

Abstract
Coronavirus disease 2019 (COVID-19), caused by a strain of coronavirus known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has become a global pandemic that has affected the lives of billions of individuals. Extensive studies have revealed that SARS-CoV-2 shares many biological features with SARS-CoV, the zoonotic virus that caused the 2002 outbreak of severe acute respiratory syndrome, including the system of cell entry, which is triggered by binding of the viral spike protein to angiotensin-converting enzyme 2. Clinical studies have also reported an association between COVID-19 and cardiovascular disease. Pre-existing cardiovascular disease seems to be linked with worse outcomes and increased risk of death in patients with COVID-19, whereas COVID-19 itself can also induce myocardial injury, arrhythmia, acute coronary syndrome and venous thromboembolism. Potential drug–disease interactions affecting patients with COVID-19 and comorbid cardiovascular diseases are also becoming a serious concern. In this Review, we summarize the current understanding of COVID-19 from basic mechanisms to clinical perspectives, focusing on the interaction between COVID-19 and the cardiovascular system. By combining our knowledge of the biological features of the virus with clinical findings, we can improve our understanding of the potential mechanisms underlying COVID-19, paving the way towards the development of preventative and therapeutic solutions.
Key points
-
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that causes coronavirus disease 2019 (COVID-19), shares many biological features with SARS-CoV, the virus that causes severe acute respiratory syndrome, owing to 80% genomic sequence identity.
-
The interaction between the viral spike (S) protein and angiotensin-converting enzyme 2, which triggers entry of the virus into host cells, is likely to be involved in the cardiovascular manifestations of COVID-19.
-
The presence of underlying cardiovascular comorbidities in patients with COVID-19 is associated with high mortality.
-
COVID-19 can cause cardiovascular disorders, including myocardial injury, arrhythmias, acute coronary syndrome and venous thromboembolism.
-
Several medications used for the treatment of COVID-19 have uncertain safety and efficacy profiles.
Similar content being viewed by others


Introduction
Coronavirus disease 2019 (COVID-19) was first reported in Wuhan, China, in late December 2019 (refs1,2,3). Since then, COVID-19 has spread rapidly worldwide and has become a global pandemic affecting >200 countries and territories, with an unprecedented effect not only on public health, but also social and economic activities. The exponential increase in the number of patients with COVID-19 in the past 6 months has overwhelmed health-care systems in numerous countries across the world. At present, preventive vaccines and prophylactic therapies for COVID-19 are not available.
COVID-19 is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is a member of the genus Betacoronavirus like the two other coronaviruses that have caused pandemic diseases (severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV))1,2,3,4. As with SARS-CoV and MERS-CoV, SARS-CoV-2 causes a respiratory infection, which leads to viral pneumonia and acute respiratory distress syndrome (ARDS) in some patients1. However, in addition to respiratory symptoms, uncontrolled SARS-CoV-2 infection can trigger a cytokine storm, whereby pro-inflammatory cytokines and chemokines such as tumour necrosis factor-α, IL-1β and IL-6 are overproduced by the immune system, resulting in multiorgan damage5. Furthermore, COVID-19 causes coagulation abnormalities in a substantial proportion of patients, which can lead to thromboembolic events6,7. The genomic sequence1,2,3,8 and viral protein structure9,10,11 of SARS-CoV-2 have been studied intensively since its emergence. To date, research shows that SARS-CoV-2 shares many biological features with SARS-CoV owing to 79.6% genomic sequence identity1,2. In particular, both SARS-CoV and SARS-CoV-2 use the same system of cell entry, which is triggered by binding of the viral spike (S) protein to angiotensin-converting enzyme 2 (ACE2) on the surface of the host cell4.
Understanding the biological features of the virus will contribute to the development of diagnostic tests, vaccines and pharmacological therapies and can further our knowledge of tissue tropism. Early clinical data indicate that both the susceptibility to and the outcomes of COVID-19 are strongly associated with cardiovascular disease (CVD)12,13,14,15,16. A high prevalence of pre-existing CVD has been observed among patients with COVID-19, and these comorbidities are associated with increased mortality17,18,19,20,21,22. Furthermore, COVID-19 seems to promote the development of cardiovascular disorders, such as myocardial injury, arrhythmias, acute coronary syndrome (ACS) and venous thromboembolism23,24,25. Children with COVID-19 have also been reported to develop hyperinflammatory shock with features akin to Kawasaki disease, including cardiac dysfunction and coronary vessel abnormalities26. Together, these data indicate the presence of a bidirectional interaction between COVID-19 and the cardiovascular system, but the mechanisms underlying this interaction remain elusive. The high burden of systemic inflammation associated with COVID-19 has been proposed to accelerate the development of subclinical disorders or cause de novo cardiovascular damage5,12,13,14. ACE2, which is a key surface protein for virus entry and part of the renin–angiotensin–aldosterone system (RAAS), is also thought to be involved in this interaction on the basis of findings from animal models12,13,14,15.
The fast-moving nature of this research field necessitates the integration of available biological data with clinical findings of COVID-19 to improve our understanding of the pathophysiology of the disease and to contribute to the development of potential therapies. In this Review, we summarize our current knowledge of SARS-CoV-2 from a biological viewpoint, with an emphasis on the interaction between the viral S protein and human ACE2. Furthermore, we provide an overview of the clinical findings related to the effects of COVID-19 on the cardiovascular system. Finally, we discuss the possible link between common cardiovascular drugs and susceptibility to COVID-19 and the potential cardiovascular effects of drugs used to treat COVID-19.
Of note, several limitations of this Review need to be acknowledged. First, given the fast-moving nature of this research field, we will discuss and cite data from preprint reports on bioRxiv or medRxiv in addition to peer-reviewed articles that have cited preprint reports. These findings need to be interpreted with care and require validation in larger studies. Second, the majority of clinical COVID-19 data mentioned in this Review are from China, given their early experience with the disease. Finally, the clinical data on COVID-19 are predominantly derived from non-randomized studies. Therefore, potential biases and confounding factors associated with observational data, such as differences in patient background, diagnostic methods and health-care systems, should be taken into account.
Biology of SARS-CoV-2
Genome, genes and proteins
Since the emergence of SARS-CoV-2, extensive efforts have been made to characterize the features of this novel coronavirus through genomic sequence studies1,2,3 and the evaluation of viral protein structure9,10,11,27,28. Coronaviruses, which are a large family of single-stranded enveloped RNA viruses, were not recognized as being highly pathogenic in humans until the outbreak of SARS caused by SARS-CoV in 2002–2003 (refs29,30). A decade after the SARS pandemic, an outbreak of MERS was detected in Saudi Arabia, caused by MERS-CoV, another highly pathogenic coronavirus29. In the ensuing years, extensive studies of SARS and MERS have contributed to our understanding of coronavirus biology. On the basis of phylogenic analyses, both SARS-CoV and MERS-CoV are thought to have originated in bats, which are likely to be a major natural reservoir of coronaviruses29. A number of genetically diverse coronaviruses that are related to SARS-CoV or MERS-CoV have been discovered in bats worldwide1,29,31. SARS-CoV-2 has been shown to have 79.6% genomic sequence identity with SARS-CoV and 96.0% with the bat coronavirus RaTG13 (refs1,2,3,8). Given this genomic sequence homology, SARS-CoV-2 is thought to share many biological features with SARS-CoV, suggesting that we can apply, at least in part, our rich knowledge of SARS-CoV biology and pathogenesis to understanding SARS-CoV-2 (refs4,29,30). For example, both SARS-CoV and SARS-CoV-2 use ACE2 as an attachment receptor to enter host cells, whereas MERS-CoV uses dipeptidyl peptidase 4 as the attachment receptor1,2,29,30.
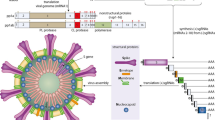
Coronaviruses have a crown-like morphology, consisting of four structural proteins known as spike (S), envelope (E), membrane (M) and nucleocapsid (N) proteins29,30,32 (Fig. 1a). The viral genome surrounded by the N protein is a positive-sense, single-stranded RNA that functions as both a genome and an mRNA29,30,32. Coronaviruses can be divided into four genera: α, β, γ and δ, of which only α and β-coronaviruses are known to infect humans. Phylogenetic studies have revealed that all three highly pathogenic coronaviruses (SARS-CoV, MERS-CoV and SARS-CoV-2) belong to the genus Betacoronavirus1,2,3,29. Like other coronaviruses, the genome of SARS-CoV-2 is approximately 30 kb in length and contains ten open reading frames (ORFs) that encode 24–27 genes1,2. The 5′-terminal two-thirds of the genome encodes polyproteins pp1a and pp1ab, which are cleaved into 16 non-structural proteins, such as RNA-dependent RNA polymerase (RdRP). The S, E, M and N structural proteins are encoded in the 3′-terminal one-third of the genome29,30,33 (Fig. 1b).

a | Coronaviruses form an enveloped spherical particle that consists of four structural proteins (spike (S), envelope (E), membrane (M) and nucleocapsid (N)) and a positive-sense, single-stranded RNA (ssRNA) genome that is 30 kb in length. b | The 5′-terminal two-thirds of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome encodes polyproteins pp1a and pp1ab, which are cleaved into 16 different non-structural proteins. Structural proteins are encoded in the 3′-terminal one-third of the genome. The S protein consists of two subunits; the S1 subunit contains a receptor-binding domain (RBD) that binds to angiotensin-converting enzyme 2 (ACE2) on the surface of host cells, whereas the S2 subunit mediates fusion between the membranes of the virus and the host cell. Compared with the S protein of SARS-CoV, the S protein of SARS-CoV-2 has two notable features. First, within the RBD of the S1 subunit, five of the six residues that are crucial for binding to human ACE2 are mutated. Second, an insertion of four amino acid residues at the boundary between the S1 and S2 subunits is present in SARS-CoV-2 but not in SARS-CoV, which introduces a novel furin cleavage site. c | SARS-CoV-2 infection is triggered by the binding of the S protein to ACE2 on the surface of host cells, and the viral complex is incorporated into the cytoplasm either by direct fusion with the cell membrane or via endocytosis with later release into the cytoplasm from the endocytic vesicle. The S protein is cleaved at the S1/S2 boundary and the S2 subunit facilitates membrane fusion. The viral genome RNA is released into the cytoplasm, and the first open reading frame (ORF) is translated into polyproteins pp1a and pp1ab, which are then cleaved by viral proteases into small, non-structural proteins such as RNA-dependent RNA polymerase (RdRP). The viral genomic RNA is replicated by RdRP. Viral nucleocapsids are assembled from genomic RNA and N proteins in the cytoplasm, whereas budding of new particles occurs at the membrane of the endoplasmic reticulum (ER)–Golgi intermediate compartment. Finally, the genomic RNA and structural proteins are assembled into new viral particles, leading to their release via exocytosis. 3CL, 3-chymotrypsin-like protease.
