

Abstract
The light-harvesting 1–reaction centre (LH1–RC) complex is a key functional component of bacterial photosynthesis. Here we present a 2.9 Å resolution cryo-electron microscopy structure of the bacteriochlorophyll b-based LH1–RC complex from Blastochloris viridis that reveals the structural basis for absorption of infrared light and the molecular mechanism of quinone migration across the LH1 complex. The triple-ring LH1 complex comprises a circular array of 17 β-polypeptides sandwiched between 17 α- and 16 γ-polypeptides. Tight packing of the γ-apoproteins between β-polypeptides collectively interlocks and stabilizes the LH1 structure; this, together with the short Mg–Mg distances of bacteriochlorophyll b pairs, contributes to the large redshift of bacteriochlorophyll b absorption. The ‘missing’ 17th γ-polypeptide creates a pore in the LH1 ring, and an adjacent binding pocket provides a folding template for a quinone, Q P, which adopts a compact, export-ready conformation before passage through the pore and eventual diffusion to the cytochrome bc 1 complex.
Similar content being viewed by others

Main
Photosynthesis provides the energy for almost all life on Earth. In the early stages of photosynthesis, light-harvesting complexes absorb solar energy, which is transferred to a membrane-bound RC, where a charge separation initiates the eventual formation of a reduced electron acceptor 1,2,– 3. The basic functional unit in purple phototrophic bacteria is LH1–RC, the complex of LH1 and the RC, in which the RC is surrounded by a ring-like oligomeric assembly of LH1 α- and β-heterodimers that bind bacteriochlorophyll (BChl) and carotenoid pigments. LH1–RC complexes in different species exhibit a variety of architectures: 16 LH1 α–β pairs completely encircle the RC in the Thermochromatium tepidum 4 and Rhodospirillum rubrum 5 complexes, whereas in Rhodopseudomonas palustris the RC is encircled by an open LH1 ring consisting of 15 α–β pairs and a W polypeptide 6. The Rhodobacter sphaeroides complex has a dimeric core 7, in which each monomer has 14 α–β pairs associated with one RC; two monomers associate through two PufX polypeptides to form an S-shaped LH1 ring 8, 9.
A high level of structural detail is required to account for the ability of LH1–RC complexes to absorb solar energy within a specific spectral range and to drive the formation of a quinol, which must traverse the confines of the LH1 ring encircling the RC. We identified the LH1–RC complex from Blastochloris (Blc.) viridis as a suitable target for a high-resolution structural study because it possesses unique architectural and spectroscopic features. Notably, the RC in the Blc. viridis photosynthetic complex yielded the first reported structure of a membrane protein complex 10, but electron microscopy has provided only low-resolution structures for the complete LH1–RC complex 11, 12. This complex accommodates BChl b rather than BChl a, and absorbs in the infrared at 1,015 nm, making it one of the most redshifted photosynthetic complexes described to date and one proposed as the basis for re-engineered photosynthesis 13. There is currently no known structural basis for this unusual in vivo absorption, which represents one of the largest redshifts observed in a photosynthetic pigment–protein complex, 220 nm from the 795 nm absorption maximum of BChl b in methanol. This property could be related to the composition of the Blc. viridis LH1 complex, which contains α-, β- and γ-polypeptides, but the position and function of the γ-subunit within the LH1 complex remain poorly understood. The Blc. viridis LH1 contains rare 1,2-dihydro-derivatives of neurosporene and lycopene as major carotenoids 14,15,– 16. The LH1–RC complex forms extensive arrays in the lamellar membranes of Blc. viridis 17,18,19,– 20, which have been hypothesized to consist of closed 16-membered LH1 rings that completely encircle each RC 20. However, such an arrangement of LH1 subunits represents a potential obstacle for quinol export from the RC to the external quinone pool in the membrane and eventual reduction of the cytochrome bc 1 complex. Here, we report a 3D cryo-electron microscopy (cryo-EM) structure at 2.9 Å resolution of this BChl b-based photosynthetic complex from Blc. viridis. Analysis of this structure shows how γ-apoproteins influence the large redshift observed in the BChl b-Q y-absorption band, reveals the position of the internal quinone channel, and identifies a third quinone binding site that prepares quinol for export through the pore in the LH1 ring.
Overall structure of the LH1–RC complex
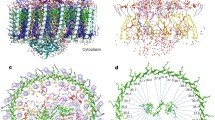
Extended Data Fig. 1 shows the absorption spectra of native photosynthetic membranes and LH1–RC complexes purified from Blc. viridis. The absorption maximum at 1,015 nm is ascribed to the Q y band of BChl b in the LH1 complex, which is slightly blueshifted to 1,008 nm after detergent solubilization and purification. Following vitrification of monodisperse complexes, we recorded 6,472 cryo-EM movies, from which 267,726 particles were picked manually for reference-free 2D classification. Further processing yielded a final resolution of 2.9 Å, enabling compilation of a colour-coded electron-density map (Fig. 1a–c) that reveals the detailed structural architecture of this LH1–RC complex and the relative locations of all pigments, cofactors and subunits. The dimensions of the LH1–RC are shown in Fig. 1c, d. The height of the core complex from the top of the periplasmic cytochrome to the bottom of the H subunit on the cytoplasmic side is 128.9 Å (Fig. 1a, d), and the diameters of this structure, which is slightly elliptical in projection, are 120.2 and 124.5 Å (Fig. 1c). The complex has a molecular weight of 414 kDa.

