

Abstract
Multiple sclerosis (MS) is an immune-mediated disease of the central nervous system that causes demyelination, axonal degeneration and astrogliosis, resulting in progressive neurological disability. Fuelled by an evolving understanding of MS immunopathogenesis, the range of available immunotherapies for clinical use has expanded over the past two decades. However, MS remains an incurable disease and even targeted immunotherapies often fail to control insidious disease progression, indicating the need for new and exceptional therapeutic options beyond the established immunological landscape. In this Review, we highlight such non-canonical targets in preclinical MS research with a focus on five highly promising areas: oligodendrocytes; the blood–brain barrier; metabolites and cellular metabolism; the coagulation system; and tolerance induction. Recent findings in these areas may guide the field towards novel targets for future therapeutic approaches in MS.
Similar content being viewed by others


Introduction
Multiple sclerosis (MS) is the most frequently occurring neuroinflammatory disease and the commonest cause of permanent disability in younger adults1. The aetiology of the disease remains elusive but a large body of evidence suggests that it is immune-mediated in nature2. Globally, some 2.8 million people are affected and its incidence and prevalence have been on the rise worldwide over the past few decades3,4. In the majority of patients, the disease takes a relapsing course with intermittent periods of neurological dysfunction that may initially completely resolve; however, as the disease advances, recovery is incomplete and disability accumulates. In addition, progression may occur independently of relapse activity. In two-thirds of cases, the transition from this relapsing course to secondary progressive MS usually occurs after 10–15 years and, for some time, superimposed relapses may occur, reflecting ongoing inflammatory activity5. 10%–15% of patients follow a primary progressive course, characterized by the continuous worsening of neurological disability from the first manifestation of disease. These descriptions of the course of the disease have recently been modified to classify MS into either relapsing forms, with disability occurring both in relation to and independently of relapses, or progressive forms that are active or inactive, classified according to clinical or magnetic resonance imaging (MRI) findings6 (Box 1).
Typical MS manifestations include visual, sensory, motor and sphincter disturbances, as well as incoordination, gait disorder and cognitive impairment1. The disease spans decades and life expectancy is shortened. MS therefore places a heavy burden on patients, their families and caregivers, healthcare systems and society at large7.
Cardinal pathological features are multifocal inflammation, primary demyelination, oligodendroglial death, neuroaxonal degeneration, and astrocytic scarring in the brain and spinal cord8,9. Both white and grey matter are affected. Axonal damage detectable early in the course of the disease foretells the development of permanent disability; tissue destruction gives rise to global and regional brain and spinal cord atrophy1,8. During the early stages of the disease, the ongoing damage may go unnoticed as compensatory functional mechanisms are recruited, involving supplementary neuronal circuitry and the capacity for remyelination of damaged dysfunctional axons by oligodendrocyte precursors10.
Early disease pathology is led by adaptive immunity outside the central nervous system (CNS). As the disease evolves, adaptive immunity loses importance and CNS-specific innate immunity, orchestrated by microglia and astroglia, takes precedence11,12. Axonal damage and neurodegeneration may be caused by collateral damage in the wake of a vigorous inflammatory response, with demyelination rendering the denuded axon susceptible to noxious mediators, lack of neurotrophic factors and retrograde degeneration1,2. Molecular effector pathways involve oxidative stress, calcium overflow, excitotoxicity and eventually mitochondrial energy failure8,13.
Although the disease remains incurable, the past three decades have witnessed the successful development of disease-modifying therapies (DMTs), predominantly for the relapsing forms1,13,14,15 (Fig. 1). DMTs predominantly curtail the migration of lymphocytes into the CNS, or deplete specific types of immune cell. Recently, siponimod (which modulates cell migration) was the first agent to provide a moderate benefit in secondary progressive MS, and ocrelizumab (which depletes B cells) was the first drug to be moderately effective in a subgroup of patients with primary progressive MS (PPMS)14.

Overview of the immunopathogenesis and targets of available disease-modifying therapies in multiple sclerosis (MS). The established therapeutic approaches have diverse mechanisms of action (pleiotropic effects, immune cell depletion, reduction of proliferation and blockade of migration) that modify or inhibit the different steps of the inflammatory process in MS within the peripheral immune system, blood–brain barrier or within the central nervous system (CNS). The therapies depicted are subdivided into monoclonal antibodies (red lines) and pharmacological agents (black lines). APC, antigen-presenting cell; OL, oligodendrocyte; S1PR, sphingosine 1-phosphate receptor; TH cell, T helper cell.
Nevertheless, there remain major unmet needs. First, for the majority of patients with relapsing disease, DMTs fail to generate sustained control of disease activity. Second, for the majority of patients with progressive disease, no sufficiently effective treatment is available. Although the conversion rate to secondary progression is reduced with DMTs16,17, roughly 10%–15% of patients develop secondary progressive disease after 5 years16; the long-term risk for conversion to SPMS (15 years) with DMTs is about 34%18. This emphasizes the urgent need for drugs fostering remyelination and repair, and thereby promoting improvement in disability.
All approved DMTs are fundamentally anti-inflammatory and/or immunomodulatory. An enhanced understanding of the immunological and neurobiological underpinnings of MS may open new avenues for therapeutic research, utilizing non-canonical pathways to improve outcomes. In this Review, we select research areas that have demonstrated substantial progress in recent years, hold great promise to identify new molecular targets in MS and may allow the design of more specific and effective therapeutic strategies in the future. We focus on five fields of research (presented in descending order of greatest future therapeutic potential): oligodendrocytes, the blood–brain barrier (BBB), metabolites and cellular metabolism, the coagulation system and tolerance induction.

Pathophysiology and therapeutics
Persistent inflammation is key to MS pathophysiology, and a better understanding of the inflammatory cascade — both peripherally and in the CNS — is critical to establishing novel therapeutic targets.
MS is considered to be an immune-mediated disease caused by the activation of T and B lymphocytes that act against CNS antigens1. Breakdown of tolerance to autoantigens allows previously dormant autoreactive T and B cells to become activated. Similar to most autoimmune diseases, the triggering event is not known in MS, but several studies indicate that genetic background plays a part by tuning the adaptive immune response, which alters immunological activation thresholds as well as the efficacy of immunoregulatory pathways. A robust genetic link has been repeatedly found between MS and certain human leukocyte antigen (HLA)-encoded class II major histocompatibility complex (MHC) molecules19. In particular, in recent ex vivo studies using samples from patients with MS, HLA-DR15 haplotypes were instrumental in orchestrating an autoimmune response against the brain and spinal cord, mediated by T cells and fuelled by B cells20.
According to these insights, which have been substantiated in variants of experimental autoimmune encephalomyelitis (EAE)21,22, activated myelin-reactive T lymphocytes and other immune cells infiltrate brain tissue by crossing the BBB. Subsequently, autoreactive T cells are locally reactivated by classical or tissue-resident antigen-presenting cells — including perivascular macrophages, dendritic cells and microglia — and by a surge of pro-inflammatory mediators, including cytokines and chemokines, that are released by immune and glial cells. This results in further immune cell recruitment and amplification of the inflammatory cascade in the CNS. Furthermore, macrophage and/or microglial activation leads to myelin destruction both directly and by antibody- or autoantibody-mediated phagocytosis of the myelin sheath, resulting in demyelination, axonal degeneration, neuronal dysfunction and consequent neurodegeneration2,23. Thus, ongoing activation of autoreactive T cells critically contributes to repeated and deleterious waves of inflammation targeting the brain and spinal cord.
Conceptually, the HLA-DR15 haplotype, which predisposes individuals to develop MS, physically interacts with both endogenous myelin autoantigens and MS-associated foreign antigens24. The latter encompasses antigens derived from gut-derived microbes, such as Akkermansia muciniphila and Acinetobacter calcoaceticus25,26,27, as well as the Epstein–Barr virus28. The Epstein–Barr virus is of particular interest as it latently infects and immortalizes B lymphocytes, driving their persistence in an activated state and possibly contributing to MS pathogenesis by generating a pro-inflammatory milieu and forming ectopic lymphoid follicles in the CNS29,30.
These insights and the unexpectedly high efficacy of B cell-depleting immunotherapies have flagged B cells as key players in MS pathophysiology31. B cells can present antigens to T cells and thereby drive their clonal proliferation and the production of pro-inflammatory cytokines32. Long-lived tissue-resident B cells are located within the meninges, and in vivo neuroinflammation augments their antigen-presenting capacity. We note that meningeal T cell infiltration has been seen before the appearance of clinical signs and even prior to dissemination into the CNS parenchyma33,34, indicating that this initial step is a potential T cell checkpoint in early disease pathology35. In addition, subpial aggregates of B cells and CD8+ T cells have been implicated as drivers of compartmentalized inflammation during the progressive stages of the disease36,37.
Currently approved DMTs primarily target the aberrant immune response in the peripheral immune system to effectively reduce episodes of inflammatory demyelination. Having fewer inflammatory episodes provides indirect — or secondary — neuroprotection by restricting subsequent neurodegeneration and thus preventing neurological disability1,38. DMTs may be categorized according to the underlying mode of action: pleiotropic effects, reduced immune cell proliferation, targeted depletion of immune cells or reduced immune cell migration (Fig. 1).
A number of established MS immunotherapies have pleiotropic effects. This is best demonstrated by mild but well established immunomodulatory agents such as interferon-β (IFNβ), glatiramer acetate and dimethylfumarate. IFNβ is an endogenous cytokine. Its potent antiviral responses include down-regulation of MHC class II expression, interference with T cell homeostasis and inhibition of adhesion molecules, thus stabilizing the BBB39. Glatiramer acetate is a complex mixture of random peptides that mimic major myelin proteins, and administration produces a mild but persistent attenuation of the pro-inflammatory phenotype, mainly by affecting the autoaggressive lymphocyte population that targets the CNS myelin sheath40. For dimethylfumarate, multiple molecular targets have been suggested, including nuclear factor erythroid 2-related factor 2 (NRF2), a transcription factor involved in both lymphogenesis41 and the oxidative stress response42. These pleiotropic therapies are effective only for relapsing forms of MS43.
Nonspecific immunosuppressants derived from classical chemotherapeutic agents, such as azathioprine, mitoxantrone and cyclophosphamide, which are not routinely used for MS, indiscriminately reduce the proliferation of all rapidly dividing cells, including immune cells. As a consequence, both physiological immune functions (such as protection from pathogens and cancer prevention) and pathological autoimmune activity are reduced; the therapeutic benefits for MS are achieved at the expense of long-term side effects including the increased risk of therapy-related secondary cancers44. Furthermore, at least for the better tolerated nonspecific immunosuppressants such as azathioprine, the therapeutic effects are rather modest45. Therefore, those therapies are generally associated with an unfavourable risk-to-benefit ratio. Although it is often considered to be a broad immunosuppressant, teriflunomide, which inhibits dihydroorotate dehydrogenase, probably has a more lymphocyte-specific mode of action, as comprehensive immunosuppressive effects have so far not been observed46.
Building on the rationale of using nonspecific immunosuppressants, depletion of select immune cell populations has been systematically explored and established in MS. A single cycle of alemtuzumab, which targets CD52, a pan-lymphocyte cell-surface molecule, removes all lymphocytes; B lymphocytes recover quickly (sometimes exceeding previous levels), whereas T lymphocytes are not detectable for up to 18 months47. A complementary approach is the use of anti-CD20 antibodies such as rituximab, ocrelizumab and ofatumumab, which deplete most cells of the B lymphocyte lineage and a small population of T cells48. A study in a PPMS cohort demonstrated moderate beneficial effects of rituximab in the inflammatory stages of PPMS49, prompting a randomized phase III placebo-controlled trial of the humanized anti-CD20 monoclonal antibody ocrelizumab. The results led to the first approval of a drug for PPMS50. In a third approach, by inhibiting lymphocyte-specific signalling cascades, pulsed oral cladribine is able to remove both B and T cells to a similar degree51. All three depletion strategies may be classified as selective immunosuppression and have superior efficacy to the milder immunotherapies described above. For alemtuzumab and cladribine, reconstitution of a normalized immune system following the initial depletion of pathogenic immune cells is thought to be the dominant mode of action52.
The ability of lymphocytes to migrate between secondary lymphoid organs and the respective target tissue, using the lymphatic and circulatory systems, is instrumental for raising an adaptive immune response. This is not only relevant for primary immune defence but also for T cell- and/or B cell-mediated autoimmune conditions such as MS, where myelin-reactive lymphocytes undergo crucial activation steps in lymph nodes prior to their transit to the CNS53,54,55. Migration of lymphocytes critically depends on sphingosine 1-phosphate (S1P) receptors for lymph node egress56 and on α4β1 integrins for transit to the CNS57. These insights paved the way for the clinical successes of the anti-α4β1 antibody natalizumab58 and the S1P receptor modulators fingolimod, ozanimod, ponesimod and siponimod, which are highly efficacious therapies for relapsing variants of MS59. Of note, siponimod was also effective in secondary progressive forms of MS, possibly independently of its anti-inflammatory activity60, supporting the hypothesized contribution of certain S1P receptors to regenerative processes in the CNS61.
Although these immunotherapies have excelled in controlling inflammation — as reflected in a marked reduction of relapse rates (Table 1) — and received regulatory approval, they generally fail to halt disease progression and to promote regeneration, with substantial residual disease burden even with treatment. The beneficial therapeutic effects of B cell-depleting agents and siponimod in progressive MS were mainly confined to a subset of patients with potentially active inflammation, visualized by MRI and/or with superimposed relapses62. Furthermore, those therapeutic effects were rather short-lived and limited63. Decelerating the insidious neurodegenerative process, especially in the progressive forms of disease, is even more challenging64. Hence, there is an urgent need for future research to re-evaluate disease pathophysiology beyond autoimmune inflammation to identify novel therapeutic targets that counteract the pathobiological consequences of the chronic stages of the disease.
Oligodendrocytes revisited
In recent years, the focus of MS research has expanded beyond immune cells and recognized the contributions of multiple glial cell types to the development, progression and amelioration of the disease. Oligodendrocytes are specialized glial cells that synthesize myelin sheaths, enable saltatory conduction and provide metabolic support to neurons.
Inflammation regularly damages oligodendrocytes, resulting in demyelination and, consequently, axonal loss65. To effect remyelination, microglia and macrophages must first clear the damaged myelin66, a process that is enhanced by activation of the triggering receptor expressed on myeloid cells 2 (TREM2)67. Next, oligodendrocyte progenitor/precursor cells (OPCs) need to be recruited to the zone of myelin loss and undergo further differentiation and maturation to become fully competent myelin-producing oligodendrocytes68,69. However, the differentiation process from OPC to mature myelin-producing oligodendrocyte is impaired in MS lesions owing to the inflammatory microenvironment and the presence of an array of inhibitory molecules, which might cause inefficient remyelination70. Such an acute inflammatory microenvironment is associated with the presence of reactive oxygen species, which may in turn affect the fate of OPCs71. Moreover, in experimental inflammatory demyelination, OPC differentiation is inhibited by effector T cells and IFNγ72. This effect is paralleled by an induction of the crucial immunoproteasome subunit PSMB8 (also known as LMP7), which increases MHC class I expression on OPCs, rendering them a more prominent target for the cytotoxic CD8+ T cells that are abundant in MS lesions73. We note that induction of immunoproteasomes in OPCs has been observed in human demyelinated MS brain lesions72. Recent evidence suggests that resident oligodendrocytes (rather than those differentiated from recently recruited OPCs) are also capable of remyelinating denuded axons74,75.
