

Abstract
In C. elegans mechanisms by which peripheral organs relay internal state information to the nervous system remain unknown, although strong evidence suggests that such signals do exist. Here we report the discovery of a peptide of the ancestral insulin superfamily called INS-7 that functions as an enteroendocrine peptide and is secreted from specialized cells of the intestine. INS-7 secretion is stimulated by food withdrawal, increases during fasting and acts as a bona fide gut-to-brain peptide that attenuates the release of a neuropeptide that drives fat loss in the periphery. Thus, INS-7 functions as a homeostatic signal from the intestine that gates the neuronal drive to stimulate fat loss during food shortage. Mechanistically, INS-7 functions as an antagonist at the canonical DAF-2 receptor and functions via FOXO and AMPK signaling in ASI neurons. Phylogenetic analysis suggests that INS-7 bears greater resemblance to members of the broad insulin/relaxin superfamily than to conventional mammalian insulin and IGF peptides. The discovery of an endogenous insulin antagonist secreted by specialized intestinal cells with enteroendocrine functions suggests unexpected and important properties of the intestine and its role in directing neuronal functions.
Similar content being viewed by others


Introduction
The central nervous system is known to play a major role in governing systemic lipid homeostasis across species1,2,3. It is also now understood that endocrine hormones signal from organs in the periphery to relay fed- and fasted state information across the body4,5, including to the nervous system6,7. For example, the intestine has emerged as a preeminent sensory and metabolic organ that relays internal state information via gut hormones to the brain and other important organs in the body8,9,10,11. It is plausible that many additional endocrine signals relaying interoceptive information to the nervous system remain undiscovered. In the roundworm C. elegans, the nervous system plays important roles in regulating fat homeostasis in the intestine12, the predominant organ in which lipids are stored and metabolized13. Several sensory neurons and the circuits in which they operate are known to regulate the rate and extent of fat utilization. Because these neurons detect and respond to distinct sources of sensory information from the environment, it has become possible to ascribe roles for individual sensory modalities in regulating lipid homeostasis. Previous work from our group has shown that oxygen-sensing (in the normoxic range) via the URX and BAG neurons14, population density-sensing via the ADL neurons15, and bacterial food sensing via the serotonergic ADF neurons strongly influence lipid stores16. These and other studies17,18 have shown that C. elegans calibrates lipid metabolism to the sensory environment in order to optimize behavior, physiology, and lifespan.
An interesting feature of the C. elegans nervous system is that sensory neurons do not directly innervate the intestine16,19 although some neurons do access the coelomic fluid, allowing a conduit by which information can transfer across tissues. To address the question of how sensory information from the nervous system is relayed to the intestine, in previous efforts we had conducted a genetic screen that uncovered a brain-to-gut neuroendocrine peptide called FLP-7, the ancestral ortholog of the mammalian tachykinin family of peptides. The FLP-7 neuropeptide is secreted from neurons and is detected by the G protein-coupled receptor NPR-22, ortholog of the mammalian Neurokinin 2 Receptor (NK2R), which resides in the intestine in which it is necessary and sufficient to trigger fat loss20. We developed an assay to measure FLP-7 secretion in living animals and showed that FLP-7 is released by neurosecretory cells called ASI in proportion to fluctuations in the functions of serotonergic and octopaminergic neurons20. Additionally, FLP-7 secretion is also modulated by the oxygen-sensing URX neurons and the population density-sensing ADL neurons (Supplementary Fig. 1a–d). Thus, the FLP-7/NPR-22 neuroendocrine axis represents the final common brain-to-gut pathway by which the sensory nervous system relays information to the intestine for the regulation of lipid metabolism12.
Over the course of these studies, we contemplated the possibility that the intestine relayed metabolic information back to the nervous system. Tantalizing evidence from us and others suggested that the C. elegans intestine communicates with distinct organs including regulation of pathogen avoidance21, avoidance of aversive stimuli22,23, regulation of rhythmic behaviors24, serotonin production and signaling25, and oxygen sensing26. However, no clear gut-to-brain molecules regulating metabolic functions had been found, and it was not clear whether the C. elegans gut itself secreted enteroendocrine peptides. One barrier to identifying such molecules was the lack of readouts of neuronal function and output that are amenable to genetic screens. To test whether the FLP-7 secretion assay could prove to be a metabolically relevant experimental handle that could circumvent these limitations, we measured the extent to which FLP-7 secretion from ASI neurons (hereafter called FLP-7ASI) could be modulated by depleting intestinal fat stores. Inactivation of genes across distinct lipid synthesis pathways previously shown to deplete intestinal fats26,27,28 (pod-2, acyl CoA Carboxylase; sbp-1, Sterol Regulatory Element Binding Protein; elo-2, Fatty Acid Elongase) resulted in increased steady-state FLP-7ASI secretion (Supplementary Fig. 1e). Prompted by this observation, we undertook an investigation to identify and define the molecular features underlying gut-to-brain information relay.
Results
Discovery of INS-7 as a gut peptide
We reasoned that in the context of lipid metabolism, a molecule that relays internal state information might originate in the intestine since it is the major somatic depot for stored lipids in C. elegans. To find such a signal, we conducted an intestine-specific RNAi-mediated screen of the family of genes encoding small peptides for alterations in FLP-7 secretion from ASI neurons (Fig. 1a). The gene encoding a peptide called ins-7 emerged as the most potent hit from the screen (Fig. 1b, c); a mutant allele tm1907 that lacks the bulk of the gene including the F peptide domain, and our newly generated CRISPR null allele ssr1532 (Fig. 1d–f) recapitulated the effect of the RNAi: absence of ins-7 increased FLP-7ASI secretion nearly 2-fold (Fig. 1g, h). INS-7 belongs to the broad insulin/relaxin superfamily of peptides; in C. elegans there are 40 members (10 in humans) that are thought to be distinguished by their developmental stage- and tissue-specific expression patterns29,30. C. elegans ins-7 has been reported to be one of the few ins genes expressed selectively in adults30; a second gene called ins-19 that has adult-specific expression did not yield a phenotype in the FLP-7ASI secretion assay.

a Model depicting the C. elegans tachykinin neuroendocrine axis and the hypothesized intestine-to-neuron signal. b Representative images of sid-1;Pvha-6::sid-1 animals bearing integrated ASI::FLP-7mCherry and CLM::GFP transgenes treated with vector or ins-7 RNAi. Left panels, GFP expression in coelomocytes; center panels, secreted FLP-7mCherry in coelomocytes; right panels, merge. Scale bar, 5 μm. c The intensity of FLP-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression. Assay validation parameters are given in Supplementary Fig. 1a, b. Data are expressed as a percentage of the normalized FLP-7mCherry fluorescence intensity of animals treated with vector RNAi ± SEM. n = 17 for vector RNAi, n = 16 for ins-7 RNAi. **p = 0.0064 by unpaired t-test (two-tailed). d The strategy of CRISPR-Cas9 mediated genome editing, depicting the genomic region of ins-7 and locations of the Cas9 cut sites for ins-7(ssr1532). Location of introduced EcoRI restriction site (Green) and double-stop codons (Red) after cleavage and repair are as indicated. The deletion region of the ins-7(tm1907) allele is marked. e qPCR of ins-7 mRNA in the indicated strains. Pan-actin (actin-1, −3, −4) mRNA was used as a control. Data are presented as fold change relative to wild-type ± SEM. n = 3 biological replicates for wild-type and ins-7(ssr1532). **p = 0.0029 by unpaired t-test (two-tailed). f Structure prediction for INS-7 peptides in wild-type or ins-7(ssr1532) animals by AlphaFold (DeepMind)89,90. The order of amino acids and specific regions are labeled; the ins-7(ssr1532) null allele is a truncation of the ins-7 gene that results in the expression of only the first 18 of 116 amino acids. g Representative images of wild-type, ins-7(tm1907) and ins-7(ssr1532) animals bearing integrated ASI::FLP-7mCherry and CLM::GFP transgenes. Left panels, GFP expression in coelomocytes; center panels, secreted FLP-7mCherry in coelomocytes; right panels, merge. Scale bar, 5 μm. h The intensity of FLP-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression for each genotype. Data are expressed as a percentage of the normalized FLP-7mCherry fluorescence intensity of wild-type animals ± SEM. n = 29, 18, 30 from left to right. p values were calculated using one-way ANOVA and Tukey’s post hoc test. Wild-type vs. ins-7(tm1907); ***p < 0.0001. Wild-type vs. ins-7(ssr1532); ***p < 0.0001. Source data are provided as a Source Data file.