a– c, Views of the colour-coded LH1–RC density map. LH1-α (yellow), LH1-β (dark blue), LH1-γ (red), BChl b (light sea green), carotenoid (orange red), RC-C (green), RC-H (cyan), RC-L (orange) and RC-M (magenta). Detergent and other disordered molecules are in grey. a, View in the plane of the membrane; two dashed lines indicate the likely position of the membrane bilayer. b, Forty-five-degree rotation of a. c, Perpendicular view from the periplasmic side. Densities outside the membrane region have been truncated for clarity. d– f, Ribbon models corresponding to a, b and c but without the truncations in f. The LH1 subunits are numbered in f. Subunits 1 and 17 are outlined with dashed lines in f.
The RC in the cryo-EM map is similar to the one in the X-ray structure (for example, PDB: 1PRC) 21. The RC consists of H, M, L and cytochrome (C) subunits. Structural differences, indicated by residue–residue distance deviation 22, are small in subunits C, M and L (Extended Data Fig. 2b–d). However, interaction with the LH1 complex constrains a loop region on RC subunit H (RC-H; residues 47–54), resulting in a larger deviation from the RC-only structure (Extended Data Fig. 2a, e). There is also a small displacement of subunits RC-C and RC-H, which is likely to be caused by interaction with the LH1 complex, which bends the RC via a hinge point near the interface between the RC-C and RC-M–RC-L subunits (Extended Data Fig. 2a). The LH1 complex encircles the RC, including subunits C, H, M and L, the structures of which are in agreement with previous studies 23 (Fig. 1b, c, e, Extended Data Fig. 2).
The LH1 complex surrounds the RC to form a closed elliptical ring. The lengths of the major and minor axes of the elliptical ring measured from centre to centre of the transmembrane helices are 75.2 and 78.7 Å for the α-ring, 107.5 and 111.7 Å for the β-ring, and 109.6 and 114.8 Å for the γ-ring, respectively. The LH1 ring consists of 17 components, with 16 heterotrimers of α–β–γ-polypeptides and one α–β-heterodimer (Fig. 1c, f). Each α-, β- and γ-polypeptide contains a single transmembrane helix. A short N-terminal helix in α-polypeptides runs parallel to the membrane surface, whereas the C-terminal region contains a loop structure. No helical structures are observed in the C- and N-terminal regions of the β-polypeptide. The N termini of the α- and β-polypeptides are on the cytoplasmic side of the membrane, but the γ-subunit has the opposite topology, with its N terminus on the periplasmic side (Extended Data Fig. 3). This arrangement of LH1 polypeptides creates a triple-ring LH1 complex, consisting of an inner circle of 17 α-polypeptides, an outer ring of 16 γ-polypeptides, and a 17 β-polypeptide ring sandwiched between them (Fig. 1c, f). Each of the 16 γ-polypeptides sits between two β-polypeptides, and the gap where the ‘missing’ 17th γ-polypeptide would otherwise be located creates an opening in the LH1 ring (Fig. 1c, f) for quinol exchange.
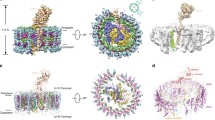
Two BChl b molecules and one carotenoid, an all- trans-1,2-dihydro-derivative of neurosporene ( n = 9) or lycopene ( n = 11), are non-covalently bound between each α–β-pair, and no pigment molecules are bound to the γ-polypeptide (Fig. 3). Major cofactors bound within the RC are as previously reported 21 with the addition of the ubiquinone-9 Q P (Fig. 2), and are arranged in the expected local pseudo-two-fold rotational symmetry (Fig. 2).

a, Pigment molecules viewed from the periplasmic side by tilting 45° in the plane of the membrane. b, RC pigment molecules viewed from the membrane plane. A local pseudo-C2 symmetry axis is shown as a dashed line. Car, carotenoid; HEM, haem cofactors of the cytochrome subunit; SP, special pair of BChl b pigments; Fe, non-haem iron; BPhe b, bacteriopheophytin b.

a, LH1 subunits 1–3 (Fig. 1f) illustrate inter-subunit interactions. Colours as in Fig. 1 except BChl b molecules in medium blue and all- trans-1,2-dihydroneurosporene in orange. Hydrogen bonds are indicated by dashed lines. b, A single LH1 α–β–γ-subunit, with the polypeptides shown in loop representation for clarity. The red arrow indicates a putative direction of approach of the γ-polypeptide to the α–β-pair during assembly of the complex. c, Projection view to show interactions made by a carotenoid with nearby pigments and polypeptides.
Interactions that stabilize the LH1 ring
The cryo-EM model of the LH1–RC from Blc. viridis reveals a complicated interconnecting series of protein–protein, pigment–protein and pigment–pigment associations within the LH1 ring. For simplicity, the LH1 heterotrimer subunits 1, 2 and 3 are used to demonstrate the stabilizing intra- and inter-subunit interactions in the LH1 complex. Inter-subunit hydrogen bonds on the periplasmic side are α( n)-Arg44 to β( n − 1)-Val55 (bond length 3.0 Å); and β( n)-Arg44 to β( n − 1)-Ala48 (3.3 Å) (Fig. 3a). There is an intra-subunit hydrogen bond between α-Arg44 on the periplasmic side and the carboxyl group of β-Trp46 (3.0 Å), which stabilizes the C-terminal loops of both the α- and β-polypeptides (Fig. 3b). The γ( n)-polypeptide forms two hydrogen bonds with the α( n)- and β( n)-polypeptides: γ-Asp14 to β-Trp41 (3.0 Å) and γ-Arg36 to the carboxyl group of α-Thr6 (3.1 Å) (Fig. 3b). Thus, an LH1 heterotrimer subunit is formed from α( n)–β( n)–γ( n), and not α( n + 1)–β( n + 1)–γ( n). This arrangement suggests an assembly sequence for the LH1 complex. It is likely that once an α(1)–β(1) subunit is formed, it interacts with RC-H to form an anchor point through the hydrogen bond between α(1)-Arg19 and RC-H-Ser256. Then, the γ(1)-polypeptide binds to the α–β-subunit to form the first LH1 subunit α(1)–β(1)–γ(1). To do so, the γ-polypeptide needs space to access the α–β-subunit by rotating and translating to achieve the correct angle of approach and the correct orientation. This procedure continues until the 17th α–β-subunit is assembled. At this point, there is no space for a correct direction of approach and orientation that would allow the 17th γ-polypeptide to dock with the 17th α–β-subunit, resulting in a gap in the LH1 ring.