Among the structural proteins, the S protein has pivotal roles in virus attachment and entry and disease pathogenesis9,10,11,29,30. In SARS-CoV and SARS-CoV-2, the binding of the viral S protein to ACE2 triggers virus entry into the host cell. Therefore, the interaction between the S protein and ACE2 has been considered to be a promising therapeutic target for the development of vaccines34,35, neutralizing antibodies36,37 and antiviral compounds38,39 for SARS-CoV and SARS-CoV-2. The sequence similarity between the S protein of SARS-CoV and that of SARS-CoV-2 is approximately 76% for the whole protein, 73% for the receptor-binding domain and 50% for the receptor-binding motif11.
The S protein of SARS-CoV-2 contains two distinctive features that are not present in the S protein of SARS-CoV4,8. First, the S protein of SARS-CoV-2 maintains a high binding affinity to human ACE2 even though five of the six residues present in the SARS-CoV receptor binding motif that are critical for binding to human ACE2 are mutated in the S protein of SARS-CoV-2 (L455, F486, Q493, S494 and N501)4,8. Cryogenic electron microscopy studies have demonstrated that the S protein of SARS-CoV-2 can bind directly to human ACE2 with a similar or even higher affinity than that of SARS-CoV9,10,11. This high binding affinity is likely to be related to the high transmissibility of SARS-CoV-2 and the severity of COVID-19. Second, the S protein of SARS-CoV-2 has an insertion of four amino acid residues (12 nucleotides) at the boundary between the S1 and S2 subunits, which introduces a novel furin cleavage site4,8. This novel cleavage site has not been observed in SARS-CoV or other SARS-related coronaviruses originating from bats and seems to facilitate the processing of S protein at the S1 and S2 subunit boundary by ubiquitously expressed furin-like proteases for preliminary activation4. Although the function of this novel cleavage site is unknown, similar cleavage sites have been described in highly pathogenic avian influenza viruses and the Newcastle disease virus4,8. This notable feature has been proposed to have a role in expanding cell or tissue tropism of SARS-CoV-2 (ref.4), contributing to the multiorgan effects of COVID-19 (Fig. 1b).
Life cycle of the virus
Infection with either SARS-CoV or SARS-CoV-2 involves binding of the viral S protein to ACE2 on the surface of the host cell. The receptor-binding domain on the surface subunit S1 of the S protein is responsible for attachment of the virus to ACE2. After binding, the S protein is cleaved at the S1/2 and S2′ regions (in a process known as S protein priming) by the transmembrane serine protease TMPRSS2, which in turn facilitates the fusion of the viral membrane with the membrane of the host cell and direct entry of the virus into the cytoplasm4,29,30,40 (Fig. 1c). Respiratory tract epithelial cells express both ACE2 and TMPRSS2 on their surface, and this direct or ‘early’ entry pathway seems to be the predominant mode of in vivo entry by SARS-CoV and, probably, SARS-CoV-2 into the respiratory tissue40. Alternatively, SARS-CoV-2 can also use an endosomal entry pathway, whereby the ACE2–virus complex is translocated to endosomes and S protein priming is performed by the endosomal cysteine proteases cathepsin B and cathepsin L, after which the virus is released from the endosome into the cytoplasm. This endosomal entry pathway, which can be blocked by either lysosomotropic agents (such as hydroxychloroquine) or cathepsin inhibitors, might be the predominant entry pathway used by coronaviruses in the infection of cells cultured in vitro40, but the importance of this pathway for infection in vivo remains unclear. After the release of the viral genomic RNA into the cytoplasm, the first ORF is translated into polyproteins pp1a and pp1ab, which are then cleaved by viral proteases into small non-structural proteins such as RdRP. The viral genomic RNA is then replicated using viral RdRP, and the four structural proteins (S, E, M and N) are translated through the endoplasmic reticulum and Golgi complex of the host cell. Finally, the genomic RNA and structural proteins are assembled into new viral particles, leading to their release through exocytosis29,30 (Fig. 1c). Each step of the viral life cycle described here is a potential therapeutic target, including S protein priming by TMPRSS2 (a target of the serine protease inhibitor camostat mesylate), membrane fusion and endocytosis (a target of the antimalarial drug chloroquine and anti-influenza drug umifenovir) and RNA replication by RdRP (a target of the antiviral agents favipiravir, remdesivir and ribavirin)41,42.
The cardiovascular system and COVID-19
Underlying cardiovascular comorbidities
CVD is a common comorbidity observed in patients infected with SARS or MERS (with a prevalence of 10% and 30%, respectively)12,13,14,43,44. A series of reports on the clinical characteristics of patients with COVID-19 have also described similar findings12,13,14,15. Early reports from China found that CVD and its risk factors, such as hypertension and diabetes mellitus, were common pre-existing conditions in patients with COVID-19, but the definition of CVD used in each study was vague17,18,21,22,45 (Table 1). In an early report from Wuhan involving 41 patients who were hospitalized with COVID-19 by 2 January 2020, the prevalence of any comorbidity was 32% and the most common underlying diseases were diabetes (20%), hypertension (15%) and other CVDs (15%)17. The high prevalence of these comorbidities was confirmed in subsequent studies18,19,20,21,22,45,46,47. Importantly, the prevalence of these pre-existing conditions was higher in critically ill patients (such as those admitted to the intensive care unit (ICU)) and in those who died. In a single-centre cohort study of 138 patients hospitalized with COVID-19 in Wuhan, 46% of patients had any comorbidity (72% of patients in the ICU), 31% of patients had hypertension (58% of patients in the ICU), 15% of patients had other CVDs (25% of patients in the ICU) and 10% of patients had diabetes (22% of patients in the ICU)18. Similarly, in a multicentre cohort study involving 191 patients hospitalized with COVID-19 in Wuhan, 48% of patients had any comorbidity (67% of those who died), 30% of patients had hypertension (48% of those who died), 19% of patients had diabetes (31% of those who died) and 8% of patients had coronary heart disease (24% of those who died)19. Furthermore, in a report involving 1,099 patients with COVID-19 from mainland China, 24% of patients had any comorbidity (39% of critically ill patients), 15% of patients had hypertension (24% of critically ill patients), 7% of patients had diabetes (16% of critically ill patients) and 3% of patients had coronary heart disease (6% of critically ill patients)20. The overall case fatality rate of COVID-19 reported by the Chinese Center for Disease Control and Prevention as of 11 February 2020 was 2.3% (1,023 deaths among 44,672 confirmed cases)21,45. The individual case fatality rate of patients with CVD was 10.5% (highest among those with any comorbidities, including chronic respiratory disease (6.3%) or cancer (5.6%)), the case fatality rate of patients with diabetes was 7.3% and that of patients with hypertension was 6.0%45. Of note, these early approximations of case fatality rate are likely to be overestimated given that the estimates did not account for the many people who had the virus but were not tested.
A similar trend in the prevalence of comorbidities has been reported by researchers in other countries46,47,48,49,50,51. In a report involving 1,591 patients with COVID-19 who were admitted to the ICU in Italy, 49% of patients had pre-existing hypertension, 21% had CVD and 17% had diabetes46. Furthermore, in a report of 393 consecutive patients hospitalized with COVID-19 in New York, USA, up to 50% of patients had hypertension (54% of ventilated patients), 36% had obesity (43% of ventilated patients), 25% of patients had diabetes (28% of ventilated patients) and 14% of patients had coronary artery disease (19% of ventilated patients)47. Of note, this study from New York highlighted the high prevalence of comorbid obesity among patients with COVID-19, which had not been reported in the studies on patients in China probably owing to differences in the background prevalence of obesity between the USA and China. Investigators in this study suggest that obesity might also be a risk factor for respiratory failure and the need for invasive mechanical ventilation47.
Diverse cardiovascular manifestations
Although the predominant clinical manifestation of COVID-19 is viral pneumonia1,2,48,49, COVID-19 can also cause cardiovascular disorders such as myocardial injury, arrhythmias, ACS and thromboembolism12,13,14,15 (Fig. 2). Some patients who present without the typical symptoms of fever or cough have cardiac symptoms as the first clinical manifestation of COVID-19 (refs52,53). Myocardial injury during the course of COVID-19 is independently associated with high mortality23. Furthermore, a possible link between COVID-19 and a Kawasaki disease-like syndrome has been described in children26.

Cardiovascular comorbidities such as hypertension and coronary artery disease are associated with high mortality in patients with coronavirus disease 2019 (COVID-19). Drugs used to reduce cardiovascular risk such as angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) have numerous effects that might influence susceptibility to or the severity of COVID-19. Furthermore, although the main presentation of COVID-19 is viral pneumonia, COVID-19 can also induce cardiovascular manifestations including myocardial injury, myocarditis, arrhythmias, acute coronary syndrome and thromboembolism. Among these cardiovascular manifestations, myocardial injury has been independently associated with high mortality among patients with COVID-19 (ref.23). Finally, medications that have been proposed as treatments for COVID-19 such as hydroxychloroquine and azithromycin have pro-arrhythmic effects. AF, atrial fibrillation; VF, ventricular fibrillation; VT, ventricular tachycardia.
Myocardial injury and myocarditis
Acute myocardial injury, as evidenced by elevated levels of cardiac biomarkers or electrocardiogram abnormalities, was observed in 7–20% of patients with COVID-19 in early studies in China17,18,19,20,21. The presence of myocardial injury was associated with a significantly worse prognosis23. In the initial report of 41 patients with COVID-19 in Wuhan, 5 patients had myocardial injury with elevated levels of high-sensitivity cardiac troponin I (>28 pg/ml), and 4 of these 5 patients were admitted to an ICU17. In a multicentre cohort study of 191 patients with COVID-19, 33 patients (17%) had acute cardiac injury, of whom 32 died19. In a subsequent study of 416 patients hospitalized with COVID-19, 82 patients (20%) had evidence of cardiac injury, which was associated with a 5-fold increase in the need for invasive mechanical ventilation and an 11-fold increase in mortality23. Of note, cardiac injury was found to be an independent risk factor for in-hospital mortality23. Another study confirmed this finding and reported that the rate of death in patients with elevated levels of cardiac troponin T was 37.5%, whereas, in patients with underlying cardiovascular comorbidities plus elevated levels of cardiac troponin T, it was almost double (69.4%)24. Furthermore, a subsequent study demonstrated that markers of myocardial injury were predictive of the risk of in-hospital mortality in patients with severe COVID-19 (ref.25). The area under the receiver operating characteristic curve of the initial cardiac troponin I level for predicting in-hospital mortality was as high as 0.92. Other predictors of myocardial injury include advanced age, presence of comorbidities and high levels of C-reactive protein.
