely at 12–14 μm. Isolated tibialis anterior muscles were immediately frozen in liquid-nitrogen-cooled isopentane and sectioned transversely at 8 μm. For PAX7 staining on adult tibialis anterior muscle, sections were post-fixed with 4%PFA, 15 min at room temperature. After 3 washes with 1× PBS, antigen retrieval was performed by incubating sections in boiling 10 mM citrate buffer pH 6 for 10 min. Sections were then blocked, permeabilized and incubated with primary and secondary antibodies as described in ‘Immunostaining on cells, sections and myofibres’.
Single myofibre isolation and siRNA transfection
Single myofibres were isolated from extensor digitorum longus muscles following the previously described protocol38. In brief, extensor digitorum longus muscles were dissected and incubated in 0.1% w/v collagenase (Sigma, C0130)/DMEM for 1 h in a 37 °C shaking water bath at 40 r.p.m. Following enzymatic digestion, mechanical dissociation was performed to release individual myofibres that were then transferred to serum-coated Petri dishes. Single myofibres were transfected with Col5a1 siRNA, Col5a3 siRNA (Dharmacon SMARTpool Col5a1 (12831) L-044167-01 and Col5a3 (53867) L-048934-01-0005) or scramble siRNA (Dharmacon ON-TARGETplus Non-targeting siRNA #2 D-001810-02-05) at a final concentration of 200 nM, using Lipofectamine 2000 (ThermoFisher, 11668) in Opti-MEM (Gibco). Four hours after transfection, 6 volumes of fresh satellite cell growth medium were added and fibres were cultured for 72 h at 37 °C, 3% O2. Myofibres were fixed for 15 min in 4% PFA before immunostaining for proliferation, differentiation and self-renewal markers39.
Immunostaining on cells, sections and myofibres
Following fixation, cells and myofibres were washed three times with PBS, then permeabilized and blocked at the same time in buffer containing 0.25% Triton X-100 (Sigma), 10% goat serum (Gibco) for 30 min at room temperature. For BrdU immunostaining, cells were unmasked with DNaseI (1,000 U/ml, Roche, 04536282001) for 30 min at 37 °C. Cells and fibres were then incubated with primary antibodies (Supplementary Table 2) for 4 h at room temperature. Samples were washed with 1× PBS three times and incubated with Alexa-conjugated secondary antibodies (Life Technologies, 1/1,000) and Hoechst 33342 (Life Technologies, 1/5,000) for 45 min at room temperature. EdU staining was chemically revealed using the Click-iT Plus kit according to manufacturer’s recommendations (Life Technologies, C10640). For collagen staining, the myofibres and the muscle sections were incubated with 0.1% Triton X-100 for 30 min at room temperature. Myofibres and sections were then washed 3 × 10 min and incubated with 10% goat serum in PBS for 30 min. After one wash, samples were incubated with primary antibodies and secondary antibodies as described in Supplementary Table 2. Confocal images were acquired with a Leica SPE microscope and Leica Application Suite or with Zeiss LSM 700 microscope and Zen Blue 2.0 software. Three-dimensional images were reconstructed from confocal Z-stacks using Imaris software. The Section view function was used to inspect the environment of the satellite cells by showing the cut in the x, y and z axes.
Reserve cell cultures
Enzymatically dissociated muscles were plated in gelatin-coated dishes (1/30 of total mouse muscles per cm2) in the satellite cell growth medium described above. When myotube formation was detected (day 7 to 10), recombination was induced by addition of 4-hydroxytamoxifen (4-OHT; Sigma, H6278) at final concentration of 1 μM every other day. Seven days later, 4-OHT-containing medium was replaced every other day with fresh medium containing elcatonin (0.1 U/ml), for an additional 10 days. To assess proliferation, cells were pulsed with 1 × 10−6M EdU for 6 h before fixation (10 min, 4% PFA). Reserve cells were defined by immunofluorescence as PAX7+EdU− cells39. For each medium change, only half of the conditioned medium was removed and replaced by an equal volume of fresh medium.