The LH1–RC from Blc. viridis reveals the basis for the stabilizing effects of carotenoids, which mainly rely on hydrophobic forces, and for excitation energy transfer from carotenoids to BChls 15. Interactions of each carotenoid with n + 1, n and n − 1 polypeptides and with bound BChls effectively crosslink one LH1 α–β-subunit to the next (Fig. 3c). One end of the carotenoid is in close proximity to the upstream neighbouring LH1 α( n + 1) near its C terminus (Phe37, 3.1 Å; Leu33, 3.7 Å; Ala32, 3.4 Å; His36, 3.9 Å); the other end approaches the downstream neighbouring LH1 α( n − 1) near its N terminus (Leu11, 4.3 Å; Lys10, 5.1 Å). In particular, this end of the carotenoid is also in close proximity to the β( n)-N terminus. The middle part of the carotenoid is close to the phytyl tails of the α- (3.2 Å) and β- (4.0 Å) BChl b molecules (Fig. 3c).
Subunits 1–16 of the LH1 complex (Fig. 1f) consist of one each of α-, β- and γ-polypeptides, two BChl b molecules and one all- trans carotenoid. The γ-polypeptide has no histidine residue and does not bind BChl b. Figure 3b illustrates this point using subunit 3; α-His36 forms a ligand with the central Mg of α-BChl b (2.5 Å) and β-His37 forms a ligand with β-BChl b (2.2 Å) (Fig. 3b, c). The C3-acetyl groups of α-BChl b and β-BChl b form a hydrogen bond with α-Trp47 (2.9 Å) and β-Trp46 (2.9 Å), respectively, to orientate the bacteriochlorin rings of BChl b. This orientation is further stabilized by a 3.0 Å hydrogen bond between β-Tyr29 and the ester group of β-BChl b on C13 2. The OH group of β-Tyr29 could form a hydrogen bond with the ester group on the phytyl tail of the α- or β-BChl b.
LH1–RC interactions
The resolution of the cryo-EM structure of the LH1–RC complex is sufficient to enable detailed analysis of the protein–protein and protein–pigment interactions within the complex. The protein–pigment interactions in the RC have previously been described in detail 24, and the relationship between the RC and its encircling LH1 can now be defined. Figure 4a shows the overall organization of the LH1–RC complex, which is divided into three zones. Zone 1 (AOC) includes a close contact between LH1 and the RC, a hydrogen bond between LH1-α1-Arg19 and RC-H-Ser256 (2.8 Å), which is likely to be the site for initiating encirclement by LH1 in a manner analogous to that in the LH1–RC–PufX complex of Rba. sphaeroides 6. This trimeric α–β–γ subunit is designated as LH1 subunit 1 (Fig. 1f). Proximity between transmembrane helix RC-L A and LH1-α2, with a centre–centre helix distance of approximately 10 Å, could facilitate the encirclement process. A third interaction in this region involves LH1-α3 and LH1-α4 on the cytoplasmic side, which constrains a loop on RC-H (Leu47–Pro54). In zone 2 (COB) there is a single point of contact between the RC and LH1, between the RC-M A helix and LH1-α9. The gap between the RC and LH1 in this region is mainly filled by the single transmembrane helix of RC-H and lipid molecules (Extended Fig. 4a). Zone 3 (BOA) is where quinol–quinone exchange occurs at the RC Q B site, and where newly released quinols and quinones arriving from outside create a dynamic quinone pool 6. Thus, the structure of the gap between the RC and LH1 in this region shows disordered densities arising from lipids and quinones 5 (Extended Data Fig. 4a). Figure 4b summarizes all intra- and inter-subunit protein–protein and protein–pigment interactions in the LH1 complex, and highlights the extent of the interactions that stabilize the LH1 complex.

a, Periplasmic side of the LH1–RC core complex; colour coding as in Fig. 1. RC-H Ser256 and LH1-α1 Arg19 are highlighted using space-fill. The RC-H-loop Leu47 to Pro54 is highlighted in orange. b, Summary of intra- and inter-subunit interactions in the LH1 complex. Only transmembrane helices of LH1 polypeptides are shown for clarity. All arrows indicate hydrogen-bonding interactions.
Structural basis for the redshift
The LH1–RC complex of Blc. viridis is able to absorb energy in the infrared region of the spectrum owing to the unusually large redshift it imposes on the BChl b pigment; its 1,015-nm absorption maximum represents, to our knowledge, the lowest energy light used by a photosynthetic bacterium. Previous studies have identified several influences on the redshift of the BChl a or BChl b–Q y absorption maximum in the bacterial light-harvesting complex 25, 26. The cryo-EM structure of the Blc. viridis LH1–RC complex shows that at least five factors contribute to the large bathochromic shift of the BChl b-Q y band.
The first factor is the chemical structure. The extra C–C double bond in BChl b relative to BChl a extends conjugation in the bacteriochlorin ring and redshifts the Q y band. The 795-nm absorption maximum of BChl b in methanol is 24 nm further towards the near infrared than that of BChl a, which directly affects the ‘site energy’ within coupled BChl b aggregates in the LH1–RC complex.