Whether typical clinical features of myocarditis were present in patients who had elevated levels of cardiac troponins during the course of COVID-19 is unclear because most of the early studies did not include echocardiography or MRI data12,13,14,54. In a cohort study involving 112 patients with COVID-19, the 14 patients with myocardial injury who had elevated high-sensitivity levels of cardiac troponin I (>0.12 ng/ml) plus abnormalities on echocardiography and/or electrocardiogram did not have typical signs of myocarditis such as segmental wall motion abnormality or reduced left ventricular (LV) ejection fraction (LVEF), suggesting that myocardial injury was secondary to systemic causes rather than a result of direct viral infection of the heart53. By contrast, several case reports have described typical signs of myocarditis in patients with COVID-19. A woman aged 53 years with myocardial injury, as evidenced by elevated levels of cardiac biomarkers and diffuse ST segment elevation on the electrocardiogram, had diffuse biventricular hypokinesis on cardiac MRI, especially in the apical segments, in addition to severe LV dysfunction (LVEF = 35%)55. MRI data also revealed marked biventricular interstitial oedema, diffuse late gadolinium enhancement and circumferential pericardial effusion, features that are consistent with acute myocarditis. Furthermore, in a man aged 37 years with chest pain and ST segment elevation, echocardiography revealed an enlarged heart (LV diastolic dimension = 58 mm) and LV dysfunction (LVEF = 27%)56. This patient was diagnosed with COVID-19-induced fulminant myocarditis and treated with methylprednisolone. Cardiac size and function recovered to normal after 1 week (LV diastolic dimension = 42 mm, LVEF = 66%).
Histological evidence of myocardial injury or myocarditis in COVID-19 is also limited. An autopsy of a patient with COVID-19 and ARDS who died of a sudden cardiac arrest showed no evidence of myocardial structural involvement, suggesting that COVID-19 did not directly impair the heart57. By contrast, another case report described a patient with low-grade myocardial inflammation and myocardial localization of coronavirus particles (outside of cardiomyocytes), as measured by endomyocardial biopsy, suggesting that SARS-CoV-2 might infect the myocardium directly58. Autopsy reports have also revealed the presence of mild inflammation and viral RNA in the hearts of patients with COVID-19 (refs59,60). However, whether these patients had myocarditis or whether the findings were a consequence of systemic inflammation remains unclear.
Our understanding of the pathophysiology underlying SARS might help to determine whether SARS-CoV-2 can infect cardiac cells directly, given that SARS-CoV and SARS-CoV-2 share the same mechanisms of entry into the host cell4,12,43 and that the heart expresses high levels of ACE2 (refs15,61). In a report that described autopsy samples from ten Canadian patients with SARS, the viral RNA of SARS-CoV was detected in 35% of the heart samples, but the infected cell types were unknown62. A marked increase in macrophage infiltration with evidence of myocardial damage was also detected, suggesting that SARS-CoV can infect the heart directly62.
Taken together, these findings suggest that myocardial injury is not only a common manifestation of COVID-19, but also a risk factor for poor prognosis. At present, we do not understand the mechanisms underlying COVID-19-related myocardial injury. However, on the basis of the available clinical evidence, myocardial injury seems to be largely attributable to advanced systemic inflammation. SARS-CoV-2 might also infect the myocardium directly, resulting in viral myocarditis in a small proportion of patients with COVID-19.
Acute coronary syndrome
As with other infectious diseases, including SARS and influenza, COVID-19 can trigger ACS44,63,64,65,66. In early studies from China, a small proportion of patients with COVID-19 presented with chest pain on admission to hospital, but the characteristics of the chest pain were not described17,18. In a case series from New York involving 18 patients with COVID-19 and ST segment elevation, which is indicative of potential acute myocardial infarction, five of the six patients with myocardial infarction required percutaneous coronary intervention66. In a case series from Italy involving 28 patients with COVID-19 and ST segment elevation myocardial infarction, assessment by coronary angiography showed that 17 patients had evidence of a culprit lesion that required revascularization52. Of note, ST segment elevation myocardial infarction was the first clinical manifestation of COVID-19 in 24 of these 28 patients who had not yet received a positive test result for COVID-19 at the time of coronary angiography. These observations suggest that COVID-19 can cause ACS even in the absence of substantial systemic inflammation. However, the incidence of ACS in patients with COVID-19 is still unknown. Considering the overwhelmed health-care facilities of many cities during the COVID-19 outbreak, the number of cases of acute myocardial infarction among patients with COVID-19 might be underestimated in early studies. The mechanisms underlying COVID-19-induced ACS might involve plaque rupture, coronary spasm or microthrombi owing to systemic inflammation or cytokine storm67,68. For example, activated macrophages secrete collagenases that degrade collagen, a major constituent of the fibrous cap on atherosclerotic plaques, which can lead to plaque rupture67. Activated macrophages are also known to secrete tissue factor, a potent procoagulant that triggers thrombus formation when the plaque ruptures67. Direct endothelial or vascular injury caused by SARS-CoV-2 infection might also increase the risk of thrombus formation and ACS69.
Despite the potential for COVID-19 to induce ACS, the number of reported cases of ACS during the COVID-19 outbreak in Italy, Spain and the USA was actually significantly lower than during pre-COVID-19 periods, with a reported 42–48% reduction in hospitalizations for ACS and a 38–40% reduction in percutaneous coronary interventions for ST segment elevation myocardial infarction70,71,72,73. By contrast, the incidence of out-of-hospital cardiac arrest increased during the COVID-19 outbreak in Italy, which was strongly associated with the cumulative incidence of COVID-19 (ref.74). This observation is in accordance with the finding that the number of patients with myocardial infarction seeking urgent hospital care declined by >50% during the peak of the COVID-19 outbreak, as reported in an extensive global survey by the ESC75.
Heart failure
In an early study from Wuhan involving 799 patients, heart failure was one of the most commonly observed complications of COVID-19, with a reported incidence of 24% in all patients and 49% in patients who died76. Elevated levels of amino-terminal pro-B-type natriuretic peptide were identified in 49% of all patients (85% of those who died)76. Similarly, in another study of 191 patients in Wuhan, heart failure was identified in 23% of all patients and in 52% of patients who died19.
The aetiology of acute or decompensated heart failure in COVID-19 has not been studied77. Given that patients with COVID-19 are likely to be older and to have pre-existing comorbidities such as coronary artery disease, hypertension and diabetes, heart failure might be the result of an exacerbation of these pre-existing conditions, whether already diagnosed or unknown, or the uncovering of subclinical cardiac dysfunction. In particular, elderly patients with reduced diastolic function might develop heart failure with preserved EF during the course of COVID-19, which can be triggered by high fever, tachycardia, excessive hydration and impaired renal function77. In patients with heart failure with preserved ejection fraction, cardiac MRI might help to detect changes induced by COVID-19 (refs78,79). Acute myocardial injury and ACS triggered by COVID-19 can also aggravate pre-existing heart disease or provoke contractile dysfunction. In the advanced stages of COVID-19, the response of the immune system to infection might trigger the development of stress-induced cardiomyopathy or cytokine-related myocardial dysfunction, as with sepsis-associated cardiac dysfunction80,81.
Given that COVID-19 primarily causes respiratory symptoms and viral pneumonia with bilateral, peripheral and lower lung distribution, the pulmonary oedema that is observed in these patients, which is usually accompanied by ARDS, is mainly regarded as non-cardiogenic. However, given that approximately 25% of patients hospitalized with COVID-19 develop heart failure, the potential contribution of pulmonary congestion by heart failure should be taken into consideration77. Additional haemodynamic data from patients with COVID-19-related respiratory failure are needed to validate this involvement.
Arrhythmias and sudden cardiac arrest
Arrhythmias and sudden cardiac arrest are common manifestations of COVID-19. Heart palpitations have been reported to be the main presenting symptom of COVID-19 in patients without a fever or cough82. In a cohort of 138 patients with COVID-19 in Wuhan, China, the presence of cardiac arrhythmia was reported in 17% of all patients (44% of patients in the ICU), but the specific types of arrhythmia were not recorded18. In another study in Wuhan involving 187 patients hospitalized with COVID-19, those with elevated levels of troponin T were more likely to develop malignant arrhythmias, such as ventricular tachycardia and fibrillation, than those with normal levels of troponin T (12% versus 5%)24. In-hospital and out-of-hospital sudden cardiac arrests have also been reported in patients with COVID-19 (refs57,66,74). However, the exact contribution of COVID-19 to cardiac arrhythmias remains uncertain given that arrhythmias, such as atrial and ventricular tachycardia and fibrillation, can be triggered by myocardial injury or other systemic causes such as fever, sepsis, hypoxia and electrolyte abnormalities24,83. Furthermore, patients with advanced COVID-19 are often treated with antiviral medications and antibiotics that are known to induce arrhythmias in some patients (described in detail below)13,41.
Coagulation abnormalities and thrombosis
COVID-19 is associated with coagulation abnormalities, which can result in thromboembolic events73. Patients with COVID-19 often have elevated levels of d-dimer, modestly reduced platelet counts and slightly prolonged prothrombin time. In an early study of 1,099 patients with COVID-19 from China, elevated levels of d-dimer (>0.5 mg/l) were observed in 46% of all patients (60% of those with severe illness)20. Similarly, another study in patients with COVID-19 in Wuhan showed that d-dimer levels were elevated (>1 mg/l) in 42% of all patients (81% of those who died), which, if detected at admission to hospital, was associated with an 18-fold increased risk of death19. By contrast, the changes in platelet counts and prothrombin time were modest. Among 41 patients with COVID-19 in Wuhan, only 5% had a low platelet count (<100 × 109 cells per litre) and the prolongation of prothrombin time was mild even in patients admitted to the ICU (11.1 s versus 12.2 s)17. Moreover, levels of fibrinogen and factor VIII were elevated in these patients, indicating a hypercoagulable state84,85. These findings show that a substantial proportion of patients with COVID-19 have coagulation abnormalities that typically do not meet the criteria of disseminated intravascular coagulation established by the International Society on Thrombosis and Haemostasis86, but nevertheless might contribute to the development of the diverse cardiovascular manifestations of COVID-19.