An extrachromosomal ins-7::GFP reporter line and an endogenous transcriptional GFP reporter we generated revealed robust expression restricted to the first quartet of intestinal cells (Fig. 2a and Supplementary Fig. 2). Called INT1, these anatomically specialized cells define the anterior-most section of the upper intestine, just posterior to the terminal bulb of the pharynx31. This intriguingly restricted expression pattern is in stark contrast to the many metabolic genes reported by us and others, which are broadly expressed throughout all the cells of the intestine (INT1-9, 20 cells)16,27,28,32. While the intestine-specific RNAi experiment suggested the necessity of ins-7 expression in INT1 cells, to determine whether ins-7 expression in INT1 cells is sufficient to rescue the aberrantly increased FLP-7ASI secretion, we generated transgenic rescue lines in which ins-7 gene expression was restored in ins-7(ssr1532) null mutants under the ins-7 endogenous promoter, the pan-intestinal promoter Pvha-6, as well as a newly developed INT1-specific promoter, Pges-1ΔB (Fig. 2b–e). We found that heterologous ins-7 expression in INT1 cells alone was sufficient to confer complete rescue of FLP-7ASI secretion to the same extent as restoring ins-7 expression more broadly across the intestine (Fig. 2b–e).

a Fluorescent image of a transgenic animal bearing the Pins-7::GFP transgene. The white arrowhead indicates GFP expression in INT1 cells. Lower panel, DIC merge; scale bar, 100 μm. Pins-7::GFP expression pattern was examined in three independent experiments with similar results. b Representative images of wild-type and ins-7(ssr1532) animals bearing integrated ASI::FLP-7mCherry and CLM::GFP transgenes, with the indicated rescuing transgenes. NT non-transgenic, TG transgenic. Upper panels, GFP expression in coelomocytes; middle panels, secreted FLP-7mCherry in coelomocytes; lower panels, merge. Scale bar, 5 μm. c–e The intensity of FLP-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression for each genotype. Data are expressed as a percentage of the normalized FLP-7mCherry fluorescence intensity of wild-type animals ± SEM. p values were calculated using one-way ANOVA and Sidak’s post hoc test. n = 21, 22, 28, 21 from left to right in (c). Wild-type vs. ins-7(ssr1532); *p = 0.0174. Wild-type vs. Pins-7::ins-7 NT; *p = 0.0183. ins-7(ssr1532) vs. Pins-7::ins-7 TG; **p = 0.0039. Pins-7::ins-7 NT vs. Pins-7::ins-7 TG; **p = 0.0037. n = 14, 15, 16, 13 from left to right in (d). Wild-type vs. ins-7(ssr1532); *p = 0.0346. Wild-type vs. Pvha-6::ins-7 NT; **p = 0.0084. Pvha-6::ins-7 NT vs. Pvha-6::ins-7 TG; *p = 0.0205. n = 25, 25, 26, 21 from left to right in (e). Wild-type vs. ins-7(ssr1532); *p = 0.0156. Wild-type vs. INT1::ins-7 NT; ***p = 0.0006. INT1::ins-7 NT vs. INT1::ins-7 TG; *p = 0.0213. f The intensity of FLP-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression. Data are expressed as a percentage of the normalized FLP-7mCherry fluorescence intensity of animals treated with vector RNAi ± SEM. n = 22 for vector RNAi, n = 24 for ins-7 RNAi. ***p < 0.0001 by unpaired t-test (two-tailed). g The intensity of FLP-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression for both wild-type and INT1::ins-7 TG animals. Data are expressed as a percentage of the normalized FLP-7mCherry fluorescence intensity of wild-type animals ± SEM. n = 19 for wild-type, n = 24 for INT1::ins-7OX. ***p < 0.0001 by unpaired t-test (two-tailed). Source data are provided as a Source Data file.
Further, INT1-specific ins-7 RNAi increased FLP-7ASI secretion (Fig. 2f and Supplementary Fig. 1f), and overexpression of ins-7 in INT1 cells led to suppression of FLP-7ASI secretion (Fig. 2g), suggesting that INS-7 functions as an instructive cue from the intestine that is necessary and sufficient to inhibit FLP-7ASI secretion from neurons. INT1 cells were first defined in the context of the developmental biology of the C. elegans intestine: they have shorter microvilli than the INT2-9 conventional epithelial enterocytes, not unlike the enteroendocrine, goblet and immune M cells of the mammalian intestinal epithelium31,33,34. Their location as the anterior-most cells of the intestine would also suggest privileged anatomic access to incoming nutrients.
We wished to define additional phenotypes of ins-7. In previous studies, we had shown that stimulating FLP-7 secretion from ASI neurons activates its cognate receptor NPR-22 in the intestine, whose activity governs fat loss and increases energy expenditure in the intestine via transcriptional activation of the triglyceride lipase ATGL-116,20,35. Thus, any condition that increases FLP-7ASI secretion would be predicted to trigger the hydrolysis of fat stored in the intestine via ATGL-1, accompanied by increased energy expenditure. Accordingly, we found that ins-7 null mutants that display increased FLP-7ASI secretion (Fig. 1g, h) had significantly decreased fat stores (Fig. 3a and Supplementary Fig. 3a). This decrease in fat stores was fully dependent on the presence of the flp-7 gene, because ins-7;flp-7 mutants suppress the effect of ins-7 on intestinal fat loss (Fig. 3a). Next, we selectively inactivated flp-7 in the ASI neurons via antisense inhibition and found that the fat phenotype of ins-7 mutants requires the presence of flp-7 in the ASI neurons (Fig. 3b). Further, ASI-specific flp-7 rescue in the ins-7;flp-7 double mutants was sufficient to drive fat loss to the extent in the ins-7 mutants (Fig. 3c). Thus, the fat phenotype of ins-7 mutants is entirely dependent on flp-7 from ASI neurons. The decreased fat stores in the intestine were corroborated by the expected increase in maximal energy expenditure in the ins-7 mutants as judged by oxygen consumption, which was also dependent on flp-7 (Fig. 3d, e). As predicted, the decreased intestinal fat stores of ins-7 nulls were dependent on atgl-1 induction (Fig. 3f, g), and this increase in atgl-1 expression was fully dependent on the presence of the flp-7 gene (Fig. 3h). Further, RNAi-mediated inactivation of atgl-1 abrogated the ins-7 fat phenotype (Fig. 3i). The effects of ins-7 on intestinal fat stores were not dependent on food intake or locomotion, which were not altered in ins-7 mutants (Supplementary Fig. 3b–d). To corroborate a role for INT1 cells in INS-7-mediated fat loss, we generated an independent set of ins-7 transgenic rescue lines using the promoters described in Fig. 2 and measured intestinal fat. We observed complete restoration of the decreased intestinal fat stores of ins-7 null mutants in all cases, again suggesting that ins-7 functions from INT1 cells to regulate fat stores across the broader intestine (Fig. 3j–l). Finally, overexpression of ins-7 in INT1 cells resulted in a small but significant increase in fat storage throughout the intestine (Fig. 3m).