The second factor is protein–pigment interactions. As already noted (Fig. 3c), the carotenoids interlink LH1 α–β-subunits (Fig. 3c) and the C3 acetyl groups of α- and β-BChls b hydrogen bond to Trp residues in LH1 (Fig. 3b), adopting an in-plane conformation similar those of the B800-850 BChls a in the LH2 complex of Rhodopseudomonas acidophila 27, 28. Experiments combining mutagenesis and Raman spectroscopy have shown that hydrogen bonds redshift the absorption of the Rba. sphaeroides LH1 complex 29, 30.
The third factor is the number of coupled BChl a and b molecules; the 17 pairs of coupled BChl b molecules in the LH1–RC complex of Blc. viridis represent the largest reported circular aggregate of pigments in light-harvesting complexes from photosynthetic bacteria 31. Increasing the oligomeric size of LH1 subunits in the LH1 complex of Rba. sphaeroides from 2 to 6 or 7 is accompanied by redshifts of 6–7 nm in absorption and fluorescence emission of the BChl a-Q y band, although larger oligomers produced no further redshifts 32.
The fourth factor is the structures of BChl a or BChl b aggregates. The Mg–Mg distances within BChl pairs reflect the degree of overlap, and therefore the electronic coupling and Q y redshifting, of BChl a and BChl b in light-harvesting complexes. Extended Data Fig. 5 shows the linear correlation of Q y-band maximum versus inter- and intra-subunit Mg–Mg distances in five different light-harvesting complexes, which shows a stronger correlation for the intra-subunit distances. The intra-subunit (8.8 Å) or inter-subunit (8.5 Å) Mg–Mg distances of BChl b in Blc. viridis are the shortest reported for a bacterial light-harvesting complex.
Finally, the shift is affected by the structural rigidity enforced by the γ-apoproteins. Sixteen γ-apoproteins pack tightly between β-polypeptides, and collectively interlock the LH1 structure through 32 hydrogen bonds to α- and β-polypeptides, constraining free movement of the LH1 ring and stabilizing the BChl b pairs in the complex and thereby contributing to the redshift of the BChl b-Q y band 33. There are parallels with the large redshift of BChl a to 915 nm in the LH1–RC complex from Tch. tepidum (Extended Data Fig. 6a). In this case, bound Ca 2+ ions constrain conformational flexibility 34 and limit disorder in site energies. Inhomogeneous narrowing is accompanied by mixing of charge transfer and lowest exciton states, which has been hypothesized to be the basis for the redshift in this complex 25.
A template for preparing quinols for export
The LH1–RC of Blc. viridis houses the RC quinones Q A and Q B, and a third quinone, Q P (Fig. 5a–c). The binding sites of Q A and Q B are similar to those reported previously, although their tail structures differ from those in isolated RCs 21, 35 (Extended Data Fig. 6b, c). Q P is located near the gap in the LH1 ring, 48.9 Å away from the Q B-binding site. The head of the Q P molecule is stabilized by π–π-interactions with RC-L-Phe40, the aromatic ring of which is roughly parallel to the plane of the quinone-head group at a distance of 3.6 Å. Q P is also in close proximity (3.0 Å) to LH1-α1-Tyr27, the aromatic ring of which is roughly perpendicular to the plane of the Q P-head ring. Unlike Q A and Q B in the RC, the tail of Q P is not free to move, and is instead conformationally constrained by a series of contacts with LH1-α(1)-Phe37 (4.7 Å), RC-L-Gln87 (2.4 Å), RC-L-Trp142 (3.5 Å) and RC-L-Val91(4.4 Å) (Extended Data Fig. 6d). This Q P-binding pocket provides a folding template such that Q P assumes a compact conformation and a suitable orientation before entering the pore in LH1 at the position of the absent 17th γ-apoprotein (Fig. 5c, d).

a, The LH1–RC complex viewed from the periplasmic side, with 80% transparency applied to the RC, and LH1 subunits 9–17. A green arrow indicates the gap between subunits 1 and 17. b, LH1–RC rotated 90° from a, with Q B and Q P viewed by removing LH1 subunits 1–8. c, Close-up of the Q P-binding pocket. d, Ribbon representation of the Q P region. Green arrow as in a. e, Close-up view of the quinone–quinol channel (dashed circle) from outside the LH1 ring. f, Electron densities of pigments adjacent to the LH1 pore.
The cryo-EM structure of the LH1–RC complex reveals the mechanism by which quinone is translocated across the LH1 ring. Of the 17 subunits, 16 are α–β–γ-heterotrimers, and only one is an α–β-heterodimer. The 16 γ-polypeptides, located outside the β-ring, pack between β-apoproteins, leaving a gap in the LH1 ring between subunits 1 and 17, and dictate the position of the pore for quinone–quinol translocation. The Q P-binding pocket is located next to the pore, and the Q P molecule appears to be folded and oriented in the binding pocket in a manner that encourages passage through the LH1 ring (Fig. 5d). There is a distinct pore measuring around 5 × 7 Å between α17 and α1 (Fig. 5e), which is created by Arg18–Phe25 (sequence, RRVLTALF) in α17 and Leu15–Leu24 (LDPRRVLTAL) in α1 (Fig. 5e). It should be noted that the electron densities of β(17)-BChl b, α(1)-BChl b and the α(1)–β(1) carotenoid are weaker than those of their counterparts in the rest of the LH1 complex. This is particularly evident for those regions of the pigments that are close to the quinone pore, for example the phytyl tails and one end of the carotenoid, as shown in Fig. 5f. This weaker density reflects the relative flexibility of this region; thus the size of this pore could fluctuate transiently, facilitating the movement of the quinone and quinol molecules through the channel.