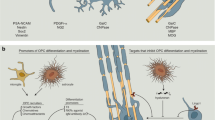
Various strategies to increase remyelination by forcing the differentiation of OPCs to mature oligodendrocytes are currently being researched76 (Fig. 2). For example, recombinant galectin 3, which regulates basic cellular functions, can promote differentiation of OPCs to oligodendrocytes by activating AKT kinase and suppressing the extracellular-signal-regulated kinase 1 (ERK1) and ERK2 (ref.77). In addition, inhibiting the ERK1–AMP-activated protein kinase (AMPK) pathway enhances oligodendrocyte generation and thereby promotes remyelination in EAE and drug-induced demyelination models78.

Oligodendrocytes differentiate from neuronal stem cells to oligodendrocyte progenitor cells (OPCs) and pre-oligodendrocytes, a process orchestrated by several pathways and factors (oligodendrocyte transcription factor 2 (OLIG2), homeobox protein NKX2.2, SRY-box transcription factor 10 (SOX10), myelin regulatory factor (MYRF), SOX5/6, HES family bHLH transcription factor 5 (HES5), inhibitor of DNA binding 2 (ID2) and ID4). The different development stages of oligodendrocytes are typified by various oligodendroglial lineage markers (OPC: platelet-derived growth factor receptor-α (PDGFRα) and neuron-glial antigen 2 (NG2); pre-oligodendrocytes: 2′,3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase) and oligodendrocyte marker 4 (O4); oligodendrocytes: galactocerebroside (GalC), CNPase, myelin-associated glycoprotein (MAG), myelin basic protein (MBP) and proteolipid protein (PLP)). In multiple sclerosis (MS), the autoimmune attack (orange arrow) drives inflammatory demyelination, which is characterized by astrocyte activation and microglia recruitment/activation. This leads to demyelination and axonal damage and is associated with astroglial proliferation and oligodendrocyte depletion, resulting in a glial scar (blue arrow). Strategies to modulate OPC differentiation to enhance remyelination (leucine-rich repeat and immunoglobin-like domain-containing protein 1 (LINGO1) and oxidative phosphorylation (OXPHOS)) are shown. To force successful regeneration (grey arrow), potential pathways are metabolic support (enhancement of glucose utilization or fatty acid and cholesterol synthesis) or promotion of OPC differentiation to oligodendrocyte (galectin 3).
Usually, the oligodendrocyte cell lineage is subclassified into oligodendrocytes and OPCs, but there is evidence for greater heterogeneity of the oligodendrocyte cell population in MS. By performing single-nucleus RNA sequencing in white matter areas of post-mortem MS brains, Jäkel et al. demonstrated an increased heterogeneity of oligodendrocytes in MS lesions and identified altered sub-clusters within normal-appearing white matter, indicating that MS is a more diffuse disease than foci suggest79. Following this approach, a recent single-cell transcriptomic analysis of oligodendrocytes from the spinal cord of EAE mice and human MS brain samples revealed that oligodendrocyte lineage sub-clusters express genes involved in antigen processing and presentation, highlighting a potential alternative role of oligodendrocytes in the context of inflammatory disease80.
We further note that in early EAE axonal damage may precede demyelination81, suggesting that loss of metabolic support from oligodendrocytes may cause demyelination. Human oligodendrocytes and OPCs use aerobic glycolysis to maintain cell function and the biosynthesis of myelin. Activation of the oligodendroglial N-methyl-d-aspartate (NMDA) receptor can support axonal energy metabolism by increasing glucose utilization in oligodendrocytes82. During times of stress (such as glucose deprivation or hypoxia), oligodendrocytes reduce their glycolytic flux and use ATP for cell survival rather than for myelin production83. Inhibiting mitochondrial oxidative phosphorylation in vitro can alter the differentiation of OPCs to oligodendrocytes and thereby affect myelin production84. Moreover, differentiation of oligodendrocytes requires reorganization of lipid metabolism, including the biosynthesis of cholesterol and sphingolipids, which are major components of myelin. Using two independent — inflammatory versus toxic — models of demyelination and remyelination, Voskuhl and colleagues showed that genes involved in cholesterol synthesis were upregulated in oligodendrocytes during the remyelination phase. Treatment with an oestrogen receptor-β ligand further increased cholesterol synthesis, indicating a potential target for enhancing remyelination85. Furthermore, fatty acid synthesis in oligodendrocytes is crucial for the correct lipid composition in myelin, and depletion of fatty acid synthase in OPCs leads to defects in remyelination in a mouse model of demyelinating spinal cord lesions86, highlighting fatty acid synthase as another potential target for remyelination.
The initial oligodendrocyte targeting therapies addressed endogenous checkpoints that otherwise inhibit remyelination, such as the leucine-rich repeat and immunoglobin-like domain-containing protein 1 (LINGO1). Opicinumab, an anti-LINGO1 monoclonal antibody that modulates OPC differentiation to enhance remyelination, was tested in pilot phase II studies in optic neuritis and relapsing-remitting multiple sclerosis (RRMS) (RENEW and SYNERGY trials)87,88. Both studies failed to meet their respective primary endpoints, but exploratory analyses suggested a potential benefit of this approach: in RRMS, participants of younger age and short disease duration responded better to opicinumab. This highlights the potential of oligodendrocyte-orientated therapies in the future87. However, a recent phase II study (AFFINITY) evaluating opicinumab as an add-on therapy to standard immunotherapy failed to reach the primary end point of improvement of the disability in comparison to placebo89,90. Remyelination starts immediately after the onset of inflammatory demyelination91, leading to the speculation that remyelination therapies need to be administered shortly after demyelination has occurred to improve the remyelination capacities of dysregulated inflammatory oligodendrocyte/OPC subtypes. Thus, remyelination-promoting therapies such as opicinumab may fail if they do not target the inflammatory oligodendrocyte/OPC phenotype in the early remyelination phase.
Another remyelination approach recently explored in experimental and clinical settings targets the histamine and muscarine receptor systems. The histamine H1 receptor antagonist clemastine, established as an allergy therapy in clinical practice, was identified in an unbiased drug repurposing screen for compounds with remyelination capacities92. Clemastine, previously shown to induce differentiation of OPCs and promote remyelination in experimental demyelination models, was further examined in a pilot trial in optic neuritis, with confirmatory but rather modest clinical effects93. We note that clemastine antagonizes not only the H1 receptor, but also muscarinic receptors in a nonselective manner. Another nonselective muscarinic receptor antagonist, benzatropine, enhanced remyelination in experimental rodent models94. In-depth analysis showed that signalling via the muscarinic M3 receptor is pivotal for inhibition of efficient remyelination by both mouse and human OPCs95. In an independent screening approach for small molecules with remyelinating properties, clobetasol and miconazole were identified as possible candidates, owing to their effects on glucocorticoid receptor signalling or mitogen-activated protein kinases (MAPKs), respectively96. While clinical validation is awaited, the discovery of these pathways reflects the ongoing and systematic search for ‘druggable’ inhibitory checkpoints of remyelination.
In summary, our knowledge of oligodendrocyte differentiation and the contribution of these cells to myelin production in health and demyelinating disease is evolving. However, the complexity of the tightly regulated remyelination process, particularly in the context of chronic neuroinflammation, produces challenges. Obviously, a better understanding of the net clinical effects of the relevant pathways in an individual patient is required, taking into consideration the extent of chronic inflammation, the capacity for endogenous remyelination, the magnitude of axonal loss and neuronal damage, as well as age and sex, to successfully use remyelination approaches that target oligodendrocytes. Furthermore, whether global remyelination is able to restore the network functions of the CNS in patients with MS remains an open question97.
The blood–brain barrier as an early target
The BBB consists of specialized endothelial cells (ECs), which communicate with other cells (including astrocytes, pericytes, neurons, smooth muscle cells, microglia and other immune cells) of the CNS to form the neurovascular unit98. The breakdown of the BBB is an early hallmark and key pathophysiological event in MS and can be visualized by the leakage of contrast agents during MRI. Gadolinium enhancement is observed in active lesions but other — more sophisticated — MRI measures demonstrate subtle BBB disruption in normal-appearing white matter. This suggests that covert BBB disruption may precede neuroinflammatory processes99.
RNA sequencing in mouse brain ECs revealed that during BBB dysfunction a similar gene expression pattern occurred in different disease models (stroke, multiple sclerosis, traumatic brain injury and seizure) when the BBB was at its most dysfunctional. Within those disease models EAE showed the most unique changes in brain ECs, with a specific gene expression pattern — including leukocyte adhesion molecules and histocompatibility loci, as well as interferon-induced, interleukin and complement pathway genes100. The integrity of the BBB is tightly regulated, and preserving this integrity may be a promising protective strategy in several neurological diseases, but especially in MS, in which BBB disturbance actively participates in initiating the pathophysiological neuroinflammatory process101.
In MS, autoreactive leukocytes enter the CNS after peripheral activation of cellular migration molecules together with chemokine and adhesion receptors23,102 (Fig. 3). The functional phenotype of infiltrating immune cells depends on where priming occurred; for example, cells can be primed in the skin or gut103. Brain ECs control the transmigration of leukocytes by expressing adhesion molecules and producing chemokines104. Selectins capture and induce rolling of immune cells; however, their absence or pharmacological blockade had no impact on EAE development in mice105,106. Next, firm adhesion is mediated by adhesion receptors on ECs and their counterpart ligands (such as integrins) on leukocytes. Natalizumab, an antibody that targets the leukocyte ligand α4β1 integrin and is approved for the treatment of RRMS, impairs the adhesion of leukocytes to vascular cell adhesion molecule 1 (VCAM1) on brain ECs104. However, treatment with natalizumab may be associated with side effects that are probably due to widespread functions of α4 integrin in haematopoietic cells, indicating a need for alternative therapeutic strategies that inhibit only the migration of pathogenic lymphocytes. For this purpose, other approaches to brain-specific inhibition of leukocyte-endothelial interaction could be a promising strategy. Additional brain EC-specific adhesion molecules, such as activated leukocyte cell adhesion molecule (ALCAM; also known as CD166), have been implicated in EAE and MS pathogenesis and may be useful therapeutic targets107,108. However, leukocytes can bypass the blockade of adhesion receptor–ligand interaction by using alternative adhesion molecules109. Therefore, strategies targeting transmigration directly by inhibition of cell adhesion molecule biosynthesis/expression on ECs or indirectly through reduction of the inflammatory EC phenotype (thereby reducing adhesiveness), might circumvent this shortcoming.

In physiological conditions, blood–brain barrier integrity is tightly regulated. In multiple sclerosis (MS), the tight barrier can be dysfunctional, leading to increased leukocyte recruitment and transmigration. In addition, autoreactive T cells can enter the CNS through peripheral activation of cellular locomotion molecules, together with chemokine and adhesion receptors. T cell trans-endothelial migration proceeds in several steps (steps 1 to 5) that are controlled by various adhesion molecules (selectins, lymphocyte function-associated antigen 1 (LFA1) and α4β1 integrin) and their receptors (selectin ligands, intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion protein 1 (VCAM1)) expressed by endothelial cells (ECs). Furthermore, tissue-resident macrophages can secrete factors (cytokines, chemokines and growth factors) that lead to EC activation, characterized by increased expression of selectin and adhesion molecules. ALCAM, activated leukocyte cell adhesion molecule; ANG, angiopoietin; CCL2, CC-chemokine ligand 2; CCL21, CC-chemokine ligand 21; CXCL12, CXC-chemokine ligand 12; LXRα, liver X receptor-α; MCAM, melanoma cell adhesion molecule; MMP2/9, matrix metalloproteinase 2/9; PSGL1, P-selectin glycoprotein ligand 1; TGFβ, transforming growth factor-β; TIE2, tyrosine-protein kinase receptor TIE2; VAP1, vascular adhesion protein 1.
Different pathways have been identified that control the morphology and adhesive capacity of ECs. For example, the kallikrein–kinin system regulates the expression of VCAM1 and intercellular cell adhesion molecule 1 (ICAM1) on brain ECs via a PAR2-receptor-mediated pathway110. Further unexpected targets might also be involved in the regulation of adhesive capacity, given that the potassium channel TREK1 was shown to modulate VCAM1 and ICAM1 expression on brain ECs111.
The WNT–β-catenin pathway, known to be involved in BBB formation and maintenance, is activated in brain ECs in human MS lesions and the EAE model112. Inhibition of the endothelial WNT–β-catenin pathway prior to disease onset leads to a more severe disease course in EAE, accompanied by BBB disruption and increased immune cell infiltration into the CNS112, indicating that endothelial WNT–β-catenin pathway reactivation could be a strategy to maintain BBB integrity in inflammatory conditions. However, WNT activation in perivascular OPCs in white matter lesions leads to secretion of WIF1, which counteracts the effects of WNT ligands in ECs and leads to endothelial dysfunction113. Furthermore, inhibition of WNT signalling in oligodendrocytes leads to regenerative myelination114, perhaps because this pathway is involved in oligodendrocyte maturation and myelination115,116. Overall, pharmacological enhancement of WNT signalling may be considered as a strategy to preserve BBB integrity, but those therapies need to be highly cell-specific to avoid potential side effects.
Another potential target, liver X receptor-α (LXRα), is a nuclear receptor involved in cholesterol and lipid metabolism. Endothelial LXRα is involved in maintaining BBB integrity. EC-specific knockdown of LXRα increases BBB permeability in vitro and in vivo and is associated with reduced tight junctions, increased VCAM1 expression and leukocyte infiltration. Moreover, EC-specific LXRα-deficient mice show exaggerated disease progression in the EAE model, indicating that LXRα could be a potential new target for improving BBB function117.
Adherent and junction molecules stabilize and tighten the BBB. The platelet/endothelial cell adhesion molecule 1 (PECAM1) maintains EC integrity and is abundant at cell–cell junctions118. PECAM1-deficient mice exhibit early onset of symptoms and leukocyte infiltration in EAE119. In vitro, PECAM1 stabilizes BBB integrity and may thereby have neuroprotective functions in neuroinflammatory conditions120. A leukocyte transmigration inhibitor (trioxotetrahydropyrimidine scaffold, compound 12) improved the clinical score in EAE without any toxic side effects. This compound blocks PECAM1 in an in silico model, although the observed effects may have been transmitted by a different mechanism121. Therefore, more studies are needed to evaluate the potential role of PECAM1 in MS.
BBB dysfunction in MS is associated with decreased levels of tight junction proteins. Downregulation of claudin 5 correlates with BBB breakdown in EAE, and recombinant expression of claudin 5 protects brain microvascular ECs from vascular endothelial cell growth factor-α (VEGFα)-induced barrier dysfunction122. In EAE, a subpopulation of claudin 5-positive leukocytes was observed in close apposition to inflamed vessels. Claudin 5 may be transferred via extracellular vesicles from ECs to leukocytes to facilitate transendothelial leukocyte migration via claudin 5 bridges in EAE123. In addition, peripheral blood leukocytes in MS patients experiencing a clinical relapse show increased claudin 5 levels124. Additional pathways (including PDGFβ–PDGFβR, TGFβ–TGFβR, SHH–PTC1, ANG1–TIE2, ANG II–AT1 and APOE–LRP1) have been implicated in tight junction formation and may therefore be interesting targets for further research in MS therapy125,126,127,128.
To take the final step into the CNS parenchyma, leukocytes have to breach the endothelial basement membrane and the glia limitans. Matrix metalloproteinases (MMPs) are essential for this step because they digest tight junction and basal membrane proteins129. MMP9 protein and RNA levels are increased in serum, mononuclear cells and cerebrospinal fluid (CSF) and correlate with disease progression129. In addition, MMP activity, detected by MMP inhibitor-positron emission tomography (MMPi-PET), is a unique feature of early MS lesions130. Young mice deficient in MMP9 are relatively resistant to EAE induction131.