a wild-type, ins-7(ssr1532), flp-7(ok2625), and ins-7(ssr1532);flp-7(ok2625) animals were fixed and stained with Oil Red O. Fat content was quantified for each genotype and expressed as a percentage of wild-type animals ± SEM. n = 16, 20, 18, 20 from left to right. p values were calculated using one-way ANOVA and Sidak’s post hoc test. Wild-type vs. ins-7(ssr1532); ***p < 0.0001. ins-7(ssr1532) vs. ins-7(ssr1532);flp-7(ok2625); ***p < 0.0001. b, c Wild-type, ins-7(ssr1532) and ins-7(ssr1532);flp-7(ok2625) animals with the indicated transgenes were fixed and stained with Oil Red O. NT non-transgenic, TG transgenic. Fat content was quantified for each genotype and expressed as a percentage of wild-type animals ± SEM. p values were calculated using one-way ANOVA and Sidak’s post hoc test. n = 20, 20, 20, 18 from left to right in (b). Wild-type vs. ins-7(ssr1532); wild-type vs. ins-7(ssr1532);ASI::flp-7 antisense NT; ins-7(ssr1532) vs. ins-7(ssr1532);ASI::flp-7 antisense TG; ins-7(ssr1532);ASI::flp-7 antisense NT vs. ins-7(ssr1532);ASI::flp-7 antisense TG; ***p < 0.0001, respectively. n = 20, 20, 20, 20 from left to right in (c). Wild-type vs. ins-7(ssr1532); wild-type vs. ins-7(ssr1532);flp-7(ok2625);ASI::flp-7 TG; ins-7(ssr1532) vs. ins-7(ssr1532);flp-7(ok2625);ASI::flp-7 NT; ins-7(ssr1532);flp-7(ok2625);ASI::flp-7 NT vs. ins-7(ssr1532);flp-7(ok2625);ASI::flp-7 TG; ***p < 0.0001, respectively. d, e Oxygen consumption rate (OCR) of wild-type, ins-7(ssr1532), flp-7(ok2625), and ins-7(ssr1532);flp-7(ok2625) animals. Basal OCR was quantified before the addition of FCCP (50 mM), and maximal OCR was measured following FCCP stimulation. Data are presented as pmol/min/worm ± SEM. The n numbers for each genotype at each time point in (d) are given in the Source Data file. n = 36, 21, 24, 36 from left to right in (e) Basal. n = 38, 27, 19, 19 from left to right in (e) Maximal. p values were calculated using one-way ANOVA and Sidak’s post hoc test. Wild-type vs. ins-7(ssr1532); *p = 0.0197. ins-7(ssr1532) vs. ins-7(ssr1532);flp-7(ok2625); **p = 0.0014. f Representative images of wild-type and ins-7(ssr1532) animals bearing the integrated Patgl-1::GFP transgene. Scale bar, 100 μm. g Patgl-1::GFP fluorescence was measured and expressed as a percentage of wild-type animals ± SEM. n = 29 for wild-type, n = 30 for ins-7(ssr1532). ***p < 0.0001 by unpaired t-test (two-tailed). h qPCR of atgl-1 mRNA in the indicated strains. act-1 mRNA was used as a control. Data are presented as fold change relative to wild-type ± SEM. n = 3 biological replicates for wild-type, ins-7(ssr1532), flp-7(ok2625) and ins-7(ssr1532);flp-7(ok2625). p values were calculated using one-way ANOVA and Sidak’s post hoc test. wild-type vs. ins-7(ssr1532); ***p < 0.0001. ins-7(ssr1532) vs. ins-7(ssr1532);flp-7(ok2625); ***p < 0.0001. i Wild-type and ins-7(ssr1532) animals treated with vector or atgl-1 RNAi were fixed and stained with Oil Red O. Fat content was quantified for each genotype and condition and expressed as a percentage of wild-type animals treated with vector RNAi ± SEM. n = 30, 26, 34, 26 from left to right. p values were calculated using two-way ANOVA and Sidak’s post hoc test. wild-type on vector RNAi vs. ins-7(ssr1532) on vector RNAi; ***p < 0.0001. wild-type on atgl-1 RNAi vs. ins-7(ssr1532) on atgl-1 RNAi; *p = 0.0482. ins-7(ssr1532) on vector RNAi vs. ins-7(ssr1532) on atgl-1 RNAi; ***p < 0.0001. j–l Wild-type and ins-7(ssr1532) animals with the indicated rescuing transgenes were fixed and stained with Oil Red O. NT non-transgenic; TG transgenic. Fat content was quantified for each genotype and expressed as a percentage of wild-type animals ± SEM. p values were calculated using one-way ANOVA and Tukey’s post hoc test. n = 26, 29, 20, 21 from left to right in (j). Wild-type vs. ins-7(ssr1532); wild-type vs. Pins-7::ins-7 NT; ins-7(ssr1532) vs. Pins-7::ins-7 TG; Pins-7::ins-7 NT vs. Pins-7::ins-7 TG; ***p < 0.0001, respectively. n = 23, 20, 13, 7 from left to right in (k). Wild-type vs. ins-7(ssr1532); wild-type vs. Pvha-6::ins-7 NT; ins-7(ssr1532) vs. Pvha-6::ins-7 TG; Pvha-6::ins-7 NT vs. Pvha-6::ins-7 TG; ***p < 0.0001, respectively. n = 23, 20, 10, 9 from left to right in (l). Wild-type vs. ins-7(ssr1532); wild-type vs. INT1::ins-7 NT; ins-7(ssr1532) vs. INT1::ins-7 TG; INT1::ins-7 NT vs. INT1::ins-7 TG; ***p < 0.0001, respectively. m Fat content was quantified for both wild-type and INT1::ins-7OX animals and expressed as a percentage of wild-type animals ± SEM. n = 20 for wild-type and INT1::ins-7OX. ***p = 0.0002 by unpaired t-test (two-tailed). Source data are provided as a Source Data file.
Mechanism of INS-7 action
Our data support a model in which ins-7 functions in the INT1 cells of the intestine to negatively regulate FLP-7ASI secretion from neurons, to regulate fat storage throughout the intestine (Fig. 4a). This model of interorgan communication led us to consider the mechanism of INS-7 action. Phylogenetic analysis of the ins-7 gene suggests that it belongs to an entirely different clade than that of the conventional mammalian insulin genes, IGF1 and IGF2, which encode proteins that function as agonist ligands for the insulin and IGF receptors36 (Fig. 4b). To investigate the mechanism of ins-7 action, we examined its relationship to daf-2, the sole insulin receptor of C. elegans37 which single-cell RNAseq efforts reveal to be clearly expression in ASI neurons38. Using the canonical e1370 allele, we found that daf-2 mutants had a dramatic reduction in FLP-7ASI secretion, a phenotype in opposition to that of ins-7 and ins-7;daf-2 mutants phenocopied daf-2 (Fig. 4c, d). Absence of DAF-16/FOXO, the major downstream target of insulin signaling, did not appreciably alter FLP-7ASI secretion, however it completely suppressed the effect of the daf-2 mutation (Fig. 4c, d). It was possible that the daf-2 phenotype of reduced FLP-7ASI secretion resulted from daf-2 expression elsewhere in the nervous system. To address this concern, we conducted antisense inhibition of daf-2 in ASI neurons alone, which phenocopied the global daf-2 mutation (Fig. 4e, f). To prevent the RNAi from spreading to other neurons39, we also performed the daf-2 antisense experiments in a sid-1(qt9) background and found a similar phenotype to that observed in the wild-type background (Fig. 4g). Thus, the loss of daf-2ASI reduced FLP-7ASI secretion. In addition, ASI-specific transgenic rescue of daf-2 restored FLP-7 secretion to wild-type levels (Fig. 4h, i). Together, these data were most consistent with ins-7 functioning in opposition to daf-2, that is, as an endogenous antagonist to the DAF-2 receptor in ASI neurons. Previous studies of local effects of INS-7 in the nervous system have also defined its action at DAF-2 as antagonistic40, although it may function as an agonist in the periphery41,42 (see “Discussion” section).

a Model depicting the role of INT1-expressed INS-7 in modulating FLP-7 secretion from ASI neurons. INS-7 is secreted from INT1 cells, which inhibits FLP-7ASI secretion. FLP-7 acts on INT2-9, where the majority of fat is stored. b Phylogenetic tree of INS-7 and members of the human Insulin/Relaxin superfamily by Clustal Omega91. The values after each protein name indicate the bootstrap values. c Representative images of wild-type, ins-7(ssr1532), daf-2(e1370), ins-7(ssr1532);daf-2(e1370), daf-16(mu86), and daf-2(e1370);daf-16(mu86) animals bearing integrated ASI::FLP-7mCherry and CLM::GFP transgenes. Upper panels, GFP expression in coelomocytes; middle panels, secreted FLP-7mCherry in coelomocytes; lower panels, merge. Scale bar, 5 μm. d The intensity of FLP-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression for each genotype. Data are expressed as a percentage of the normalized FLP-7mCherry fluorescence intensity of wild-type animals ± SEM. n = 30, 24, 35, 24, 18, 23 from left to right. p values were calculated using one-way ANOVA and Dunnett’s T3 post hoc test. Wild-type vs. ins-7(ssr1532); wild-type vs. daf-2(e1370); wild-type vs. ins-7(ssr1532);daf-2(e1370); ins-7(ssr1532) vs. daf-2(e1370); ins-7(ssr1532) vs. ins-7(ssr1532);daf-2(e1370); daf-2(e1370) vs. daf-16(mu86); daf-2(e1370) vs. daf-2(e1370);daf-16(mu86); ***p < 0.0001, respectively. e Representative images of wild-type FLP-7ASI animals bearing antisense-mediated inactivation of daf-2 expression in ASI neurons using the str-3 promoter. NT non-transgenic, TG transgenic. Scale bar, 5 μm. f, g The intensity of FLP-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression for each genotype. Data are expressed as a percentage of the normalized FLP-7mCherry fluorescence intensity of wild-type or sid-1(qt9) animals ± SEM. p values were calculated using one-way ANOVA and Dunnett’s T3 post hoc test. n = 23, 24, 22 from left to right in (f). Wild-type vs. ASI::daf-2 antisense TG; *p = 0.0136. ASI::daf-2 antisense NT vs. ASI::daf-2 antisense TG; **p = 0.0026. n = 16, 16, 10 from left to right in (g). sid-1(qt9) vs. sid-1(qt9);ASI::daf-2 antisense TG; ***p = 0.0003. sid-1(qt9);ASI::daf-2 antisense NT vs. sid-1(qt9);ASI::daf-2 antisense TG; ***p < 0.0001. h Representative images of wild-type and daf-2(e1370) animals bearing integrated ASI::FLP-7mCherry and CLM::GFP with the indicated rescuing transgenes. NT non-transgenic, TG transgenic. Scale bar, 5 μm. i The intensity of FLP-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression for each genotype. Data are expressed as a percentage of the normalized FLP-7mCherry fluorescence intensity of wild-type animals ± SEM. n = 27, 28, 28, 28 from left to right. p values were calculated using one-way ANOVA and Dunnett’s T3 post hoc test. Wild-type vs. daf-2(e1370); ***p < 0.0001. wild-type vs. daf-2(e1370);ASI::daf-2 NT; ***p < 0.0001. daf-2(e1370) vs. daf-2(e1370);ASI::daf-2 TG; ***p = 0.0008. daf-2(e1370);ASI::daf-2 NT vs. daf-2(e1370);ASI::daf-2 TG; ***p = 0.0002. Source data are provided as a Source Data file.
