

Abstract
Polyamorphic transition (PT) is a compelling and pivotal physical phenomenon in the field of glass and materials science. Understanding this transition is of scientific and technological significance, as it offers an important pathway for effectively tuning the structure and property of glasses. In contrast to the PT observed in conventional metallic glasses (MGs), which typically exhibit a pronounced first-order nature, herein we report a continuous PT (CPT) without first-order characteristics in high-entropy MGs (HEMGs) upon heating. This CPT behavior is featured by the continuous structural evolution at the atomic level and an increasing chemical concentration gradient with temperature, but no abrupt reduction in volume and energy. The continuous transformation is associated with the absence of local favorable structures and chemical heterogeneity caused by the high configurational entropy, which limits the distance and frequency of atomic diffusion. As a result of the CPT, numerous glass states can be generated, which provides an opportunity to understand the nature, atomic packing, formability, and properties of MGs. Moreover, this discovery highlights the implication of configurational entropy in exploring polyamorphic glasses with an identical composition but highly tunable structures and properties.
Similar content being viewed by others
Introduction
Polyamorphic transition (PT), a well-known amorphous analog of crystalline polymorphism, delineates the transition between amorphous states that are chemically identical but structurally distinct. In fact, PT is ubiquitous in the realm of amorphous solids, such as phosphorus1, silicon2, silica3,4, and molecular glass5. Water, often regarded as the “molecule of life” in the universe6, exhibits PT in its amorphous states, which is a phenomenon modulated by external pressure and temperature7,8, influencing its density and structure. The appearance of PT in diverse materials underscores the complexity of the inherent configuration space of glasses, providing profound insights into the fundamental nature of these disordered states. Moreover, the continuous progress of switchable glass materials, which typically have an identical composition but distinct physical properties, further explains the imperativeness to reveal the complexity of PT. The microscopic structural scenario responsible for the PT remains an intriguing mystery in the field of condensed matter physics.
In metallic glasses (MGs), temperature- and pressure-modulated PTs, such as liquid-liquid and glass-glass transitions, have been observed despite their closely packed atomic structure. Specifically, thermal-induced liquid-liquid transitions have been reported in various glass-forming metallic liquids, such as Pd-9,10,11, rare-earth- (RE-)12, and Zr-based13 MGs, which typically exhibit first-order characteristics with discontinuous changes in volume and energy. In addition, pressure-induced glass-glass transitions have also been found in some MGs, particularly in RE-based MGs, where the change in electronic structure is triggered by pressure14,15. Unlike oxide and molecular glasses, MGs are typically composed of multiple constituent elements, leading to the complexity and diversity of atomic packing arrangements. As such, configurational entropy is expected to play a nontrivial role in phase transition and related properties of MGs. In fact, high configurational entropy has been proven to be a useful parameter for regulating atomic structures16, relaxation behavior17,18,19, glass transition17,20, and crystallization behavior21. The emergency of high-entropy MGs (HEMGs) consisting of multiple elements with equimolar or near-equimolar ratios, featured by high configurational entropy, provides an opportunity to explore the high-entropy effect on PT. Recently, it has been found that the high-entropy effect can induce glass-glass transition during heating due to the abundant configuration sites in HEMGs22,23,24. Although the high-entropy effect is believed to favor the occurrence of PTs in HEMGs, the PT phenomenon in HEMGs is still considered similar to that observed in conventional MGs. In this case, the critical role of high configurational entropy in the development of PTs, particularly its physical nature and differences from conventional PTs, remains to be elucidated.
To address the aforementioned issues, in this work, we carefully scrutinize the PT process in the model TiZrHfCuNi HEMG through state-of-the-art experimental characterizations, coupled with reverse Montel Carlo simulations. We discover a PT during the reheating of the TiZrHfCuNi HEMG, which is featured by a continuous reduction in volume and energy, without first-order characteristics. Therefore, this continuous transformation produces multiple polyamorphous states with identical compositions but distinct energy states and properties. Moreover, we propose a Landau-based phenomenal model to describe and understand this continuous PT (CPT) in HEMGs. These findings not only clarify the role of high configurational entropy in the PT process but also provide opportunities to tune glassy structures and properties of HEMGs.