Construction of luciferase reporters and luciferase assays
For the generation of luciferase reporters, candidate enhancers of Col5a1, Col5a3, Col6a1 and Col6a2 (a shared enhancer), and Hey1 were amplified by PCR from genomic DNA of C2C12 cells. The enhancers were then cloned into the firefly-luciferase pGL3-Basic vector (Promega, E1751) upstream of a minimal thymidine kinase promoter. The sequences of enhancers are listed in Supplementary Table 3. Transfected cells (Lipofectamine LTX, Life Technologies, 15338030) were lysed and luciferase signal was scored using the Dual-Luciferase Reporter Assay System (Promega, E1910). For normalization, Renilla luciferase (pCMV-Renilla) was transfected at 1:20 ratio relative to firefly-luciferase constructs.
RNA isolation and RT–qPCR
Total RNA was extracted from satellite cells isolated by FACS using QIAGEN mini RNeasy kit and reverse transcribed using SuperScript III (Invitrogen, 18080093), according to manufacturer’s instructions. RT–qPCR was performed using FastStart Universal SYBR Green Master mix (Roche, 04913914001) and analysis was performed employing the 2−ΔΔCt method and using the average of the control values as a reference36. Specific forward and reverse primers used in this study are listed in Supplementary Table 1.
Stable cell line manipulations
The mouse myoblast cell line C2C12 was cultured in DMEM/ 20% FBS/ 1% PS at 37 °C, 5% CO2.
Notch activation
Notch activation was achieved by plating cells on DLL1-coated dishes or by doxycycline-inducible Notch constructs, as previously described12.
Calcr retrovirus preparation and transduction
Calcitonin receptor C1a-type (pMXs-Calcr-C1a-IRES-GFP) and mock control (pMXs-IRES-GFP) retrovirus vectors were prepared as previously described21,40. In brief, 48 h after transfection of Platinum-E cells the supernatant was recovered and used to transduce C2C12 cells. Two days later stably labelled GFP+ C2C12 cells were isolated by FACS. All stable cell lines used in this study are negative for mycoplasma contamination.
Quantification of cAMP
Transduced mock (IRES-GFP) and Calcr (CalcR-C1a-IRES-GFP) C2C12 cells were isolated by FACS based on GFP expression and seeded on 0.1% gelatin-coated, white culture 96-well plates (Falcon, 353296) at 3 × 103 cells per well. After overnight culture, the cells were incubated with the complete induction medium containing DMEM/1% PS/500 μM isobutyl-1-methylxanthine (Sigma, 17018)/100 μM 4-(3-butoxy-4-methoxy-benzyl) imidazolidone (Ro 20-1724 Sigma, B8279)/MgCl2 40 mM, collagen, solvant HOAc or elcatonin (0.1 U/ml) for 3 h. The amount of intracellular cAMP was measured using cAMP-Glo Max Assay (Promega, V1681) following the manufacturer’s protocol. Luminescence was quantified with FLUOstar OPTIMA (BMG Labtech). The EC50 value was determined with GraphPad Prism software using a sigmoid dose–response curve (variable slope).
Biotinylation of collagens
Commercial collagen proteins (COLI rat tail, BD Biosciences, 354236; COLV human placenta, Sigma, C3657) were biotinylated using the Pierce EZ-Link Biotinylation Kit, with slight modifications. In brief, 20 μl of 1 M HEPES was added to 0.5 ml of 1 mg/ml collagen dissolved in 0.5 M HOAc. Then, 20 μl of 100 mM biotin reagent were added and incubated at room temperature for 1.5 h. Biotinylated collagens were next dialysed in 25 mM HEPES, 2.5 M CaCl2, 125 mM NaCl, 0.005% Tween (Slide-A-Lyze MINI Dialysis Device, Thermo Fisher 88401) overnight at 4 °C.