Methods
Protein purification
Wild-type Blc. viridis (DSM-133) was obtained from DSMZ. Photosynthetic cultures of Blc. viridis were grown in sodium succinate medium 27 (N medium) under illumination (100 μmol photons per m 2 s −1) at 30 °C in 20-l screw-capped vessels, completely filled with N 2-sparged medium, as previously described 36. Cells were collected when the culture reached an optical density of 1.6 at 680 nm by centrifugation at 3,290 g for 30 min. Washed cells were broken by passage through a French press three times at 18,000 p.s.i. The crude cell lysate was applied to a two-step sucrose gradient (15% and 40% (w/w) in an ultracentrifugation tube). Photosynthetic membrane was collected at the interface of 15% and 40% sucrose after 5 h centrifugation at 100,000 g. Membranes were pelleted and resuspended in working buffer (20 mM HEPES, pH 7.8). The optical density of the membrane was adjusted to ~100 at 1,015 nm. For solubilization of the core complexes, the optical density at 1,015 nm of the photosynthetic membrane was adjusted to 60, and 3% (w/w) n-dodecyl β- d-maltoside was added. This mixture was then stirred in the dark at 4 °C for 30 min. Unsolubilized material was removed by centrifugation for 1 h at 211,000 g. The clarified supernatant was loaded onto an ion exchange column pre-equilibrated with working buffer solution containing 0.03% n-dodecyl β- d-maltoside. The core complexes eluted at ~250 mM NaCl and were collected and concentrated. These were further purified using a Superdex 200 gel filtration column. The fractions with an absorption ratio of A 1,008 nm/ A 280 nm higher than 1.22 were pooled together and used for cryo-EM data collection.
Cryo-EM data collection
The protein concentration was adjusted to an optical density of 40 at 1,008 nm. Three microlitres of protein solution was applied to a glow-discharged holey carbon grid (Quantifoil grid R1.2/1.3, 300 mesh Cu). The grid was plunged into liquid ethane cooled by liquid nitrogen using a Leica EM GP vitrobot. Parameters were set as follows: blotting time 4 s, humidity 99%, sample chamber temperature 5 °C. The frozen grid was stored in liquid nitrogen before use. A second grid was prepared using a Quantifoil grid R3.5/1.0 covered by a thin carbon film (EM resolution, Inc.), with the protein diluted tenfold. Vitrification conditions were the same as for the first grid. Data were recorded at eBIC on a Titan Krios electron microscope with a Gatan 968 GIF Quantum with a K2 summit detector operating at 300 kV accelerating voltage, at nominal magnification of 130 k in counting mode. Movies were collected in super-resolution mode and Fourier-cropped to give a resulting calibrated pixel size of 1.06 Å at the specimen level. An energy-selecting slit of 20 eV was used. An exposure rate of 5 electrons per pixel per s was set and a fresh super-resolution gain reference was performed at this dose rate before data acquisition. A total dose of 45 electrons per Å 2 was used for movies of 20 frames. In total, 6,472 movies were collected with defocus values from 1.0 to 3.0 μm. Two typical cryo-EM images, which are averaged from motion-corrected movie frames, are shown in Extended Data Fig. 7a, b.
Data processing
All images that were empty, contained few particles, or were contaminated with ice were discarded. Dose-fractionated images were subjected to beam-induced motion correction using MotionCorr 37. Images derived from the sum of all frames were used for further data processing using RELION 2.0 38,39,– 40. CTF parameters were determined using gctf 41. In total, 267,726 particles were picked manually. These particles were subjected to reference-free 2D classification. Those particles that categorized into poorly defined classes were rejected. This cleaning procedure using 2D classification was repeated three times, resulting in rejection of 9.45% of total particles. The resulting 2D classes were subjected to an initial 3D model calculation using EMAN2 42 for maximum-likelihood-based 3D classification. One of the four stable 3D classes accounting for 62.3% total particles was selected for high resolution refinement and 3D reconstruction without subtraction of detergent micelle from the raw micrographs. This resulted in a map at a global resolution of 3.3 Å. The density map was corrected for the modulation transfer function (MTF) of the Gatan K2 summit camera and further sharpened using the post-processing subroutine in RELION 2.0 using an estimated temperature factor and a mask was created using RELION 2.0 with a lowpass of 15 Å and a soft edge of 7 Å. The Fourier shell correlation (FSC) curve corrected for masking is shown in Extended Data Fig. 7c. The estimate of final resolution of 2.9 Å for the LH1–RC map was based on a FSC cut off of 0.143. ResMap 43 was used for calculation of the local resolution map (Extended Data Fig. 4b, c).
Modelling and refinement
Initially, the crystal structure of the Blc. viridis RC (PDB: 1PRC) was fitted to the cryo-EM map as a rigid body using the ‘fit in map’ routine in Chimera 44. COOT 45 was then used for manual adjustment and real-space refinement for both polypeptides and cofactors. All amino acid sequences of polypeptides in the RC are listed in Extended Data Fig. 8. Ubiquinone-9 molecules (Q B and Q P) were also fitted to the density map independently using COOT.