Results
Continuous PT behavior in HEMGs
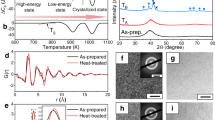
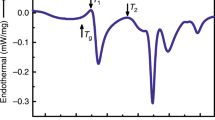
Figure 1a shows the differential scanning calorimetry (DSC) curve of the as-cast Ti20Zr20Hf20Cu20Ni20 HEMG (hereafter referred to as G0) at a heating rate of 0.17 K s−1. It can be seen that there are two exothermic peaks on the curve. To ascertain whether the first exothermic peak is related to crystallization, an analysis was conducted on the annealed sample. The annealed sample, which was obtained by reheating G0 to the end temperature (771 K) of the first peak and subsequently quenching to room temperature, was observed by high-resolution transmission electron microscopy (HRTEM). The HRTEM image, as depicted in Fig. 1b, displays a uniform maze-like pattern, accompanied by an obvious diffuse diffraction halo in the corresponding selected area electron diffraction (SAED) pattern (the inset in Fig. 1b), confirming that the annealed sample remains fully amorphous. Additionally, as illustrated in Fig. 1c, in-situ heating synchrotron X-ray diffraction (XRD) measurements of the G0 sample indicate that the sample maintains its glass state below 771 K. This finding suggests that the first exothermic peak is associated with a PT event rather than crystallization, like the phenomenon reported in other HEMGs22,24. The onset (TPT-s) and end (TPT-e) temperatures of the first exothermic peak were determined to be 697 and 771 K, respectively. Furthermore, by heating the initial glass to 713, 726, 740, 753, and 771 K, respectively, and then quenching to room temperature, multiple glass states were obtained, referred to as Gmi (where i = 1, 2, 3, and 4) and Gn. Subsequent DSC scans of these annealed samples (Fig. 1a) showed a steady decrease in the first exothermic signal from G0 to Gm4, which totally disappeared in Gn, indicating a substantial difference in their thermodynamic properties, although all samples were in a fully amorphous state. Temperature-modulated differential scanning calorimetry (TMDSC) experiments were carried out to determine the glass transition temperature (Tg) of these different glass states through analyzing the reversible heat flow signals. As depicted in Fig. 1d, Tg continuously increases from 637 to 694 K corresponding to the sample from G0 to Gn, signifying the enhanced thermal stability resulted from the PT process. It is worth noting that all samples only exhibit a single endothermic event, indicative of glass transition. This indicates that despite having the same composition, each sample represents a different glass state with its own Tg.

a DSC traces of the as-prepared TiZrHfCuNi HEMG (G0) and after pre-heat treatment at 713, 726, 740, 753, and 771 K (named as Gm1-4 and Gn, respectively) at a heating rate of 0.17 K s−1. b The HRTEM image and corresponding SEAD pattern for Gn. c Synchrotron XRD curves taken at various temperatures, where the temperature intervals for each transition are indicated by different colors. d Reverse heat flow evolution as a function of temperature obtained from TMDSC measurements for all glasses. e–h The exothermic peak evolution in calorimetric scan and corresponding characteristic temperatures during PT of the Pd42.5Ni42.5P15 MG and the TiZrHfCuNi HEMG. In (f), red, blue, and yellow colors correspond to Tg1, TPT-s, and Tg2, respectively. In (h), red and blue colors correspond to Tg and TPT-s, respectively. Coordinates represent different intrinsic structures in configuration spaces. Source data are provided as a Source Data file.
As a typical example of conventional MGs with PT, Pd42.5Ni42.5P15 undergoes a transition to the second glass state, resulting in two distinct glass transition events11. Likewise, analogous interruption experiments have been conducted to analyze the thermodynamic behaviors during PT25. In Fig. 1e–h, we compared the PT behavior between the HEMG and Pd-based MG. Although the peak area (enthalpy) caused by PT decreased in both cases, the changes in peak height and position were visibly different between the two glasses. Specifically, as shown in Fig. 1e, the intensity of the exothermic peak in Pd-based MG obviously decreased, accompanied by the stabilization in the onset temperature TPT-s. In addition, the values of Tg1 and Tg2, representing the glass transition temperatures of the as-cast and as-PT Pd-based glasses, remain constant (see Fig. 1f), confirming the co-existence of initial and final glasses during heating. This analysis indicates that the transformation in conventional MGs is primarily governed by the nucleation and growth of the second glass, which is a typical characteristic of first-order transitions.
In contrast, appreciable increases in TPT-s and Tg were observed during the heating of the TiZrHfCuNi HEMG (Fig. 1h). In fact, this distinct behavior is a common feature of PTs observed in HEMGs, as evident in Supplementary Fig. 1, suggesting a different PT mechanism between HEMGs and conventional MGs. In particular, it seems to involve multiple intermediate states (such as Gm1-4) in the CPT process of HEMGs, rather than just two coexisting states observed in conventional MGs11. In HEMGs, slight heat perturbation can lead to the disappearance of the parent glass, forming a glass through the CPT process. Consequently, the continuous PT in HEMGs results in an expansive array of glassy variants, each with its own atomic configuration and intricate thermodynamic characteristics. This intriguing phenomenon not only underscores the inherent complexity in glassy materials but also offers a model material to further explore the intricate transformation process and underlying mechanisms of PT.
Structural evolution during continuous PT
Figure 2a illustrates the evolution of the total structure factor curves, S(q), at several representative temperatures. After PT, the full width at half maximum (FWHM) of the first peak in S(q) decreased from 0.388 Å−1 (697 K) to 0.365 Å−1 (766 K), signifying a more ordered atomic structure induced by the CPT process. The position of first diffraction peak (q1) was extracted to estimate the volume density via a power-law function26,27, with the slope reflecting the expansion coefficient. As shown in the lower panel of Fig. 2b, the linear expansion coefficient of (2.45 ± 0.03) × 10−5 K−1 was determined below 575 K, which is a characteristic of normal thermal expansion. Nevertheless, above 697 K, a gradual decrease in volume is noted without any abrupt change during the CPT process. By meticulously probing the evolution of the S(q) curve26,28 and the length variation29 during the heating process, we determined a density change of ≈0.25% after the CPT (Supplementary Fig. 2). Previous studies have revealed that the first-order PT is accompanied with significant volume shrinkage rates, as demonstrated in Pd42.5Ni42.5P1511, Pd41.5Ni41.5P179, La32.5Ce32.5Co25Al1012, and Zr46Cu38Al8Ag813. In the case of HEMGs, however, the high-entropy effect leads to continuous and sluggish densification during CPT beyond the first order (Supplementary Fig. 2).