Clinical observations of increased thromboembolic events in patients with COVID-19 suggest the presence of a hypercoagulable state. Venous thromboembolism, which includes deep vein thrombosis and pulmonary embolism, is a common complication in critically ill patients with COVID-19. An autopsy study revealed that deep vein thrombosis was present in 7 of 12 patients who died with COVID-19 in whom venous thromboembolism was not suspected before death, whereas pulmonary embolism was identified in 4 of the 12 patients59. Arterial thrombotic events have also been reported. A case series from New York described five patients aged ≤50 years who presented to the same hospital with large-vessel ischaemic stroke and who all tested positive for SARS-CoV-2 infection87. Furthermore, acute limb ischaemia was also reported in 20 patients with COVID-19 (90% men, mean age 75 ± 9 years) in a case series from Italy88. All 20 patients were diagnosed with COVID-19-related pneumonia before acute limb ischaemia was detected.
The mechanisms underlying these coagulation abnormalities, particularly hypercoagulation, in the setting of COVID-19 are unclear. One hypothesis is that the severe inflammatory response and endothelial damage induced by COVID-19 in combination with underlying comorbidities might predispose patients to a hypercoagulable state6. Of note, certain antiviral medications and investigational therapies given to these patients might promote thrombosis or bleeding events through drug–drug interactions with antiplatelet agents and anticoagulants41.
A retrospective study in New York showed that systemic anticoagulation was associated with prolonged survival in patients hospitalized with COVID-19 (ref.89). Among 2,773 patients, 786 (28%) received systemic anticoagulation. The median survival time of patients who were treated with anticoagulation was longer than in those who were not treated (21 days versus 14 days), although overall mortality between the two groups remained similar (22.5% versus 22.8%)89. The differences in median survival time and mortality were more pronounced among patients who required mechanical ventilation (21 days versus 9 days and 29.1% versus 62.7%, respectively)89. Another retrospective study in China also showed reduced mortality in patients with COVID-19-associated coagulopathy who were treated with prophylactic heparin90. Importantly, findings from these retrospective studies are limited by potential selection bias, the presence of confounding factors and the undefined indication of anticoagulation treatment. In addition, the optimal anticoagulation agent to prevent thromboembolic events in these patients is not known (for example, low-molecular-weight heparin, unfractionated heparin, direct oral anticoagulants or others). Prospective, randomized trials are needed to validate the protective effect of anticoagulation therapy in patients with COVID-19.
Kawasaki disease
Children are thought to be less susceptible than adults to COVID-19, and the majority of children with COVID-19 are asymptomatic or present with only mild symptoms91. However, COVID-19 has been reported to cause severe inflammatory symptoms in a small proportion of paediatric patients26,92. A case series from the UK reported an unprecedented cluster of eight children (aged 4–14 years) presenting with a hyperinflammatory syndrome with features of Kawasaki disease, five of whom tested positive for SARS-CoV-2 or were potentially exposed to SARS-CoV-2 from family members26. Clinical presentations included fever, variable rash, conjunctivitis, peripheral oedema, extremity pain and severe gastrointestinal symptoms. A common finding on echocardiography was echo-bright coronary vessels, which progressed to a giant coronary aneurysm in one patient. Furthermore, researchers in Bergamo, Italy, found a 30-fold increase in the incidence of Kawasaki-like disease among children during the peak of the pandemic92. These paediatric patients were older and had a higher rate of cardiac involvement than patients diagnosed with Kawasaki disease before the pandemic. Together, these early clinical findings are suggestive of a new phenomenon caused by SARS-CoV-2 infection in children that can lead to a hyperinflammatory syndrome with features that are similar to those of Kawasaki disease, including coronary artery abnormalities.
Immunocompromised patients
In general, patients with depressed immunity are at a higher risk of infectious diseases. The effect of COVID-19 on the cardiovascular system in immunocompromised patients, such as those with cancer or those who have undergone organ transplantation, is largely unknown. Heart transplantation recipients might be at higher risk of COVID-19 owing to their immunosuppressed state combined with their baseline cardiovascular disorders14,93. COVID-19 has been reported in two heart transplantation recipients from China, both of whom made a full recovery94. The clinical presentations of these two patients were not distinct from those of non-immunosuppressed patients. In a retrospective case series of heart transplantation recipients with COVID-19 admitted to hospitals in Michigan, USA, between 21 March 2020 and 22 April 2020, 13 patients were identified, all of whom were African American men95. Six patients required admission to the ICU and two patients died during hospitalization. Of note, the clinical presentation and laboratory markers of disease severity of these patients were not distinct from those of the general population, despite immunosuppression use to preserve allograft function. However, another case series involving 28 heart transplantation recipients with COVID-19 in New York reported higher mortality and an increased incidence of severe complications in these individuals than in the general population96. A total of 22 patients were hospitalized, of whom 7 patients required mechanical ventilation and 7 patients died (case fatality rate 25%). Moreover, 13 out of 17 patients had myocardial injury, as evidenced by elevated levels of troponin T (>0.022 ng/ml).
These findings suggest that recipients of heart transplantation are at high risk of severe complications from COVID-19. Whether organ transplantation recipients are more susceptible to COVID-19 and whether immunosuppressive treatments have harmful (or protective) effects on disease progression needs to be assessed in large-scale studies. Another important point regarding heart transplantation is the need to screen for COVID-19. Given that individuals with SARS-CoV-2 infection might be asymptomatic, routine screening of donor tissues is necessary during the pandemic. Screening of recipients before transplantation will also be required to avoid worsening of subclinical infection after starting immunosuppression.
As with patients with CVD, patients with cancer are thought to be at higher risk of severe COVID-19 symptoms than the general population97,98,99. This high risk of poor outcomes might be attributable to suppressed immunity by chemotherapy, the presence of cardiovascular risk factors (such as hypertension and diabetes), cardiotoxicity of cancer treatment and/or cardiovascular damage by COVID-19, combined with their impaired baseline condition97. Therefore, a major consideration in the delivery of care to these patients during the pandemic is to balance the risk of SARS-CoV-2 infection with the need to provide timely cancer treatment. Clinicians need to determine the optimal timing of treatment in patients with cancer and cardiovascular comorbidities, especially if they are infected with or exposed to SARS-CoV-2. Routine COVID-19 screening might be necessary before cancer treatment to avoid worsening of a subclinical infection.
ACE2 and cardiovascular manifestations
The mechanisms underlying the development of COVID-19-related cardiovascular injury are not known. ACE2 expression is thought to be one of the major factors involved in the biological mechanism underlying tissue-specific infection. As with SARS-CoV, SARS-CoV-2 infection is triggered by binding of viral S protein to human ACE2, whereas TMPRSS2 induces S protein priming4. The interaction between S protein and ACE2 has gained much research interest given that ACE2 is known to have crucial roles in both the cardiovascular system and the immune system61,100. ACE2 is a part of the RAAS and is involved in the development of diabetes, hypertension and heart failure. At the tissue level, ACE2 is highly expressed in the lungs, kidneys, heart and blood vessels61,100. According to bulk RNA sequencing data in the Genotype-Tissue Expression (GTEx) project V8, the expression of ACE2 in the heart and coronary arteries is even higher than in the lungs101. At the single-cell level, ACE2 is highly expressed in pericytes of adult human hearts102. Single-cell RNA sequencing data have also revealed that cardiomyocytes (especially those in the right ventricle) express ACE2 at a lower level than pericytes and that neither pericytes nor cardiomyocytes express TMPRSS2 (ref.103). However, both cell types have a high expression of cathepsin B and cathepsin L, which facilitate S protein priming and might promote entry of the virus into the cell via the endocytic pathway. Therefore, SARS-CoV-2 might be capable of directly infecting multiple cardiovascular cell types, including cardiomyocytes, endothelial cells and pericytes. Importantly, however, expression of ACE2 is not in itself sufficient for entry of the virus into a cell, and the efficiency of viral replication and release might also have a role in host cell infection. To date, clinical evidence of direct viral infection of cardiomyocytes has not been found. Given that myocarditis related to SARS-CoV-2 infection is rare23,53, the interaction between SARS-CoV-2 and ACE2 might affect the cardiovascular system in an indirect manner61,100. Genome-wide association studies would help to facilitate the identification of novel pathways involved in SARS-CoV-2 pathogenesis104,105.
Potential downregulation of ACE2 by SARS-CoV-2
SARS-CoV entry into cells has been shown to downregulate ACE2 expression62,106,107. In a mouse model of SARS, ACE2 levels in the heart were significantly reduced after SARS-CoV infection62. In addition, a separate study reported that knockout of Ace2 in mice resulted in a significant reduction in cardiac contractility108. Furthermore, Ace2–/–Apoe–/– mice had greater atherosclerotic plaque accumulation and upregulated expression of genes encoding adhesion molecules and inflammatory cytokines such as IL-6 and CCL2 compared with Apoe–/– mice109. These results support a cardioprotective role of ACE2 (ref.61).
Similarly, ACE2 has a protective effect in the lungs. ACE2 is expressed primarily in alveolar epithelial type II cells in the normal adult lung110,111,112. These cells produce surfactant proteins that reduce surface tension, preventing the alveoli from collapsing. In a mouse model of ARDS, Ace2 knockout exacerbated acute lung injury, whereas treatment with recombinant ACE2 rescued lung damage106. Therefore, like SARS-CoV, SARS-CoV-2 infection might result in the downregulation of ACE2, which can lead to cardiac dysfunction and progression of atherosclerosis, as well as exacerbated lung damage.
ACE2 as a therapeutic target
Angiotensin II, the main effector molecule in the RAAS, is upregulated in many diseases and is a common treatment target for various cardiovascular disorders61,100,113. ACE2 inactivates angiotensin II by converting angiotensin II to angiotensin (1–7)61,100. Cryogenic electron microscopy studies have demonstrated that the S protein of SARS-CoV-2 can directly bind to human ACE2 with a similar or even higher affinity than the binding of the S protein of SARS-CoV to human ACE2 (refs9,10,11). In this context, a study has shown that exogenous administration of recombinant human ACE2 (rhACE2) can prevent SARS-CoV-2 infection by acting as a decoy114. The investigators demonstrated that clinical-grade rhACE2 can reduce SARS-CoV-2 infection in cell culture and in engineered human blood vessel organoids and kidney organoids114. Given that rhACE2 has been shown to be protective against various CVDs61,115,116, rhACE2 therapy might be a promising approach to treat patients with COVID-19-related cardiovascular disorders114.