For LH1, the electron density of LH1 subunit 3 was selected for modelling first. On the basis of structural similarity compared with the LH1 of Tch. tepidum 5 and LH2 of Rhodospirillum molischianum 46, the locations of His residues, which ligate BChl b molecules in the α–β-polypeptides (Extended Data Fig. 9), were located in the density map. The fitted RC was used as a reference to determine the orientation of the α–β-polypeptides. Their amino acid sequences, taken from previous work 47, were fitted into an electron-density map using COOT. Two BChl b molecules and one all- trans carotenoid were added into the model based on their densities. Analysis of pigment composition shows that the major carotenoid in the core complex is all- trans-1,2-dihydroneurosporene 15; this carotenoid was therefore modelled into the density map. Having no His residues, the γ-polypeptide does not bind BChl b molecules. No 3D structural information of the γ-subunit was available, but the 2.9 Å resolution allows assignment of the larger amino acid side chains such as Trp and Tyr. By matching three Trps and one Tyr residue in the γ-polypeptide, its orientation was determined, and all other residues were traced based on the density map using COOT. Comparison with the sequence of the γ-polypeptide 47 leaves 12 N-terminal residues unaccounted for. The structure of the LH1 α–β–γ-subunit was then used as a rigid body to fit into the density map for other LH1 subunits. For the LH1 subunit 17, only α–β and pigments were used. All of the LH1 subunits then underwent real-space refinement using COOT. The final model was subjected to global refinement and minimization using REFMAC5 48. The final refinement statistics are summarized in Extended Data Table 1. The quality of fit for the structural model within the electron-density map was validated using EMRinger 49.
Data availability
The cryo-EM density map has been deposited in the World Wide Protein Data Bank (wwPDB) under accession code EMD-3951 and the coordinates have been deposited in the Protein Data Bank (PDB) under accession number 6ET5.
Change history
05 April 2018
The Extended Data Figures and Tables section originally published with this article was missing Table 1. This has now been corrected.
References
Blankenship, R. E. Molecular Mechanisms of Photosynthesis 2nd edn (John Wiley & Sons, Oxford, 2014).
Sundström, V., Pullerits, T. & van Grondelle, R. Photosynthetic light-harvesting: Reconciling dynamics and structure of purple bacterial LH2 reveals function of photosynthetic unit. J. Phys. Chem. B 103, 2327–2346 (1999).
Cogdell, R. J., Gall, A. & Köhler, J. The architecture and function of the light-harvesting apparatus of purple bacteria: from single molecules to in vivo membranes. Q. Rev. Biophys. 39, 227–324 (2006).
Niwa, S. et al. Structure of the LH1–RC complex from Thermochromatium tepidum at 3.0 Å. Nature 508, 228–232 (2014).
Jamieson, S. J. et al. Projection structure of the photosynthetic reaction centre-antenna complex of Rhodospirillum rubrum at 8.5 A resolution. EMBO J. 21, 3927–3935 (2002).
Roszak, A. W. et al. Crystal structure of the RC–LH1 core complex from Rhodopseudomonas palustris. Science 302, 1969–1972 (2003).
Jungas, C., Ranck, J. L., Rigaud, J. L., Joliot, P. & Verméglio, A. Supramolecular organization of the photosynthetic apparatus of Rhodobacter sphaeroides. EMBO J. 18, 534–542 (1999).
Qian, P., Hunter, C. N. & Bullough, P. A. The 8.5 Å projection structure of the core RC–LH1–PufX dimer of Rhodobacter sphaeroides. J. Mol. Biol. 349, 948–960 (2005).
Qian, P. et al. Three-dimensional structure of the Rhodobacter sphaeroides RC–LH1–PufX complex: dimerization and quinone channels promoted by PufX. Biochemistry 52, 7575–7585 (2013).
Deisenhofer, J., Epp, O., Miki, K., Huber, R. & Michel, H. X-ray structure analysis of a membrane protein complex. Electron density map at 3 Å resolution and a model of the chromophores of the photosynthetic reaction center from Rhodopseudomonas viridis. J. Mol. Biol. 180, 385–398 (1984).
Miller, K. R. & Jacob, J. S. The Rhodopseudomonas viridis photosynthetic membrane: arrangement in situ. Arch. Microbiol. 142, 333–339 (1985).
Ikeda-Yamasaki, I., Odahara, T., Mitsuoka, K., Fujiyoshi, Y. & Murata, K. Projection map of the reaction center-light harvesting 1 complex from Rhodopseudomonas viridis at 10 Å resolution. FEBS Lett. 425, 505–508 (1998).
Blankenship, R. E. et al. Comparing photosynthetic and photovoltaic efficiencies and recognizing the potential for improvement. Science 332, 805–809 (2011).
Magdaong, N. C. M., Niedzwiedzki, D. M., Goodson, C. & Blankenship, R. E. Carotenoid-to-bacteriochlorophyll energy transfer in the LH1–RC core complex of a bacteriochlorophyll b containing purple photosynthetic bacterium Blastochloris viridis. J. Phys. Chem. B 120, 5159–5171 (2016).
Thornber, J. P., Cogdell, R. J., Seftor, R. E. B. & Webster, G. D. Further studies on the composition and spectral properties of the photochemical reaction centers of bacteriochlorophyll b-containing bacteria. Biochim. Biophys. Acta 593, 60–75 (1980).
Malhotra, H. C., Britton, G. & Goodwin, T. W. Occurrence of 1,2-dihydro-carotenoids in Rhodopseudomonas viridis. J. Chem. Soc. Chem. Comm. 127, 127–128 (1970).
Konorty, M. et al. Photosynthetic system in Blastochloris viridis revisited. Biochemistry 48, 4753–4761 (2009).
Welte, W. & Kreutz, W. Structure of thylakoids in cells of Rhodopseudomonas viridis as influenced by growth conditions. Arch. Microbiol. 141, 325–329 (1985).
Engelhardt, H., Baumeister, W. & Saxton, W. O. Electron microscopy of photosynthetic membranes containing bacteriochlorophyll b. Arch. Microbiol. 135, 169–175 (1983).
Scheuring, S. et al. Nanodissection and high-resolution imaging of the Rhodopseudomonas viridis photosynthetic core complex in native membranes by AFM. Proc. Natl Acad. Sci. USA 100, 1690–1693 (2003).
