

Abstract
Many antimicrobial peptides directly disrupt bacterial membranes yet can also damage mammalian membranes. It is therefore central to their therapeutic use that rules governing the membrane selectivity of antimicrobial peptides be deciphered. However, this is difficult even for short peptides owing to the large combinatorial space of amino acid sequences. Here we describe a method for measuring the loss or maintenance of antimicrobial-peptide activity for thousands of peptide-sequence variants simultaneously, and its application to Protegrin-1, a potent yet toxic antimicrobial peptide, to determine the positional importance and flexibility of residues across its sequence while identifying variants with changes in membrane selectivity. More bacterially selective variants maintained a membrane-bound secondary structure while avoiding aromatic residues and cysteine pairs. A machine-learning model trained with our datasets accurately predicted membrane-specific activities for over 5.7 million Protegrin-1 variants, and identified one variant that showed substantially reduced toxicity and retention of activity in a mouse model of intraperitoneal infection. The high-throughput methodology may help elucidate sequence–structure–function relationships in antimicrobial peptides and inform the design of peptide-based synthetic drugs.






Similar content being viewed by others
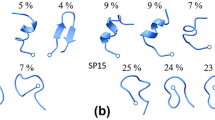
Data availability
Raw sequencing data from dmSLAY are available from the SRA database under accession number PRJNA1022479. The raw and analysed datasets generated during the study are available for research purposes from the corresponding author on reasonable request. Source data for the figures are provided with this paper.
Code availability
Coding materials for sequence analysis and machine learning are available on GitHub at https://github.com/facebookresearch/esm#main-models ref. 40 and https://github.com/ziul-bio/DMS_ML_AMP ref. 41.
References
Mookherjee, N., Anderson, M. A., Haagsman, H. P. & Davidson, D. J. Antimicrobial host defence peptides: functions and clinical potential. Nat. Rev. Drug Discov. 19, 311–332 (2020).
Huang, Y., Huang, J. & Chen, Y. Alpha-helical cationic antimicrobial peptides: relationships of structure and function. Protein Cell 1, 143–152 (2010).
Fowler, D. M. & Fields, S. Deep mutational scanning: a new style of protein science. Nat. Methods 11, 801–807 (2014).
Koch, P. et al. Optimization of the antimicrobial peptide Bac7 by deep mutational scanning. BMC Biol. 20, 114 (2022).
Tucker, A. T. et al. Discovery of next-generation antimicrobials through bacterial self-screening of surface-displayed peptide libraries. Cell 172, 618–628.e13 (2018).
Khabbaz, H., Karimi-Jafari, M. H., Saboury, A. A. & BabaAli, B. Prediction of antimicrobial peptides toxicity based on their physico-chemical properties using machine learning techniques. BMC Bioinformatics 22, 549 (2021).
Huang, J. et al. Identification of potent antimicrobial peptides via a machine-learning pipeline that mines the entire space of peptide sequences. Nat. Biomed. Eng. 7, 797–810 (2023).
Randall, J. R. et al. Designing and identifying β-hairpin peptide macrocycles with antibiotic potential. Sci. Adv. 9, eade0008 (2023).
Fahrner, R. L. et al. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 3, 543–550 (1996).
Panteleev, P. V., Bolosov, I. A., Balandin, S. V. & Ovchinnikova, T. V. Structure and biological functions of β-hairpin antimicrobial peptides. Acta Nat. 7, 37–47 (2015).
Steinberg, D. A. et al. Protegrin-1: a broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob. Agents Chemother. 41, 1738–1742 (1997).
Edwards, I. A. et al. Contribution of amphipathicity and hydrophobicity to the antimicrobial activity and cytotoxicity of β-hairpin peptides. ACS Infect. Dis. 2, 442–450 (2016).
Soundrarajan, N. et al. Protegrin-1 cytotoxicity towards mammalian cells positively correlates with the magnitude of conformational changes of the unfolded form upon cell interaction. Sci. Rep. 9, 11569 (2019).
Díez-Aguilar, M. et al. Murepavadin antimicrobial activity against and resistance development in cystic fibrosis Pseudomonas aeruginosa isolates. J. Antimicrob. Chemother. 76, 984–992 (2021).