As suggested by our data, we tested the hypothesis that INS-7 acts as an antagonist for neuronal DAF-2 by conducting a direct test of DAF-2 function: we examined DAF-16 localization within the ASI neurons in various genetic contexts. DAF-16 activity as measured by the ratio of its cytoplasmic-to-nuclear localization (abbreviated as C:N ratio) has long been established as an accurate and sensitive hallmark of DAF-2 function43,44,45. In wild-type well-fed animals (in which ins-7 and daf-2 are present), DAF-16 resides predominantly in the cytoplasm, such that the DAF-16 C:N ratio is approximately 1.2 (Fig. 5a, b). In daf-2(e1370) mutants in which insulin signaling is diminished, as expected, DAF-16/FOXO translocates to the nucleus such that the C:N ratio drops to 0.5 (Fig. 5a, b). However, in the ins-7(ssr1532) and ins-7OX worms, we also did not observe any deviation in the C:N ratio of DAF-16 localization relative to wild-type animals (Fig. 5a, b).

a, c Representative images of control, daf-2(e1370), ins-7(ssr1532), and INT1::ins-7OX animals bearing integrated DAF-16::GFP transgenes stained with DiI under fed and 3-h fasted conditions. Upper panels, DiI staining; middle panels, DAF-16::GFP; lower panels, merge. Neurons are indicated with closed arrowheads (ASI) and open arrowheads (ASK and ADL). Scale bar, 5 μm. b, d The mean intensity of DAF-16::GFP in the cytoplasm was divided by the mean intensity of DAF-16::GFP in the nucleus for control, daf-2(e1370), ins-7(ssr1532), and INT1::ins-7OX animals. Data are expressed as a ratio ± SEM. p values were calculated using one-way ANOVA and Dunnett’s post hoc test. n = 6, 6, 7, 7 from left to right in (b). Control vs. daf-2(e1370); ***p < 0.0001. Control vs. ins-7(ssr1532); nsp = 0.8409. Control vs. INT1::ins-7OX; nsp > 0.9999. n = 6, 7, 6, 6 from left to right in (d). Control vs. daf-2(e1370); ***p < 0.0001. Control vs. ins-7(ssr1532); nsp = 0.9851. Control vs. INT1::ins-7OX; ***p < 0.0001. e Representative images of INT1::ins-7OX animals bearing endogenous GFP-tagged DAF-2 stained with DiD under 3-h fasted conditions. First panel, DiD staining; second panel, DAF-2::GFP; third panel, INS-7mCherry; last panel, DIC. Scale bar, 10 μm. The fluorescence of DAF-2::GFP, INS-7mCherry, and DiD was examined in three independent experiments with similar results. f Insets: magnified view of the region enclosed by the dashed line from (e). Neurons are indicated with closed arrowheads (ASI) and open arrowheads (ASK and ADL). The colocalization of DAF-2::GFP and INS-7mCherry signals are indicated with arrows. g Colocalization of DAF-2::GFP and INS-7mCherry signals on the DiD labeled membrane area. Left panel, Merge of DiD, DAF-2::GFP, and INS-7mCherry channels. Right panel, Merge of DAF-2::GFP and INS-7mCherry channels. The region indicated by the dashed line was used for making the intensity profile. Lower panel, intensity profile generated in Fiji. Note that peaks of GFP and mCherry overlap at the same locations. The colocalization of DAF-2::GFP and INS-7mCherry signals was examined in three independent experiments with similar results. Source data are provided as a Source Data file.
This result prompted a deeper investigation into the conditions in which ins-7 normally functions. Conventional insulin, IGF1 and IGF2 are peptides secreted during abundant food availability and during growth and development. Because the phylogenetic tree suggested that INS-7 belongs to a different clade (Fig. 4b), and the functional role of INS-7 was in opposition to DAF-2, we postulated that as a physiological antagonist to the insulin receptor, INS-7 may function preferentially in the fasted state, and that its temporal functions may not be fully revealed in well-fed animals. Thus, we tested the effects of a 3-h fast (a duration sufficient to deplete about 80% of the fat stores in the intestine26) on DAF-16 localization. Interestingly, in the fasted condition DAF-16 localization in the ASI neurons did not shift from the previously noted fed state ratios between the cytoplasm and the nucleus in wild-types, daf-2(e1370) or ins-7(ssr1532) mutants. However, in the fasted state, worms bearing transgenic overexpression of ins-7 from the INT1 cells (INS-7OX) showed a significant shift in DAF-16 localization to the nucleus (C:N ratio 0.8; Fig. 5c, d). This result resembles the effect of the daf-2(e1370)-mediated reduction C:N DAF-16 localization (0.5). Furthermore, we found the colocalization of endogenous DAF-2GFP and INT1-expressed INS-7mCherry on the cell surface of ASI neurons in wild-type animals fasted for 3 h, suggesting a direct interaction between the ligand and the receptor under these conditions (Fig. 5e–g). Together, these experiments show that INS-7 functions in an antagonistic capacity in the context of regulating ASI function and output via DAF-2, preferentially in the fasted state.
Multiple lines of evidence show that hundreds of genes containing DAF-16 cis-binding sites function as effectors of the insulin signaling pathway42,46,47,48,49,50. Of these, the AMP-activated protein kinase (AMPK) occupies a central role51,52, and are also expressed in ASI neurons based on single-cell RNAseq studies38. As noted in our previous work on the role of ASI neurons in regulating FLP-7 secretion20, we found that AMPK α subunit aak-2(ok524) mutants had significantly increased FLP-7ASI secretion (Fig. 6a, b). To exclude the possibility that the increased FLP-7ASI secretion in aak-2 mutants resulted from its absence elsewhere in the nervous system, we used antisense inhibition of aak-2 solely in the ASI neurons. Antisense inhibition of ASI-specific aak-2 resembled the global aak-2 mutation (Fig. 6c, d), that is, loss of aak-2ASI increased FLP-7ASI secretion in a manner indistinguishable from the global absence of aak-2. Additionally, ASI-specific antisense inhibition of aak-2 increased FLP-7ASI secretion in daf-2(e1370) mutants to the same extent as it did in the wild-type animals (Fig. 6c, d), thus establishing a mechanistic link between DAF-2, DAF-16 and AMPK in the ASI neurons for the modulation of FLP-7ASI secretion (Fig. 6e). Together, our results show that INS-7 from specialized INT1 intestinal cells inhibits the functional output of ASI neurons to dampen FLP-7 secretion, which drives fat loss throughout the intestine (INT2-9). This effect occurs via INS-7-mediated antagonism of DAF-2 and its downstream actions via DAF-16/FOXO and AMPK signaling in ASI neurons.