In recent decades, pharmacological MMP inhibitors have showed efficacy in experimental animal models but have failed in clinical trials, with limited beneficial effects and serious adverse events. Since MMPs are involved in several important biological pathways (including tissue morphogenesis, angiogenesis and cell migration), inhibiting all MMP family members leads to adverse effects132. Treatment with more-specific inhibitors, such as triple-helical peptide inhibitors, which target MMP9 and MMP2, reduces EAE severity133. Furthermore, the monoclonal anti-MMP9 antibody andecaliximab has been tested in initial phase I clinical trials for other autoimmune diseases such as rheumatoid arthritis or ulcerative colitis and was found to be safe and well tolerated132, so MMP9 inhibition may be safe in MS. Some of the effects of IFNβ, a standard treatment for MS, may be through MMP9-mediated BBB regulation, because IFNβ downregulates MMP9 expression to reduce the migratory capacity of immune cells134,135,136.
Chemokines produced by ECs are involved in all stages of the transmigratory process. Brain ECs induce firm adhesion of T cells in EAE and MS through CC-chemokine ligand 19 (CCL19) and CCL21 (ref.137). CXC-chemokine ligand 12 (CXCL12) also has a role in EAE and MS pathogenesis as it mediates T cell arrest on brain ECs and the basolateral release of inflammatory cells138,139,140. Thus, chemokine-targeted therapies can be exploited to regulate dysfunctional chemokine production in MS.
Other cells of the neurovascular unit critically influence BBB integrity and might therefore be therapeutically targeted98. Pericytes ensheath the endothelial monolayer of the BBB and regulate BBB function141. Pericytes contribute to MS pathogenesis by expressing adhesion molecules, producing pro-inflammatory mediators (including cytokines, chemokines and MMPs), presenting antigens and producing reactive oxygen species142,143. Therefore, pericytes might be interesting therapeutic targets in MS; however, specifically targeting pericytes is challenging. Lipid and protein carriers with pericyte-targeting motifs have been developed and may be instrumental for future pericyte-directed therapies144,145.
Cell-based strategies have also been proposed to treat BBB breakdown. In rodent models, systemically administered mesenchymal stem cells reduce leukocyte transmigration and regulate the production of MMPs, reactive oxygen species and pro-inflammatory cytokines, as well as stabilizing the cellular components of the neurovascular unit146,147. Small-scale clinical trials in MS supported the feasibility and safety of mesenchymal stem cell administration. However, larger studies are needed to evaluate the efficacy of those approaches146.
Numerous interactions have been demonstrated between the gut microbiome and the BBB148. Metabolic products, cytokines or other immune-active substances can alter BBB integrity, transport rates and phenotypes of barrier cells149. Further, certain bacterial factors promote CNS penetration of T cells149. Thus, the gut microbiome might be another target to influence BBB function and immune cell infiltration to the CNS in the context of MS.
In summary, the preservation or restoration of BBB integrity is a promising new target for MS therapy and may be used to treat additional CNS disorders. However, most approaches are far from clinical development and targeted therapies are needed to specifically address dysregulation of brain ECs or other components of the neurovascular unit without affecting physiological function. The benefits of targeting BBB integrity are mostly confined to early stages of MS pathogenesis, whereas chronic neuroinflammation and subsequent neurodegeneration are unlikely to be affected by this treatment strategy. Furthermore, alternative routes to the CNS such as the plexus epithelium might circumvent BBB-targeting therapies and need to be considered in the clinical development of new treatment strategies150.
Is multiple sclerosis a metabolic disease?
New technologies (such as metabolomics) have provided useful insights into the cellular metabolism of cancer and inflammatory diseases. In this section, we focus on metabolic alterations occurring in patients with MS and describe the metabolic profile of cells — T cells and neurons — that are primarily involved in MS pathogenesis (Fig. 4).

a | A variety of metabolic changes indicated by the different levels of metabolites in multiple sclerosis (MS) are known to occur. It is assumed that the MS brain metabolism has increased glycolysis and therefore a reduction of the flux through the tricarboxylic acid cycle (TCA) and oxidative phosphorylation (OXPHOS). Furthermore, other pathways such as the urea cycle, the kynurenine pathway and glutamine metabolism are also affected in MS. Red upwards and downwards arrows indicate changes in the metabolite level or metabolic pathways (grey circular arrows, changes in red) in MS. b | T cell subtypes show different metabolic properties. In MS, T cells seem to increase their metabolic fluxes (indicated by red upwards and downwards arrows); however, after differentiation, they display a different metabolic profile. T memory (Tmem) cells rely on fatty acid oxidation (FAO) and OXPHOS, whereas T helper 17 (TH17) cells display increased fatty acid synthesis (FAS), glycolysis and glutaminolysis. c | Metabolic active pathways of neurons (yellow) and astrocytes (blue). Neurons rely on the astrocyte–neuron lactate shuttle for their energy supply. Astrocytes take up glucose and metabolize it to pyruvate using glycolysis, and, finally, lactate is generated through lactate dehydrogenase (LDH). Lactate can exit the cell and the extracellular lactate can be shuttled into neurons to either fuel neuronal ATP synthesis or to generate reducing agents such as NADPH to maintain redox homeostasis. In MS, this mechanism seems to be diminished (indicated by red upwards and downwards arrows). ADMA, asymmetric dimethylarginine; BCAA, branched-chain amino acid; GLS, glutamine synthetase; iNOS, inducible nitric oxide synthetase; KA, kynurenic acid; NO, nitric oxide; PPP, pentose phosphate pathway; QA, quinolinic acid.
Metabolites in MS
Metabolites are intermediate or end products of numerous physiological and pathological cellular processes and can be detected within cells as well as biological samples available in clinical practice — specifically, CSF, serum, urine and tissue. Several metabolic changes are reported in MS (Supplementary Table). Here, we focus on metabolites that have a role in MS disease progression and could unveil new biomarkers or therapeutic targets151 (Fig. 4a).
Several studies report alterations in metabolite levels of amino acids in MS. The amino acid glutamate is a neurotransmitter, and excessive levels are excitotoxic. Glutamate and glutamine levels are elevated in the plasma of patients with RRMS (and other neurological diseases), and glutamate concentration correlates with disease severity in RRMS152,153. In addition, levels of branched-chain amino acids (leucine, isoleucine and valine), which are substrates for glutamate synthesis and have an important role in transporting amino acids through the BBB, are decreased in patients with RRMS152.
Recently, Fitzgerald et al. identified abnormalities in aromatic amino acid (AAA) metabolites using a multi-omic approach in patients with MS. A reduced quantity of AAA metabolites correlated with higher disability, and altered AAA metabolism was found in CSF- and serum-derived monocytes of patients with MS. These AAA metabolites may come from the gut microbiota154. Levels of butyrate- or indolelactate-producing bacteria are reduced in patients with MS155. Indolelactate is an intermediate product of tryptophan degradation in bacteria and tryptophan metabolism is involved in inflammatory processes. High levels of microbiota-derived indolelactate are also associated with a lower risk of developing paediatric MS156. Butyrate influenced T cell differentiation and suppressed demyelination in vivo157. However, the gut microbiome profile did not differ among the different forms of MS or in response to treatment with DMT155.
In accordance with this altered gut microbiota, bile acid metabolism is altered in patients with MS158,159. Endogenous bile acid supplementation is neuroprotective, ameliorates disease severity in an EAE model and is currently being evaluated in a phase 1 clinical trial158. Although not within the scope of this Review, there is an evolving understanding of the interaction between the gut microbiome and the pathophysiology of MS; influencing the gut microbiota might be another therapeutic avenue (reviewed elsewhere160,161,162,163).
Modulating the metabolism of the AAA tryptophan, which is reduced in the serum and CSF of patients with MS154,164,165, could have therapeutic value. In vivo models showed reduced levels of tryptophan metabolites on both sides of the BBB (cortex and serum) during demyelination166. Furthermore, the ratio of tryptophan to kynurenine, a key tryptophan metabolite, in urine negatively correlates with the disability score of patients with RRMS167. Kynurenine can be metabolized to either quinolinic acid, which is neurotoxic, or kynurenic acid, which is neuroprotective168,169. The balance of these neurotoxic and neuroprotective kynurenine metabolites is disturbed during MS progression170. Increased levels of kynurenic acid were observed in patients with RRMS but not in those with SPMS or PPMS, whereas neurotoxic quinolinic acid concentrations were progressively raised in both SPMS and PPMS171,172. The upregulated levels of kynurenic acid may compensate for quinolinic acid-induced excitotoxicity in the early stage of the disease, whereas neurotoxicity dominates in progressive stages. Interestingly, quinolinic acid levels are positively associated with the gut microbiota Akkermansia spp., which is known to be altered in patients with MS26,27,173.
Lipid metabolism is also altered in MS: sphingolipid levels are decreased and phospholipid levels are increased in active MS lesions174. Levels of phospholipids, particularly lysophosphatidylcholine (LPC), are also elevated in the CSF of patients with MS175. LPC is cleaved from a major component of the membrane, phosphatidylcholine, by phospholipase A2 (PLA2)176. Increased PLA2 activity is associated with neuroinflammatory diseases and dysfunctional BBB177. Thus, the high LPC levels in the CSF of patients with MS indicate augmented PLA2 activity178, which may be important for initiating membrane breakdown. Interestingly, inhibition of PLA2 protects mice from acute relapse in the EAE model, preventing membrane breakdown and reducing potential pathological effects of LPC and other phospholipid metabolites179,180.
When considered together, these studies indicate a distinct metabolic profile in MS. Differentiating between the origin of the samples (CSF versus blood) and the knowledge that some metabolites, such as lactate and fructose, cannot pass the BBB is important for future analysis. Furthermore, the use of different techniques (nuclear magnetic resonance (NMR) versus mass spectroscopy)181, and differences in sample handling and other factors (such as storage conditions) can limit comparability between metabolic studies. Further investigations with larger cohorts and standardized methods are needed to validate the metabolic signature of MS derived from blood or CSF.
T cell metabolism
T cells are highly adaptive and require energy and metabolites for proliferation, activation and differentiation into specific cell subsets. To fulfill these manifold functions, their metabolism adapts. Master transcription factors and immune signals orchestrate T cell fate, and cell metabolism can also dictate this decision.
Quiescent T cells fuel their energy demand through mitochondrial respiration and fatty acid oxidation182. In contrast, proliferating T cells have a dynamic metabolism and rely mainly on glycolysis, which provides energy quickly, and increase glucose influx by increasing expression of the glucose transporter GLUT1 (ref.183) (Fig. 4b).
Activated CD4+ T cells differentiate into effector CD4+ T cells, including T helper 1 cells (TH1 cells) and TH2 cells, IL-17-producing TH17 cells, and regulatory T (Treg) cells. TH17 cells can induce MS-like pathology in experimental models, and these cells are the first encephalitogenic T cells to infiltrate the CNS, which leads to secondary immune cell infiltration184. In contrast, Treg cells suppress the activity of TH17 and TH1 cells and thereby reduce neuroinflammation in MS.
The metabolism of all of these cells could be targets for therapies. The metabolism of CD4+ T cells is dysregulated in MS, and recent studies attempted to decipher their metabolic properties to identify new potential drug targets185. In peripheral immune cells from patients with RRMS, glycolysis and oxidative phosphorylation were impaired during T cell activation186. However, the study did not distinguish between T cell subsets, and the patient cohort was small. In another study, CD4+ T cells activated in vitro from patients with RRMS showed increased oxidative phosphorylation and glycolysis if isolated from patients during relapses, but not from those in remission187. Inhibiting the mitochondrial enzyme dihydroorotate dehydrogenase (DHODH), which affects complex III of the respiratory chain, reduced the number of high-affinity T cells produced in patients with RRMS, probably by altering the metabolic properties of these cells during relapse187.
Cell metabolism can determine T cell fate, making it a favourable target for counteracting the deregulated T cell balance in MS. T cell activation is accompanied by a rapid increase in mitochondrial oxidative phosphorylation during lineage specification towards pathogenic TH17 cells188. Differentiated TH17 cells mainly rely on glycolysis and fatty acid synthesis (FAS) to fulfill their energy and biosynthesis demands189. Inhibiting glycolysis with either 2-deoxy-d-glucose or inhibitors of pyruvate kinase slows EAE progression190,191,192. Moreover, dimethylfumarate, a drug approved for the treatment of relapsing MS, acts at least in part by blocking glycolysis in TH1 and TH17 cells193. In addition, inhibiting the glucose transporter GLUT1 suppresses TH17 differentiation and increases Treg cell induction194. Furthermore, blockade of acetyl-CoA carboxylase 1 (ACC1), which catalyses the first step in FAS, decreases the TH17 cell population and promotes the development of Treg cells, and thus attenuates inflammation in the EAE model189,195. TH17 cells also depend on glutaminolysis for energy and upregulate glutaminase 1 (GLS1). Genetic disruption of GLS1 or pharmacological inhibition of either GLS1 or the glutaminolytic pathway enzyme glutamic oxaloacetic transaminase 1 (GOT1) reduces initial T cell proliferation and impairs TH17 differentiation, and thereby ameliorates disease progression in EAE196,197,198. Further, mice deficient in the neutral amino acid transporter B(0) (also known as ASCT2), a Na+-dependent transporter that regulates glutamine uptake upon T cell activation, were protected from EAE initiation via impaired TH1 and TH17 cell induction199,200.
Treg cells and T memory (Tmem) cells have historically been thought to use fatty acid oxidation (FAO) to generate reducing agents for oxidative phosphorylation and intermediates for the TCA cycle to sustain their energy needs, but the role of FAO in some T cell subsets has been called into question189,201. The rate-limiting enzyme for FAO is carnitine palmitoyl-transferase 1a (CPT1A), which transports fatty acids into the mitochondria, where they undergo β-oxidation201. Inhibition of mTOR or activation of AMP-activated protein kinase (AMPK) pathways can inhibit glycolysis in TH17 cells and favour T cell differentiation to Treg cells with increased CPT1A expression and lipid oxidation202,203. Treatment with the pharmacological CPT1A inhibitor etomoxir in vitro did not alter TH17 differentiation but suppressed Treg formation203. Counterintuitively, mice with a specific mutation in CPT1A, which is associated with low susceptibility to MS in the Inuit population and reduced CPTa1 activity, have reduced disease severity in EAE compared to wild-type mice204. Inhibiting FAO via etomoxir reduced CNS inflammation and demyelination in EAE205,206, indicating that overall reduction of CPT1A activity (and consequently FAO) can ameliorate neuroinflammation.
Mechanistically, etomoxir induces apoptosis of activated myelin oligodendrocyte glycoprotein (MOG)-specific T cells (CD8+) in vitro, and thereby reduces cytokine production. However, Raud et al. demonstrated that etomoxir in higher dose presents an off-target effect with inhibition of T cell proliferation and differentiation207. Recent studies in mice lacking CPT1A in T cells question the role of FAO in Tmem and Treg cells, given that they showed that CPT1A is largely dispensable for the formation of Tmem cell or Treg cell differentiation207, suggesting an alternative mechanism for the effects of etomoxir in the EAE model.
Overall, fatty acid metabolism represents a promising target to counteract neuroinflammation. However, it is questionable whether an altered T cell metabolism underlies the observed effects in vivo and more studies are needed to clarify the role of FAO in contributing cell types in the pathophysiology of MS.