Broad tissue tropism of SARS-CoV-2
Although SARS-CoV-2 preferentially infects the lungs and respiratory tract like other respiratory viruses, COVID-19 can cause a diverse range of extrapulmonary manifestations including CVD, stroke, seizures, liver damage, renal dysfunction and gastrointestinal symptoms117. Systemic hyperinflammation induced by viral pneumonia is likely to have an important role in the development of these varied manifestations of COVID-19, but numerous studies have also reported histological evidence of direct viral infection in non-respiratory organs such as the heart, brain, liver and kidney59,69,118.
In an autopsy case series involving 12 patients with COVID-19, in which SARS-CoV-2 RNA was detected by quantitative reverse transcription PCR in the lungs at high concentrations in all patients, 5 patients also had high viral RNA titres in the heart, liver or kidney59. Similarly, SARS-CoV-2 RNA was detected in the lungs, heart, brain, liver or kidneys in an autopsy analysis of 27 patients who died with COVID-19 (ref.118), indicating a broad organotropism of SARS-CoV-2. Although the specific cell types infected by SARS-CoV-2 in each organ are unknown, these preliminary data support the possibility that the extrapulmonary manifestations and multiorgan failure observed in patients who died with COVID-19 are not just a consequence of systemic inflammation or cytokine storm, but might also be caused by direct infection of numerous organ systems by SARS-CoV-2. Mechanistically, the broad tissue tropism of SARS-CoV-2 might be a result of the instability of the S protein of SARS-CoV-2 related to the presence of the novel furin cleavage site, as mentioned above4.
Viral targeting of endothelial cells
Preliminary histological data from a case series of three patients with COVID-19 have shown that endothelial cells might be a direct target of SARS-CoV-2 infection69. In a patient with a history of renal transplantation who died from COVID-19-induced multiorgan failure, viral particles were detected by electron microscopy in endothelial cells in the kidney69. A prominent endotheliitis (inflammation within the endothelium) with recruitment of inflammatory cells was identified on histological assessment, as well as an unusually high concentration of apoptotic bodies in numerous organs including the lungs, small bowel and heart69. In another patient who died from COVID-19-related multiorgan failure and ST segment elevation myocardial infarction, lymphocytic endotheliitis was observed in the lungs, heart, liver and kidneys69. In the third patient who had mesenteric ischaemia and underwent resection of the small intestine, histological assessment of the small intestine revealed prominent endotheliitis of the submucosal vessels in addition to a large concentration of apoptotic bodies69. Findings from this case series suggest that SARS-CoV-2 can infect endothelial cells directly, which can lead to inflammation in the endothelium69. Given that endothelial cells are an important component of every organ and have a high level of ACE2 expression, inflammation in the endothelium caused by infection with SARS-CoV-2 might underlie the diverse clinical manifestations of COVID-19.
Potential drug–disease interactions
The potential drug–disease interactions in patients with COVID-19 have become a highly researched topic12,13,14 (Fig. 2). First, whether antihypertensive agents such as ACE inhibitors and angiotensin II receptor blockers (ARBs) are involved in the progression or prevention of COVID-19 is unknown113,119. Second, some of the potential antiviral drugs used to treat patients with COVID-19 are known to induce cardiotoxicity41.
Effect of RAAS inhibitors on COVID-19
Given that ACE2 is a receptor for SARS-CoV-2, clinicians have expressed concern that medications that upregulate the cell surface expression of ACE2 might be harmful113,119,120. ACE inhibitors and ARBs have been shown to increase the expression of ACE2 in animal models121. Given that both ACE inhibitors and ARBs are commonly used worldwide for the treatment of hypertension and other CVDs, whether these drugs should be discontinued during the COVID-19 pandemic has become a pertinent question. Importantly, indiscriminate withdrawal of these drugs could harm high-risk patients. Several medical societies including the ACC, AHA, Chinese Society of Cardiology, ESC and the Heart Failure Society of America have issued statements recommending continuation of RAAS antagonists for those who are currently prescribed these agents122,123,124. Findings from clinical studies support this recommendation. In four studies from Spain (n = 1,139)125, Italy (n = 6,772)126 and the USA (n = 5,894 and 1,735)127,128, the use of RAAS inhibitors was not associated with a positive COVID-19 test, suggesting that these agents do not affect susceptibility to SARS-CoV-2 infection. Furthermore, the use of these agents was not associated with a substantial increase in the risk of severe or fatal illness among patients with COVID-19 (refs126,127).
Two main points need to be discussed on this topic. First, the effect of RAAS inhibitors on ACE2 is controversial113. As shown in Fig. 3, the targets of ACE and ACE2 are different despite high structural similarity between the two enzymes. ACE converts angiotensin I to angiotensin II, whereas ACE2 degrades angiotensin II to angiotensin (1–7). ACE inhibitors prevent the conversion from angiotensin I to angiotensin II, whereas ARBs inhibit the angiotensin II receptor type 1, which is downstream of angiotensin II61,113. Therefore, neither class of drugs affects ACE2 directly. Although some animal studies have shown that treatment with ACE inhibitors or ARBs can increase the expression of Ace2 (refs121,129), findings from other preclinical studies were more mixed113,119,120,130,131,132. In clinical studies, long-term exposure to the ACE inhibitor captopril133 or the ARB olmesartan134 was associated with increased plasma levels of angiotensin (1–7) or urinary levels of ACE2, respectively, indicating increased ACE2 activity. However, other studies have not shown evidence of upregulated ACE2 by RAAS inhibitors in patients with heart failure135, aortic stenosis136, atrial fibrillation137 or coronary artery disease138. These inconsistent results are likely to be attributable to the indirect effects of ACE inhibitors or ARBs on ACE2, which depend on conditions such as baseline expression levels of ACE2, dosing and treatment periods. Second, whether potential upregulation of ACE2 is harmful or protective is uncertain. Even if ACE inhibitors or ARBs can upregulate ACE2, no direct evidence has been found to show that upregulation of ACE2 affects susceptibility to viral infection113. Furthermore, as shown in Ace2-knockout mice106,107,108,109 and rhACE2 studies114,115,116, ACE2 is considered to be protective rather than harmful in settings of lung injury and CVDs61,100. Together, these findings indicate no benefit of withdrawal of ACE inhibitors or ARBs in protecting against COVID-19.

Angiotensin II, the main effector molecule in the renin–angiotensin–aldosterone system (RAAS), is upregulated in many pathological conditions, for which inhibition of angiotensin II by RAAS inhibitors is a common therapeutic strategy.Angiotensin-converting enzyme (ACE) produces angiotensin II from angiotensin I, whereas ACE2 inactivates angiotensin II by converting it to angiotensin (1–7). Therefore, ACE2 has a protective effect against cardiovascular disease and lung injury. In the setting of coronavirus disease 2019, downregulation of ACE2 by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection might be involved in mediating cardiovascular damage. ARB, angiotensin II receptor blocker.
Cardiovascular effects of antiviral drugs
At present, many research teams worldwide are focused on the development of drugs for the prevention and treatment of COVID-19 (ref.41). Of note, the development and testing of new drugs are time-consuming processes139 and not a viable strategy during this COVID-19 pandemic. Drug repurposing139, in which existing medications that have already been approved for a disease are tested for a new condition, is currently the main approach in the search for new drugs for COVID-19 (refs41,42,140,141). However, some of the drugs under investigation have known or unknown cardiovascular adverse effects142 or might be involved in drug–drug or drug–disease interactions41,140 (Table 2).
Hydroxychloroquine and azithromycin
Chloroquine and hydroxychloroquine have been widely touted as potential treatment strategies for COVID-19 (refs143,144,145). Chloroquine and hydroxychloroquine can potentially block virus entry into cells, particularly via the endosomal pathway, by inhibiting the glycosylation of host receptors, proteolytic processing and endosomal acidification41,145. These agents can also mediate immunomodulatory effects through attenuation of cytokine production and inhibition of autophagy and lysosomal activity41. Both drugs are used in the treatment of malaria and chronic inflammatory diseases such as systemic lupus erythematosus and rheumatoid arthritis41,146. A French observational study involving 36 patients with COVID-19 reported improved virus clearance with hydroxychloroquine treatment144. Furthermore, combining hydroxychloroquine with azithromycin in six patients resulted in even better virus clearance than with the use of hydroxychloroquine alone144. Importantly, however, several concerns have been raised regarding the characteristics of the control group (which consisted of patients recruited from a separate hospital) and ethics approval147. In a randomized study involving 62 patients with COVID-19 in China, patients in the treatment group received hydroxychloroquine (400 mg per day) for 5 days, whereas patients in the control group received standard treatment (oxygen therapy, antiviral agents, antibacterial agents and immunoglobulin, with or without corticosteroids)143. Hydroxychloroquine improved the time to clinical recovery, body temperature recovery time, cough remission time and pneumonia-related symptoms compared with standard treatment alone. Furthermore, in a retrospective study from Wuhan involving 550 critically ill patients with COVID-19, mortality was significantly lower among patients treated with hydroxychloroquine plus standard treatment (which included other antiviral drugs and antibiotics) compared with standard treatment alone (18.8% versus 47.4%)148. However, in a large observational study involving 1,376 patients from New York City, hydroxychloroquine treatment did not alter the risk of the composite end point of intubation or death149. These inconsistent results show that the efficacy of hydroxychloroquine is still controversial and needs to be validated in large, randomized studies150.
Although chloroquine and hydroxychloroquine have a long history of clinical use for numerous conditions, these agents are also known to induce arrhythmias41,142,151. Azithromycin, which has been assessed in combination with hydroxychloroquine as a treatment for COVID-19, is also known to prolong the QT interval152. In a cohort study of 90 patients with COVID-19 who received hydroxychloroquine (with or without azithromycin), those who received a combination of hydroxychloroquine and azithromycin had greater QT interval prolongation than those taking hydroxychloroquine alone151. Furthermore, in a retrospective cohort study of 1,438 patients hospitalized with COVID-19 in New York, treatment with hydroxychloroquine, azithromycin or both was compared with neither treatment153. None of the groups had an increase in in-hospital mortality, but the secondary outcome of cardiac arrest was more likely in patients receiving both hydroxychloroquine and azithromycin than in patients receiving neither drug153. Given that some patients with COVID-19 might have impaired renal function owing to systemic illness, frequent electrocardiographic evaluation should be strongly considered in patients treated with hydroxychloroquine and/or azithromycin.