On-cell enzyme-linked immunosorbent assay
Transduced mock and Calcr C2C12 cells were seeded on a clear-bottom 96-well plate (TPP, 92096) at a density of 3 × 103 cells per well. After overnight culture, cells were treated with 50 μg/ml of biotinylated collagens for 2 h and fixed with 4% PFA/PBS for 15 min. After 3× PBS washes, cells were blocked with a solution containing 10% goat serum, 2% BSA, PBS for 1 h at room temperature, washed and incubated for 1 h at room temperature with goat anti-mouse biotin–HRP antibody (Jackson, 1/1000e, 115-035-003). After 3× PBS washes, the HRP signal was developed by addition of 3,3′,5,5′ tetramethylbenzidine (1-Step Ultra TMB-ELISA, Sigma, 34028). HRP substrate and absorbance at 650 nm was measured once every 30 s for 30 min with FLUOstar OPTIMA (BMG Labtech). The signal was normalized to the background signal (no secondary antibody) and to the number of cells assessed by Janus green staining (Abcam, ab111622).
Statistical analysis
No statistical methods were used to predetermine sample size. The investigators were not blinded to allocation during experiments and outcome assessment. No animal has been excluded from analysis and no randomization method has been applied in this study. For comparison between two groups, two-tailed paired and unpaired Student’s t-tests were performed to calculate P values and to determine statistically significant differences (see legends of Figs. 1–4). Additional specific statistical tests are detailed in legends of Figs. 1–4. All experiments have been done twice with the same results. All statistical analyses were performed with Excel software or GraphPad Prism software; Kruskal–Wallis test was performed in R.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All data that support the findings of this study are available from the corresponding authors upon request.
Change history
29 May 2018
In the originally published article, two panel labels were missing in the legend for Extended Data Fig. 1. This has now been corrected.
References
Raymond, K., Deugnier, M. A., Faraldo, M. M. & Glukhova, M. A. Adhesion within the stem cell niches. Curr. Opin. Cell Biol. 21, 623–629 (2009).
Moore, K. A. & Lemischka, I. R. Stem cells and their niches. Science 311, 1880–1885 (2006).
Watt, F. M. & Huck, W. T. Role of the extracellular matrix in regulating stem cell fate. Nat. Rev. Mol. Cell Biol. 14, 467–473 (2013).
Mourikis, P. et al. A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells 30, 243–252 (2012).
Bjornson, C. R. et al. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30, 232–242 (2012).
Rozo, M., Li, L. & Fan, C. M. Targeting β1-integrin signaling enhances regeneration in aged and dystrophic muscle in mice. Nat. Med. 22, 889–896 (2016).
Cheung, T. H. et al. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 482, 524–528 (2012).
Zismanov, V. et al. Phosphorylation of eIF2α is a translational control mechanism regulating muscle stem cell quiescence and self-renewal. Cell Stem Cell 18, 79–90 (2016).
Chakkalakal, J. V., Jones, K. M., Basson, M. A. & Brack, A. S. The aged niche disrupts muscle stem cell quiescence. Nature 490, 355–360 (2012).
Shen, H. et al. The Notch coactivator, MAML1, functions as a novel coactivator for MEF2C-mediated transcription and is required for normal myogenesis. Genes Dev. 20, 675–688 (2006).
Buas, M. F., Kabak, S. & Kadesch, T. The Notch effector Hey1 associates with myogenic target genes to repress myogenesis. J. Biol. Chem. 285, 1249–1258 (2010).
Castel, D. et al. Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 27, 1059–1071 (2013).
Vasyutina, E. et al. RBP-J (Rbpsuh) is essential to maintain muscle progenitor cells and to generate satellite cells. Proc. Natl Acad. Sci. USA 104, 4443–4448 (2007).
Sun, M. et al. Targeted deletion of collagen V in tendons and ligaments results in a classic Ehlers–Danlos syndrome joint phenotype. Am. J. Pathol. 185, 1436–1447 (2015).
Gilbert, P. M. et al. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science 329, 1078–1081 (2010).
Yennek, S., Burute, M., Théry, M. & Tajbakhsh, S. Cell adhesion geometry regulates non-random DNA segregation and asymmetric cell fates in mouse skeletal muscle stem cells. Cell Reports 7, 961–970 (2014).