Moreno-Morales, J., Guardiola, S., Ballesté-Delpierre, C., Giralt, E. & Vila, J. A new synthetic protegrin as a promising peptide with antibacterial activity against MDR Gram-negative pathogens. J. Antimicrob. Chemother. 77, 3077–3085 (2022).
Polyphor Ltd. Pivotal study in nosocomial pneumonia suspected or confirmed to be due to Pseudomonas (PRISM-UDR). Study Record. Beta ClinicalTrials.gov https://beta.clinicaltrials.gov/study/NCT03582007 (2019).
Aumelas, A. et al. Synthesis and solution structure of the antimicrobial peptide protegrin-1. Eur. J. Biochem. 237, 575–583 (1996).
Avitabile, C., D’Andrea, L. D. & Romanelli, A. Circular dichroism studies on the interactions of antimicrobial peptides with bacterial cells. Sci. Rep. 4, 4293 (2014).
Greenfield, N. & Fasman, G. D. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 8, 4108–4116 (1969).
Feng, X. et al. The critical role of tryptophan in the antimicrobial activity and cell toxicity of the duck antimicrobial peptide DCATH. Front. Microbiol. 11, 1146 (2020).
Wei, S. Y. et al. Solution structure of a novel tryptophan-rich peptide with bidirectional antimicrobial activity. J. Bacteriol. 188, 328–334 (2006).
Subbalakshmi, C., Bikshapathy, E., Sitaram, N. & Nagaraj, R. Antibacterial and hemolytic activities of single tryptophan analogs of indolicidin. Biochem. Biophys. Res. Commun. 274, 714–716 (2000).
Azad, M. A. K. et al. Significant accumulation of polymyxin in single renal tubular cells: a medicinal chemistry and triple correlative microscopy approach. Anal. Chem. 87, 1590–1595 (2015).
Sales, G. T. M. & Foresto, R. D. Drug-induced nephrotoxicity. Rev. Assoc. Med. Bras. 66, 82–90 (2020).
Poirel, L., Jayol, A. & Nordmanna, P. Polymyxins: antibacterial activity, susceptibility testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin. Microbiol. Rev. 30, 557–596 (2017).
Bolosov, I. A. et al. Design of protegrin-1 analogs with improved antibacterial selectivity. Pharmaceutics 15, 2047 (2023).
Cherkasov, A. et al. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 4, 65–74 (2009).
Guralp, S. A., Murgha, Y. E., Rouillard, J. M. & Gulari, E. From design to screening: a new antimicrobial peptide discovery pipeline. PLoS ONE 8, e59305 (2013).
Hilpert, K., Winkler, D. F. H. & Hancock, R. E. W. Peptide arrays on cellulose support: SPOT synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nat. Protoc. 2, 1333–1349 (2007).
Bobone, S. & Stella, L. Selectivity of antimicrobial peptides: a complex interplay of multiple equilibria. Adv. Exp. Med. Biol. 1117, 175–214 (2019).
Lai, J. R., Epand, R. F., Weisblum, B., Epand, R. M. & Gellman, S. H. Roles of salt and conformation in the biological and physicochemical behavior of protegrin-1 and designed analogues: correlation of antimicrobial, hemolytic, and lipid bilayer-perturbing activities. Biochemistry 45, 15718–15730 (2006).
Harwig, S. S. L. Intramolecular disulfide bonds enhance the antimicrobial and lytic activities of protegrins at physiological sodium chloride concentrations. Eur. J. Biochem. 240, 352–357 (1996).
Lai, J. R., Huck, B. R., Weisblum, B. & Gellman, S. H. Design of non-cysteine-containing antimicrobial β-hairpins: structure–activity relationship studies with linear protegrin-1 analogues. Biochemistry 41, 12835–12842 (2002).
Chen, J. et al. Development of protegrins for the treatment and prevention of oral mucositis: structure–activity relationships of synthetic protegrin analogues. Biopolymers 55, 88–98 (2000).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE https://doi.org/10.1371/journal.pone.0163962 (2016).
Dodt, M., Roehr, J. T., Ahmed, R. & Dieterich, C. FLEXBAR—flexible barcode and adapter processing for next-generation sequencing platforms. Biology 1, 895–905 (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science https://doi.org/10.1126/science.ade2574 (2023).
Detlefsen, N. S., Hauberg, S. & Boomsma, W. Learning meaningful representations of protein sequences. Nat. Commun. 13, 1914 (2022).