a Representative images of wild-type and aak-2(ok524) animals bearing ASI::FLP-7mCherry and CLM::GFP transgenes. Upper panels, GFP expression in coelomocytes; middle panels, secreted FLP-7mCherry in coelomocytes; lower panels, merge. Scale bar, 5 μm. b The intensity of FLP-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression for each genotype. Data are expressed as a percentage of the normalized FLP-7mCherry fluorescence intensity of wild-type animals ± SEM. n = 25 for wild-type, n = 23 for aak-2(ok524). ***p = 0.0006 by unpaired t-test (two-tailed). c Representative images of wild-type and daf-2(e1370) FLP-7ASI animals bearing antisense-mediated inactivation of aak-2 expression in ASI neurons using the str-3 promoter. NT non-transgenic, TG transgenic. Scale bar, 5 μm. d The intensity of FLP-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression for each genotype. Data are expressed as a percentage of the normalized FLP-7mCherry fluorescence intensity of wild-type animals ± SEM. n = 42, 27, 23, 21, 46 from left to right. p values were calculated using one-way ANOVA and Sidak’s post hoc test. Wild-type vs. ASI::aak-2 antisense TG; ***p < 0.0001. Wild-type vs. daf-2(e1370);ASI::aak-2 antisense NT; **p = 0.0017. Wild-type vs. daf-2(e1370);ASI::aak-2 antisense TG; ***p < 0.0001. ASI::aak-2 antisense NT vs. ASI::aak-2 antisense TG; ***p < 0.0001. daf-2(e1370);ASI::aak-2 antisense NT vs. daf-2(e1370);ASI::aak-2 antisense TG; ***p < 0.0001. e Model depicting the role of INS-7 as a DAF-2 antagonist in modulating FLP-7 secretion from ASI neurons via DAF-16/FOXO and AMPK signaling. f–i FLP-7 secretion dynamics during fasting and re-feeding were determined at the indicated time points. For wild-type and ins-7(ssr1532) animals, the intensity of FLP-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression for each time point. Data are expressed as a percentage of the normalized FLP-7mCherry fluorescence intensity of wild-type fed animals ± SEM. n = 22, 18, 15, 20 from left to right in (f). p values were calculated using one-way ANOVA and Dunnett’s post hoc test. Fed vs. 180 min fasted; **p = 0.0041. n = 27, 17, 23 from left to right in (g). p values were calculated using one-way ANOVA and Tukey’s post hoc test. Fed vs. 180 min fasted; 180 min fasted vs. 30 min refed; ***p < 0.0001, respectively. n = 20, 17, 23, 24 from left to right in (h). p values were calculated using one-way ANOVA and Dunnett’s post hoc test. Fed vs. 30 min fasted; nsp = 0.264. Fed vs. 90 min fasted; nsp = 0.4779. Fed vs. 180 min fasted; nsp = 0.8616. n = 24, 22, 16 from left to right in (i). p values were calculated using one-way ANOVA and Tukey’s post hoc test. Fed vs. 180 min fasted; nsp = 0.8814. 180 min fasted vs. 30 min refed; nsp = 0.8771. Source data are provided as a Source Data file.
Dynamics of INS-7 function
The experiments delineating a role for INS-7 as a gut-derived signal were designed to reveal steady-state differences in homeostatic mechanisms in well-fed animals. However, our results from the DAF-16 localization experiment suggested that INS-7INT1 may regulate FLP-7ASI differentially in the fed and fasted states. Additionally, a role for INS-7 as an antagonist might indicate a function in opposition to canonical insulin which is a peptide secreted in the post-prandial state53. To test the idea that INS-7 may regulate FLP-7 secretion differentially in the fed and fasted states, we measured the dynamics of FLP-7 secretion in the presence and absence of ins-7. In wild-type animals subjected to food deprivation, an increase in FLP-7 secretion is not discernable until 180 min (3 h; Fig. 6f and Supplementary Fig. 4b), a time point at which greater than 80% of the intestinal fat stores of the animal have been depleted26. Re-exposure to food after 3 h of fasting restores FLP-7 secretion back to baseline levels (Fig. 6g), suggesting that the rise in FLP-7 secretion during the fasting regimen is reset by re-feeding. Notably, in ins-7 null animals (Fig. 6h), the feeding-state-dependent regulation of FLP-7 is abrogated such that FLP-7 secretion levels are chronically high, regardless of feeding or fasting status (Fig. 6h,i) as per the original observation for ins-7 nulls (Fig. 1). Thus, gut-derived INS-7 provides a brake on FLP-7ASI secretion to inhibit the neuronal drive to trigger fat loss during food shortage.
We investigated whether INS-7 itself is a secreted peptide by generating a transgenic line expressing an INS-7mCherry fusion protein solely from INT1 (pseudocolored yellow; Fig. 7a, b). To our interest, we found that INT1-expressed INS-7 also accumulates in the coelomocytes, the characteristic hallmark of a secreted peptide in C. elegans20,54,55. Importantly, INS-7 accumulation in coelomocytes does not correlate with their GFP intensity, suggesting that these two parameters are independent of one another (Fig. 7c). Extensive validation of the coelomocyte secretion assay by us and others20,54 has shown that it faithfully represents the steady-state levels of a given secreted peptide under a broad range of genetic and physiological conditions. Intestine-specific RNAi of aex-4, an intestinal SNARE protein required for vesicular protein secretion56 greatly diminished INS-7 secretion, as did asna-1, an ATP hydrolase known to specifically regulate insulin secretion57 (Fig. 7d, e). Next, to test whether INS-7, as an antagonist, is secreted under conditions in opposition to canonical insulin, we measured the dynamics of INS-7 secretion in wild-type animals subjected to food deprivation. INS-7 secretion increased within 30 min off food and further increased until 3 h off food (Fig. 7f). Re-feeding restores INS-7 secretion back to baseline levels (Fig. 7g). Thus, INS-7 is a bona fide gut peptide secreted by the INT1 cells of the intestine and acts as a homeostatic signal to restrict FLP-7ASI secretion during food deprivation. Together, our results define a novel gut-brain pathway that relays intestinal state information during acute food shortage.

a Model depicting the coelomocyte uptake assay for INS-7 secretion. The INS-7mCherry fusion protein (marked in yellow) is expressed in INT1 cells of the intestine, and GFP is expressed in the coelomocytes (marked in green). b Fluorescent image of a transgenic animal bearing ins-7mCherry under the control of the INT1-specific promoter. INS-7mCherry signals were observed in INT1 (pseudocolored yellow; closed arrowhead) and coelomocytes (open arrowhead). Upper panel, GFP expression in coelomocytes; middle panel, secreted INS-7mCherry in coelomocytes (pseudocolored in yellow); lower panels, DIC. Scale bar, 50 μm. The secretion of INS-7mCherry was examined in three independent experiments with similar results. c mCherry fluorescence intensity values are plotted against GFP fluorescence intensity values for each animal across representative experimental conditions (n = 341). Scale bar, 5 μm. d Representative images of sid-1;INT1::sid-1 animals bearing integrated INT1::INS-7mCherry and CLM::GFP transgenes treated with vector, aex-4 or asna-1 RNAi. Scale bar, 5 μm. e The intensity of INS-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression for each RNAi treatment. Data are expressed as a percentage of the normalized INS-7mCherry fluorescence intensity of animals treated with vector RNAi ± SEM. n = 36, 14, 19 from left to right. p values were calculated using one-way ANOVA and Dunnett’s T3 post hoc test. Vector RNAi vs. aex-4 RNAi; ***p < 0.0001. Vector RNAi vs. asna-1 RNAi; **p = 0.0019. f, g INS-7 secretion dynamics during fasting and re-feeding were determined at the indicated time points. The intensity of INS-7mCherry fluorescence within a single coelomocyte was quantified and normalized to the area of CLM::GFP expression for each time point. Data are expressed as a percentage of the normalized INS-7mCherry fluorescence intensity of wild-type fed animals ± SEM. p values were calculated using one-way ANOVA and Dunnett’s T3 post hoc test. n = 23, 23, 25, 25, 23 from left to right in (f). Fed vs. 30 min fasted; fed vs. 60 min fasted; fed vs. 90 min fasted; fed vs. 180 min fasted; ***p < 0.0001, respectively. n = 26, 23, 23 from left to right in (g). Fed vs. 180 min fasted; 180 min fasted vs. 180 min refed; ***p < 0.0001, respectively. Source data are provided as a Source Data file.
Discussion
Here, we report that a peptide of the insulin superfamily called INS-7 is released from specialized INT1 enteroendocrine cells of the C. elegans intestine. INS-7 functions as an antagonist at the canonical DAF-2 receptor in the ASI neurons to inhibit FLP-7 secretion. Because FLP-7ASI secretion is a signal that integrates neuronal information to promote fat loss, our results show that the INS-7 gut-to-brain inhibitory peptide serves to limit this signal in the absence of incoming food to the intestine. Work presented here describes the molecular features of our previously postulated hypothesis that homeostatic mechanisms must exist to ensure that neural signals that stimulate fat loss in the intestine are only deployed when there are sufficient fat reserves to do so26. These findings uncover a hitherto unknown mechanism of gut-to-brain homeostatic communication in which C. elegans lipid metabolism balances between external sensory cues and internal metabolic states.