a Selected structure factor S(q) curves at several representative temperatures. b 2π/q1 (lower panel) and the FWHM (upper panel) of the first peak in S(q) as a function of temperature, and their corresponding derivative traces. The shaded region indicates the temperature interval in which the polyamorphic transition occurs. c Typical G(r) curves obtained by Fourier transformation of structure factors. d Difference in G(r), ΔG(r) = G(r)T–G(r)636K, where T is from 697 to 766 K. The purple and orange dotted frames show the nearest and next nearest neighbor range scales, respectively. Source data are provided as a Source Data file.
To understand the CPT behavior, we scrutinized the atomic structural evolution in real space through analyzing the evolution of pair distribution function, G(r), with temperature, as depicted in Fig. 2c. Taking the G(r) curve at 636 K as a reference, we calculated the ΔG(r) from 697 to 766 K (Fig. 2d). It can be seen that the intensity obviously increases within the interatomic distances range of 2.2–4.0 Å. This enhancement is strong evidence of an ongoing and progressive process of atomic structural ordering within the first atomic shell of the HEMG. Specifically, we noted a decrease in ΔG(r) for atomic pairs with interatomic distances below 2.9 Å or exceeding 3.3 Å, while an increase was observed within the distance range between 2.9 and 3.3 Å. These distinctive trends in atomic rearrangement during heating vividly demonstrate the dynamic reorganization of the atomic structure, with atoms repositioning at specific preferred atomic distances. To better classify the components, we divided them into two categories based on their atomic radius and mixing enthalpy, namely Cu/Ni as A and Ti/Zr/Hf as B. Our supplementary analysis, presented in Supplementary Note 1 and Supplementary Fig. 3, delves into the realm of short-range orders (SROs) and reveals that thermal agitation facilitates the dissociation of A-A and B-B bonds, as well as the formation of additional A–B bonds during CPT. The intriguing binding phenomenon between small and large atoms not only helps to improve stacking efficiency but also enhances chemical ordering. Extending our investigation to medium-range orders (MROs) beyond the first nearest neighbors, we elucidated the intricate intensity changes characterizing these broader atomic arrangements (see Supplementary Note 2 and Supplementary Fig. 4). Based on these results, we conclude that the heat release above Tg on the DSC curve of TiZrHfCuNi HEMG is related to the ongoing structural ordering process between the length scale of SRO and MRO.
Atomic rearrangement mechanism responsible for continuous PT
To further understand the atomic mechanism underlying the CPT, we carefully analyzed local chemical fluctuation (i.e., chemical heterogeneity), which may serve as the driving force for the CPT process. Firstly, we performed small-angle neutron scattering (SANS) measurements to assess chemical heterogeneity at the nanoscale. The SANS profiles of the initial state (G0), two intermediate states (Gm2, Gm4), and the final state (Gn) are depicted in Fig. 3a. As can be seen, G0 displays a typical feature of monolithic glass state, and its scattering intensity rapidly declines with the increase of q until reaching a plateau value of 0.019. However, the scattering intensities of Gm2, Gm4, and Gn exhibit a weak shoulder peak in the q range above 0.02 Å−1, which become more pronounced with increasing annealing temperature. This observation strongly implies an enhancement in electron density heterogeneity, resulted from chemical fluctuations during the CPT process. Furthermore, the distribution of electron density in real space, P(r)30, was obtained through the inverse Fourier transformation of the scattering function, as shown in the inset of Fig. 3a. Although G0 did not show a detectable particle size distribution, P(r), the rest samples exhibited visible scattered size distributions with a correlation length less than 2.6 nm. This result implies that the newly generated glass has a significantly different scattering density from the as-prepared amorphous state, leading to an increase in the maximum of P(r) and the corresponding distance between local heterogeneous structures. These observations collectively indicate that the length and extent of chemical heterogeneities in the glass state were enhanced with the CPT process.

a SANS results of G0, Gm2, Gm4 and Gn. The inset shows the distribution of electron density in real space P(r) derived from SANS data by indirect Fourier transformation. Tomographic maps showing the elemental distribution of b G0 and c Gn. d 3D APT tip reconstruction of G0. The threshold for the isocomposition surface is 15.1 at.%. e, f 2D Ni concentration map over 40 × 40 × 5 nm3 volume of data for G0 and Gn. The HAADF-EDS mappings of each component in g G0 and h Gn. White and yellow dashed circles represent the regions with large and small elemental density, respectively. Source data are provided as a Source Data file.