Differentiation into pro- and anti-inflammatory T cell subsets is also influenced by epigenetic mechanisms, including chromatin modelling. Generally, the activity of chromatin-modifying enzymes is regulated by the availability of substrates or co-factors. Methionine is essential for synthesizing the methyl donor S-adenosylmethionine (SAM), which is a cofactor for regulating gene expression in T cells. Dietary methionine reduction slowed EAE disease onset and progression via impaired TH cell proliferation208. Furthermore, dietary serine restriction can influence pathogen-driven T cell expansion in vivo as serine supplies glycine and one-carbon units for de novo nucleotide biosynthesis in proliferating T cells209. Consistent with this observation, T cells show altered serine metabolic pathways in the murine EAE model210. T cell differentiation can be influenced by post-translational modification via citrullination, a conversion of peptidyl arginine into peptidyl citrulline, which is catalysed by the peptidylarginine deiminases, including PAD2 (ref.211). Inhibition of PAD2-mediated citrullination can attenuate the TH17 response, and pharmacological peptidylarginine deiminase inhibitors prevent disease progression in EAE212,213.
A further epigenetic mechanism is involved in controlling T cell fate decisions. (Aminooxy)-acetic acid (AOA) reprograms TH17 differentiation towards Treg cells. AOA inhibits GOT1, which transfers glutamate to α-ketoglutarate198. This decreased levels of 2-hydroxyglutarate, which usually inhibits transcription of FOXP3, an essential transcription factor for Treg cell determination. Since the balance of TH17 and Treg cells is important in MS, the inhibition of GOT1 via AOA also mitigated EAE pathogenesis by reducing the proportion of CNS-infiltrating TH17cells by more than 70%198.
In summary, manipulating the metabolic program or the substrates of T cell metabolism may be a new tool to develop therapeutic strategies that orchestrate T cell differentiation and thereby influence their function in MS.
Neuronal metabolism
Neurodegeneration is a major challenge in the therapeutic management of MS. In healthy individuals, neurons require large amounts of ATP to sustain membrane potential and mitochondrial homeostasis over long-ranging axonal projections. To sustain the production of ATP while minimizing oxidative stress, neurons metabolize lactate through oxidative metabolism. Lactate is shuttled via glucose to the pentose phosphate pathway, resulting in the production of NADPH, an essential cofactor for the synthesis of glutathione — an important antioxidative molecule in the CNS. Accordingly, the glycolysis rate in healthy neurons is low and the activation of glycolysis leads to neuronal death through oxidative stress, potentially via a reduced availability of glucose for the pentose phosphate pathway214.
To meet the requirements for ATP and reducing agents, neurons rely on the support of glia to provide metabolic intermediates such as lactate via the astrocyte–neuron lactate shuttle215 (Fig. 4c).
In MS, neuronal death results from an energy imbalance: increased energy demand coupled with dysfunctional energy supply. Single-nucleus RNA sequencing in cortical and subcortical white matter lesions of human brain samples of patients with MS revealed that upper-layer excitatory neurons in MS lesions upregulate genes involved in oxidative stress, mitochondrial dysfunction and cell death216. From a metabolic perspective, MS neurons exhibit reduced oxidative phosphorylation, further indicating mitochondrial dysregulation217. The increased energy demand is caused by sodium/potassium pumps, which need to increase their activity in order to propagate action potentials after the myelin sheath has been damaged218. Energy supply via astrocytes is also affected in MS. During inflammation, astrocytes have a lower activity of hexokinase 2, an enzyme that catalyses the initial step in glycolysis, resulting in reduced glycolysis and impaired lactate release. Pharmacological suppression of pathogenic astrocyte metabolic reprogramming using miglustat, a drug approved for Niemann–Pick disease type C, is beneficial in EAE219.
Neuronal — as well as oligodendroglial — death in MS is also associated with oxidative stress220. Reactive oxygen species are critically involved in neurodegeneration221, particularly during an acute immune attack targeting the myelin sheath222, and the co-factors NADPH and NADH are pivotal for neuronal redox homeostasis. A recent analysis showed decreased NADH levels within the retinas of patients with MS223 and an altered NAD+/NADH ratio in the serum of patients with MS, indicating chronic oxidative stress224. However, it remains to be determined whether this is a product of a defective defence mechanism or increased production of reactive oxygen species.
In parallel, established concepts of oxidative stress have recently been challenged and expanded42, as subtle redox alterations may modulate intracellular signalling cascades and thus the course of autoimmune neuroinflammation. We note that ion imbalance also seems to occur in neurons of patients with MS. Specifically, elevated sodium levels were detected within lesions and high sodium levels were observed in individuals with SPMS and greater disability, indicating an imbalance in ion channelling225,226. Finally, increased intracellular sodium levels can lead to reverse operation of the sodium/calcium exchanger, causing high intracellular calcium levels, resulting in mitochondrial damage and finally axonal degeneration227. Thus, manipulating the ion balance might serve as a potential target to counteract mitochondrial damage and prevent neuronal death.
The coagulation system
Several studies have highlighted an important link between the blood coagulation cascade and neuroinflammation. Dysregulation of coagulation factors can contribute to inflammatory neurodegeneration in MS, so these factors may be new therapeutic targets (Fig. 5).

Several components of the coagulation pathway are involved in the neuroinflammatory process in multiple sclerosis (MS) and could serve as potential therapeutic targets. This overview demonstrates the classical coagulation pathway, including the extrinsic pathway (ePW), the intrinsic pathway (iPW), the common pathway (cPW) and fibrinolysis. In MS lesions the coagulation factors protein C inhibitor (PCI) and tissue factor (TF) are expressed, and the interaction of TF and factor VII initiates the ePW. Protein C levels are increased in MS patients, and recombinant activated protein C can reduce severity in autoimmune encephalomyelitis (EAE). The exposure of collagen and other intracellular molecules following cell damage can activate the iPW via factor XII. Both the ePW and iPW converge in the common pathway, which includes the cascade from factor X to thrombin and finally fibrin strands. Fibrin deposits accumulate within the brain tissue and can thereby activate inflammatory pathways. Tissue plasminogen activator (tPa) and urokinase plasminogen activator (uPa) activate plasminogen to plasmin, which can degrade the fibrin strands. The highlighted components are known to be altered in MS patients. Several known inhibitors (coloured red), including approved drugs (rivaroxaban, dabigatran and hirudin) and preclinical components (infestin 4 and monoclonal antibody 5B8), are known to inhibit the cascade at certain steps, which has potentially protective effects in in vitro models of MS.
Coagulation factors such as tissue factor (TF) and protein C inhibitor (PCI), which are part of the proximal extrinsic coagulation cascade, are expressed in chronic active plaque samples from patients with MS228. Furthermore, protein C plasma levels are associated with neurodegenerative MRI outcomes in MS — specifically, low grey matter volume229. In addition, thrombin activity has been shown to precede the onset of neurological signs in an EAE model, and increased thrombin activity was observed at the peak of the course of the disease230.
Alterations in proximal parts of the intrinsic coagulation pathway have been implicated in MS pathogenesis. Patients with MS show high plasma levels of factor XII (FXII) during relapse. Pharmacological blockade of FXII leads to reduced susceptibility in an EAE model — an effect that is mediated by a shift in the cytokine profile of dendritic cells that reduces immune activation231. In addition, factor X (FX) serum levels are also increased in individuals with MS232, and inhibition of FX with rivaroxaban, a clinically approved anticoagulant therapy, can reduce EAE severity233.
Inhibition of more distal parts of the coagulation system using hirudin, a thrombin inhibitor, or recombinant activated protein C reduced EAE severity. EAE amelioration via thrombin inhibition is associated with decreased immune cell proliferation and cytokine production228. Dabigatran, a clinically approved anticoagulant drug, suppresses the thrombin-induced activation of astrocytes and thereby effectively recovers neurological function and protects against demyelination in EAE234. We note that thrombin is a well known activator of pro-inflammatory proteinase activated receptor 1 (PAR1) signalling, so some of the effects of thrombin inhibition may be mediated through reduced PAR1 activity234.
During BBB disruption, the terminal blood coagulation factor fibrinogen is able to enter the CNS and be converted to fibrin, which then activates an immune response235. BBB disruption is one of the earliest hallmarks of MS pathology, with fibrin deposition observed throughout the course of the disease and detected in the CSF of patients with MS236. Pre-demyelinating lesions demonstrate BBB disruption and increased fibrin deposition with local microglial activation237. In progressive MS, fibrin is detected in active and chronic lesions and fibrin deposition is associated with neuronal loss238. Recently, Ryu et al. developed a monoclonal antibody (5B8) that specifically targets the cryptic fibrin epitope γ377–395 and selectively inhibits fibrin-induced inflammation without altering clotting. Application of this antibody suppressed the innate immune response, oxidative stress, demyelination, and axonal damage and, in aggregate, ameliorated the course of the disease in multiple experimental models of MS239.
Plasminogen is converted to plasmin, which cleaves fibrin networks240. Interestingly, tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA), which cleave plasminogen to plasmin, have heightened activity in MS lesions and in the CSF of patients with MS. Deficiency of tPA or uPA leads to an earlier onset and more severe EAE course241,242, but neuronal tPA overexpression failed to alter the disease242. In contrast, treating mice with tPA variant proteins or plasminogen activator inhibitor 1-derived peptide (PAI1-dp) — which block the intrinsic binding of PAI1 and thereby increase the activity of tPA and uPA — significantly ameliorates disease severity in EAE. This effect might be mediated, at least in part, through an increased number of Treg cells and diminished T cell reactivity241,243. Recombinant tPA is used as a thrombolytic agent in ischaemic stroke; however, owing to bleeding caused by its catalytic activity, it can be administered only once and is therefore not useful in chronic disease. Other preclinical anticoagulative therapies such as infestin-4, a highly specific FXII inhibitor, can improve disease progression in EAE without compromising haemostasis, as seen in other animal models231,244. To overcome possible bleeding or thrombotic side effects, more-specific therapies are needed that target only the potentially immunomodulatory aspect of the coagulation system.
Tolerance induction
The breakdown of immunological tolerance mechanisms is a hallmark of MS pathogenesis. Autoreactive T cells are eliminated by central tolerance mechanisms in the thymus. However, this process is imperfect and some autoreactive T cells are released into the circulation. Under physiological conditions, those autoreactive cells are controlled by peripheral immune tolerance, which is mainly mediated by Treg cells. In MS, autoreactive T cells and autoantibodies against CNS antigens as well as impaired Treg cell function can be detected and have been implicated as central drivers of pathology245,246.
Therapeutic approaches addressing those pathogenic factors with antigen-specific tolerization (AST) or antigen-unspecific tolerization strategies have already been tested at early stages of clinical development in MS (Fig. 6). The rationale behind AST is to silence and/or remove autoreactive CD4+ T cells that recognize CNS antigens in an HLA-DR-restricted manner. Previous therapeutic approaches focused mainly on myelin proteins such as myelin basic protein (MBP), myelin proteolipid protein (PLP) or MOG, because they are encephalitogenic in EAE and are immunodominant in patients with MS23,247,248. However, a plethora of antigens, such as GDP l-fucose synthase and RAS guanyl-releasing protein 2, may also have a decisive role249. Consistent with this, patients with MS demonstrate high T cell receptor (TCR) repertoire diversity and interindividual variability250. With increasing disease duration, epitope spreading further complicates AST strategies251,252. To circumvent epitope spreading, autoantigen cocktails or coupling of peptides to cells have been proposed as potential mitigation strategies253,254,255,256. The multitude of administration routes (oral, nasal, transdermal, intramuscular, intravenous, coupled to cells, or coupled to nanoparticles) and modalities (whole protein, peptides, DNA, T cell or TCR vaccinations, and tolerogenic dendritic cells) further increase the complexity of AST257. Moreover, it is also difficult to assess treatment efficacy owing to the rarity of autoreactive T cells and a lack of specific markers for them. Despite success in animal models, these challenges and other limitations, such as insufficient patient stratification, have led to many discouraging results in clinical tolerization trials258,259,260,261,262,263,264,265. We note that attempted tolerization with an altered peptide ligand of MBP83-99 has induced MS disease activity266. The use of autoantigen cocktails provided the first promising results as transdermal administration of three myelin peptides (MBP85-99, MOG35-55 and PLP139-155) reduced clinical and MRI disease activity in patients with RRMS267. However, this trial included only 30 patients and thus larger studies are required.

Immune tolerance induction aims to treat multiple sclerosis (MS) at the initial stages of pathogenesis. Various approaches are of interest to induce immune tolerance and can be separated into antigen-specific (labelled 1 to 8) and unspecific tolerization strategies (labelled 10 to 13). The mechanisms (labelled 9) of immune tolerance are driven by tolerogenic antigen-presenting cells (APCs). Application of the different potential auto-pathogen factors can lead to the development of those cells. APC interaction with T cells leads to several changes in the immune response: the induction of regulatory T (Treg) cells; reduction of effector T cells; an increase in exhausted T cells and Treg cells with strong bystander immunosuppression capacity; apoptosis of autoreactive T cells; and anergy in T cells. Ag, antigen; AHR, aryl hydrocarbon receptor; CAR, chimeric antigen receptor; CTLA4, cytotoxic T lymphocyte antigen 4; DC, dendritic cell; GAL9, galectin 9; MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; PB, peripheral blood; PBMC, peripheral blood mononuclear cell; PD1, programmed cell death 1; PDL1, programmed cell death 1 ligand; RBC, red blood cell; TH, T helper; TCR, T cell receptor; TGFβ, transforming growth factor-β; TIM3, T cell immunoglobulin mucin receptor 3; UCB, umbilical cord blood.
Nevertheless, recent advances in AST have created enthusiasm in the field. The advent of mRNA vaccines during the COVID-19 pandemic has generated great interest in exploiting this vaccination strategy to treat other conditions. Krienke et al. vaccinated mice with EAE with a nanoparticle-formulated 1-methylpseudouridine-modified mRNA (m1ψ-mRNA) that codes for multiple disease-related autoantigens (such as MOG35-55 and PLP139-151). The m1ψ modification and subsequent removal of double-stranded mRNA contaminants prevented an innate immune response to the mRNA. M1ψ-mRNA vaccination resulted in autoantigen presentation by APCs without inducing costimulatory signals or cytokines (such as CD86 and IFN), which reduced the number of effector T cells, increased the number of exhausted T cells, and induced Treg cells with strong bystander immunosuppression capacity. Furthermore, the vaccine either abrogated disease development or stopped disease progression in the MOG35-55 EAE model, and controlled relapses in PLP139-151 EAE. Non-MOG antigen-specific immune responses were not affected, indicating that other functions of the immune system are not compromised. M1ψ-mRNA coding for autoantigens suppressed EAE induced by other autoantigens almost as effectively as if both antigens were identical. The observed strong bystander immunosuppression by Treg cells might therefore — at least in part — be able to compensate for epitope spreading, interindividual antigen variability and polyclonality of autoimmunity in MS. Moreover, repeat administration of M1ψ-mRNA was not compromised by induction of autoantigen-specific antibody responses. mRNA vaccines can encode any antigen and thus allow for tailored AST therapies268.
Another approach with which to address polyclonality and individual antigen variability is the use of oligodendrocyte-derived extracellular vesicles that contain multiple myelin antigens. Intravenous administration of oligodendrocyte-derived extracellular vesicles induced immunosuppressive monocytes and apoptosis of autoreactive CD4+ T cells, and ameliorated the course of the disease in chronic and relapsing-remitting EAE models. Human oligodendrocyte-derived extracellular vesicles contained relevant myelin peptides, thereby providing the basis for human translation269. However, it remains unclear how durable monocyte-mediated tolerization is and whether there is sufficient bystander immunosuppression to counteract epitope spreading. Extracellular vesicle generation is complex, which may also interfere with large-scale production and personalized approaches. In addition to myelin antigens, several neuroaxonal antigens have been implicated in the pathogenesis of MS270 and T cells display antigen-specific immune responses to these molecules271. Therefore, future tolerization approaches might be tailored to axonal antigens, potentially providing neuroprotection.