The identification of INT1 cells as specialized secretory cells of the intestine is intriguing. As the foremost cells of the intestine immediately adjoining the pharynx in the alimentary canal31, they have privileged access to incoming nutrient information before food absorption has begun. Sulston et al. showed that INT1 cells have shorter microvilli than the rest of the intestinal cells (INT2-9)31 and form a collapsible conical reservoir at the intestine anterior, suggesting that INT1 cells may possess unique properties in the context of enteroendocrine INS-7 secretion that remain to be discovered. Based on our results, we propose that the INT1 quartet functions as enteroendocrine cells that receive information not only from the lumen but perhaps also from the rest of the intestine.
Amongst the many peptides we screened for gut-to-brain signals, INS-7 emerged as the primary suppressor of FLP-7ASI secretion. Initial reports describing a role for INS-7 in longevity regulation had suggested an agonist function at the DAF-2 receptor41,42, whereas later reports suggested antagonistic functions in olfactory learning40,58. It is possible that an explanation lies within the expression features of the various DAF-2 isoforms. In the C. elegans nervous system, mutually exclusive expression of DAF-2a and DAF-2c isoforms has been noted. Multiple lines of evidence show that neuronal DAF-2c expression in sensory neurons including the ASI neurons is further induced by fasting and drives the antagonistic effects of DAF-2 signaling in the nervous system59,60,61. Our results indicate a clear role for INS-7 as an antagonist for DAF-2 in ASI neurons; we postulate that agonism and antagonism of insulin peptides at the DAF-2 receptor is determined in a cell-type-specific manner. In contrast to gut-derived INS-7, neuronally-derived INS-7 has been implicated in innate immunity and pathogen avoidance62.
The increase in INS-7 secretion upon acute food withdrawal and fasting is of great interest. During short-term fasting, two additional molecular events occur: the colocalization of INS-7mCherry and DAF-2GFP in ASI neurons and the nuclear translocation of DAF-16GFP in the ASI neurons. These observations suggest that the action of INS-7 on DAF-2 in ASI occurs preferentially in the short-term fasted state. In this way, INS-7 secretion from the intestine could be the origin of the fascinating FOXO-to-FOXO signaling paradigm described in the literature41,63,64. In this scenario, we would postulate that the origin of the signaling loop would be the secretion event of INS-7 from INT1 cells upon food deprivation. Further experimentation is needed to delve into the effects of INS-7 secretion across the nervous system and the rest of the body, particularly with respect to the opposite effects of DAF-16 and PQM-1 and their key target genes65 and across time scales66.
Phylogenetic analysis shows that INS-7 may be more orthologous to the mammalian insulin-like peptides and to the relaxin superfamily, rather than the canonical insulin/IGF peptides. Most of the mammalian insulin superfamily members remain poorly characterized, however the insulin-like peptide INSL5 is secreted by L cells of the colon and is a gut hormone thought to be secreted in response to dietary fat intake, and modulates food intake via the hypothalamus67,68,69. In mammals, gut hormones that play critical roles in the regulation of energy and glucose metabolism or in the modulation of food intake via the gut-brain axis have been identified70,71. For example, glucagon-like peptide 1 (GLP-1) and cholecystokinin (CCK) are produced in enteroendocrine cells in the intestine and released after nutrient ingestion72 and target various brain regions to reduce food intake73,74,75; long-acting GLP-1 variants are now the basis of medications that effect substantial fat loss via the suppression of food intake and restoring glucose homeostasis in humans76,77.
In Drosophila, gut-secreted peptide CCHa1 modulates the activity of dopaminergic neurons to suppress the responsiveness to vibration during sleep78, and in addition, gut-secreted Hedgehog (Hh) targets taste sensory neurons and suppresses sweet sensation and preference79. Interestingly, most gut peptides to date do not directly target fat oxidation in the periphery but instead regulate animal behavior such as food intake, thus indirectly affecting fat metabolism. Instead, we report here that INS-7 acts as a gut-to-brain signal to suppress neuronally stimulated fat loss by inhibiting the secretion of FLP-7 peptide without modulating feeding behaviors. The discovery of intestinal INS-7 as an insulin antagonist that targets neurons that directly regulate fat metabolism defines a new mode of interorgan communication regulated by insulin signaling.
The many uncharacterized insulin family peptides across species suggest that new features of interorgan communication regulated by insulin signaling remain to be discovered. For example, an insulin antagonist ortholog to INS-7 may be secreted and function primarily in the fasted state, in opposition to canonical insulin which signals the fed state. We speculate that such a ‘food absence’ signal from the intestine may help animals optimize their physiology while retaining energy reserves during short-term fasts or between meals. Finally, the discovery of a specialized intestinal cell with enteroendocrine functions suggests that much remains to be learned about the C. elegans intestine and its role in directing neuronal functions.
Methods
C. elegans maintenance and strains
Worms were cultured on nematode growth medium (NGM) agar plates with Escherichia coli strain OP50 at 20 °C as described80. The N2 Bristol strain was obtained from the Caenorhabditis Genetic Center (CGC) and used as wild-type. All mutant and transgenic strains used in this study are listed in Supplementary Table 1. Worms were synchronized for Oil Red O staining and oxygen consumption by hypochlorite treatment, after which hatched L1 larvae were seeded on plates with the appropriate bacteria; worms were synchronized for neuropeptide secretion assays and behavioral assays by letting gravid adult worms lay eggs on plates with the appropriate bacteria for 1.5 h. For fasting experiments, young gravid adult worms were transferred with platinum wire without bacteria, to intermediate unseeded NGM plates first after which they were transferred to new unseeded NGM plates for the fasted conditions. All experiments were performed on Day 1 adults. C. elegans studies did not require IRB or IACUC approvals at The Scripps Research Institute.
Cloning and transgenic strain construction
Promoters and cDNAs were amplified using standard PCR techniques from N2 genomic DNA or cDNA and subcloned into expression vectors using Gateway Cloning Technology (Life Technologies). Primers used for cloning in this study are listed in Supplementary Table 2. All transgenic rescue constructs were generated using polycistronic GFP. Transgenic strains were generated by injecting plasmids into the germline of wild-type or mutant worms followed by visual selection of co-injection markers (Punc-122::GFP or Plin-44::GFP) under the fluorescence microscope. To inhibit the expression of daf-2 or aak-2 specifically in ASI neurons, we generated plasmids for antisense-mediated inhibition81 with the str-3 promoter. Sense and antisense sequences targeting daf-2 or aak-2 were amplified from N2 lysates and subcloned into donor vectors using Gateway Cloning Technology (Life Technologies). The final plasmids for the sense and antisense expression of daf-2 or aak-2 under the str-3 promoter were generated using Gateway Cloning Technology (Life Technologies) and injected into FLP-7 secretion line (SSR1164) at 5 ng/μL (daf-2) or 1 ng/μL (aak-2) each. The FLP-7 secretion line was previously developed and validated20.
For the INS-7 secretion line, N2 worms were injected with 10 ng/μL of the INT1::ins-7mCherry plasmid, 15 ng/μL of a Punc-122::GFP plasmid, and 75 ng/μL of an empty vector to bring the final concentration of injection mix to 100 ng/μL. A transgenic line with high transmission rate and consistent expression was integrated using the Stratalinker UV Crosslinker 2400 (Stratagene) and backcrossed six times before experimentation. For other microinjections, we injected the animals with 5–25 ng/μL of the desired plasmid, 25 ng/μL of Punc-122::GFP or 10 ng/μL of Plin-44::GFP and empty vector to maintain a final injection mix concentration of 100 ng/μL. Two lines were selected for experimentation based on the transmission rate and consistency of expression.
RNAi and qPCR
RNAi experiments were performed as described82,83. Carbenicillin-IPTG plates were seeded with HT115 bacteria containing the empty vector or the relevant RNAi clone and allowed to grow for four days before seeding larvae. Total RNA was extracted from Day 1 adults using TRIzol reagent (Invitrogen). Genomic DNA was isolated using an RNase-free DNase kit (QIAGEN). cDNA was prepared using iScript Reverse Transcription Supermix for RT-qPCR kit (Bio-Rad) according to the manufacturer’s instructions. Quantitative RT-PCR was performed using the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) following the manufacturer’s instructions. Data were normalized to actin mRNA. Primer sequences used in this study are listed in Supplementary Table 2.