Remdesivir
Remdesivir is a promising investigational nucleotide analogue for the treatment of COVID-19 that has broad-spectrum antiviral activity and functions by targeting RdRP27,154,155. Remdesivir was originally developed for the treatment of Ebola virus disease. Prophylactic and therapeutic administration of remdesivir has been shown to improve pulmonary function and to decrease viral load in a mouse model of MERS156. In a randomized, double-blind, placebo-controlled trial involving 237 patients with COVID-19 in China, remdesivir was associated with a numerically (but not statistically significant) faster time to clinical improvement than was the placebo155. Preliminary results from a double-blind, randomized, multicentre, placebo-controlled trial involving 1,063 patients with COVID-19 indicate that those who received remdesivir had a 31% faster time to recovery than those who received placebo (median time to recovery 11 days versus 15 days)157. Of note, in light of these preliminary findings, the FDA granted emergency use of remdesivir for COVID-19 in May 2020 to meet the urgent demand for treatment of hospitalized patients158. The optimal dosing and duration of treatment is still under investigation. Under the emergency use authorization, a 10-day treatment regimen (200 mg on day 1 followed by 100 mg per day for 9 days) is suggested for patients requiring invasive mechanical ventilation and/or extracorporeal membrane oxygenation, and a 5-day treatment course is suggested for patients with milder symptoms. Although prominent cardiovascular adverse effects associated with remdesivir have not been reported so far, these might become apparent with more widespread use158.
Lopinavir–ritonavir
Lopinavir–ritonavir is a fixed-dose drug combination used for the prevention and treatment of HIV infection and works by inhibiting protease activity159. Lopinavir is available only in combination with ritonavir, which functions to slow the breakdown of lopinavir by inhibiting cytochrome P450 3A4 (ref.41). In a randomized, controlled, open-label trial involving 199 patients with COVID-19, no benefit was observed with lopinavir–ritonavir treatment compared with standard care159. Gastrointestinal adverse effects were more frequently reported in the lopinavir–ritonavir treatment group than in the standard group, but adverse cardiovascular effects were not reported in either group159. Nevertheless, lopinavir–ritonavir should be used with caution in patients with COVID-19 because this drug combination might interact with common cardiovascular drugs that are metabolized by cytochrome P450 3A4, including antiarrhythmic agents, antiplatelet drugs and anticoagulants41,160.
Conclusions
Given that numerous studies have demonstrated that SARS-CoV-2 shares many biological features with SARS-CoV, our knowledge of the pathophysiological mechanisms underlying SARS can be used to understand the disease processes involved in COVID-19. Mechanistically, the interaction between the S protein and ACE2 is likely to have a central role in disease pathogenesis, especially in cardiovascular manifestations of this disease, and this interaction is a potential target for the prevention and treatment of COVID-19.
Several hurdles need to be overcome in the study of the mechanisms underlying COVID-19. First, biological experiments using SARS-CoV-2 can be performed only in laboratories with a biosafety level 3 rating161,162. Second, the use of animal models to mimic the disease process is associated with numerous challenges163,164,165. Given that cellular or tissue tropism is likely to be an important factor contributing to the diverse phenotypes of COVID-19 (ref.4), mouse or rat models are not ideal to study host tropism because they are not as susceptible to SARS-CoV-2 as humans owing to differences in the amino acid sequence of ACE2 (ref.166). To use mice or rats, human ACE2 needs to be introduced artificially. Transgenic mice expressing ACE2 infected with SARS-CoV-2 have been reported to show signs of pneumonia, but the overall symptoms experienced by these mice are much milder than those in humans163. Therefore, alternative platforms might involve genome-edited mouse or rat models in which Ace2 is replaced by human ACE2, other animal species that are naturally susceptible to SARS-CoV-2 infection (such as ferrets, hamsters and non-human primates)164,165,167,168,169,170 or in vitro models such as induced pluripotent stem cells171,172,173 and organoids114,174,175.
The COVID-19 pandemic is changing our lives in unprecedented ways. Given the lack of safe and effective vaccines or proven treatments for COVID-19, our main strategy to combat the pandemic is social distancing. The capacity of health-care systems globally has been severely tested (and in some countries completely overwhelmed), and the effect of this pandemic on social interactions, health-care delivery and the global economy continues to mount. Reduced physical activity owing to lockdown measures might also contribute to poor control of cardiovascular risk factors. Vaccine development is expected to take 12–18 months34. To meet the urgent need for effective treatment and preventative strategies, a concerted effort must be made by researchers globally to investigate and integrate biological and clinical findings related to COVID-19 (Box 1).
References
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Tay, M. Z., Poh, C. M., Renia, L., MacAry, P. A. & Ng, L. F. P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 20, 363–374 (2020).
Bikdeli, B. et al. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up: JACC state-of-the-art review. J. Am. Coll. Cardiol. 75, 2950–2973 (2020).
Connors, J. M. & Levy, J. H. Thromboinflammation and the hypercoagulability of COVID-19. J. Thromb. Haemost. 18, 1559–1561 (2020).
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C. & Garry, R. F. The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452 (2020).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292 (2020).
Wan, Y., Shang, J., Graham, R., Baric, R. S. & Li, F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus.J. Virol. 94, e00127-20 (2020).
Madjid, M., Safavi-Naeini, P., Solomon, S. D. & Vardeny, O. Potential effects of coronaviruses on the cardiovascular system: a review. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.1286 (2020).
Clerkin, K. J. et al. COVID-19 and cardiovascular disease. Circulation 141, 1648–1655 (2020).
Driggin, E. et al. Cardiovascular considerations for patients, health care workers, and health systems during the COVID-19 pandemic. J. Am. Coll. Cardiol. 75, 2352–2371 (2020).
Zheng, Y. Y., Ma, Y. T., Zhang, J. Y. & Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 17, 259–260 (2020).
Han, Y. et al. CSC expert consensus on principles of clinical management of patients with severe emergent cardiovascular diseases during the COVID-19 epidemic. Circulation 141, e810–e816 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Wang, D. et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA https://doi.org/10.1001/jama.2020.1585 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Guan, W. J. et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 382, 1708–1720 (2020).
Wu, Z. & McGoogan, J. M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA https://doi.org/10.1001/jama.2020.2648 (2020).
Ruan, Q., Yang, K., Wang, W., Jiang, L. & Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 46, 846–848 (2020).
Shi, S. et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.0950 (2020).
Guo, T. et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.1017 (2020).
Shi, S. et al. Characteristics and clinical significance of myocardial injury in patients with severe coronavirus disease 2019. Eur. Heart J. 41, 2070–2079 (2020).
Riphagen, S., Gomez, X., Gonzalez-Martinez, C., Wilkinson, N. & Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 395, 1607–1608 (2020).
Yin, W. et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 368, 1499–1504 (2020).
Dai, W. et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368, 1331–1335 (2020).
Cui, J., Li, F. & Shi, Z. L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192 (2019).
Du, L. et al. The spike protein of SARS-CoV — a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 7, 226–236 (2009).
Ge, X. Y. et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538 (2013).
Bar-On, Y. M., Flamholz, A., Phillips, R. & Milo, R. SARS-CoV-2 (COVID-19) by the numbers. Elife https://doi.org/10.7554/eLife.57309 (2020).
Masson, P. et al. ViralZone: recent updates to the virus knowledge resource. Nucleic Acids Res. 41, D579–D583 (2013).
Lurie, N., Saville, M., Hatchett, R. & Halton, J. Developing COVID-19 vaccines at pandemic speed. N. Engl. J. Med. 382, 1969–1973 (2020).
Callaway, E. The race for coronavirus vaccines: a graphical guide. Nature 580, 576–577 (2020).
Suthar, M. S. et al. Rapid generation of neutralizing antibody responses in COVID-19 patients. Cell Rep. Med. 1, 100040 (2020).
Wang, C. et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 11, 2251 (2020).
Ho, T. Y., Wu, S. L., Chen, J. C., Li, C. C. & Hsiang, C. Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antivir. Res. 74, 92–101 (2007).
Robson, B. COVID-19 coronavirus spike protein analysis for synthetic vaccines, a peptidomimetic antagonist, and therapeutic drugs, and analysis of a proposed Achilles’ heel conserved region to minimize probability of escape mutations and drug resistance. Comput. Biol. Med. 121, 103749–103749 (2020).
Perlman, S. & Masters, P. S. in Fields Virology: Emerging Viruses (eds Howley, P. M & knipe, D. M.) 410–448 (Lippincott Williams & Wilkins, 2020).
Sanders, J. M., Monogue, M. L., Jodlowski, T. Z. & Cutrell, J. B. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): a review. JAMA https://doi.org/10.1001/jama.2020.6019 (2020).
Li, G. & De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 19, 149–150 (2020).
Li, S. S. et al. Left ventricular performance in patients with severe acute respiratory syndrome: a 30-day echocardiographic follow-up study. Circulation 108, 1798–1803 (2003).
Peiris, J. S. et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361, 1767–1772 (2003).
The Novel Coronavirus Pneumonia Emergency Response Epidemiology Team. Vital surveillances: the epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) — China, 2020. China CDC Wkly 2, 113–122 (2020).
Grasselli, G. et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy region, Italy. JAMA https://doi.org/10.1001/jama.2020.5394 (2020).
Goyal, P. et al. Clinical characteristics of COVID-19 in New York City. N. Engl. J. Med. 382, 2372–2374 (2020).
Arentz, M. et al. Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington state. JAMA https://doi.org/10.1001/jama.2020.4326 (2020).
Bhatraju, P. K. et al. COVID-19 in critically ill patients in the Seattle region — case series. N. Engl. J. Med. 382, 2012–2022 (2020).
Onder, G., Rezza, G. & Brusaferro, S. Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. JAMA https://doi.org/10.1001/jama.2020.4683 (2020).
Richardson, S. et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA https://doi.org/10.1001/jama.2020.6775 (2020).
Stefanini, G. G. et al. ST-elevation myocardial infarction in patients with COVID-19: clinical and angiographic outcomes. Circulation 141, 2113-2116 (2020).
Deng, Q. et al. Suspected myocardial injury in patients with COVID-19: evidence from front-line clinical observation in Wuhan, China. Int. J. Cardiol. 311, 116–121 (2020).
Wang, D. et al. Chinese Society of Cardiology expert consensus statement on the diagnosis and treatment of adult fulminant myocarditis. Sci. China Life Sci. 62, 187–202 (2019).
Inciardi, R. M. et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.1096 (2020).