Antigen-unspecific tolerization strategies may also be valuable therapeutic options in MS. Their main advantages are their potential broad applicability and efficacy in several autoimmune diseases, in a range of patient cohorts. In contrast to AST, these unspecific approaches do not affect the underlying cause of the dysregulation and thus have to be administered repeatedly. Moreover, the unspecific effects might affect immune system function.
Antigen-unspecific strategies might include modulating immune checkpoints, inhibitors of which have revolutionized cancer therapy272. Immune checkpoints control immune homeostasis by shifting T cell cytokine profiles from pro-inflammatory to anti-inflammatory, and by promoting and maintaining Treg cells. For some of these pathways, activators have been developed and tested in autoimmunity.
The fusion protein containing cytotoxic T lymphocyte antigen 4 (CTLA4) fused to immunoglobulin, abatacept, inhibits T cell activation and is approved for the treatment of rheumatoid arthritis; however, a pilot study including 20 patients with RRMS was prematurely terminated because of increased disease activity in the low-dose abatacept group273. Interestingly, unblinding revealed a higher relapse rate in this group prior to investigational treatment; the negative outcome could therefore be related to a randomization error. Other coinhibitory receptors such as T cell immunoglobulin mucin receptor 3 (TIM3, also known as HAVCR2) and programmed cell death 1 (PD1) and its ligand PDL1 are potential targets in MS therapy. Dysfunction of these pathways plays a part in the pathogenesis of EAE and MS274,275, and activation of these pathways downregulates T cell responses276. Conversely, PD1/PDL1 blockade may result in hyperactivation of T and B cells, and this has been invoked as the mechanism underlying the demyelinating adverse effects of this class of cancer drugs277.
Additional, unspecific tolerization strategies exploit the tolerance-inducing abilities of parasites. Patients with MS who were infected with Ascaris lumbricoides demonstrated low MS disease activity, and treatment with Trichuris suis reduced the number of gadolinium-enhancing lesions278,279,280,281. However, the studies had considerable limitations — small sample size, no control group, unknown mechanism of action, and adverse effects. Identifying the tolerance-inducing mechanisms used by those parasites may lead to new therapeutic strategies that do not require parasite administration. The ligand-activated transcription factor aryl hydrocarbon receptor (AHR) integrates signals from environmental stressors to produce immune responses. AHR activation induces functional Treg cells and various other immune pathways282. Activation of AHR suppresses the development of EAE and patients with MS show lower levels of circulating AHR ligands compared to healthy controls, indicating an important role in MS pathology283,284. Therefore, AHR-activating ligands may also be used as an MS therapy. Among other effects, laquinimod activates AHR and demonstrated beneficial effects in initial clinical trials in MS285,286,287. In a phase III study (CONCERTO), laquinimod protected nervous tissue (as measured by brain volume), but this treatment was not able to reduce the risk of progressive disability288. Interestingly, the combination of AHR agonists and MOG35-55 in nanoliposomes induced antigen-specific tolerance and strong bystander immunosuppression, which abrogated EAE289. These data suggest a strategy combining AST with nonspecific tolerance induction. Furthermore, dietary intake affects the production of gut microbiome metabolites, such as short-chain fatty acids, that act as tolerance-inducing signals290.
Another possible way to restore peripheral tolerance is to administer tolerogenic cells. These strategies mainly utilize Treg cells, but can use other cell types such as myeloid-derived suppressor cells291. Treg cells engineered with high-affinity TCRs or a chimeric antigen receptor also allow for antigen-specific approaches292. However, many challenges, such as stability, functional activity, cost and delivery are yet to be overcome to allow cell-based strategies in clinical practice293. Nevertheless, a phase Ib/IIa clinical trial using autologous Treg cells in patients with MS has been completed with good safety outcomes294.
Recently, the potassium channel K2P18.1 has been identified as a critical regulator of thymic Treg cell differentiation. Loss of K2P18.1 function reduced Treg cell numbers and worsened EAE295. Furthermore, patients with MS who had a dominant-negative missense K2P18.1 variant had lower Treg cell numbers and worse clinical outcomes compared to non-carriers. Interestingly, pharmacologic activation of K2P18.1 rapidly and reversibly increased Treg cell numbers in humans, thus presenting a new potential therapeutic strategy to exploit tolerance induction by Treg cells for the treatment of autoimmune disorders295.
Additional targets for antigen-specific and antigen-unspecific tolerization strategies are found in the gut-associated lymphoid tissue and the gut microbiome. Microbiota–immune system interactions are essential for immune homeostasis and imbalances are involved in a multitude of immune-mediated disorders296. The gut microbiome has been implicated in providing a pro-inflammatory environment that allows the emergence of activated myelin-specific T cells through bystander activation297. Through the release of metabolic products such as tryptophan derivatives that engage AHR, bacteria can regulate the cytokine milieu in the CNS and the function of neurons and glial cells154,282. Patients with MS have evidence of gut dysbiosis but studies have yielded heterogeneous results regarding overrepresentation and underrepresentation of bacterial species155,298. Frequently, the concentration of the bacterial short-chain fatty acids butyrate and propionic acid are reduced155,163. Supplementation with propionate, AHR ligands and orally delivered antigens can induce Treg cells via gut dendritic cells that are specifically conditioned by gut epithelial cells and microbiota299,300. Probiotics, prebiotics, specific diets, microbiota supplementation and faecal transplantation have been tested in clinical trials to evaluate their effects on the gut microbiome and MS disease outcomes301,302,303.
Thus, immune tolerance strategies offer promising opportunities in MS treatment. However, several challenges remain, including epitope spreading, interindividual antigen variability and polyclonality as well as infrastructural and cost limitations for cell-based therapies. Those pitfalls need to be addressed prior to their introduction to clinical practice.
Conclusions and perspectives
Remarkable progress has been made in our understanding of MS. This knowledge should facilitate the development of therapeutics that focus on the prevention of active lesion formation and early neuroinflammation. However, clinical progression, characterized by increased physical disability and cognitive impairment over time, places a great burden on affected patients. Because the long-term prevention of neurodegeneration remains a major therapeutic challenge, focusing on the mechanisms underlying neurodegeneration in MS is important. Moreover, a deeper understanding of the processes leading to neuronal and axonal degeneration would benefit the treatment of MS, but also of other diseases associated with neurodegeneration, such as Alzheimer disease or Parkinson disease.
Studying aberrations of cellular metabolism is a promising and evolving novel approach in MS. Modulating or fuelling different metabolic pathways could lead to new therapeutics. Moreover, metabolism is linked to environmental cues (such as exposure to pollutants or nutrition) and changes in the redox equilibrium in both the immune and the nervous system. However, most metabolic alterations have instantaneous effects, and metabolism can adapt if needed. This is particularly relevant in patients with co-morbidities such as diabetes or arterial hypertension, which frequently occur in the elderly and interact with MS-specific damage pathways and normal ageing processes.
Currently approved DMTs for MS focus mainly on the inflammatory aspects of the disease and target immune cell populations. However, glial cells such as oligodendrocytes maintain brain homeostasis by supporting neuronal health and contributing to endogenous regenerative processes. Therefore, new therapies improving the function of oligodendrocytes and other glial cells may aid in preventing neurodegeneration and reversing structural damage.
Several coagulation factors may prove to be suitable therapeutic targets in MS, and there are already clinically approved and well characterized drugs available for long-term use. Nevertheless, more research is needed to evaluate the potential therapeutic utility of these drugs in MS and to weigh that utility against the known detrimental side effects, such as bleeding.
Preserving the integrity of the BBB could prevent immune cell infiltration into the CNS, and ECs have a unique position next to the bloodstream, which makes them potential therapeutic targets. However, BBB dysfunction may be a prerequisite for, but not the sole cause of, MS. Therefore, inhibiting BBB breakdown could have beneficial effects but may not prevent all aspects of the disease.
Inducing immune tolerance might address the underlying causes of MS. However, many challenges remain prior to their reaching clinical practice.
Another important factor to be considered when developing CNS therapeutics is the challenge of drug delivery. New pharmacological technologies are required to precisely target specific components within the CNS tissue and must be able to cross the BBB without altering its protective function.
In conclusion, innovative MS therapies may combine strategies of promoting immunomodulation, fostering remyelination and providing neuroprotection, and future trials should pursue such a multifaceted approach to improve the long-term prognosis for this crippling disease.
References
Reich, D. S., Lucchinetti, C. F. & Calabresi, P. A. Multiple sclerosis. N. Engl. J. Med. 378, 169–180 (2018).
Baecher-Allan, C., Kaskow, B. J. & Weiner, H. L. Multiple sclerosis: mechanisms and immunotherapy. Neuron 97, 742–768 (2018).
Walton, C. et al. Rising prevalence of multiple sclerosis worldwide: insights from the Atlas of MS, third edition. Mult. Scler. 26, 1816–1821 (2020).
Sorensen, P. S. et al. The apparently milder course of multiple sclerosis: changes in the diagnostic criteria, therapy and natural history. Brain 143, 2637–2652 (2020).
Cree, B. A. C. et al. Secondary progressive multiple sclerosis: new insights. Neurology 97, 378–388 (2021).
Lublin, F. D., Coetzee, T., Cohen, J. A., Marrie, R. A. & Thompson, A. J. The 2013 clinical course descriptors for multiple sclerosis. Neurology 94, 1088–1092 (2020).
Mitchell, T. W. et al. Global, regional, and national burden of multiple sclerosis 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 269–285 (2019).
Lassmann, H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front. Immunol. 9, 3116 (2019).
Pardini, M., Brown, J. W. L., Magliozzi, R., Reynolds, R. & Chard, D. T. Surface-in pathology in multiple sclerosis: a new view on pathogenesis? Brain 144, 1646–1654 (2021).
Vollmer, T. L., Nair, K. V., Williams, I. M. & Alvarez, E. Multiple sclerosis phenotypes as a continuum: the role of neurologic reserve. Neurol. Clin. Pract. 11, 342–351 (2021).
Bar-Or, A. & Li, R. Cellular immunology of relapsing multiple sclerosis: interactions, checks, and balances. Lancet Neurol. 20, 470–483 (2021).
Linnerbauer, M., Wheeler, M. A. & Quintana, F. J. Astrocyte crosstalk in CNS inflammation. Neuron 108, 608–622 (2020).
Thompson, A. J., Baranzini, S. E., Geurts, J., Hemmer, B. & Ciccarelli, O. Multiple sclerosis. Lancet 391, 1622–1636 (2018).
McGinley, M. P., Goldschmidt, C. H. & Rae-Grant, A. D. Diagnosis and treatment of multiple sclerosis: a review. JAMA 325, 765–779 (2021).
Tintore, M., Vidal-Jordana, A. & Sastre-Garriga, J. Treatment of multiple sclerosis — success from bench to bedside. Nat. Rev. Neurol. 15, 53–58 (2019).
Brown, J. W. L. et al. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA 321, 175–187 (2019).
Amato, M. P. et al. Disease-modifying drugs can reduce disability progression in relapsing multiple sclerosis. Brain 143, 3013–3024 (2020).
Rollot, F. et al. Cumulative effects of therapies on disability in relapsing multiple sclerosis. Mult. Scler. J. 27, 1760–1770 (2021).
Martin, R., Sospedra, M., Eiermann, T. & Olsson, T. Multiple sclerosis: doubling down on MHC. Trends Genet 37, 784–797 (2021).
Jelcic, I. et al. Memory B cells activate brain-homing, autoreactive CD4+ T cells in multiple sclerosis. Cell 175, 85–100.e23 (2018).
Wekerle, H. B cells in multiple sclerosis. Autoimmunity 50, 57–60 (2017).
Mundt, S., Greter, M., Flügel, A. & Becher, B. The CNS immune landscape from the viewpoint of a T cell. Trends Neurosci. 42, 667–679 (2019).
Dendrou, C. A., Fugger, L. & Friese, M. A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15, 545–558 (2015).
Wang, J. et al. HLA-DR15 molecules jointly shape an autoreactive T cell repertoire in multiple sclerosis. Cell 183, 1264–1281.e20 (2020).
Berer, K. et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl Acad. Sci. USA 114, 10719–10724 (2017).
Cekanaviciute, E. et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl Acad. Sci. USA 114, 10713–10718 (2017).
Jangi, S. et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 7, 12015 (2016).
Bar-Or, A. et al. Epstein–Barr virus in multiple sclerosis: theory and emerging immunotherapies. Trends Mol. Med. 26, 296–310 (2020).
Absinta, M. et al. Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis. Neurology 85, 18–28 (2015).
Magliozzi, R. et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 130, 1089–1104 (2007).
Li, R., Patterson, K. R. & Bar-Or, A. Reassessing B cell contributions in multiple sclerosis. Nat. Immunol. 19, 696–707 (2018).
Michel, L. et al. B cells in the multiple sclerosis central nervous system: trafficking and contribution to CNS-compartmentalized inflammation. Front. Immunol. 6, 636 (2015).
Louveau, A. et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci. 21, 1380–1391 (2018).
Rustenhoven, J. et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell 184, 1000–1016.e27 (2021).
Alves de Lima, K., Rustenhoven, J. & Kipnis, J. Meningeal immunity and its function in maintenance of the central nervous system in health and disease. Annu. Rev. Immunol. 38, 597–620 (2020).
Graf, J. et al. Targeting B cells to modify MS, NMOSD, and MOGAD. Neurol. Neuroimmunol. Neuroinflamm. 8, e918 (2020).
Machado-Santos, J. et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 141, 2066–2082 (2018).
Pardo, G. & Jones, D. E. The sequence of disease-modifying therapies in relapsing multiple sclerosis: safety and immunologic considerations. J. Neurol. 264, 2351–2374 (2017).
Jakimovski, D., Kolb, C., Ramanathan, M., Zivadinov, R. & Weinstock-Guttman, B. Interferon β for multiple sclerosis. Cold Spring Harb. Perspect. Med. 8, 165–172 (2018).
Prod’homme, T. & Zamvil, S. S. The evolving mechanisms of action of glatiramer acetate. Cold Spring Harb. Perspect. Med. 9, a029249 (2019).
Tsai, J. J. et al. Nrf2 regulates haematopoietic stem cell function. Nat. Cell Biol. 15, 309–316 (2013).
Sies, H., Berndt, C. & Jones, D. P. Oxidative stress. Annu. Rev. Biochem. 86, 715–748 (2017).
Lee, D.-H., Stangel, M., Gold, R. & Linker, R. A. The fumaric acid ester BG-12: a new option in MS therapy. Expert Rev. Neurother. 13, 951–958 (2013).
Lebrun, C. & Rocher, F. Cancer risk in patients with multiple sclerosis: potential impact of disease-modifying drugs. CNS Drugs 32, 939–949 (2018).
Rae-Grant, A. et al. Practice guideline recommendations summary: disease-modifying therapies for adults with multiple sclerosis: report of the guideline development, dissemination, and implementation subcommittee of the American Academy of Neurology. Neurology 90, 777–788 (2018).
Vermersch, P. et al. Teriflunomide vs injectable disease modifying therapies for relapsing forms of MS. Mult. Scler. Relat. Disord. 43, 102158 (2020).