Deisenhofer, J., Epp, O., Sinning, I. & Michel, H. Crystallographic refinement at 2.3 Å resolution and refined model of the photosynthetic reaction centre from Rhodopseudomonas viridis. J. Mol. Biol. 246, 429–457 (1995).
Chen, J. E., Huang, C. C. & Ferrin, T. E. RRDistMaps: a UCSF Chimera tool for viewing and comparing protein distance maps. Bioinformatics 31, 1484–1486 (2015).
Deisenhofer, J., Epp, O., Miki, K., Huber, R. & Michel, H. Structure of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3 Å resolution. Nature 318, 618–624 (1985).
Deisenhofer, J. & Michel, H. Nobel lecture. The photosynthetic reaction centre from the purple bacterium Rhodopseudomonas viridis. EMBO J. 8, 2149–2170 (1989).
Ma, F., Yu, L. J., Wang-Otomo, Z. Y. & van Grondelle, R. The origin of the unusual Q y red shift in LH1–RC complexes from purple bacteria Thermochromatium tepidum as revealed by Stark absorption spectroscopy. Biochim. Biophys. Acta 1847, 1479–1486 (2015).
Cogdell, R. J., Howard, T. D., Isaacs, N. W., McLuskey, K. & Gardiner, A. T. Structural factors which control the position of the Q y absorption band of bacteriochlorophyll a in purple bacterial antenna complexes. Photosynth. Res. 74, 135–141 (2002).
McDermott, G. et al. Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature 374, 517–521 (1995).
McLuskey, K., Prince, S. M., Cogdell, R. J. & Isaacs, N. W. The crystallographic structure of the B800–820 LH3 light-harvesting complex from the purple bacteria Rhodopseudomonas acidophila strain 7050. Biochemistry 40, 8783–8789 (2001).
Olsen, J. D., Sockalingum, G. D., Robert, B. & Hunter, C. N. Modification of a hydrogen bond to a bacteriochlorophyll a molecule in the light-harvesting 1 antenna of Rhodobacter sphaeroides. Proc. Natl Acad. Sci. USA 91, 7124–7128 (1994).
Sturgis, J. N., Olsen, J. D., Robert, B. & Hunter, C. N. Functions of conserved tryptophan residues of the core light-harvesting complex of Rhodobacter sphaeroides. Biochemistry 36, 2772–2778 (1997).
Cogdell, R. J. & Roszak, A. W. Structural biology: the purple heart of photosynthesis. Nature 508, 196–197 (2014).
Westerhuis, W. H. J., Sturgis, J. N., Ratcliffe, E. C., Hunter, C. N. & Niederman, R. A. Isolation, size estimates, and spectral heterogeneity of an oligomeric series of light-harvesting 1 complexes from Rhodobacter sphaeroides. Biochemistry 41, 8698–8707 (2002).
Yu, L. J., Kawakami, T., Kimura, Y. & Wang-Otomo, Z. Y. Structural basis for the unusual Q y red-shift and enhanced thermostability of the LH1 complex from Thermochromatium tepidum. Biochemistry 55, 6495–6504 (2016).
Jakob-Grun, S., Radeck, J. & Braun, P. Ca 2+-binding reduces conformational flexibility of RC–LH1 core complex from thermophile Thermochromatium tepidum. Photosynth. Res. 111, 139–147 (2012).
Roszak, A. W. et al. New insights into the structure of the reaction centre from Blastochloris viridis: evolution in the laboratory. Biochem. J. 442, 27–37 (2012).
Lang, F. S. & Oesterhelt, D. Microaerophilic growth and induction of the photosynthetic reaction center in Rhodopseudomonas viridis. J. Bacteriol. 171, 2827–2834 (1989).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Scheres, S. H. W. Semi-automated selection of cryo-EM particles in RELION-1.3. J. Struct. Biol. 189, 114–122 (2015).
Scheres, S. H. W. Processing of structurally heterogeneous cryo-EM data in RELION. Methods Enzymol. 579, 125–157 (2016).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Koepke, J., Hu, X., Muenke, C., Schulten, K. & Michel, H. The crystal structure of the light-harvesting complex II (B800-850) from Rhodospirillum molischianum. Structure 4, 581–597 (1996).
Brunisholz, R. A., Jay, F., Suter, F. & Zuber, H. The light-harvesting polypeptides of Rhodopseudomonas viridis—the complete amino acid sequences of B1015-α, B1015-β and B1015-γ. Biol. Chem. 366, 87–98 (1985).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367 (2011).
Barad, B. A. et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015).
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand and contrast loss in single particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003).
Acknowledgements
C.N.H. and P.Q. acknowledge support from the Biotechnology and Biological Sciences Research Council (BBSRC) UK, award number BB/M000265/1, the European Research Council Advanced Award 338895 and the Sheffield University Imagine programme. C.N.H. was also partially supported by the Photosynthetic Antenna Research Center (PARC), an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences (Award number DE-SC0001035). D.P.C. acknowledges funding from the European Commission (Marie Skłodowska-Curie Global Fellowship 660652).
Reviewer information
Nature thanks R. Cogdell, R. A. Niederman and J. Rubinstein for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
P.Q. and C.N.H. conceived the study. P.Q. and C.N.H. designed the experiments. P.Q., C.A.S., D.P.C. and P.W. performed the experiments. P.Q. analysed the results and generated structural models. P.Q. and C.N.H. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Absorption spectra of photosynthetic membranes and the purified LH1–RC core complex from Blc. viridis.