To quantitatively probe chemical heterogeneities at the atomic scale, we conducted atom probe tomography (APT) analyses for the initial state G0 and the final state Gn. The samples exhibit a uniform distribution of elements without any visible phase separation (see Fig. 3b, c). To further assess the randomness, the frequency distribution of experimental data was compared with that from a binomial distribution with the same mean concentration for each constituent, as shown in Supplementary Fig. 5a, b. After careful examination, we found that for G0, the experimental concentration distributions are in agreement with the hypothesized binomial distribution, with the μ values, indicative of deviation from the binomial distribution, approximating zero (Supplementary Table 1). This result indicates that all elements are randomly distributed within the material. However, when conducting the same analysis on Gn, it was found that the distribution of Ni deviated from the binomial curve, whilst the other elements still exhibited random distribution (Supplementary Table 1). This divergence is exemplified by a pair of 2D cross-sections in the form of Ni distribution maps provided in Fig. 3d–f. Clearly, significant fluctuations in Ni density were observed in Gn (with more red and dark blue regions).
Figure 3g displays scanning transmission electron microscopy-energy dispersive spectrometer (STEM-EDS) mappings of G0, revealing homogeneous elemental distributions in the HEMG with inconspicuous chemical fluctuations at the nanometer scale, which is consistent with ref. 17. However, Fig. 3h demonstrates that although the contrast fluctuations of Zr, Ti, Hf, and Cu in Gn are similar to those in the as-prepared glass, the Ni distribution becomes heterogeneous, with larger contrast and domain size. This observation provides conclusive evidence that the rearrangement of Ni and heterogeneous interactions between atoms are indeed involved in the CPT process, as supported by the SANS results (Fig. 3a) and APT data (Fig. 3d–f). It is important to note that these glasses display the same crystallization behavior, as evidenced by the consistent second exothermic peak in Fig. 1a, indicating the same chemical composition of the glassy phase. Consequently, the CPT process during reheating the as-cast HEMG leads to multiple glass states with continuous structural ordering, accompanied by the modified thermodynamic behavior and chemical inhomogeneity.
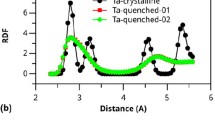
To elucidate the process of structural ordering during the CPT event, we utilized the extended X-ray absorption fine structure (EXAFS) technique to examine the evolution of SROs from G0, Gm2, Gm4 to Gn. The resulting Ni, Cu, and Zr K-edge EXAFS spectra in real space are displayed in Supplementary Fig. 6. The FT(χ(k)k3) curve showed larger intensity after PT, indicating a higher ordering degree in the local atomic structures. To further comprehend the atomic structural evolution during CPT, we performed reverse Monte Carlo (RMC) modeling on the structure factors and EXAFS (Ni, Cu, and Zr K-edge) data. The fitting results of RMC modeling agree well with the experimental measurements (Fig. 4a, b, and Supplementary Fig. 7), indicating a reasonable reconstruction of the real amorphous TiZrHfCuNi structure. Based on the atomic configuration reconstructed by the RMC modeling (e.g., Fig. 4c), we extracted partial pair distribution functions (PPDFs). The PPDFs around Ni atoms exhibit the most significant alterations after the ordering process, as illustrated in Fig. 4e, which displays typical PPDFs of Ni-Hf, Ni-Ni, and Ni-Zr pairs. The distance between Ni and Zr in the first nearest neighbor progressively decreases, at the expense of a gradual decomposition of Ni-Ni bonds. Also, to evaluate chemical SROs, the Warren-Cowley parameter31, αij, was employed (Supplementary Fig. 8) to analyze the resultant RMC configurations. The αij value of the Ni-Zr pair gradually decreased from −0.029 to −0.048, denoting an enhanced Ni-Zr chemical SRO. This result can be understood by the large negative heat of mixing between Ni and Zr and the smallest atomic size of Ni in the TiZrHfCuNi HEMG, which is expected to facilitate its diffusion upon heating.

Experimental and RMC modeled a Ni K-edge EXAFS spectra and b structure factors. Black, green, red, and yellow empty circles are experimental data for G0, Gm2, Gm4, and Gn, respectively. The curves of the corresponding colors indicate the RMC fitting data of these samples. c Typical 3D atomic configuration obtained from the RMC simulation of G0. Red, green, blue, orange, and yellow colors correspond to Zr, Ti, Hf, Cu, and Ni atoms, respectively. d The schematic illustration of the atomic structural evolution involved in CPT. e Partial pair distribution functions of the Ni-Hf, Ni-Ni, and Ni-Zr pairs. Source data are provided as a Source Data file.
Voronoi tessellation analyses on the RMC-derived atomic configurations are shown in Supplementary Fig. 9. Specifically, the enhanced frequency of the ten most prevalent Voronoi polyhedra after CPT suggests that certain characteristic structures were progressively stabilized locally. The atomic mechanism involved in the CPT is schematically outlined in Fig. 4d. The high-entropy effect tends to homogenize the atomic structure. In typical Ni-centered Voronoi polyhedra, such as <0 4 4 3>, <0 3 6 3>, and <0 2 8 1>, the types of coordinating atoms are random without appreciable local chemical ordering. Upon heating, avoidance of identical atoms is enhanced to favor shorter Ni-Zr bonds, thereby allowing more Zr atoms to surround the Ni center. Consequently, locally favored structures with a well-defined ordering degree are generated during CPT. In addition, the excess configurational entropy gradually decreases during the CPT, indicating the structural ordering32,33 as observed in Fig. 2b. Also, the distribution of excess configurational entropy becomes wider after CPT, due to the development of locally favorable structures and the enhanced structural heterogeneity (see details in Supplementary Note 3 and Supplementary Fig. 10). These findings provide critical insights into the evolution of atomic structure and SRO during CPT in HEMGs.