Ruck, T., Bittner, S., Wiendl, H. & Meuth, S. G. Alemtuzumab in multiple sclerosis: mechanism of action and beyond. Int. J. Mol. Sci. 16, 16414–16439 (2015).
Comi, G. et al. Role of B cells in multiple sclerosis and related disorders. Ann. Neurol. 89, 13–23 (2021).
Hawker, K. et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 66, 460–471 (2009).
Montalban, X. et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 376, 209–220 (2017).
Derfuss, T. et al. Advances in oral immunomodulating therapies in relapsing multiple sclerosis. Lancet Neurol. 19, 336–347 (2020).
Sellner, J. & Rommer, P. S. Immunological consequences of “immune reconstitution therapy” in multiple sclerosis: a systematic review. Autoimmun. Rev. 19, 102492 (2020).
Wekerle, H., Linington, C., Lassmann, H. & Meyermann, R. Cellular immune reactivity within the CNS. Trends Neurosci. 9, 271–277 (1986).
Zamvil, S. S. & Steinman, L. The T lymphocyte in experimental allergic encephalomyelitis. Annu. Rev. Immunol. 8, 579–621 (1990).
Stern, J. N. H. et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl Med. 6, 248ra107 (2014).
Matloubian, M. et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427, 355–360 (2004).
Yednock, T. A. et al. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4βl integrin. Nature 356, 63–66 (1992).
Polman, C. H. et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 354, 899–910 (2006).
Roy, R., Alotaibi, A. A. & Freedman, M. S. Sphingosine 1-phosphate receptor modulators for multiple sclerosis. CNS Drugs 35, 385–402 (2021).
Cree, B. A. et al. Siponimod: disentangling disability and relapses in secondary progressive multiple sclerosis. Mult. Scler. J. 27, 1564–1576 (2020).
Aktas, O., Küry, P., Kieseier, B. & Hartung, H.-P. Fingolimod is a potential novel therapy for multiple sclerosis. Nat. Rev. Neurol. 6, 373–382 (2010).
Faissner, S. & Gold, R. Progressive multiple sclerosis: latest therapeutic developments and future directions. Ther. Adv. Neurol. Disord. 12, 1756286419878323 (2019).
Burman, J. Delaying the inevitable: are disease modifying drugs for progressive MS worthwhile? Mult. Scler. Relat. Disord. 54, 103134 (2021).
Ciotti, J. R. & Cross, A. H. Disease-modifying treatment in progressive multiple sclerosis. Curr. Treat. Options Neurol. 20, 12 (2018).
Philips, T. & Rothstein, J. D. Oligodendroglia: metabolic supporters of neurons. J. Clin. Invest. 127, 3271–3280 (2017).
Mishra, M. K. et al. Harnessing the benefits of neuroinflammation: generation of macrophages/microglia with prominent remyelinating properties. J. Neurosci. 41, 3366–3385 (2021).
Cignarella, F. et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 140, 513–534 (2020).
Lubetzki, C., Zalc, B., Williams, A., Stadelmann, C. & Stankoff, B. Remyelination in multiple sclerosis: from basic science to clinical translation. Lancet Neurol. 19, 678–688 (2020).
Franklin, R. J. M., Frisén, J. & Lyons, D. A. Revisiting remyelination: towards a consensus on the regeneration of CNS myelin. Semin. Cell Dev. Biol. 116, 3–9 (2020).
Kuhlmann, T. et al. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131, 1749–1758 (2008).
Prozorovski, T., Schneider, R., Berndt, C., Hartung, H.-P. & Aktas, O. Redox-regulated fate of neural stem progenitor cells. Biochim. Biophys. Acta 1850, 1543–1554 (2015).
Kirby, L. et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 10, 3887 (2019).
Babbe, H. et al. Clonal expansions of CD8+ T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 192, 393–404 (2000).
Göttle, P. et al. Teriflunomide promotes oligodendroglial differentiation and myelination. J. Neuroinflamm. 15, 76 (2018).
Yeung, M. S. Y. et al. Dynamics of oligodendrocyte generation in multiple sclerosis. Nature 566, 538–542 (2019).
Kremer, D., Göttle, P., Hartung, H.-P. & Küry, P. Pushing forward: remyelination as the new frontier in CNS diseases. Trends Neurosci. 39, 246–263 (2016).
Thomas, L. & Pasquini, L. A. Galectin-3 exerts a pro-differentiating and pro-myelinating effect within a temporal window spanning precursors and pre-oligodendrocytes: insights into the mechanisms of action. Mol. Neurobiol. 57, 976–987 (2019).
Suo, N., Guo, Y., He, B., Gu, H. & Xie, X. Inhibition of MAPK/ERK pathway promotes oligodendrocytes generation and recovery of demyelinating diseases. Glia 67, 1320–1332 (2019).
Jäkel, S. et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543–547 (2019).
Falcão, A. M. et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 24, 1837–1844 (2019).
Nikić, I. et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 17, 495–499 (2011).
Saab, A. S. et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 91, 119–132 (2016).
Rone, M. B. et al. Oligodendrogliopathy in multiple sclerosis: low glycolytic metabolic rate promotes oligodendrocyte survival. J. Neurosci. 36, 4698–4707 (2016).
Ziabreva, I. et al. Injury and differentiation following inhibition of mitochondrial respiratory chain complex IV in rat oligodendrocytes. Glia 58, 1827–1837 (2010).
Voskuhl, R. R. et al. Gene expression in oligodendrocytes during remyelination reveals cholesterol homeostasis as a therapeutic target in multiple sclerosis. Proc. Natl Acad. Sci. USA 116, 10130–10139 (2019).
Dimas, P. et al. CNS myelination and remyelination depend on fatty acid synthesis by oligodendrocytes. eLife 8, e44702 (2019).
Cadavid, D. et al. Safety and efficacy of opicinumab in patients with relapsing multiple sclerosis (SYNERGY): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 18, 845–856 (2019).
Cadavid, D. et al. Safety and efficacy of opicinumab in acute optic neuritis (RENEW): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 16, 189–199 (2017).
Calabresi, P. et al. Efficacy and safety of opicinumab in participants with relapsing multiple sclerosis: a randomized, placebo-controlled, phase 2 trial (AFFINITY part 1). Presented at the European Committee for Treatment and Research in Multiple Sclerosis Conference (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03222973 (2022).
Stangel, M., Kuhlmann, T., Matthews, P. M. & Kilpatrick, T. J. Achievements and obstacles of remyelinating therapies in multiple sclerosis. Nat. Rev. Neurol. 13, 742–754 (2017).
Mei, F. et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 20, 954–960 (2014).
Green, A. J. et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial. Lancet 390, 2481–2489 (2017).
Deshmukh, V. A. et al. A regenerative approach to the treatment of multiple sclerosis. Nature 502, 327–332 (2013).
Welliver, R. R. et al. Muscarinic receptor M3R signaling prevents efficient remyelination by human and mouse oligodendrocyte progenitor cells. J. Neurosci. 38, 6921–6932 (2018).
Najm, F. J. et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 522, 216–220 (2015).
Cerina, M. et al. The quality of cortical network function recovery depends on localization and degree of axonal demyelination. Brain Behav. Immun. 59, 103–117 (2017).
Banks, W. A. From blood–brain barrier to blood–brain interface: new opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 15, 275–292 (2016).
Wuerfel, J. et al. Changes in cerebral perfusion precede plaque formation in multiple sclerosis: a longitudinal perfusion MRI study. Brain J. Neurol. 127, 111–119 (2004).
Munji, R. N. et al. Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood–brain barrier dysfunction module. Nat. Neurosci. 22, 1892–1902 (2019).
Sweeney, M. D., Zhao, Z., Montagne, A., Nelson, A. R. & Zlokovic, B. V. Blood–brain barrier: from physiology to disease and back. Physiol. Rev. 99, 21–78 (2018).
Odoardi, F. et al. T cells become licensed in the lung to enter the central nervous system. Nature 488, 675–679 (2012).
Hiltensperger, M. et al. Skin and gut imprinted helper T cell subsets exhibit distinct functional phenotypes in central nervous system autoimmunity. Nat. Immunol. 22, 880–892 (2021).
Engelhardt, B. & Ransohoff, R. M. Capture, crawl, cross: the T cell code to breach the blood–brain barriers. Trends Immunol. 33, 579–589 (2012).
Brocke, S., Piercy, C., Steinman, L., Weissman, I. L. & Veromaa, T. Antibodies to CD44 and integrin α4, but not l-selectin, prevent central nervous system inflammation and experimental encephalomyelitis by blocking secondary leukocyte recruitment. Proc. Natl Acad. Sci. USA 96, 6896–6901 (1999).
Döring, A., Wild, M., Vestweber, D., Deutsch, U. & Engelhardt, B. E- and P-selectin are not required for the development of experimental autoimmune encephalomyelitis in C57BL/6 and SJL mice. J. Immunol. 179, 8470–8479 (2007).
Cayrol, R. et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat. Immunol. 9, 137–145 (2008).
Wagner, M. et al. ALCAM — novel multiple sclerosis locus interfering with HLA-DRB1*1501. J. Neuroimmunol. 258, 71–76 (2013).
Schneider-Hohendorf, T. et al. VLA-4 blockade promotes differential routes into human CNS involving PSGL-1 rolling of T cells and MCAM-adhesion of TH17 cells. J. Exp. Med. 211, 1833–1846 (2014).
Göbel, K. et al. Plasma kallikrein modulates immune cell trafficking during neuroinflammation via PAR2 and bradykinin release. Proc. Natl Acad. Sci. USA 116, 271–276 (2019).
Bittner, S. et al. Endothelial TWIK-related potassium channel-1 (TREK1) regulates immune-cell trafficking into the CNS. Nat. Med. 19, 1161–1165 (2013).
Lengfeld, J. E. et al. Endothelial Wnt/β-catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc. Natl Acad. Sci. USA 114, E1168–E1177 (2017).
Niu, J. et al. Aberrant oligodendroglial-vascular interactions disrupt the blood–brain barrier, triggering CNS inflammation. Nat. Neurosci. 22, 709–718 (2019).
Lee, H. K. et al. Apcdd1 stimulates oligodendrocyte differentiation after white matter injury. Glia 63, 1840–1849 (2015).
Niu, J. et al. Oligodendroglial ring finger protein Rnf43 is an essential injury-specific regulator of oligodendrocyte maturation. Neuron 109, 3104–3118.e6 (2021).
Chavali, M. et al. Wnt-dependent oligodendroglial-endothelial interactions regulate white matter vascularization and attenuate injury. Neuron 108, 1130–1145.e5 (2020).
Wouters, E. et al. Liver X receptor alpha is important in maintaining blood–brain barrier function. Front. Immunol. 10, 1811 (2019).
Lertkiatmongkol, P., Liao, D., Mei, H., Hu, Y. & Newman, P. J. Endothelial functions of PECAM-1 (CD31). Curr. Opin. Hematol. 23, 253–259 (2016).
Graesser, D. et al. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J. Clin. Invest. 109, 383–392 (2002).
Wimmer, I. et al. PECAM-1 stabilizes blood-brain barrier integrity and favors paracellular T-cell diapedesis across the blood–brain barrier during neuroinflammation. Front. Immunol. 10, 711 (2019).
Getter, T. et al. Novel inhibitors of leukocyte transendothelial migration. Bioorg. Chem. 92, 103250 (2019).
Argaw, A. T., Gurfein, B. T., Zhang, Y., Zameer, A. & John, G. R. VEGF-mediated disruption of endothelial CLN-5 promotes blood–brain barrier breakdown. Proc. Natl Acad. Sci. USA 106, 1977–1982 (2009).
Paul, D. et al. Appearance of claudin-5+ leukocytes in the central nervous system during neuroinflammation: a novel role for endothelial-derived extracellular vesicles. J. Neuroinflammation 13, 292 (2016).
Mandel, I. et al. Tight junction proteins expression and modulation in immune cells and multiple sclerosis. J. Cell. Mol. Med. 16, 765–775 (2012).
Daneman, R., Zhou, L., Kebede, A. A. & Barres, B. A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 468, 562–566 (2010).
Dohgu, S. et al. Brain pericytes contribute to the induction and up-regulation of blood–brain barrier functions through transforming growth factor-β production. Brain Res. 1038, 208–215 (2005).
Lee, S.-W. et al. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 9, 900–906 (2003).
Wosik, K. et al. Angiotensin II controls occludin function and is required for blood–brain barrier maintenance: relevance to multiple sclerosis. J. Neurosci. 27, 9032–9042 (2007).
Rempe, R. G., Hartz, A. M. & Bauer, B. Matrix metalloproteinases in the brain and blood–brain barrier: versatile breakers and makers. J. Cereb. Blood Flow Metab. 36, 1481–1507 (2016).
Gerwien, H. et al. Imaging matrix metalloproteinase activity in multiple sclerosis as a specific marker of leukocyte penetration of the blood–brain barrier. Sci. Transl Med. 8, 364ra152 (2016).
Dubois, B. et al. Resistance of young gelatinase B-deficient mice to experimental autoimmune encephalomyelitis and necrotizing tail lesions. J. Clin. Invest. 104, 1507–1515 (1999).
Fields, G. B. The rebirth of matrix metalloproteinase inhibitors: moving beyond the dogma. Cells 8, 984 (2019).
Bar-Or, A. et al. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain 126, 2738–2749 (2003).
Yen, J.-H., Kong, W. & Ganea, D. IFN-β inhibits dendritic cell migration through STAT-1-mediated transcriptional suppression of CCR7 and matrix metalloproteinase 9. J. Immunol. 184, 3478–3486 (2010).
Ma, Z., Qin, H. & Benveniste, E. N. Transcriptional suppression of matrix metalloproteinase-9 gene expression by IFN-γ and IFN-β: critical role of STAT-1α. J. Immunol. 167, 5150–5159 (2001).
Stüve, O. et al. Interferon β-1b decreases the migration of T lymphocytes in vitro: effects on matrix metalloproteinase-9. Ann. Neurol. 40, 853–863 (1996).
Alt, C., Laschinger, M. & Engelhardt, B. Functional expression of the lymphoid chemokines CCL19 (ELC) and CCL 21 (SLC) at the blood–brain barrier suggests their involvement in G-protein-dependent lymphocyte recruitment into the central nervous system during experimental autoimmune encephalomyelitis. Eur. J. Immunol. 32, 2133–2144 (2002).
McCandless, E. E., Wang, Q., Woerner, B. M., Harper, J. M. & Klein, R. S. CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J. Immunol. 177, 8053–8064 (2006).
McCandless, E. E. et al. Pathological expression of CXCL12 at the blood–brain barrier correlates with severity of multiple sclerosis. Am. J. Pathol. 172, 799–808 (2008).
Cruz-Orengo, L. et al. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J. Exp. Med. 208, 327–339 (2011).
Winkler, E. A., Bell, R. D. & Zlokovic, B. V. Central nervous system pericytes in health and disease. Nat. Neurosci. 14, 1398–1405 (2011).
Cheng, J. et al. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 136, 507–523 (2018).
Rustenhoven, J., Jansson, D., Smyth, L. C. & Dragunow, M. Brain pericytes as mediators of neuroinflammation. Trends Pharmacol. Sci. 38, 291–304 (2017).
Padel, T. et al. Platelet-derived growth factor-BB has neurorestorative effects and modulates the pericyte response in a partial 6-hydroxydopamine lesion mouse model of Parkinson’s disease. Neurobiol. Dis. 94, 95–105 (2016).