Absorption spectra of isolated membranes (dashed line) and the purified LH1–RC complex (solid line) were recorded at room temperature and normalized at their Q y bands at 1,015 nm and 1,008 nm. The peak at 831 nm together with a shoulder at ~970 nm arise from BChl b in the RC. Bacteriopheophytin appears as a poorly resolved peak at about 810 nm. The Q x bands give rise to a composite peak at 602 nm. The minor peak at about 558 nm arises from the cytochromes, the Soret band of which contributes in the approximately 410-nm region. Absorption features at 482, 450 and 420 nm correspond to carotenoids and the 399-nm maximum corresponds to the Soret band of BChl b in the core complex. No oxidized BChl b is observed which, if present, would cause an absorption peak at about 685 nm.
Extended Data Fig. 2 Residue–residue distance deviation between cryo-EM and X-ray structures of the RC from Blc. viridis.
a, Superposition of the X-ray structure (PDB: 1PRC, grey) and the cryo-EM structure (colour-coded as in Fig. 1) of the RC. A putative hinge point is indicated with a red dot. The bending direction of the cryo-EM structure is indicated with two green arrows. A red arrow points to a flexible RC-H loop. b– e, Residue–residue (RR) distance deviation maps 22 of the individual RC subunits C, M, L and H, respectively, comparing the structures from cryo-EM and X-ray crystallography (PDB: 1PRC) 21. Each vertical scale shows the standard deviation (s.d.) in Å. The flexible loop of RC-H is indicated with a red perpendicular arrow in e.
Extended Data Fig. 3 Cryo-EM densities and structural models of polypeptides and pigments in the Blc. viridis LH1–RC complex.
The colour code is the same as in Fig. 1. The contour levels of the density maps were adjusted to mirror their molecular weights.
Extended Data Fig. 4 Electron densities between and outside the LH1 and RC complexes, and local resolution maps of the LH1–RC core complex.
a, The LH1–RC complex as shown in Fig. 1f, but displayed at 70% transparency. Electron densities belonging to detergent, lipid and other disordered molecules are in grey. b, Side view of the core complex with the periplasmic side uppermost. c, View of the periplasmic side. All membrane-extrinsic parts of the complex were truncated for clarity. The coloured bar chart on the right shows the local structural resolution in Å.
Extended Data Fig. 5 Relationship between BChl a and BChl b Mg–Mg distances and Q y-band absorption in bacterial light harvesting complexes.
a, Correlation of Q y-band maximum and inter-subunit BChl a and BChl b Mg–Mg distances in five bacterial light-harvesting complexes. b, As in a, but for intra-subunit Mg–Mg distances. c, Values for the linear correlation coefficient R, calculated using least-squares linear regression ( n = 5 biologically independent samples in each case; one-sided significance test).
Extended Data Fig. 6 Structural comparisons of selected cofactors and details of the Q P binding site.
a, The LH1-B1008 BChl b pair from Blc. viridis (blue) compared with the LH1-B915 BChl a pair (green) from the X-ray structure of the Tch. tepidum LH1–RC complex (PDB: 3WMM). b, Comparison of the Q A menaquinone-9 (blue) from the cryo-EM model of the Blc. viridis LH1–RC with the Q A (green) from the X-ray structure of the Blc. viridis RC (PDB: 3T6E). c, As in b, but comparing Q B. d, The Q P binding site. Only LH1-α1 and part of RC-L are shown for clarity. Yellow, LH1-α1; orange, RC-L; blue, Q P; wheat, Q B. Amino acid residues making close contacts around Q P are numbered and listed accordingly.
Extended Data Fig. 7 Cryo-EM micrographs of the LH1–RC complex from Blc. viridis and calculation of the cryo-EM map resolution.
a, Protein particles embedded in vitrified ice. Examples of LH1–RC complexes are circled. 6,472 cryo-EM movies were recorded, from which 267,726 particles were picked manually for reference-free two-dimensional classification. During data processing, datasets of around 100,000 and around 167,000 particles were used independently for 3D reconstruction. They generated very similar 3D maps for the LH1–RC complex, so they were combined. b, The LH1–RC particles are covered by a thin layer of vitrified ice on a supported carbon film. Each image measures 393.2 × 406.8 nm. c, Gold-standard refinement was used for estimation of the final map resolution. The global resolution of 2.9 Å was calculated using an FSC cut off of 0.143.
Extended Data Fig. 8 Amino acid sequence of polypeptides in the LH1–RC complex from Blc. viridis.
Black, genome sequence; red, protein sequence; blue, missing in protein sequence.
Extended Data Fig. 9 Amino acid sequence alignment of LH1 α- and β-polypeptides in LH1–RC core complexes from purple photosynthetic bacteria.
All sequences have been aligned relative to the His residue that ligates BChls in the LH1 complexes. The α- and β-polypeptides of the P. molischianum LH2 complex are included for comparison. The sequence alignment was performed using CLUSTAL O v.1.2.4.
Supplementary information
Rights and permissions
About this article

Cite this article
Qian, P., Siebert, C.A., Wang, P. et al. Cryo-EM structure of the Blastochloris viridis LH1–RC complex at 2.9 Å. Nature 556, 203–208 (2018). https://doi.org/10.1038/s41586-018-0014-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-018-0014-5
- Springer Nature Limited
This article is cited by
-
Bioinspired polymeric supramolecular columns as efficient yet controllable artificial light-harvesting platform
Nature Communications (2024)
-
De novo design of proteins housing excitonically coupled chlorophyll special pairs
Nature Chemical Biology (2024)
-
Atomic force microscopic analysis of the light-harvesting complex 2 from purple photosynthetic bacterium Thermochromatium tepidum
Photosynthesis Research (2023)
-
Structural basis for the assembly and quinone transport mechanisms of the dimeric photosynthetic RC–LH1 supercomplex
Nature Communications (2022)
-
An LH1–RC photocomplex from an extremophilic phototroph provides insight into origins of two photosynthesis proteins
Communications Biology (2022)