Leitinger, B. Transmembrane collagen receptors. Annu. Rev. Cell Dev. Biol. 27, 265–290 (2011).
Vogel, W., Gish, G. D., Alves, F. & Pawson, T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1, 13–23 (1997).
Paavola, K. J., Sidik, H., Zuchero, J. B., Eckart, M. & Talbot, W. S. Type IV collagen is an activating ligand for the adhesion G protein-coupled receptor GPR126. Sci. Signal. 7, ra76 (2014).
Luo, R. et al. G protein-coupled receptor 56 and collagen III, a receptor–ligand pair, regulates cortical development and lamination. Proc. Natl Acad. Sci. USA 108, 12925–12930 (2011).
Yamaguchi, M. et al. Calcitonin receptor signaling inhibits muscle stem cells from escaping the quiescent state and the niche. Cell Reports 13, 302–314 (2015).
Evans, B. N., Rosenblatt, M. I., Mnayer, L. O., Oliver, K. R. & Dickerson, I. M. CGRP-RCP, a novel protein required for signal transduction at calcitonin gene-related peptide and adrenomedullin receptors. J. Biol. Chem. 275, 31438–31443 (2000).
Rocheteau, P., Gayraud-Morel, B., Siegl-Cachedenier, I., Blasco, M. A. & Tajbakhsh, S. A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell 148, 112–125 (2012).
Mourikis, P. & Tajbakhsh, S. Distinct contextual roles for Notch signalling in skeletal muscle stem cells. BMC Dev. Biol. 14, 2 (2014).
Machado, L. et al. In situ fixation redefines quiescence and early activation of skeletal muscle stem cells. Cell Reports 21, 1982–1993 (2017).
Mourikis, P., Gopalakrishnan, S., Sambasivan, R. & Tajbakhsh, S. Cell-autonomous Notch activity maintains the temporal specification potential of skeletal muscle stem cells. Development 139, 4536–4548 (2012).
Haldar, M., Karan, G., Tvrdik, P. & Capecchi, M. R. Two cell lineages, myf5 and myf5-independent, participate in mouse skeletal myogenesis. Dev. Cell 14, 437–445 (2008).
Murphy, M. M., Lawson, J. A., Mathew, S. J., Hutcheson, D. A. & Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 138, 3625–3637 (2011).
Murtaugh, L. C., Stanger, B. Z., Kwan, K. M. & Melton, D. A. Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl Acad. Sci. USA 100, 14920–14925 (2003).
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. & Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007).
Han, H. et al. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int. Immunol. 14, 637–645 (2002).
Sun, M. et al. Collagen V is a dominant regulator of collagen fibrillogenesis: dysfunctional regulation of structure and function in a corneal-stroma-specific Col5a1-null mouse model. J. Cell Sci. 124, 4096–4105 (2011).
Sambasivan, R. et al. Distinct regulatory cascades govern extraocular and pharyngeal arch muscle progenitor cell fates. Dev. Cell 16, 810–821 (2009).
Hicks, C. et al. A secreted Delta1-Fc fusion protein functions both as an activator and inhibitor of Notch1 signaling. J. Neurosci. Res. 68, 655–667 (2002).
Vasconcelos, F. F. et al. MyT1 counteracts the neural progenitor program to promote vertebrate neurogenesis. Cell Reports 17, 469–483 (2016).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC t method. Methods 25, 402–408 (2001).
Gao, M. et al. Discovery and optimization of 3-(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)ethynyl)benzamides as novel selective and orally bioavailable discoidin domain receptor 1 (DDR1) inhibitors. J. Med. Chem. 56, 3281–3295 (2013).
Shinin, V., Gayraud-Morel, B., Gomes, D. & Tajbakhsh, S. Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat. Cell Biol. 8, 677–687 (2006).
Yoshida, N., Yoshida, S., Koishi, K., Masuda, K. & Nabeshima, Y. Cell heterogeneity upon myogenic differentiation: down-regulation of MyoD and Myf-5 generates ‘reserve cells’. J. Cell Sci. 111, 769–779 (1998).
Morita, S., Kojima, T. & Kitamura, T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7, 1063–1066 (2000).