Kang, E. & Shin, J. W. Pericyte-targeting drug delivery and tissue engineering. Int. J. Nanomed. 11, 2397–2406 (2016).
Gugliandolo, A., Bramanti, P. & Mazzon, E. Mesenchymal stem cells in multiple sclerosis: recent evidence from pre-clinical to clinical studies. Int. J. Mol. Sci. 21, 8662 (2020).
Do, P. T., Wu, C.-C., Chiang, Y.-H., Hu, C.-J. & Chen, K.-Y. Mesenchymal stem/stromal cell therapy in blood–brain barrier preservation following ischemia: molecular mechanisms and prospects. Int. J. Mol. Sci. 22, 10045 (2021).
Braniste, V. et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl Med. 6, 263ra158 (2014).
Logsdon, A. F., Erickson, M. A., Rhea, E. M., Salameh, T. S. & Banks, W. A. Gut reactions: how the blood–brain barrier connects the microbiome and the brain. Exp. Biol. Med. 243, 159–165 (2018).
Fleischer, V. et al. Translational value of choroid plexus imaging for tracking neuroinflammation in mice and humans. Proc. Natl Acad. Sci. USA 118, e2025000118 (2021).
Bhargava, P. & Calabresi, P. A. Metabolomics in multiple sclerosis. Mult. Scler. J. 22, 451–460 (2016).
Kasakin, M. F. et al. Targeted metabolomics approach for identification of relapsing–remitting multiple sclerosis markers and evaluation of diagnostic models. Medchemcomm 10, 1803–1809 (2019).
Tisell, A. et al. Increased concentrations of glutamate and glutamine in normal-appearing white matter of patients with multiple sclerosis and normal MR imaging brain scans. PLoS ONE 8, e61817 (2013).
Fitzgerald, K. C. et al. Multi-omic evaluation of metabolic alterations in multiple sclerosis identifies shifts in aromatic amino acid metabolism. Cell Rep. Med. 2, 100424 (2021).
Levi, I. et al. Potential role of indolelactate and butyrate in multiple sclerosis revealed by integrated microbiome-metabolome analysis. Cell Rep. Med. 2, 100246 (2021).
Nourbakhsh, B. et al. Altered tryptophan metabolism is associated with pediatric multiple sclerosis risk and course. Ann. Clin. Transl. Neurol. 5, 1211–1221 (2018).
Chen, T., Noto, D., Hoshino, Y., Mizuno, M. & Miyake, S. Butyrate suppresses demyelination and enhances remyelination. J. Neuroinflammation 16, 165 (2019).
Bhargava, P. et al. Bile acid metabolism is altered in multiple sclerosis and supplementation ameliorates neuroinflammation. J. Clin. Invest. 130, 3467–3482 (2020).
Crick, P. J. et al. Reduced plasma levels of 25-hydroxycholesterol and increased cerebrospinal fluid levels of bile acid precursors in multiple sclerosis patients. Mol. Neurobiol. 54, 8009–8020 (2017).
Parodi, B. & Kerlero de Rosbo, N. The gut–brain axis in multiple sclerosis. is its dysfunction a pathological trigger or a consequence of the disease? Front. Immunol. 12, 718220 (2021).
Kadowaki, A. & Quintana, F. J. The gut–CNS axis in multiple sclerosis. Trends Neurosci. 43, 622–634 (2020).
Dopkins, N., Nagarkatti, P. S. & Nagarkatti, M. The role of gut microbiome and associated metabolome in the regulation of neuroinflammation in multiple sclerosis and its implications in attenuating chronic inflammation in other inflammatory and autoimmune disorders. Immunology 154, 178–185 (2018).
Takewaki, D. & Yamamura, T. Gut microbiome research in multiple sclerosis. Neurosci. Res. 168, 28–31 (2021).
Cocco, E. et al. 1H-NMR analysis provides a metabolomic profile of patients with multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 3, e185 (2015).
Monaco, F., Fumero, S., Mondino, A. & Mutani, R. Plasma and cerebrospinal fluid tryptophan in multiple sclerosis and degenerative diseases. J. Neurol. Neurosurg. Psychiat. 42, 640–641 (1979).
Polyák, H. et al. Cuprizone markedly decreases kynurenic acid levels in the rodent brain tissue and plasma. Heliyon 7, e06124 (2021).
Gaetani, L. et al. Host and microbial tryptophan metabolic profiling in multiple sclerosis. Front. Immunol. 11, 157 (2020).
Huang, Y.-S., Ogbechi, J., Clanchy, F. I., Williams, R. O. & Stone, T. W. IDO and kynurenine metabolites in peripheral and CNS disorders. Front. Immunol. 11, 388 (2020).
Majláth, Z., Annus, Á. & Vécsei, L. Kynurenine system and multiple sclerosis, pathomechanism and drug targets with an emphasis on laquinimod. Curr. Drug Targets 19, 1873–5592 (2018).
Lovelace, M. D. et al. Current evidence for a role of the kynurenine pathway of tryptophan metabolism in multiple sclerosis. Front. Immunol. 7, 246 (2016).
Lim, C. K. et al. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci. Rep. 7, 1–9 (2017).
Rejdak, K. et al. Decreased level of kynurenic acid in cerebrospinal fluid of relapsing-onset multiple sclerosis patients. Neurosci. Lett. 331, 63–65 (2002).
Leclercq, S., Schwarz, M., Delzenne, N. M., Stärkel, P. & de Timary, P. Alterations of kynurenine pathway in alcohol use disorder and abstinence: a link with gut microbiota, peripheral inflammation and psychological symptoms. Transl. Psychiat. 11, 1–9 (2021).
Nogueras, L. et al. Lipid profile of cerebrospinal fluid in multiple sclerosis patients: a potential tool for diagnosis. Sci. Rep. 9, 11313 (2019).
Pieragostino, D. et al. An integrated metabolomics approach for the research of new cerebrospinal fluid biomarkers of multiple sclerosis. Mol. Biosyst. 11, 1563–1572 (2015).
Law, S.-H. et al. An updated review of lysophosphatidylcholine metabolism in human diseases. Int. J. Mol. Sci. 20, 1149 (2019).
Chalbot, S. et al. Cerebrospinal fluid secretory Ca2+-dependent phospholipase A2 activity: a biomarker of blood–cerebrospinal fluid barrier permeability. Neurosci. Lett. 478, 179–183 (2010).
Trotter, A. et al. The role of phospholipase A2 in multiple sclerosis: a systematic review and meta-analysis. Mult. Scler. Relat. Disord. 27, 206–213 (2019).
Thakker, P. et al. Cytosolic phospholipase A2α blockade abrogates disease during the tissue-damage effector phase of experimental autoimmune encephalomyelitis by its action on APCs. J. Immunol. 187, 1986–1997 (2011).
Qiao, J. et al. Lysophosphatidylcholine impairs endothelial barrier function through the G protein-coupled receptor GPR4. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L91–L101 (2006).
Emwas, A.-H. et al. NMR spectroscopy for metabolomics research. Metabolites 9, 123 (2019).
Konjar, Š. & Veldhoen, M. Dynamic metabolic state of tissue resident CD8 T cells. Front. Immunol. 10, 1683 (2019).
Macintyre, A. N. et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20, 61–72 (2014).
Reboldi, A. et al. C–C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 10, 514–523 (2009).
Puniya, B. L. et al. Integrative computational approach identifies drug targets in CD4+ T-cell-mediated immune disorders. NPJ Syst. Biol. Appl. 7, 4 (2021).
La Rocca, C. et al. Immunometabolic profiling of T cells from patients with relapsing-remitting multiple sclerosis reveals an impairment in glycolysis and mitochondrial respiration. Metabolism 77, 39–46 (2017).
Klotz, L. et al. Teriflunomide treatment for multiple sclerosis modulates T cell mitochondrial respiration with affinity-dependent effects. Sci. Transl Med. 11, eaao5563 (2019).
Shin, B. et al. Mitochondrial oxidative phosphorylation regulates the fate decision between pathogenic Th17 and regulatory T cells. Cell Rep. 30, 1898–1909.e4 (2020).
Cluxton, D., Petrasca, A., Moran, B. & Fletcher, J. M. Differential regulation of human Treg and Th17 cells by fatty acid synthesis and glycolysis. Front. Immunol. 10, 115 (2019).
Shi, L. Z. et al. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376 (2011).
Kono, M. et al. Pyruvate kinase M2 is requisite for Th1 and Th17 differentiation. JCI Insight 4, e127395 (2019).
Angiari, S. et al. Pharmacological activation of pyruvate kinase M2 inhibits CD4+ T cell pathogenicity and suppresses autoimmunity. Cell Metab. 31, 391–405.e8 (2020).
Kornberg, M. D. et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 360, 449–453 (2018).
Li, W. et al. Targeting T cell activation and lupus autoimmune phenotypes by inhibiting glucose transporters. Front. Immunol. 10, 833 (2019).
Berod, L. et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 20, 1327–1333 (2014).
Johnson, M. O. et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 175, 1780–1795.e19 (2018).
Kono, M., Yoshida, N., Maeda, K. & Tsokos, G. C. Transcriptional factor ICER promotes glutaminolysis and the generation of Th17 cells. Proc. Natl Acad. Sci. USA 115, 2478–2483 (2018).
Xu, T. et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 548, 228–233 (2017).
Nakaya, M. et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40, 692–705 (2014).
Stathopoulou, C., Nikoleri, D. & Bertsias, G. Immunometabolism: an overview and therapeutic prospects in autoimmune diseases. Immunotherapy 11, 813–829 (2019).
Bogie, J. F. J., Haidar, M., Kooij, G. & Hendriks, J. J. A. Fatty acid metabolism in the progression and resolution of CNS disorders. Adv. Drug Deliv. Rev. 159, 198–213 (2020).
Zhang, J., Jin, H., Xu, Y. & Shan, J. Rapamycin modulate Treg/Th17 balance via regulating metabolic pathways: a study in mice. Transplant. Proc. 51, 2136–2140 (2019).
Michalek, R. D. et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303 (2011).
Mørkholt, A. S. et al. CPT1A plays a key role in the development and treatment of multiple sclerosis and experimental autoimmune encephalomyelitis. Sci. Rep. 9, 1–11 (2019).
Shriver, L. P. & Manchester, M. Inhibition of fatty acid metabolism ameliorates disease activity in an animal model of multiple sclerosis. Sci. Rep. 1, 79 (2011).
Trabjerg, M. S. et al. Dysregulation of metabolic pathways by carnitine palmitoyl-transferase 1 plays a key role in central nervous system disorders: experimental evidence based on animal models. Sci. Rep. 10, 15583 (2020).
Raud, B. et al. Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. 28, 504–515.e7 (2018).
Roy, D. G. et al. Methionine metabolism shapes T helper cell responses through regulation of epigenetic reprogramming. Cell Metab. 31, 250–266.e9 (2020).
Ma, E. H. et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. 25, 345–357 (2017).
Andrejeva, G. et al. Metabolomics analysis reveals differential T cell serine metabolism as a target in autoimmunity. J. Immunol. 200, 167.7 (2018).
Bruggeman, Y. et al. Targeting citrullination in autoimmunity: insights learned from preclinical mouse models. Expert Opin. Ther. Targets 0, 1–13 (2021).
Sarswat, A. et al. Inhibitors of protein arginine deiminases and their efficacy in animal models of multiple sclerosis. Bioorg. Med. Chem. 25, 2643–2656 (2017).
Sun, B. et al. Reciprocal regulation of Th2 and Th17 cells by PAD2-mediated citrullination. JCI Insight 4, e129687 (2019).
Robinson, R. R., Dietz, A. K., Maroof, A. M., Asmis, R. & Forsthuber, T. G. The role of glial–neuronal metabolic cooperation in modulating progression of multiple sclerosis and neuropathic pain. Immunotherapy 11, 129–147 (2019).
Pellerin, L. & Magistretti, P. J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl Acad. Sci. USA 91, 10625–10629 (1994).
Schirmer, L. et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573, 75–82 (2019).
Vallée, A., Lecarpentier, Y., Guillevin, R. & Vallée, J.-N. Demyelination in multiple sclerosis: reprogramming energy metabolism and potential PPARγ agonist treatment approaches. Int. J. Mol. Sci. 19, 1212 (2018).
Alizadeh, A., Dyck, S. M. & Karimi-Abdolrezaee, S. Myelin damage and repair in pathologic CNS: challenges and prospects. Front. Mol. Neurosci. 8, 35 (2015).
Chao, C.-C. et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 179, 1483–1498.e22 (2019).
Michaličková, D., Šíma, M. & Slanař, O. New insights in the mechanisms of impaired redox signaling and its interplay with inflammation and immunity in multiple sclerosis. Physiol. Res. 69, 1–19 (2020).
Zipp, F. & Aktas, O. The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci. 29, 518–527 (2006).
Lepka, K. et al. Iron-sulfur glutaredoxin 2 protects oligodendrocytes against damage induced by nitric oxide release from activated microglia. Glia 65, 1521–1534 (2017).
Alba-Arbalat, S. et al. In vivo molecular changes in the retina of patients with multiple sclerosis. Invest. Ophthalmol. Vis. Sci. 62, 11–11 (2021).
Braidy, N., Lim, C. K., Grant, R., Brew, B. J. & Guillemin, G. J. Serum nicotinamide adenine dinucleotide levels through disease course in multiple sclerosis. Brain Res. 1537, 267–272 (2013).
Biller, A. et al. Sodium MRI in multiple sclerosis is compatible with intracellular sodium accumulation and inflammation-induced hyper-cellularity of acute brain lesions. Sci. Rep. 6, 31269 (2016).
Paling, D. et al. Sodium accumulation is associated with disability and a progressive course in multiple sclerosis. Brain 136, 2305–2317 (2013).
Dutta, R. et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 59, 478–489 (2006).
Han, M. H. et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451, 1076–1081 (2008).
Ziliotto, N. et al. Plasma levels of protein C pathway proteins and brain magnetic resonance imaging volumes in multiple sclerosis. Eur. J. Neurol. 27, 235–243 (2020).
Davalos, D. et al. Early detection of thrombin activity in neuroinflammatory disease. Ann. Neurol. 75, 303–308 (2014).
Göbel, K. et al. Blood coagulation factor XII drives adaptive immunity during neuroinflammation via CD87-mediated modulation of dendritic cells. Nat. Commun. 7, 11626 (2016).
Göbel, K. et al. Prothrombin and factor X are elevated in multiple sclerosis patients. Ann. Neurol. 80, 946–951 (2016).
Merker, M. et al. Rivaroxaban ameliorates disease course in an animal model of multiple sclerosis. J. Neuroimmunol. 313, 125–128 (2017).
Chen, R. et al. Dabigatran suppresses PAR-1/SphK/S1P activation of astrocytes in experimental autoimmune encephalomyelitis model. Front. Mol. Neurosci. 13, 114 (2020).
Akassoglou, K. & Strickland, S. Nervous system pathology: the fibrin perspective. Biol. Chem. 383, 37–45 (2002).
Magliozzi, R. et al. Iron homeostasis, complement, and coagulation cascade as CSF signature of cortical lesions in early multiple sclerosis. Ann. Clin. Transl. Neurol. 6, 2150–2163 (2019).
Marik, C., Felts, P. A., Bauer, J., Lassmann, H. & Smith, K. J. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain J. Neurol. 130, 2800–2815 (2007).
Petersen, M. A., Ryu, J. K. & Akassoglou, K. Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat. Rev. Neurosci. 19, 283–301 (2018).
Ryu, J. K. et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat. Immunol. 19, 1212–1223 (2018).