Hu, H., Ma, F., Wei, X. & Fang, Y. Coronavirus fulminant myocarditis saved with glucocorticoid and human immunoglobulin. Eur. Heart J. https://doi.org/10.1093/eurheartj/ehaa190 (2020).
Xu, Z. et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 8, 420–422 (2020).
Tavazzi, G. et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur. J. Heart Fail. 22, 911–915 (2020).
Wichmann, D. et al. Autopsy findings and venous thromboembolism in patients with COVID-19. Ann. Intern. Med. https://doi.org/10.7326/M20-2003 (2020).
Schaller, T. et al. Postmortem examination of patients with COVID-19. JAMA https://doi.org/10.1001/jama.2020.8907 (2020).
Jiang, F. et al. Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nat. Rev. Cardiol. 11, 413–426 (2014).
Oudit, G. Y. et al. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Invest. 39, 618–625 (2009).
Kwong, J. C. et al. Acute myocardial infarction after laboratory-confirmed influenza infection. N. Engl. J. Med. 378, 345–353 (2018).
Madjid, M. et al. Influenza epidemics and acute respiratory disease activity are associated with a surge in autopsy-confirmed coronary heart disease death: results from 8 years of autopsies in 34,892 subjects. Eur. Heart J. 28, 1205–1210 (2007).
Chong, P. Y. et al. Analysis of deaths during the severe acute respiratory syndrome (SARS) epidemic in Singapore: challenges in determining a SARS diagnosis. Arch. Pathol. Lab. Med. 128, 195–204 (2004).
Bangalore, S. et al. ST-segment elevation in patients with COVID-19 – a case series. N. Engl. J. Med. 382, 2478-2480 (2020).
Libby, P., Tabas, I., Fredman, G. & Fisher, E. A. Inflammation and its resolution as determinants of acute coronary syndromes. Circ. Res. 114, 1867–1879 (2014).
Bentzon, J. F., Otsuka, F., Virmani, R. & Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 114, 1852–1866 (2014).
Varga, Z. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418 (2020).
Garcia, S. et al. Reduction in ST-segment elevation cardiac catheterization laboratory activations in the United States during COVID-19 pandemic. J. Am. Coll. Cardiol. 75, 2871–2872 (2020).
De Filippo, O. et al. Reduced rate of hospital admissions for ACS during COVID-19 outbreak in Northern Italy. N. Engl. J. Med. 383, 88–89 (2020).
Rodríguez-Leor, O. et al. Impact of the COVID-19 pandemic on interventional cardiology activity in Spain. Rec. Interventional Cardiol. Engl. Ed. https://doi.org/10.24875/recice.M20000123 (2020).
De Rosa, S. et al. Reduction of hospitalizations for myocardial infarction in Italy in the COVID-19 era. Eur. Heart J. 41, 2083–2088 (2020).
Baldi, E. et al. Out-of-hospital cardiac arrest during the COVID-19 outbreak in Italy. N. Engl. J. Med. https://doi.org/10.1056/NEJMc2010418 (2020).
Pessoa-Amorim, G. et al. Admission of patients with STEMI since the outbreak of the COVID-19 pandemic. A survey by the European Society of Cardiology. Eur. Heart J. Qual. Care Clin. Outcomes https://doi.org/10.1093/ehjqcco/qcaa046 (2020).
Chen, T. et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ 368, m1091 (2020).
Mehra, M. R. & Ruschitzka, F. COVID-19 illness and heart failure: a missing link? JACC Heart Fail. 8, 512–514 (2020).
Dewey, M. et al. Clinical quantitative cardiac imaging for the assessment of myocardial ischaemia. Nat. Rev. Cardiol. 17, 427–450 (2020).
Manka, R. et al. Myocardial edema in COVID-19 on cardiac MRI. J. Heart Lung Transplant. 39, 730–732 (2020).
Fried, J. A. et al. The variety of cardiovascular presentations of COVID-19. Circulation 141, 1930–1936 (2020).
Prabhu, S. D. Cytokine-induced modulation of cardiac function. Circ. Res. 95, 1140–1153 (2004).
Liu, K. et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei province. Chin. Med. J. 133, 1025–1031 (2020).
Lakkireddy, D. R. et al. Guidance for cardiac electrophysiology during the COVID-19 pandemic from the Heart Rhythm Society COVID-19 task force; electrophysiology section of the American College of Cardiology; and the electrocardiography and arrhythmias committee of the council on clinical cardiology, American Heart Association. Circulation 141, e823–e831 (2020).
Panigada, M. et al. Hypercoagulability of COVID-19 patients in intensive care unit. A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 18, 1738–1742 (2020).
Ranucci, M. et al. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J. Thromb. Haemost. 18, 1747–1751 (2020).
Taylor, F. B. Jr. et al. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb. Haemost. 86, 1327–1330 (2001).
Oxley, T. J. et al. Large-vessel stroke as a presenting feature of COVID-19 in the young. N. Engl. J. Med. 382, e60 (2020).
Bellosta, R. et al. Acute limb ischemia in patients with COVID-19 pneumonia. J. Vasc. Surg. https://doi.org/10.1016/j.jvs.2020.04.483 (2020).
Paranjpe, I. et al. Association of treatment dose anticoagulation with in-hospital survival among hospitalized patients with COVID-19. J. Am. Coll. Cardiol. 76, 122–124 (2020).
Tang, N. et al. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 18, 1094–1099 (2020).
Qiu, H. et al. Clinical and epidemiological features of 36 children with coronavirus disease 2019 (COVID-19) in Zhejiang, China: an observational cohort study. Lancet Infect. Dis. 20, 689–696 (2020).
Verdoni, L. et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet 395, 1771–1778 (2020).
Aslam, S. & Mehra, M. R. COVID-19: yet another coronavirus challenge in transplantation. J. Heart Lung Transplant. 39, 408–409 (2020).
Li, F., Cai, J. & Dong, N. First cases of COVID-19 in heart transplantation from China. J. Heart Lung Transplant. 39, 496–497 (2020).
Ketcham, S. W. et al. Coronavirus disease-2019 in heart transplant recipients in southeastern Michigan: a case series. J. Card. Fail. 26, 457-461(2020).
Latif, F. et al. Characteristics and outcomes of recipients of heart transplant with coronavirus disease 2019. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.2159 (2020).
Gosain, R. et al. COVID-19 and cancer: a comprehensive review. Curr. Oncol. Rep. 22, 53 (2020).
Ganatra, S., Hammond, S. P. & Nohria, A. The novel coronavirus disease (COVID-19) threat for patients with cardiovascular disease and cancer. JACC CardioOncol. https://doi.org/10.1016/j.jaccao.2020.03.001 (2020).
Liang, W. et al. Cancer patients in SARS-CoV-2 infection: a nationwide analysis in China. Lancet Oncol. 21, 335–337 (2020).
Turner, A. J., Hiscox, J. A. & Hooper, N. M. ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol. Sci. 25, 291–294 (2004).
GTEx Portal (ACE2). Gene expression for ACE2 https://www.gtexportal.org/home/gene/ACE2 (2020).
Chen, L., Li, X., Chen, M., Feng, Y. & Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 116, 1097–1100 (2020).
Litvinukova, M. et al. Cells and gene expression programs in the adult human heart. Preprint at bioRxiv https://doi.org/10.1101/2020.04.03.024075 (2020).
Kaiser, J. How sick will the coronavirus make you? The answer may be in your genes. Science https://doi.org/10.1126/science.abb9192 (2020).
The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28, 715–718 (2020).
Imai, Y. et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436, 112–116 (2005).
Kuba, K. et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 11, 875–879 (2005).
Crackower, M. A. et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417, 822–828 (2002).
Thomas, M. C. et al. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ. Res. 107, 888–897 (2010).
Zhao, Y. et al. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. Preprint at bioRxiv https://doi.org/10.1101/2020.01.26.919985 (2020).
Qi, F., Qian, S., Zhang, S. & Zhang, Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun. 526, 135–140 (2020).
Sungnak, W. et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 26, 681–687 (2020).
Vaduganathan, M. et al. Renin–angiotensin–aldosterone system inhibitors in patients with COVID-19. N. Engl. J. Med. 382, 1653–1659 (2020).
Monteil, V. et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181, 905–913 (2020).
Haschke, M. et al. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin. Pharmacokinet. 52, 783–792 (2013).
Khan, A. et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care 21, 234 (2017).
Wadman, M., Couzin-Frankel, J., Kaiser, J. & Matacic, C. A rampage through the body. Science 368, 356–360 (2020).
Puelles, V. G. et al. Multiorgan and renal tropism of SARS-CoV-2. N. Engl. J. Med. https://doi.org/10.1056/NEJMc2011400 (2020).
Kuster, G. M. et al. SARS-CoV-2: should inhibitors of the renin–angiotensin system be withdrawn in patients with COVID-19? Eur. Heart J. 41, 1801–1803 (2020).
Sommerstein, R., Kochen, M. M., Messerli, F. H. & Grani, C. Coronavirus disease 2019 (COVID-19): do angiotensin-converting enzyme inhibitors/angiotensin receptor blockers have a biphasic effect? J. Am. Heart Assoc. 9, e016509 (2020).
Ferrario, C. M. et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 111, 2605–2610 (2005).
Bozkurt, B., Kovacs, R. & Harrinton, B. HFSA/ACC/AHA statement addresses concerns re: using RAAS antagonists in COVID-19. AHA Professional Heart Daily https://professional.heart.org/professional/ScienceNews/UCM_505836_HFSAACCAHA-statement-addresses-concerns-re-using-RAAS-antagonists-in-COVID-19.jsp (2020).
de Simone, G. Position statement of the ESC council on hypertension on ACE-inhibitors and angiotensin receptor blockers. ESC escardio https://www.escardio.org/Councils/Council-on-Hypertension-(CHT)/News/position-statement-of-the-esc-council-on-hypertension-on-ace-inhibitors-and-ang (2020).
Chinese Society of Cardiology. Scientific statement on using renin–angiotensin system blockers in patients with cardiovascular disease and COVID-19. Chin. J. Cardiol. 48, E014 (2020).
de Abajo, F. J. et al. Use of renin–angiotensin–aldosterone system inhibitors and risk of COVID-19 requiring admission to hospital: a case-population study. Lancet 395, 1705–1714 (2020).
Mancia, G., Rea, F., Ludergnani, M., Apolone, G. & Corrao, G. Renin–angiotensin–aldosterone system blockers and the risk of COVID-19. N. Engl. J. Med. 382, 2431–2440 (2020).
Reynolds, H. R. et al. Renin–angiotensin–aldosterone system inhibitors and risk of COVID-19. N. Engl. J. Med. 382, 2441–2448 (2020).