Acknowledgements
We thank H. Stunnenberg for the ChIP–seq and RNA sequencing data; D. Castro for the RBPJ ChIP protocol; D. Greenspan for the anti-a3-COLV antibody and Col5a3-knockout muscle samples; C. Moali for the SPR assay; F. Auradé and the Protein Core Facility, Institut Curie, for the production of CalcR proteins; K. Ding for the 7rh DDR1 inhibitor; F. Ruggiero for suggesting the on-cell enzyme-linking immunosorbent assay experiment; and the Cytometry platforms of Institut Pasteur and IMRB, Inserm U955, Creteil. F.R. was funded by the Association Française contre les Myopathies via TRANSLAMUSCLE (PROJECT 19507), Agence Nationale pour la Recherche grant Satnet (ANR-15-CE13-0011-01) and RHU CARMMA (ANR-15-RHUS-0003). S.T. was funded by Institut Pasteur, Centre National pour la Recherche Scientific and the Agence Nationale de la Recherche (Laboratoire d’Excellence Revive, Investissement d’Avenir; ANR-10-LABX- 73) and the European Research Council (Advanced Research Grant 332893). M.B.B. was funded by the Doctoral School grant and Fondation pour la Recherche Médicale.
Reviewer information
Nature thanks I. Conboy, G. Kardon and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
M.B.B., S.T. and P.M. proposed the concept, designed experiments and wrote the manuscript, F.R. oversaw revisions, and S.T. funded most of the study. P.M. and D.C. conducted initial experiments on enhancer analysis. D.C. and L.M. performed and analysed ChIP experiments. M.B.B. performed the remaining experiments and, together with P.M., analysed the data. S.F. and D.E.B. provided mouse models.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Identification of NICD/RBPJ-bound enhancers and response to activation of Notch signalling.
a, Gene expression microarray data show that satellite cells express a specific subset of collagen types, which include the fibrillar COLI (Col1a1 and Col1a2), COLIII (Col3a1, possibly as (α1(III))3 homodimer) and COLV (Col5a1, Col5a2 and Col5a3) and the non-fibrillar COLIV (Col4a1 and Col4a2), COLVI (Col6a1 and Col6a2) and COLXV (Col15a1, possibly as (α1(XV))3 homodimer). Data are shown as a heat map of normalized collagen transcripts expressed at different developmental time points (E12.5, E17.5 and post-natal day (P)8; Tg-Pax7-nGFP, Gene Expression Omnibus (GEO) accession number GSE52192), quiescent and post-injury (t = 60 h after BaCl2 injury). b, ChIP–seq tracks indicating NICD/RBPJ-occupied enhancers, associated with mouse Col5a1, Col5a3, Col6a1 and Col6a2 loci. H3K4me1, H3K27ac, p300 and NICD are shown. Orange rectangles indicate RBPJ binding positions and asterisks indicate the enhancers used for transcriptional activity assays in c. c, Core sequences of the selected NICD/RBPJ-bound enhancers (asterisked orange rectangle in Fig. 1a and in b). The RBPJ consensus binding motif is highlighted in yellow. d, Transcriptional response of isolated enhancers to activation of Notch signalling in C2C12 cells. Firefly luciferase signal was measured in cells with doxycycline-inducible expressed human Notch1–GFP (NICD) and GFP control cells treated with (2S)-N-[(3,5-difluorophenyl)acetyl]-l-alanyl-2-phenyl]glycine 1,1-dimethylethyl ester (DAPT) and were normalized to internal control (pCMV-Renilla). Data are expressed as relative luminescence units (n = 3 independent experiments). Data are mean ± s.d.; two-sided paired t-test. e, Expression measurements, based on RNA sequencing, of collagen genes in myogenic C2C12 cells, with active (treated with Delta-like 1) or inhibited (treated with DAPT) Notch signalling for 6 or 24 h (data available at GEO, accession number GSE37184). Data are shown as Delta-like 1-to-DAPT ratios of average reads per kilobase of exon model per million mapped reads (RPKMs). Genes with low expression (RPKM < 2) were eliminated. HeyL and Hey1 transcripts indicate Notch pathway activation. Red line designates no change (ratio = 1).