Facebook Research. Evolutionary scale modeling. GitHub https://github.com/facebookresearch/esm#main-models (2023).
Vieira, L. C. Deep mutational analysis and machine learning uncover antimicrobial peptide features driving membrane selectivity. GitHub https://github.com/ziul-bio/DMS_ML_AMP (2024).
Randall, J. R. et al. Synthetic antibacterial discovery of symbah-1, a macrocyclic β-hairpin peptide antibiotic. iScience 25, 103611 (2021).
Acknowledgements
We thank the Targeted Therapeutic Drug Discovery and Development Program at the University of Texas for access to circular dichroism training and equipment. The authors disclose support for the research described in this study from the National Institutes of Health (grant numbers AI125337, AI148419 and AI159203), the Welch Foundation (grant number F-2137), the Defense Threat Reduction Agency (grant number ADTRA1-17-C0008) and Tito’s Handmade Vodka.
Author information
Authors and Affiliations
Contributions
J.R.R., L.C.V., B.W.D. and C.O.W. conceptualized this work. J.R.R. and L.C.V. were responsible for methodology, investigation and data visualization. B.W.D. and C.O.W. supervised the work and acquired funding. J.R.R. wrote the original draft and L.C.V., B.W.D. and C.O.W. helped with editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Biomedical Engineering thanks Jian Ji and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Protegin-1 dmSLAY library diversity and sequencing analysis.
a, an alanine scan performed on the native Protegrin-1 (PG-1.0) amino acid sequence showing antibacterial activity (MIC) in µg/ml. Reported MIC is the median of triplicate reactions. b, Chart of the sequence variance found within the Protegrin-1 dmSLAY library. The native Protegrin-1 sequence is shown at the top with mutations observed at each location within the library below. Amino acids are color coded by side chain similarity. Brackets represent where disulfide bonds are present. c, Principal Component Analysis of the overall induced and uninduced triplicate sample variance. d, ROC curve with different MIC cut offs for active and inactivity for log2-fold change cut off across a range of log2-fold change scores (L2FC).
Extended Data Fig. 2 Selectivity of serine and histidine containing PG-1 variants.
a, Scatter plot of the log2-fold change in MIC versus %Hemolysis for dmSLAY active PG-1 variants from Fig. 3. Dotted line represents PG-1.0 selectivity score. b, Table showing the biochemical characteristics of serine and histidine containing Protegrin-1 variants from dmSLAY. MIC is the median of triplicate assays and %Hemolysis is the mean of triplicate assays. c, Bar chart showing the selectivity score of serine and histidine containing variants on a log2 scale. Residue changes are shown below. Brackets show where disulfide bonds are formed in the native structure.
Extended Data Fig. 3 Comparing Protegrin-1 variant activity in mixed cultures.
a-d, Graphs of PG-1 (top left), PG-1.1 (bottom left), PG-1.20 (top right), and PG1-37 (bottom right) percentage of bacterial killing (green) and % hemolysis (purple) with 1 × 109 red blood cells (RBC), 1 × 106 E. coli W3110 cells (Bacteria) or both at various concentrations shown on a log2 scale. Each data point is the mean of triplicate reactions and error bars are one standard deviation.
Extended Data Fig. 4 Training of machine learning models and specific attribute mutational profiles.
a, Precision and recall of for predicting PG-1 variants with an MIC > or < 8 µg/ml. b, predicted versus true hemolysis for trained and test data c, or predicted versus true log10-selectivity score. All models were trained on 80% of data and validated with 20%. Bottom panels: Mutational profiles of variants from 5.7 million candidates with a predicted MIC ≤ 8 µg/ml (a) % hemolysis ≤ 2 (b) or log10-Selectivity score ≤ 0.5 (c).
Supplementary information
Supplementary Information
Supplementary Methods, Figures and Tables.
Data
Machine-learning-identified Protegin-1 variants.
Source data
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Randall, J.R., Vieira, L.C., Wilke, C.O. et al. Deep mutational scanning and machine learning for the analysis of antimicrobial-peptide features driving membrane selectivity. Nat. Biomed. Eng 8, 842–853 (2024). https://doi.org/10.1038/s41551-024-01243-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-024-01243-1
- Springer Nature Limited