Property modulation by continuous PT
In contrast to the first-order PT reported in conventional MGs, which only produces two glass states, the CPT observed in HEMGs can create an infinite number of glass states with different local structures. This special behavior presents an opportunity to explore the structure-property relationship using the products of CPT. As a proof of concept, we measured the hardness and modulus of different glass states. Given that each glass state produced by CPT possesses its own Tg (Fig. 1d), which increases as the CPT proceeds, we plotted the hardness and modulus values as a function of Tg (as shown in Fig. 5). The results indicate an increase of ≈25% in both hardness and modulus. It is worth noting that the increment in hardness and modulus induced by the CPT is much more pronounced, as compared to that caused by relaxation34. This evidence underscores that the CPT treatment is a more effective approach for tuning the properties of MGs. Furthermore, our findings suggest that the thermodynamic or mechanical properties of MGs can be tailored by modulating the extent of the CPT process without altering the chemical composition. These implications furnish a compelling avenue for the deliberate manipulation of MG properties.

Ten independent property tests were performed for every sample. The colored area enclosed by the dashed line indicates the standard deviation of the mean value. Source data are provided as a Source Data file.
Discussion
In conventional MGs, the central solute atom is usually enveloped by surrounding solvent atoms35,36, resulting in predictable coordination relationships. However, in highly concentrated HEMGs without clearly defined solvent or solute atoms, the inherent diversity of clusters and inevitable chemical interaction between constituent elements challenge conventional concepts. This diversity of this type of bonding facilitates more efficient atomic packing of available space, and thermodynamically, high configurational entropy expands the potential atomic configurations20,24, providing a prerequisite for CPT. The rich structural selectivity in HEMGs also ensures that the glass undergoes changes as a whole, rather than gradually transforming into another by increasing its volume fraction. Specifically, in high-temperature melts, high configurational entropy results in low viscosity and promotes atomic diffusion37, thus leading to a homogeneous chemical potential17 and the absence of locally favorable structures after freezing. Upon reheating, HEMGs exhibit a propensity to attain compositional and structural undulations by adjusting local structures, thereby narrowing the gap between MGs and their crystalline counterparts (Supplementary Note 4 and Supplementary Fig. 11). This means that introducing high-entropy effects in MGs enhances the flexibility and tunability of atomic structures. As a pivotal conduit for bridging the gap between as-prepared MGs and their crystalline counterparts24, CPT in the current HEMGs plays a critical role. It should be noted that in addition to the configurational entropy, atomic size misfit22, and enthalpy also have influences on CPT (Supplementary Note 5 and Supplementary Fig. 12).
PT in conventional MGs is difficult to be kinetically suppressed unless atomic motion is sluggish enough (i.e., the temperature is low enough)38. In HEMGs, however, even within the supercooled liquid region20, atoms have limited translational degrees of freedom, thereby limiting the distance and frequency of atomic diffusion. Consequently, elemental redistributions only take place within the short-to-medium range during CPT. Eventually, the atomic structure has a weak dependence on temperature, which leads to the confined PT. Through a series of isothermal annealing experiments, we determined the characteristic time of the CPT by measuring the decay rate of the exothermic peak. As illustrated in Supplementary Fig. 13, the kinetic time scale of CPT is relatively larger compared to that of conventional PTs10, suggesting that high configurational entropy slows down the kinetics of atomic rearrangements during the transition. Even at temperatures above TPT-s, the CPT process will continue for ≈6.4 × 104 s. This behavior suggests that CPT in HEMGs is essentially kinetically frustrated.
Figure 6 illustrates the difference of PTs between HEMGs and conventional MGs in terms of the potential energy landscape (PEL). In conventional MGs, a typical PT is characterized by a direct transformation from one glass state to another glass state with lower energy and involves atomic rearrangement from short-to-medium range scale. High-entropy effects, however, introduce novel characteristics to this PT scenario. In the case of HEMGs, the increased mixing entropy results in the formation of a deep local minimum of energy, i.e., G0, within the PEL20. When the system is heated beyond the critical transition temperature TTP-s, atoms will undergo continuous rearrangement towards a more stable state, characterized by a successive drop in potential energy. This dynamic is reflected by a decrease in the proportion of alike atomic bonds and a corresponding increase in dislike bond types. Therefore, CPT is mainly controlled by atomic shuffling within the nearest neighbor shell, i.e., the short-range order, while atomic rearrangements at larger length scales are suppressed. The higher-order characteristics and the increased number of available states in HEMGs suggest a more complex and rougher energy landscape after CPT. Thus, the continuous and higher-order PT provides a model system for revealing the intrinsic nature of glass states (e.g., glass transition and glass-forming ability) and the structure-property relationships in MGs. This phenomenon can be phenomenologically described by a modified Landau theory-based model (see details in Supplementary Note 6 and Supplementary Figs. 14, 15).

Coordinates represent different intrinsic structures in configuration spaces.