Extended Data Fig. 2 Notch signalling regulates Col5 and Col6 expression in vivo.
a, Satellite cells isolated by FACS at day 10 after tamoxifen injections, from resting tibialis anterior muscle from control (Tg:Pax7-CT2;Rbpj+/−;R26mTmG/+) and Rbpj-null (Tg:Pax7-CT2;Rbpjflox/−;R26mTmG/+) mice immunostained for RBPJ. b, RT–qPCR of collagen genes in Rbpj cKO and control satellite cells. Hey1 used as control for Notch signalling (n = 3 mice per genotype). c, Induction of collagen genes in E17.5 control (Myf5Cre/+;R26mTmG/+) and Myf5Cre-NICD (Myf5Cre/+;R26stop-NICD-nGFP/+) cells isolated by FACS. RT–qPCR was normalized to Gapdh, n = 3 fetuses per genotype. HeyL reports Notch activity. d, FACS plots showing fractionation of GFP+ cells from E17.5 Tg:Pax7-nGFP fetuses into Pax7high (20% of population), Pax7mid (40%), and Pax7low (20%). The intensity of the GFP signal reflects the activity of the Pax7 promoter. e, Transcript levels of GFP+ cells isolated by FACS show a tight correlation between lineage progression, Notch signalling activity and collagen gene expression (n = 3 fetuses per genotype). f, Specificity of α3-COLV antibody assessed by immunostaining of tibialis anterior muscle transverse section from wild-type and Col5a3 cKO P14 postnatal pups (n = 3 mice per genotype). g, Time course of gene expression performed by RT–qPCR on freshly isolated satellite cells (Quiescent), 48 h or 60 h after cardiotoxin injury of tibialis anterior muscle (48 hours post injury (hpi), 60 hpi), and isolated single myofibres from extensor digitorum longus muscle of Tg:Pax7-nGFP mice. Col5a1 and Col5a3 were strongly downregulated in activated and differentiated cells. Quiescence (Pax7, Calcr) and differentiation (Myog) markers are indicated. Col4a2, a major component of the basement membrane, is expressed mainly by myofibres (n = 3 mice per condition). Data are mean ± s.d.; one-sided unpaired t-test. Scale bars, 50 μm.
Extended Data Fig. 3 COLV delays proliferation and differentiation of satellite cells.
a, Experimental scheme: isolated Tg:Pax7-nGFP satellite cells cultured overnight (o/n) before collagen treatment. b, Myosin heavy chain (MyHC) and EdU staining of satellite cells treated with COLI or COLV. Fusion index: 82%, 86% and 33% for HOAc solvent, COLI and COLV, respectively (n = 3 mice, ≥250 cells, 2 wells per condition). c, Percentage of EdU+ primary myogenic cells after ten days of culture with indicated collagens. EdU: 2.6%, 1.3% and 18.2% for COLI, COLVI and COLV, respectively (n = 3 mice, ≥250 cells, 2 wells per condition). d, Experimental scheme for control and cKO mice. Satellite cells were plated overnight before collagen treatment. e, GFP and MyHC immunostaining of Rbpj cKO satellite cells (n = 3 mice per condition) incubated 60 h in presence of COLI or COLV, or with HOAc control (n = 3 mice, ≥200 cells, 2 wells per condition). f, Percentage of EdU+ cells (2 h pulse) of Rbpj-null primary myogenic cells, after ten days of culture with HOAc or indicated collagens. EdU: 1.0% and 7.6% for COLI and COLV, respectively (n = 3 mice, ≥150 cells, 2 wells per condition). g, RT–qPCR on GFP+ Rbpj-null satellite cells isolated by FACS and cultured for 72 h in the presence of COLI or COLV. Results are normalized to Tbp. Data are mean ± s.d.; two-sided paired t-test; P value: two-sided unpaired t-test. Scale bars, 50 μm.