Mehta, N. et al. Association of use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with testing positive for coronavirus disease 2019 (COVID-19). JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.1855 (2020).
Ishiyama, Y. et al. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 43, 970–976 (2004).
Soler, M. J. et al. Localization of ACE2 in the renal vasculature: amplification by angiotensin II type 1 receptor blockade using telmisartan. Am. J. Physiol. Ren. Physiol 296, F398–F405 (2009).
Burrell, L. M. et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur. Heart J. 26, 369–375; discussion 322–324 (2005).
Ocaranza, M. P. et al. Enalapril attenuates downregulation of angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension 48, 572–578 (2006).
Luque, M. et al. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1–7) in essential hypertension. J. Hypertens. 14, 799–805 (1996).
Furuhashi, M. et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am. J. Hypertens. 28, 15–21 (2015).
Epelman, S. et al. Soluble angiotensin-converting enzyme 2 in human heart failure: relation with myocardial function and clinical outcomes. J. Card. Fail. 15, 565–571 (2009).
Ramchand, J. et al. Plasma ACE2 activity predicts mortality in aortic stenosis and is associated with severe myocardial fibrosis. JACC Cardiovasc. Imaging 13, 655–664 (2020).
Walters, T. E. et al. Angiotensin converting enzyme 2 activity and human atrial fibrillation: increased plasma angiotensin converting enzyme 2 activity is associated with atrial fibrillation and more advanced left atrial structural remodelling. Europace 19, 1280–1287 (2017).
Ramchand, J., Patel, S. K., Srivastava, P. M., Farouque, O. & Burrell, L. M. Elevated plasma angiotensin converting enzyme 2 activity is an independent predictor of major adverse cardiac events in patients with obstructive coronary artery disease. PLoS One 13, e0198144 (2018).
Pushpakom, S. et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 18, 41–58 (2019).
Andersen, P. I. et al. Discovery and development of safe-in-man broad-spectrum antiviral agents. Int. J. Infect. Dis. 93, 268–276 (2020).
Guy, R. K., DiPaola, R. S., Romanelli, F. & Dutch, R. E. Rapid repurposing of drugs for COVID-19. Science 368, 829–830 (2020).
Roden, D. M., Harrington, R. A., Poppas, A. & Russo, A. M. Considerations for drug interactions on QTc in exploratory COVID-19 treatment. Circulation 141, e906–e907 (2020).
Chen, Z. et al. Efficacy of hydroxychloroquine in patients with COVID-19: results of a randomized clinical trial. Preprint at medRxiv https://doi.org/10.1101/2020.03.22.20040758 (2020).
Gautret, P. et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents, 105949, https://doi.org/10.1016/j.ijantimicag.2020.105949 (2020).
Wang, M. et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 30, 269–271 (2020).
Savarino, A., Boelaert, J. R., Cassone, A., Majori, G. & Cauda, R. Effects of chloroquine on viral infections: an old drug against today’s diseases? Lancet Infect. Dis. 3, 722–727 (2003).
ISAC/Elsevier statement. Joint ISAC and Elsevier statement on Gautret et al. paper. International Society of Antimicrobial Chemotherapy https://www.isac.world/news-and-publications/isac-elsevier-statement (2020).
Yu, B. et al. Low dose of hydroxychloroquine reduces fatality of critically ill patients with COVID-19. Sci. China Life. Sci. https://doi.org/10.1007/s11427-020-1732-2 (2020).
Geleris, J. et al. Observational study of hydroxychloroquine in hospitalized patients with COVID-19. N. Engl. J. Med. 382, 2411–2418 (2020).
Fihn, S. D., Perencevich, E. & Bradley, S. M. Caution needed on the use of chloroquine and hydroxychloroquine for coronavirus disease 2019. JAMA Netw. Open. 3, e209035 (2020).
Mercuro, N. J. et al. Risk of QT interval prolongation associated with use of hydroxychloroquine with or without concomitant azithromycin among hospitalized patients testing positive for coronavirus disease 2019 (COVID-19). JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.1834 (2020).
Hancox, J. C., Hasnain, M., Vieweg, W. V., Crouse, E. L. & Baranchuk, A. Azithromycin, cardiovascular risks, QTc interval prolongation, Torsade de Pointes, and regulatory issues: a narrative review based on the study of case reports. Ther. Adv. Infect. Dis. 1, 155–165 (2013).
Rosenberg, E. S. et al. Association of treatment with hydroxychloroquine or azithromycin with in-hospital mortality in patients with COVID-19 in New York state. JAMA https://doi.org/10.1001/jama.2020.8630 (2020).
Grein, J. et al. Compassionate use of remdesivir for patients with severe COVID-19. N. Engl. J. Med. 382, 2327–2336 (2020).
Wang, Y. et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 395, 1569–1578 (2020).
Sheahan, T. P. et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 11, 222 (2020).
Beigel, J. H. et al. Remdesivir for the treatment of COVID-19 – preliminary report. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2007764 (2020).
US FDA. Fact sheet for health care providers: emergency use authorization (EUA) of remdesivir (GS-5734™). https://www.fda.gov/media/137566/download (2020).
Cao, B. et al. A trial of lopinavir–ritonavir in adults hospitalized with severe COVID-19. N. Engl. J. Med. 382, 1787–1799 (2020).
Stockman, L. J., Bellamy, R. & Garner, P. SARS: systematic review of treatment effects. PLoS Med. 3, e343 (2006).
Centers for Disease Control and Prevention. Interim laboratory biosafety guidelines for handling and processing specimens associated with coronavirus disease 2019 (COVID-19). https://www.cdc.gov/coronavirus/2019-ncov/lab/lab-biosafety-guidelines.html (CDC, 2020).
World Health Organization. Laboratory biosafety guidance related to coronavirus disease 2019 (COVID-19). https://www.who.int/publications-detail/laboratory-biosafety-guidance-related-to-coronavirus-disease-2019-(covid-19) (2020).
Bao, L. et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature https://doi.org/10.1038/s41586-020-2312-y (2020).
Rockx, B. et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 368, 1012–1015 (2020).
Shi, J. et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 368, 1016–1020 (2020).
McCray, P. B. Jr. et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 81, 813–821 (2007).
Kim, Y. I. et al. Infection and rapid transmission of SARS-CoV-2 in ferrets. Cell Host Microbe. 27, 704–709 (2020).
Park, S. J. et al. Ferret animal model of severe fever with thrombocytopenia syndrome phlebovirus for human lethal infection and pathogenesis. Nat. Microbiol. 4, 438–446 (2019).
Chan, J. F. et al. Simulation of the clinical and pathological manifestations of coronavirus disease 2019 (COVID-19) in golden Syrian hamster model: implications for disease pathogenesis and transmissibility. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa325 (2020).
Bao, L. et al. Lack of reinfection in rhesus macaques infected with SARS-CoV-2. Preprint at bioRxiv https://doi.org/10.1101/2020.03.13.990226 (2020).
Chen, I. Y., Matsa, E. & Wu, J. C. Induced pluripotent stem cells: at the heart of cardiovascular precision medicine. Nat. Rev. Cardiol. 13, 333–349 (2016).
Shi, Y., Inoue, H., Wu, J. C. & Yamanaka, S. Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov. 16, 115–130 (2017).
Sharma, A. et al. Human iPSC-derived cardiomyocytes are susceptible to SARS-CoV-2 infection. Cell Rep. Med. https://doi.org/10.1016/j.xcrm.2020.100052 (2020).
McCauley, K. B., Hawkins, F. & Kotton, D. N. Derivation of epithelial-only airway organoids from human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 45, e51 (2018).
McCauley, K. B. et al. Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of Wnt signaling. Cell Stem Cell 20, 844–857 (2017).
Acknowledgements
The authors thank all the patients, health-care workers and other frontline workers globally for their patience, hard work and dedication during this unprecedented pandemic. The authors are supported by the AHA (17MERIT33610009), Burroughs Wellcome Foundation IRSA 1015009 (J.C.W.) and the JSPS Overseas Research Fellowship (M.N.).
Author information
Authors and Affiliations
Contributions
M.N. and J.C.W. researched data and wrote the article. All the authors contributed to the discussion of content and reviewed and edited the manuscript before submission.
Corresponding authors
Additional information
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Cardiology thanks M. Mehra, P. Zhou and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Acute respiratory distress syndrome
-
(ARDS). A type of severe, acute respiratory failure characterized by bilateral pulmonary infiltrates and severe hypoxaemia that occurs as a result of illness or injury.
- Cytokine storm
-
A form of severe immune reaction characterized by overproduction of cytokines and chemokines that can be triggered by a variety of factors such as infection and drugs.
- Tissue tropism
-
The ability of a given pathogen to infect specific cell or tissue types of a host.
- Kawasaki disease
-
An acute febrile illness of unknown cause that primarily affects children aged <5 years and can cause coronary artery aneurysms.
- Positive-sense, single-stranded RNA
-
A sequence of RNA, also known as plus-strand RNA, that can be directly translated to a sequence of amino acids to synthesize proteins without a complementary RNA intermediate.
- Open reading frames
-
ORFs. Continuous stretches of nucleotide sequences in genomic DNA or mRNA between a start codon (usually ATG in DNA) and a stop codon (usually TAA, TAG or TGA in DNA) that are potentially translated into proteins.
- Furin cleavage site
-
A specific peptide sequence in precursor proteins that can be cleaved by an enzyme furin to facilitate conversion to a biologically active state.
- Lysosomotropic agents
-
Drugs that are taken up selectively into lysosomes.
- Case fatality rate
-
The proportion of people who die from a certain disease among all individuals diagnosed with the disease over a certain period of time.
Rights and permissions
About this article

Cite this article
Nishiga, M., Wang, D.W., Han, Y. et al. COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol 17, 543–558 (2020). https://doi.org/10.1038/s41569-020-0413-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-020-0413-9
- Springer Nature Limited
This article is cited by
-
COVID-19-related cardiovascular disease risk due to weight gain: a nationwide cohort study
European Journal of Medical Research (2024)
-
Syndecan-1 as a prognostic biomarker in COVID-19 patients: a retrospective study of a Japanese cohort
Thrombosis Journal (2024)
-
Cohort study of cardiovascular safety of different COVID-19 vaccination doses among 46 million adults in England
Nature Communications (2024)
-
Organoids in virology
npj Viruses (2024)
-
Research progress of post-acute sequelae after SARS-CoV-2 infection
Cell Death & Disease (2024)