Plantone, D., Inglese, M., Salvetti, M. & Koudriavtseva, T. A perspective of coagulation dysfunction in multiple sclerosis and in experimental allergic encephalomyelitis. Front. Neurol. 9, 1175 (2019).
Gur-Wahnon, D. et al. The plasminogen activator system: involvement in central nervous system inflammation and a potential site for therapeutic intervention. J. Neuroinflamm. 10, 891 (2013).
Dahl, L. C. et al. The influence of differentially expressed tissue-type plasminogen activator in experimental autoimmune encephalomyelitis: implications for multiple sclerosis. PLoS ONE 11, e0158653 (2016).
Mizrachi, T., Gur-Wahnon, D., Al-Roof Higazi, A. & Brenner, T. Role of tissue plasminogen activator in clinical aggravation of experimental autoimmune encephalomyelitis and its therapeutic potential. Cell. Immunol. 348, 104040 (2020).
Xu, Y. et al. Factor XIIa inhibition by Infestin-4: in vitro mode of action and in vivo antithrombotic benefit. Thromb. Haemost. 111, 694–704 (2014).
Hohlfeld, R., Dornmair, K., Meinl, E. & Wekerle, H. The search for the target antigens of multiple sclerosis. Part 2: CD8+ T cells, B cells, and antibodies in the focus of reverse-translational research. Lancet Neurol. 15, 317–331 (2016).
Hohlfeld, R., Dornmair, K., Meinl, E. & Wekerle, H. The search for the target antigens of multiple sclerosis. Part 1: autoreactive CD4+ T lymphocytes as pathogenic effectors and therapeutic targets. Lancet Neurol. 15, 198–209 (2016).
Sospedra, M. & Martin, R. Immunology of multiple sclerosis. Semin. Neurol. 36, 115–127 (2016).
Baxter, A. G. The origin and application of experimental autoimmune encephalomyelitis. Nat. Rev. Immunol. 7, 904–912 (2007).
Lutterotti, A., Hayward-Koennecke, H., Sospedra, M. & Martin, R. Antigen-specific immune tolerance in multiple sclerosis — promising approaches and how to bring them to patients. Front. Immunol. 12, 640935 (2021).
Alves Sousa, A. et al. Comprehensive analysis of TCR-β repertoire in patients with neurological immune-mediated disorders. Sci. Rep. 9, 344 (2019).
Vanderlugt, C. L. & Miller, S. D. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat. Rev. Immunol. 2, 85–95 (2002).
Goebels, N. et al. Repertoire dynamics of autoreactive T cells in multiple sclerosis patients and healthy subjects: epitope spreading versus clonal persistence. Brain 123, 508–518 (2000).
Lutterotti, A. et al. Antigen-specific tolerance by autologous myelin peptide–coupled cells: a phase 1 trial in multiple sclerosis. Sci. Transl Med. 5, 188ra75 (2013).
Miller, S. D., Turley, D. M. & Podojil, J. R. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat. Rev. Immunol. 7, 665–677 (2007).
Walker, L. S. K. & Abbas, A. K. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2, 11–19 (2002).
Turley, D. M. & Miller, S. D. Peripheral tolerance induction using ethylenecarbodiimide-fixed APCs uses both direct and indirect mechanisms of antigen presentation for prevention of experimental autoimmune encephalomyelitis. J. Immunol. 178, 2212–2220 (2007).
Serra, P. & Santamaria, P. Antigen-specific therapeutic approaches for autoimmunity. Nat. Biotechnol. 37, 238–251 (2019).
Garren, H. et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann. Neurol. 63, 611–620 (2008).
Weiner, H. L. et al. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science 259, 1321–1324 (1993).
Goodkin, D. E. et al. A phase I trial of solubilized DR2:MBP84-102 (AG284) in multiple sclerosis. Neurology 54, 1414–1420 (2000).
Freedman, M. S. et al. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology 77, 1551–1560 (2011).
Hohol, M. J. et al. Three-year open protocol continuation study of oral tolerization with myelin antigens in multiple sclerosis and design of a phase III pivotal trial. Ann. NY Acad. Sci. 778, 243–250 (1996).
Medaer, R., Stinissen, P., Raus, J., Zhang, J. & Truyen, L. Depletion of myelin-basic-protein autoreactive T cells by T-cell vaccination: pilot trial in multiple sclerosis. Lancet 346, 807–808 (1995).
Warren, K. G., Catz, I., Ferenczi, L. Z. & Krantz, M. J. Intravenous synthetic peptide MBP8298 delayed disease progression in an HLA class II-defined cohort of patients with progressive multiple sclerosis: results of a 24-month double-blind placebo-controlled clinical trial and 5 years of follow-up treatment. Eur. J. Neurol. 13, 887–895 (2006).
Bar-Or, A. et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch. Neurol. 64, 1407–1415 (2007).
Bielekova, B. et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat. Med. 6, 1167–1175 (2000).
Walczak, A., Siger, M., Ciach, A., Szczepanik, M. & Selmaj, K. Transdermal application of myelin peptides in multiple sclerosis treatment. JAMA Neurol. 70, 1105–1109 (2013).
Krienke, C. et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 371, 145–153 (2021).
Casella, G. et al. Oligodendrocyte-derived extracellular vesicles as antigen-specific therapy for autoimmune neuroinflammation in mice. Sci. Transl Med. 12, eaba0599 (2020).
Vyshkina, T. & Kalman, B. Autoantibodies and neurodegeneration in multiple sclerosis. Lab. Invest. 88, 796–807 (2008).
Sauer, B., Schmalstieg, W. & Howe, C. Axons are injured by antigen-specific CD8+ T cells through a MHC class I-and granzyme B-dependent mechanism. Neurobiol. Dis. 59, 194–205 (2013).
Oliveira, M. C. B., de Brito, M. H. & Simabukuro, M. M. Central nervous system demyelination associated with immune checkpoint inhibitors: review of the literature. Front. Neurol. 11, 538695 (2020).
Khoury, S. J. et al. ACCLAIM: a randomized trial of abatacept (CTLA4-Ig) for relapsing-remitting multiple sclerosis. Mult. Scler. J. 23, 686–695 (2017).
Saresella, M. et al. A role for the TIM-3/GAL-9/BAT3 pathway in determining the clinical phenotype of multiple sclerosis. FASEB J. 28, 5000–5009 (2014).
Salama, A. D. et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med. 198, 71–78 (2003).
Li, H. et al. PD-1/PD-L1 axis as a potential therapeutic target for multiple sclerosis: a T cell perspective. Front. Cell. Neurosci. 15, 267 (2021).
Rimkus, C. M. et al. Drug-related demyelinating syndromes: understanding risk factors, pathophysiological mechanisms and magnetic resonance imaging findings. Mult. Scler. Relat. Disord. 55, 103146 (2021).
Fleming, J. et al. Probiotic helminth administration in relapsing–remitting multiple sclerosis: a phase 1 study. Mult. Scler. 17, 743–754 (2011).
Summers, R. W., Elliott, D. E., Urban, J. F., Thompson, R. & Weinstock, J. V. Trichuris suis therapy in Crohn’s disease. Gut 54, 87–90 (2005).
Correale, J. & Farez, M. Association between parasite infection and immune responses in multiple sclerosis. Ann. Neurol. 61, 97–108 (2007).
Voldsgaard, A. et al. Trichuris suis ova therapy in relapsing multiple sclerosis is safe but without signals of beneficial effect. Mult. Scler. J. 21, 1723–1729 (2015).
Rothhammer, V. & Quintana, F. J. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 19, 184–197 (2019).
Quintana, F. J. et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl Acad. Sci. USA 107, 20768–20773 (2010).
Rothhammer, V. et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22, 586–597 (2016).
Vollmer, T. L. et al. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J. Neurol. 261, 773–783 (2014).
Filippi, M. et al. Placebo-controlled trial of oral laquinimod in multiple sclerosis: MRI evidence of an effect on brain tissue damage. J. Neurol. Neurosurg. Psychiatry 85, 851–858 (2014).
Comi, G. et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N. Engl. J. Med. 66, 1000–9 (2012).
Comi, G. et al. CONCERTO: a randomized, placebo-controlled trial of oral laquinimod in relapsing-remitting multiple sclerosis. Mult. Scler. J. 28, 608–619 (2021).
Kenison, J. E. et al. Tolerogenic nanoparticles suppress central nervous system inflammation. Proc. Natl Acad. Sci. USA 117, 32017–32028 (2020).
Haase, S., Haghikia, A., Wilck, N., Müller, D. N. & Linker, R. A. Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology 154, 230–238 (2018).
Park, M.-J. et al. Myeloid-derived suppressor cells therapy enhance immunoregulatory properties in acute graft versus host disease with combination of regulatory T cells. J. Transl. Med. 18, 483 (2020).
Eggenhuizen, P. J., Ng, B. H. & Ooi, J. D. Treg enhancing therapies to treat autoimmune diseases. Int. J. Mol. Sci. 21, 7015 (2020).
Raffin, C., Vo, L. T. & Bluestone, J. A. Treg cell-based therapies: challenges and perspectives. Nat. Rev. Immunol. 20, 158–172 (2020).
Selck, C. & Dominguez-Villar, M. Antigen-specific regulatory T cell therapy in autoimmune diseases and transplantation. Front. Immunol. 12, 661875 (2021).
Ruck, T. et al. K2P18.1 translates T cell receptor signals into thymic regulatory T cell development. Cell Res. 32, 72–88 (2021).
Zheng, D., Liwinski, T. & Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 30, 492–506 (2020).
Wekerle, H. Brain autoimmunity and intestinal microbiota: 100 trillion game changers. Trends Immunol. 38, 483–497 (2017).
Cox, L. M. et al. Gut microbiome in progressive multiple sclerosis. Ann. Neurol. 89, 1195–1211 (2021).
Weiner, H. L., Cunha, A. P., da, Quintana, F. & Wu, H. Oral tolerance. Immunol. Rev. 206, 232–259 (2005).
Duscha, A. et al. Propionic acid shapes the multiple sclerosis disease course by an immunomodulatory mechanism. Cell 180, 1067–1080.e16 (2020).
Schepici, G., Silvestro, S., Bramanti, P. & Mazzon, E. The gut microbiota in multiple sclerosis: an overview of clinical trials. Cell Transpl. 28, 1507–1527 (2019).
Zhu, W., Dykstra, K., Zhang, L. & Xia, Z. Gut microbiome as potential therapeutics in multiple sclerosis. Curr. Treat. Options Neurol. 23, 37 (2021).
Tankou, S. K. et al. A probiotic modulates the microbiome and immunity in multiple sclerosis. Ann. Neurol. 83, 1147–1161 (2018).
The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology 43, 655–661 (1993).
Johnson, K. P. et al. (The Copolymer 1 Multiple Sclerosis Study Group) Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. Neurology 45, 1268–1276 (1995).
Jacobs, L. D. et al. (The Multiple Sclerosis Collaborative Research Group, MSCRG) Intramuscular interferon β-1a for disease progression in relapsing multiple sclerosis. Ann. Neurol. 39, 285–294 (1996).
Ebers, G. C. Randomised double-blind placebo-controlled study of interferon β-1a in relapsing/remitting multiple sclerosis. Lancet 352, 1498–1504 (1998).
Hartung, H.-P. et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet 360, 2018–2025 (2002).
Kappos, L. et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 362, 387–401 (2010).
Cohen, J. A. et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 362, 402–415 (2010).
Calabresi, P. A. et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 13, 545–556 (2014).
Giovannoni, G. et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N. Engl. J. Med. 362, 416–426 (2010).
O’Connor, P. et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N. Engl. J. Med. 365, 1293–1303 (2011).
Miller, A. E. et al. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 13, 977–986 (2014).
Confavreux, C. et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 13, 247–256 (2014).
Fox, R. J. et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 367, 1087–1097 (2012).
Gold, R. et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 367, 1098–1107 (2012).
CAMMS223 Trial Investigators. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N. Engl. J. Med. 359, 1786–1801 (2008).
Cohen, J. A. et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet 380, 1819–1828 (2012).
Kappos, L. et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet 391, 1263–1273 (2018).
Hauser, S. L. et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 376, 221–234 (2017).
Comi, G. et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 18, 1009–1020 (2019).
Kappos, L. et al. Ponesimod compared with teriflunomide in patients with relapsing multiple sclerosis in the active-comparator phase 3 OPTIMUM study: a randomized clinical trial. JAMA Neurol. 78, 558 (2021).
Tremlett, H. & Marrie, R. A. The multiple sclerosis prodrome: emerging evidence, challenges, and opportunities. Mult. Scler. J. 27, 6–12 (2021).
Lublin, F. D. et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 83, 278–286 (2014).
Author information
Authors and Affiliations
Contributions
L.B., H.-P.H., O.A., T.R. and S.G.M. researched data, discussed the content, wrote the article and edited/reviewed the manuscript before submission. M.R. edited and reviewed the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Drug Discovery thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Excitotoxicity
-
Neuronal damage or death that is caused by excessive release of neurotransmitters such as glutamate or aspartate.
- Experimental autoimmune encephalomyelitis
-
(EAE). An inflammatory, autoimmune demyelinating disease of the central nervous system in rodents that has high pathological and clinical similarities to human multiple sclerosis and is the most used experimental model for the disease.
- Glia limitans
-
Also called the glia limiting membrane; defined as a barrier that surrounds the brain and spinal cord and is formed by astrocytic endfeet processes that limit the perivascular space.
- T helper 1 cells
-
(TH1 cells). A subgroup of T helper cells (also known as CD4+ cells) that is mainly involved in the cell-mediated immune response against intracellular pathogens such as bacteria by maximizing the efficacy of macrophages and cytotoxic T cells. The main effector cytokines of TH1 cells are IFNγ and IL-2.
- TH2 cells
-
A subgroup of T helper cells that is involved in the humoral immune system. TH2 cells secrete IL-4 and IL-10 (among others), are involved in the recognition of extracellular pathogens and activate the B cell-mediated antibody response.
- TH17 cells
-
A subgroup of T helper cells that is developmentally distinct from TH1 and TH2 cells and is defined by production of IL-17. TH17 cells are involved in host defence against extracellular pathogens, but also contribute to the pathogenesis of immune-mediated diseases such as MS.
- Regulatory T (Treg) cells
-
A subpopulation of CD4+ T cells, also known as suppressor T cells, characterized by the expression of CD4 and CD25. Treg cells have a critical role in preventing autoimmunity by controlling the immune response to self-antigens and can inhibit T cell proliferation and cytokine production.
- Epitope spreading
-
The development/expansion of the immune response against the initial dominant epitope to include a secondary epitope over time.
Rights and permissions
About this article

Cite this article
Bierhansl, L., Hartung, HP., Aktas, O. et al. Thinking outside the box: non-canonical targets in multiple sclerosis. Nat Rev Drug Discov 21, 578–600 (2022). https://doi.org/10.1038/s41573-022-00477-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-022-00477-5
- Springer Nature Limited
This article is cited by
-
Mitochondrial and metabolic dysfunction of peripheral immune cells in multiple sclerosis
Journal of Neuroinflammation (2024)
-
The neuropathobiology of multiple sclerosis
Nature Reviews Neuroscience (2024)
-
Microbiota–gut–brain axis and its therapeutic applications in neurodegenerative diseases
Signal Transduction and Targeted Therapy (2024)
-
Increased level of GATA3-AS1 long non-coding RNA is correlated with the upregulation of GATA3 and IL-4 genes in multiple sclerosis patients
Molecular Biology Reports (2024)
-
A new perspective on therapies involving B-cell depletion in autoimmune diseases
Molecular Biology Reports (2024)