CRISPR-Cas9 gene editing
Guide RNAs for generating the ins-7 null allele were designed using the CRISPR guide RNA selection tool84. The dpy-10 guide RNA and repair template were used as reported85. All sequences of guide RNAs and repair templates used in this study are listed in Supplementary Table 2. Steps for CRISPR-Cas9 gene editing are briefly described as follows: 0.88 μL tracrRNA (200 μM, IDT), 0.82 μL ins-7 guide RNAs (100 μM, IDT), and 0.12 μL dpy-10 guide RNA (100 μM, IDT) were mixed and incubated at 95 °C for 5 min. 2.52 μL Cas9 protein (IDT, Catalog# 1081058) was added to the mix and incubated at 25 °C for 5 min. 0.6 μL ins-7 repair template (100 μM, IDT), 0.5 μL dpy-10 repair template (10 μM, IDT), and 3.74 μL nuclease-free water were added to the mix and incubated at 25 °C for 60 min. The final injection mix was loaded using a pulled capillary needle (1B100F-4, World Precision Instruments) and injected into the germline of N2 young adults. Screening strategy for isolating ins-7 null mutants was followed as previously described for C. elegans CRISPR allele isolation86. The final ins-7 null mutant (ins-7(ssr1532)) used for experiments was backcrossed four times before experimentation.
Guide RNAs for generating the ins-7 endogenous transcriptional GFP reporter (ins-7(ssr1732)) were designed using the CRISPR guide RNA selection tool84. The second and third exons of ins-7 were replaced with GFP, which was inframe with the first exon. Steps for CRISPR-Cas9 gene editing are briefly described as follows: 0.88 μL tracrRNA (200 μM, IDT), 0.82 μL ins-7 guide RNAs (100 μM, IDT), and 0.12 μL dpy-10 guide RNA (100 μM, IDT) were mixed and incubated at 95 °C for 5 min. 2.52 μL Cas9 protein (IDT, Catalog# 1081058) was added to the mix and incubated at 25 °C for 5 min. 2.5 μL GFP repair template (1 μM, IDT), 0.5 μL dpy-10 repair template (10 μM, IDT), and 1.84 μL nuclease-free water were added to the mix and incubated at 25 °C for 60 min. The final injection mix was loaded using a pulled capillary needle (1B100F-4, World Precision Instruments) and injected into the germline of N2 young adults.
Oil Red O staining
Oil Red O staining was performed as described16. Briefly, worms were harvested with phosphate-buffered saline (PBS) and incubated on ice for 10 min before fixation. Worms were then stained in filtered Oil Red O (Thermo Scientific) working solution (60% Oil Red O in isopropanol: 40% water) overnight. Approximately 2000 worms were fixed and stained for all genotypes within a single experiment. For each experimental condition, we visually observed about 100 worms on slides, after which 15–20 worms were randomly chosen for imaging. All experiments were repeated at least three times.
Lipid extraction and quantification
Lipid extraction was performed as described14. For each group, 2000 worms per 10 cm plate were grown at 25 °C until the worms reached the young adult stage. After washing with PBS twice, the worms were flash-frozen in liquid nitrogen. Worms were homogenized in PBS containing 5% TritonX-100 and a protease inhibitor cocktail (Roche), and lipid was extracted using the TissueLyser II (QIAGEN) or a Dounce homogenizer. Triglyceride content was measured using the EnzyChrom Triglyceride Assay Kit (BioAssay Systems), and triglyceride levels were normalized to the total protein level determined by the Pierce BCA Protein Assay (Thermo Scientific).
Oxygen consumption
Oxygen consumption rates (OCR) were measured using the Seahorse XFe96 Analyzer (Agilent) as described14. Adult worms were washed with M9 buffer and transferred into a 96-well plate with approximately 10 worms per well. Five basal measurements were taken and then FCCP (50 mM) was injected into each well to measure maximal OCR. Lastly, sodium azide (40 mM) was injected to measure residual OCR. Values were normalized to the number of worms per well. Basal OCR was the average of all measured values prior to the addition of FCCP (50 mM); maximal OCR was the average of the first two measured values after FCCP injection.
Food intake
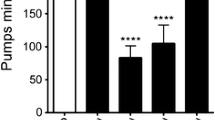
Food intake was measured by counting pharyngeal pumping, as described83. For each worm, the rhythmic contractions of the pharyngeal bulb were counted over 10 s under a Zeiss M2 Bio Discovery microscope. For each genotype, 25 worms were assessed, and the experiment was repeated at least three times.
Enhanced slowing response
The enhanced slowing response was measured as described87. Day 1 adults were washed off OP50 plates with PBS, washed five times to remove bacteria, and placed on NGM agar plates without OP50. After 30 min off food, worms were collected with PBS and seeded onto NGM plates with OP50. Worms were allowed to acclimatize for 5 min, after which the number of body bends per 20 s was counted.
Thrashing assay
Thrashing rate was measured as described88. For each worm, a movement in which the head and/or tail swung to the other side and back to the original position was counted as one thrash. 15–20 Day 1 adults were assessed for each phenotype.
DiI and DiD staining
The stock solution containing 2 mg/mL DiI or DiD was prepared in dimethylformamide. L4 animals were incubated overnight in a 1:200 dilution of DiI or DiD stock solution on the OP50 bacterial lawn. The next day, Day 1 adult worms were selected for imaging.
Image acquisition and quantitation
Oil Red O-stained worms were imaged using 10× objective on a Zeiss Axio Imager microscope. Images were acquired with the Software AxioVision (Zeiss). Lipid droplet staining over the whole body of each worm was quantified using ImageJ software (NIH). All reported results were consistent across biological replicates. Fluorescent images of reporters for FLP-7 and INS-7 secretion were acquired with the software AxioVision (Zeiss) using a 20× objective on a Zeiss Axio Imager microscope. The first pair of coelomocytes was imaged. mCherry fluorescence intensity in one of the two imaged coelomocytes was quantified and normalized to the surface area of the coelomocyte (unc-122::GFP) as previously described and validated20. Within each experiment, at least 15 worms from each condition were quantified using ImageJ software (NIH). Fluorescent images of ins-7 expression pattern (Pins-7::GFP) were collected with the software AxioVision (Zeiss) using a 10× objective on a Zeiss Axio Imager microscope. Fluorescent images of DAF-16::GFP and DiI-stained neurons were collected with the software NIS-Elements (Nikon) using a 60× objective on a Nikon A1 confocal microscope. Fluorescent images of DAF-2::GFP, INS-7mCherry, and DiD-stained neurons were collected with software NIS-Elements (Nikon) using a 40× objective on a Nikon Crest X-Light spinning disk confocal microscope.
Statistics
Wild-type animals were included as controls for every experiment. Error bars represent the standard error of the mean (SEM). Student’s t-test, one-way ANOVA, and two-way ANOVA were used as indicated in the figure legends. All statistical analyses were performed using GraphPad Prism 10 (GraphPad Software). Appropriate multiple comparison corrections were used for ANOVAs. *p < 0.05, **p < 0.01, ***p < 0.001, ns: not significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data in the main manuscript and supplementary information are listed in the Source data file. Source data are provided with this paper.
References
Lemieux, G. A. & Ashrafi, K. Neural regulatory pathways of feeding and fat in caenorhabditis elegans. Annu. Rev. Genet 49, 413–438 (2015).
Caron, A., Lee, S., Elmquist, J. K. & Gautron, L. Leptin and brain-adipose crosstalks. Nat. Rev. Neurosci. 19, 153–165 (2018).
Musselman, L. P. & Kuhnlein, R. P. Drosophila as a model to study obesity and metabolic disease. J. Exp. Biol. 221, jeb163881 (2018).
Castillo-Armengol, J., Fajas, L. & Lopez-Mejia, I. C. Inter-organ communication: a gatekeeper for metabolic health. EMBO Rep. 20, e47903 (2019).
Priest, C. & Tontonoz, P. Inter-organ cross-talk in metabolic syndrome. Nat. Metab. 1, 1177–1188 (2019).
Kubota, N. et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 6, 55–68 (2007).
Bookout, A. L. et al. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat. Med. 19, 1147–1152 (2013).
Inagaki, T. et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2, 217–225 (2005).
Ge, X. et al. LEAP2 is an endogenous antagonist of the ghrelin receptor. Cell Metab. 27, 461–469.e466 (2018).
Batterham, R. L. et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature 418, 650–654 (2002).
Turton, M. D. et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 379, 69–72 (1996).
Srinivasan, S. Neuroendocrine control of lipid metabolism: lessons from C. elegans. J. Neurogenet. 34, 482–488 (2020).
Srinivasan, S. Regulation of body fat in Caenorhabditis elegans. Annu. Rev. Physiol. 77, 161–178 (2015).
Hussey, R. et al. Oxygen-sensing neurons reciprocally regulate peripheral lipid metabolism via neuropeptide signaling in Caenorhabditis elegans. PLoS Genet. 14, e1007305 (2018).
Hussey, R. et al. Pheromone-sensing neurons regulate peripheral lipid metabolism in Caenorhabditis elegans. PLoS Genet. 13, e1006806 (2017).