Extended Data Fig. 4 COLV—and specifically α3-COLV—is critical for satellite cell self-renewal.
a, RT–qPCR of Col5a1 in control (Ctr; Tg:Pax7-CT2;Col5a1+/+;R26mTmG), heterozygous (Het; Tg:Pax7-CT2;Col5a1flox/+;R26mTmG) and conditional knockout (cKO; Tg:Pax7CT2;Col5a1flox/flox;R26mTmG) mice two weeks after tamoxifen diet (n = 3 mice per genotype). b, Transcript levels of the different Col5 mRNA chains in C2C12 after transfection of either control scramble, Col5a1 or Col5a3 siRNA, showing the specificity of each siRNA for its given targeted mRNA. Data are normalized to Tbp gene expression (n = 3 independent assays). c, Col5a1 and Col5a3 siRNA transfection of Tg:Pax7-nGFP isolated single myofibres cultured for 72 h and immunostained for GFP and MYOD. Resident satellite cells enter the myogenic program and form clusters composed of proliferating (PAX7+MYOD+MYOG−), differentiated (PAX7−MYOG+) and self-renewed (PAX7+MYOD−) cells within 72 h. Quantification of PAX7+MYOD−, PAX7+MYOD+ and PAX7−MYOD+ populations 72 h after transfection. Scramble siRNA was used as negative control (n ≥ 15 fibres counted from 3 mice). Data are mean ± s.d.; a, two-sided unpaired t-test; b, c, two-sided paired t-test. Scale bar, 50 μm.
Extended Data Fig. 5 Screening for COLV receptor candidates identifies CALCR.
a, Screening for the COLV receptor: satellite cells from Tg:Pax7-nGFP mice were incubated for ten days with COLV and candidate receptors were targeted with respective inhibitors: 7rh for DDR1 (sub-panels C, D), the broad-spectrum Integrin-binding competitor RGDS peptide (sub-panels E, F), Obtustatin for Integrin α1β1 (sub-panels G, H), TC-I 15 for Integrin α2β1 (sub-panels I, J). DMSO solvent was used as a control for TC-I 15 and 7rh (sub-panels A, B). Satellite cell differentiation was assayed by MyHC immunostaining. b, EdU (2 h pulse) and CALCR staining of GFP+ C2C12 cells isolated by FACS and transduced with Calcr-GFP or mock-GFP retrovirus and cultured for 24 h with COLI (top) or COLV (bottom). Quantification of EdU+ Calcr-transduced C2C12 cells or mock-GFP cells treated for 24h with COLV or with the controls, COLI and HOAc (n = 5 independent experiments, ≥250 cells counted, 2 wells per condition). There was no significant difference between HOAc and COLI treated samples (data not shown). c, Experimental scheme of tamoxifen administration to control (Ctr) (Calcr+/+) and cKO (Calcrflox/flox) mice. FACS plot of satellite cells from Pax7CreERT2/+;Calcrflox/flox;R26stop-YFP and Pax7CreERT2/+;Calcr+/+;R26stop-YFP mice. Cells sorted based on YFP expression. d, Control and Calcr cKO satellite cells isolated by FACS, fixed immediately after sorting and immunostained for CALCR to confirm the absence of CALCR protein from recombined cells. For control (upper panel), two fields from the same culture dish are shown, separated by a white line. Asterisk shows a non-recombined, CALCR+ cell in the cKO sample (lower panel). e, Quantification of PAX7+, Myogenin+ and EdU+ cells in Calcr-depleted satellite cells (Pax7CT2/+;Calcrflox/flox;R26stop-YFP) isolated by FACS and treated for 32 h or 72 h with COLI or COLV (n = 3 mice, ≥250 cells counted, 2 wells per condition). f, Quantification of total PAX7+ (GFP), Myogenin+ and EdU+ myogenic cells isolated by FACS from Tg:Pax7-nGFP mice three days after cardiotoxin injury of tibialis anterior muscle, and incubated for 72 h in presence of COLI or COLV, or HOAc as a control, in the culture medium (n = 3 mice, ≥200 cells counted). g, CALCR protein in freshly isolated satellite cells, or satellite cells cultured for 12 h, from Tg:Pax7-nGFP mice, demonstrating that CALCR protein is still present when satellite cells are treated with different collagens (see Extended Data Fig. 2). h, Induction of Calcr transcript expression by RT–qPCR of Tg:Pax7-nGFP satellite cells isolated by FACS and cultured for 72 h in the presence of COLI or COLV. Results are normalized to Tbp (n = 3 mice). i, Immunostainings for CALCR protein of Tg:Pax7-nGFP satellite cells cultured for 72 h in presence of COLI or COLV (n = 3 mice, ≥50 cells, 2 wells per condition). Data are mean ± s.d.; b, two-sided unpaired t-test; c–i, two-sided paired t-test. Scale bars, 25 μm (g), 50 μm (a, b, d, i).