In this study, we have discovered a PT phenomenon driven by high configurational entropy, which induces elemental redistribution and severe short- to medium-range ordering. Unlike conventional PTs, the confined CPT in HEMGs is characterized by an asymptotic and continuous progression, exhibiting high-order-like attributes of phase transformation. This behavior, facilitated by the enhanced flexibility and adjustability of atomic arrangements in the glass state, affords a wider spectrum of accessible atomic packing configurations, thereby fostering structural adaptability. Significantly, this CPT proceeds without abrupt changes in volume and structure, leading to the discovery of multiple polyamorphous states. This positions the current type of HEMGs as an ideal model system for investigating the fundamental principles underpinning the inherent relationship between structure and properties in glassy materials. Our findings provide insights into the nature of PTs in MGs and opportunities for exploring the rich and complex behavior of these materials.
Methods
Materials preparation
The master alloys used in this study, namely Ti20Zr20Hf20Cu20Ni20, Ti20Zr20Cu20Ni20Nb20, and Ti20Zr20Pd20Cu20Ni20 with an equal atomic ratio, were prepared via vacuum arc melting of high-purity constituent elements. To ensure the chemical homogeneity of the alloys, the synthesized materials were re-melted at least four times. Using a single roller melt-spinning apparatus, 30 μm thick ribbons were produced through the melt-spinning process.
Thermodynamics and properties measurements
Crystallization experiments were carried out in a DSC using alumina pans under a flowing argon atmosphere at a heating rate of 0.17 K s−1. TMDSC was also used to detect the heat flow signals during heating. The TMDSC measurements involved continuous heating of the sample at a rate of 2 K min−1 with a temperature amplitude of 1 K and a cycle time of 40 s.
Considering the slight differences in density between these glasses, the measurement of the absolute density utilizing Archimedes’ method may bring potential errors. Therefore, we utilized two alternative methods to ascertain the relative changes in density during CPT. One method is to determine the density variations according to the power-law relationship between volume and q126,28. The second one is to obtain the density change from the quantitative correlation between volumetric and linear dimensional changes observed during linear thermal expansion measurements29.
Mechanical properties were assessed by continuous stiffness measurement using an MTS DCM (Nano Indenter II) nanoindentation system at a constant strain rate of 0.05 s−1 to a depth of 1000 nm.
Isothermal annealing was conducted on the base glass (G0) at various temperatures (Ta) for different durations (ta). Subsequently, we examined the evolution of exothermic peaks in DSC scans to determine the time scale of CPT. We quantified the area under the exothermic peak associated with the CPT as the exothermic enthalpy \(\varDelta {H}_{{T}_{{{{\rm{a}}}}},{t}_{{{{\rm{a}}}}}}\), and normalized it by the initial peak area ΔHtotal. The decay rate of the exothermic enthalpy associated with CPT against ta can be well described by the Kohlrausch-Williams-Watts (KWW) equation39:
where \({\tau }_{{{{\rm{CPT}}}}}\) represents the characteristic time of CPT and β is the exponential factor.
Structure characterization
Cs-corrected transmission electron microscopy (TEM) and HRTEM (TECNAI F30 STWIN microscopy) were used for structural identification, while EDS mappings were acquired by a JEOL ARM200F TEM under the STEM mode with the Oxford X-maxN 100TLE energy dispersive spectroscopy. For TEM analysis, the samples were thinned to electron transparency using a twin-jet electropolisher in a mixed solution of HNO3:CH4O = 1:3 (in volume ratio). The chemical inhomogeneity of the sample tip was characterized in the laser mode using a Leap-5000XR APT instrument. The optimized parameters, including temperature (50 K), detection rate (0.5%), and pulse rate (200 kHz), were used to obtain the APT data, which were reconstructed using the commercial software package CAMECA.
We conducted in-situ heating synchrotron high-energy X-ray diffraction (HEXRD) experiments at the 11-ID-C beamline of the Advanced Photon Source (APS), Argonne National Laboratory (ANL), to study the structural evolution of our samples at a heating rate of 0.17 K s−1. X-ray wavelength was set to 0.11798 Å (photon energy ≈105 keV). To prevent the oxidation of samples during heating, we used a flowing Ar atmosphere. Two-dimensional X-ray diffraction patterns were integrated using the FIT2D software package, and the total structure factor, S(q), was obtained using standard procedures described in ref. 40. We estimated the FWHMs of S(q) by fitting the data with a Voigt function. Using the program PDFgetX2, we calculated the pair distribution function, G(r) versus r, where r represents the distance between atomic pairs, via the Fourier transform of q[S(q)-1].
At the China Spallation Neutron Source (CSNS), we conducted SANS experiments to investigate structural heterogeneity at the nanoscale in the amorphous phase, using a neutron wavelength range of 1–10 Å. To prepare the SANS samples, we overlapped the ribbons.
To further examine the local atomic structure, we performed EXAFS spectroscopy in the transmission mode of Cu, Ni, and Zr K-edges of all samples. These experiments were conducted at beamline BL01C2, in the Synchrotron Radiation Research Center (SRRC) located in Taiwan. The EXAFS spectra χ(k)k3 were extracted from the total absorption spectra after pre-edge and post-edge background subtractions using Athena and Artemis software packages. The Fourier-transformed real-space data were then fitted in k-space and r-space using existing paths in the configuration from ab initio.