Extended Data Fig. 6 CALCR ligand elcatonin can substitute the depletion of the surrogate ligand COLV.
a, Intracellular levels of cAMP in Calcr-transduced C2C12 cells treated with COLV for up to 480 min (n = 4 independent assays). b, Rescue of loss of COLV by elcatonin in an ex vivo self-renewal reserve-cell model, where PAX7+ non-proliferative cells return to quiescence (see Methods). MyHC and PAX7 staining of control (Ctr: Tg:Pax7-CT2;Col5a1+/+;R26mTmG) and Col5a1-null (Tg:Pax7-CT2;Col5a1flox/flox;R26mTmG) cells, non-treated (NT) or treated with elcatonin. No GFP+EdU+ cells (12 h pulse) could be detected under any of the conditions, indicating GFP+ cells are quiescent (data not shown). c, Quantification of percentage of reserve cells (PAX7+ per total nuclei) (n = 3 mice per genotype and condition, ≥350 cells counted). Elcat, elcatonin. Data are mean ± s.d.; two-sided paired t-test; #, P value calculated by two-sided unpaired t-test. Scale bar, 50 μm.
Extended Data Fig. 7 Schematic of Notch–COLV–CALCR axis in satellite cells.
A Notch–COLV–CALCR signalling cascade actively maintains satellite cell quiescence. Satellite cells are in direct contact with the plasma membrane of the myofibre (black outline) and an overlying basement membrane (blue line). Activation of the Notch receptor is achieved by a ligand (probably DLL1 or DLL4) present on the muscle fibre. Induction of Col5a1 and Col5a3 (and also Col6a1 and Col6a2) genes occurs via distal regulatory elements (grey box). Satellite-cell-produced COLV is deposited under the basement membrane and acts as a surrogate ligand of the plasma membrane receptor CALCR, also expressed by the satellite cells, thereby propagating a cell-autonomous signalling system in the local niche. In the absence of COLV (deletion of Col5a1) the quiescent niche is disturbed, CALCR signalling is abrogated, and satellite cells spontaneously differentiate and fuse to myofibres, leading to exhaustion of the muscle stem cell pool.
Supplementary information
Supplementary Information
This file contains Supplementary Tables 1-3
Rights and permissions
About this article

Cite this article
Baghdadi, M.B., Castel, D., Machado, L. et al. Reciprocal signalling by Notch–Collagen V–CALCR retains muscle stem cells in their niche. Nature 557, 714–718 (2018). https://doi.org/10.1038/s41586-018-0144-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-018-0144-9
- Springer Nature Limited
This article is cited by
-
Neurofibromin 1 controls metabolic balance and Notch-dependent quiescence of murine juvenile myogenic progenitors
Nature Communications (2024)
-
RhoA-mediated G12-G13 signaling maintains muscle stem cell quiescence and prevents stem cell loss
Cell Discovery (2024)
-
Post-transcriptional regulation of myogenic transcription factors during muscle development and pathogenesis
Journal of Muscle Research and Cell Motility (2024)
-
Striated muscle: an inadequate soil for cancers
Cancer and Metastasis Reviews (2024)
-
Organoid culture promotes dedifferentiation of mouse myoblasts into stem cells capable of complete muscle regeneration
Nature Biotechnology (2024)