Reverse Monte Carlo modeling and structural analysis
We utilized the RMCProfile software package for RMC simulations41. The simulation commenced with a configuration of 20,000 atoms, randomly distributed within a cubic box for the G0 glass. Atomic number densities (ρ) obtained from experimental measurements were used, namely ρ equals 0.0569, 0.0573, 0.0581 and 0.0586 atom Å−3 for the typical glass states G0, Gm2, Gm4, and Gn, respectively. The simulation process involves an iterative procedure that randomly modifies the atomic coordinates, with the objective of reducing the difference between the experimental and simulation S(q) and EXAFS k-space data.
The simulation results were visualized by Atomeye42. The structural analysis data were derived from 20 RMC configurations by 20 independent simulations. Local excess entropy33,43 was calculated by the Ovito software44. Voronoi tessellation method45 was adopted to analyze local atomic structures. Top 10 kinds of polyhedra for each centered atom were analyzed. The Warren-Cowley chemical short-range order (CSRO) parameter31 was used to evaluate the degree of chemical ordering.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding author upon request. Source data are provided with this paper.
References
Katayama, Y. et al. A first-order liquid-liquid phase transition in phosphorus. Nature 403, 170–173 (2000).
Sastry, S. & Austen Angell, C. Liquid-liquid phase transition in supercooled silicon. Nat. Mater. 2, 739–743 (2003).
Hemley, R. J., Mao, H. K., Bell, P. M. & Mysen, B. O. Raman spectroscopy of SiO2 glass at high pressure. Phys. Rev. Lett. 57, 747–750 (1986).
Grimsditch, M. Polymorphism in amorphous SiO2. Phys. Rev. Lett. 52, 2379–2381 (1984).
Kobayashi, M. & Tanaka, H. The reversibility and first-order nature of liquid-liquid transition in a molecular liquid. Nat. Commun. 7, 13438 (2016).
Tielens, A. G. G. M. The Physics and Chemistry of the Interstellar Medium (Cambridge Univ. Press, Cambridge, 2005).
Mishima, O. Reversible first-order transition between two H2O amorphs at ∼0.2 GPa and ∼135 K. J. Chem. Phys. 100, 5910–5912 (1994).
Mishima, O., Calvert, L. D. & Whalley, E. An apparently first-order transition between two amorphous phases of ice induced by pressure. Nature 314, 76–78 (1985).
Lan, S. et al. Hidden amorphous phase and reentrant supercooled liquid in Pd-Ni-P metallic glasses. Nat. Commun. 8, 14679 (2017).
Shen, J. et al. The kinetics of reentrant glass transition in metallic liquids. Acta Mater. 244, 118554 (2023).
Du, Q. et al. Reentrant glass transition leading to ultrastable metallic glass. Mater. Today 34, 66–77 (2020).
Shen, J. et al. Metallic glacial glass formation by a first-order liquid-liquid transition. J. Phys. Chem. Lett. 11, 6718–6723 (2020).
Dong, W. X. et al. In situ neutron scattering studies of a liquid-liquid phase transition in the supercooled liquid of a Zr-Cu-Al-Ag glass-forming alloy. Appl. Phys. Lett. 118, 191901 (2021).
Zeng, Q. S. et al. Anomalous compression behavior in lanthanum/cerium-based metallic glass under high pressure. Proc. Natl Acad. Sci. USA 104, 13565–13568 (2007).
Li, G., Wang, Y. Y., Liaw, P. K., Li, Y. C. & Liu, R. P. Electronic structure inheritance and pressure-induced polyamorphism in lanthanide-based metallic glasses. Phys. Rev. Lett. 109, 125501 (2012).
Zhang, X. et al. Pressure-induced local structural crossover in a high-entropy metallic glass. Phys. Rev. B 105, 224201 (2022).
Jiang, J. et al. Decoupling between calorimetric and dynamical glass transitions in high-entropy metallic glasses. Nat. Commun. 12, 3843 (2021).
Zhang, L. T. et al. Sluggish dynamics of homogeneous flow in high-entropy metallic glasses. Scr. Mater. 214, 114673 (2022).
Duan, Y. J. et al. Intrinsic correlation between the fraction of liquidlike zones and the β relaxation in high-entropy metallic glasses. Phys. Rev. Lett. 129, 175501 (2022).
Yang, M. et al. Configurational entropy effects on glass transition in metallic glasses. J. Phys. Chem. Lett. 13, 7889–7897 (2022).
Yang, M. et al. High thermal stability and sluggish crystallization kinetics of high-entropy bulk metallic glasses. J. Appl. Phys. 119, 245112 (2016).
Luan, H. W. et al. High-entropy induced a glass-to-glass transition in a metallic glass. Nat. Commun. 13, 2183 (2022).
Zhang, X. et al. Highly variable chemical short-range order in a high-entropy metallic glass. Mater. Today Phys. 27, 100799 (2022).
Yang, Q., Yang, X. M., Zhang, T., Liu, X. W. & Yu, H. B. Structure and entropy control of polyamorphous transition in high-entropy metallic glasses. Acta Mater. 266, 119701 (2024).
Cao, Y. H. et al. Unravel unusual hardening behavior of a Pd-Ni-P metallic glass in its supercooled liquid region. Appl. Phys. Lett. 118, 121902 (2021).
Ma, D., Stoica, A. D. & Wang, X. L. Power-law scaling and fractal nature of medium-range order in metallic glasses. Nat. Mater. 8, 30–34 (2009).
Zeng, Q. et al. General 2.5 power law of metallic glasses. Proc. Natl Acad. Sci. USA 113, 1714–1718 (2016).
Yavari, A. R. et al. Excess free volume in metallic glasses measured by X-ray diffraction. Acta Mater. 53, 1611–1619 (2005).
Russew, K. & Sommer, F. Length and density changes of amorphous Pd40Cu30Ni10P20 alloys due to structural relaxation. J. Non-Cryst. Solids 319, 289–296 (2003).
Glatter, O. Determination of particle-size distribution functions from small-angle scattering data by means of the indirect transformation method. J. Appl. Crystallogr. 13, 7–11 (1980).
Cowley, J. M. An approximate theory of order in alloys. Phys. Rev. 77, 669–675 (1950).
Spieckermann, F. et al. Structure-dynamics relationships in cryogenically deformed bulk metallic glass. Nat. Commun. 13, 127 (2022).
Şopu, D., Moitzi, F., Mousseau, N. & Eckert, J. An atomic-level perspective of shear band formation and interaction in monolithic metallic glasses. Appl. Mater. Today 21, 100828 (2020).
Castellero, A., Moser, B., Uhlenhaut, D. I., Dalla Torre, F. H. & Löffler, J. F. Room-temperature creep and structural relaxation of Mg-Cu-Y metallic glasses. Acta Mater. 56, 3777–3785 (2008).
Sheng, H. W., Luo, W. K., Alamgir, F. M., Bai, J. M. & Ma, E. Atomic packing and short-to-medium-range order in metallic glasses. Nature 439, 419–425 (2006).
Miracle, D. B. A structural model for metallic glasses. Nat. Mater. 3, 697–702 (2004).
Yang, M. et al. Unusual relation between glass-forming ability and thermal stability of high-entropy bulk metallic glasses. Mater. Res. Lett. 6, 495–500 (2018).
Shen, J., Sun, Y. H., Orava, J., Bai, H. Y. & Wang, W. H. Liquid-to-liquid transition around the glass-transition temperature in a glass-forming metallic liquid. Acta Mater. 225, 117588 (2022).
Qiao, J. C. & Pelletier, J. M. Kinetics of structural relaxation in bulk metallic glasses by mechanical spectroscopy: determination of the stretching parameter βKWW. Intermetallics 28, 40–44 (2012).
Waseda, Y. The Structure of Non-crystalline Materials: Liquids and Amorphous Solids (McGraw-Hill Press, New York 1980).
McGreevy, R. L. & Pusztai, L. Reverse monte Carlo Simulation: a new technique for the determination of disordered structures. Mol. Simula. 1, 359–367 (1988).
Li, J. AtomEye: an efficient atomistic configuration viewer. Model. Simul. Mater. Sci. Eng. 11, 173–177 (2003).
Piaggi, P. M. & Parrinello, M. Entropy based fingerprint for local crystalline order. J. Chem. Phys. 147, 114112 (2017).
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the open visualization tool. Model. Simul. Mater. Sci. Eng. 18, 015012 (2010).
Finney, J. L. Random packings and the structure of simple liquids. I. The geometry of random close packing. Proc. R. Soc. A Math. Phys. 319, 479–493 (1970).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Nos. 52071024, 52271003, and 52101188), the Funds for Creative Research Groups of NSFC (51921001), Projects of International Cooperation and Exchanges of NSFC (51961160729, and 52061135207), and 111 Project (BP0719004), Program for Changjiang Scholars and Innovative Research Team in University of China (IRT_14R05). Q.S.Z. acknowledges the support from the Shanghai Key Laboratory of Material Frontiers Research in Extreme Environments (MFree), China (No. 22dz2260800) and the Shanghai Science and Technology Committee, China (No. 22JC1410300). The synchrotron XRD was performed at the beamline 11-ID-C, APS, USA. APS is supported by the DOE Office of Science (DE-AC02-06CH11357).
Author information
Authors and Affiliations
Contributions
X.J.L. and Z.P.L. supervised the project. Y.H.C. carried out materials fabrication, mechanical characterizations, thermodynamical measurements, and RMC modeling. F.Z., H.B.L. and Q.S.Z. performed the synchrotron radiation X-ray experiments, and M.Y. analyzed the data. Q.D. participated in data interpretation and manuscript correction. F.K.C. and Y.J.Z. performed the TEM characterization. S.W.C. conducted EXAFS experiments. Y.B.K. conducted the SANS experiments. Y.H.C., X.J.L. and Z.P.L. wrote and revised the manuscript. All authors including Y.W., H.W., S.H.J. and X.B.Z. discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks J.C. Qiao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, Y., Yang, M., Du, Q. et al. Continuous polyamorphic transition in high-entropy metallic glass. Nat Commun 15, 6702 (2024). https://doi.org/10.1038/s41467-024-51080-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51080-8
- Springer Nature Limited