Noble, T., Stieglitz, J. & Srinivasan, S. An integrated serotonin and octopamine neuronal circuit directs the release of an endocrine signal to control C. elegans body fat. Cell Metab. 18, 672–684 (2013).
Greer, E. R., Perez, C. L., Van Gilst, M. R., Lee, B. H. & Ashrafi, K. Neural and molecular dissection of a C. elegans sensory circuit that regulates fat and feeding. Cell Metab. 8, 118–131 (2008).
Mak, H. Y., Nelson, L. S., Basson, M., Johnson, C. D. & Ruvkun, G. Polygenic control of Caenorhabditis elegans fat storage. Nat. Genet. 38, 363–368 (2006).
Altun, Z. F. & Hall, D. H. Alimentary system, intestine. WormAtlas (2009).
Palamiuc, L. et al. A tachykinin-like neuroendocrine signalling axis couples central serotonin action and nutrient sensing with peripheral lipid metabolism. Nat. Commun. 8, 14237 (2017).
Lee, K. & Mylonakis, E. An intestine-derived neuropeptide controls avoidance behavior in Caenorhabditis elegans. Cell Rep. 20, 2501–2512 (2017).
Matty, M. A. et al. Intestine-to-neuronal signaling alters risk-taking behaviors in food-deprived Caenorhabditis elegans. PLoS Genet. 18, e1010178 (2022).
Ryu, L. et al. Feeding state regulates pheromone-mediated avoidance behavior via the insulin signaling pathway in Caenorhabditis elegans. EMBO J. 37, e98402 (2018).
Wang, H. et al. Neuropeptide secreted from a pacemaker activates neurons to control a rhythmic behavior. Curr. Biol. 23, 746–754 (2013).
Bouagnon, A. D. et al. Intestinal peroxisomal fatty acid beta-oxidation regulates neural serotonin signaling through a feedback mechanism. PLoS Biol. 17, e3000242 (2019).
Witham, E. et al. C. elegans body cavity neurons are homeostatic sensors that integrate fluctuations in oxygen availability and internal nutrient reserves. Cell Rep. 14, 1641–1654 (2016).
McKay, R. M., McKay, J. P., Avery, L. & Graff, J. M. C elegans: a model for exploring the genetics of fat storage. Dev. Cell 4, 131–142 (2003).
Kniazeva, M. et al. Suppression of the ELO-2 FA elongation activity results in alterations of the fatty acid composition and multiple physiological defects, including abnormal ultradian rhythms, in Caenorhabditis elegans. Genetics 163, 159–169 (2003).
Ritter, A. D. et al. Complex expression dynamics and robustness in C. elegans insulin networks. Genome Res. 23, 954–965 (2013).
Baugh, L. R., Kurhanewicz, N. & Sternberg, P. W. Sensitive and precise quantification of insulin-like mRNA expression in Caenorhabditis elegans. PLoS ONE 6, e18086 (2011).
Sulston, J. E., Schierenberg, E., White, J. G. & Thomson, J. N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119 (1983).
Walker, A. K. et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 24, 1403–1417 (2010).
Jang, M. H. et al. Intestinal villous M cells: an antigen entry site in the mucosal epithelium. Proc. Natl Acad. Sci. USA 101, 6110–6115 (2004).
Shearman, D. J. C. & Muir, A. R. Observations on the secretory cycle of goblet cells. Q. J. Exp. Physiol. Cogn. Med. Sci. 45, 337–342 (1960).
Littlejohn, N. K., Seban, N., Liu, C. C. & Srinivasan, S. A feedback loop governs the relationship between lipid metabolism and longevity. Elife 9, e58815 (2020).
Hakuno, F. & Takahashi, S. I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 61, T69–T86 (2018).
Kimura, K. D., Tissenbaum, H. A., Liu, Y. & Ruvkun, G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942–946 (1997).
Taylor, S. R. et al. Molecular topography of an entire nervous system. Cell 184, 4329–4347.e4323 (2021).
Jose, A. M., Smith, J. J. & Hunter, C. P. Export of RNA silencing from C. elegans tissues does not require the RNA channel SID-1. Proc. Natl Acad. Sci. USA 106, 2283–2288 (2009).
Chen, Z. et al. Two insulin-like peptides antagonistically regulate aversive olfactory learning in C. elegans. Neuron 77, 572–585 (2013).
Murphy, C. T., Lee, S. J. & Kenyon, C. Tissue entrainment by feedback regulation of insulin gene expression in the endoderm of Caenorhabditis elegans. Proc. Natl Acad. Sci. USA 104, 19046–19050 (2007).
Murphy, C. T. et al. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283 (2003).
Henderson, S. T. & Johnson, T. E. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 11, 1975–1980 (2001).
Lin, K., Hsin, H., Libina, N. & Kenyon, C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 28, 139–145 (2001).
Lee, R. Y., Hench, J. & Ruvkun, G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr. Biol. 11, 1950–1957 (2001).
Furuyama, T., Nakazawa, T., Nakano, I. & Mori, N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem. J. 349, 629–634 (2000).
Oh, S. W. et al. Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nat. Genet. 38, 251–257 (2006).
McElwee, J., Bubb, K. & Thomas, J. H. Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell 2, 111–121 (2003).
Lee, S. S., Kennedy, S., Tolonen, A. C. & Ruvkun, G. DAF-16 target genes that control C. elegans life-span and metabolism. Science 300, 644–647 (2003).
Schuster, E. et al. DamID in C. elegans reveals longevity-associated targets of DAF-16/FoxO. Mol. Syst. Biol. 6, 399 (2010).
Greer, E. L. et al. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 17, 1646–1656 (2007).
Tullet, J. M. et al. DAF-16/FoxO directly regulates an atypical AMP-activated protein kinase gamma isoform to mediate the effects of insulin/IGF-1 signaling on aging in Caenorhabditis elegans. PLoS Genet. 10, e1004109 (2014).
Campbell, J. E. & Newgard, C. B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 22, 142–158 (2021).
Sieburth, D., Madison, J. M. & Kaplan, J. M. PKC-1 regulates secretion of neuropeptides. Nat. Neurosci. 10, 49–57 (2007).
Speese, S. et al. UNC-31 (CAPS) is required for dense-core vesicle but not synaptic vesicle exocytosis in Caenorhabditis elegans. J. Neurosci. 27, 6150–6162 (2007).
Mahoney, T. R. et al. Intestinal signaling to GABAergic neurons regulates a rhythmic behavior in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA 105, 16350–16355 (2008).
Kao, G. et al. ASNA-1 positively regulates insulin secretion in C. elegans and mammalian cells. Cell 128, 577–587 (2007).
Zheng, S. et al. A functional study of all 40 Caenorhabditis elegans insulin-like peptides. J. Biol. Chem. 293, 16912–16922 (2018).
Ohno, H. et al. Role of synaptic phosphatidylinositol 3-kinase in a behavioral learning response in C. elegans. Science 345, 313–317 (2014).
Tomioka, M., Naito, Y., Kuroyanagi, H. & Iino, Y. Splicing factors control C. elegans behavioural learning in a single neuron by producing DAF-2c receptor. Nat. Commun. 7, 11645 (2016).
Cheng, D. et al. Insulin/IGF signaling regulates presynaptic glutamate release in aversive olfactory learning. Cell Rep. 41, 111685 (2022).
Kawli, T. & Tan, M. W. Neuroendocrine signals modulate the innate immunity of Caenorhabditis elegans through insulin signaling. Nat. Immunol. 9, 1415–1424 (2008).
Uno, M. et al. Neuronal DAF-16-to-intestinal DAF-16 communication underlies organismal lifespan extension in C. elegans. iScience 24, 102706 (2021).
Libina, N., Berman, J. R. & Kenyon, C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115, 489–502 (2003).
Tepper, R. G. et al. PQM-1 complements DAF-16 as a key transcriptional regulator of DAF-2-mediated development and longevity. Cell 154, 676–690 (2013).
Honjoh, S., Yamamoto, T., Uno, M. & Nishida, E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature 457, 726–730 (2009).
Grosse, J. et al. Insulin-like peptide 5 is an orexigenic gastrointestinal hormone. Proc. Natl Acad. Sci. USA 111, 11133–11138 (2014).
Lewis, J. E. et al. Relaxin/insulin-like family peptide receptor 4 (Rxfp4) expressing hypothalamic neurons modulate food intake and preference in mice. Mol. Metab. 66, 101604 (2022).
Lee, Y. S. et al. Insulin-like peptide 5 is a microbially regulated peptide that promotes hepatic glucose production. Mol. Metab. 5, 263–270 (2016).
Wachsmuth, H. R., Weninger, S. N. & Duca, F. A. Role of the gut-brain axis in energy and glucose metabolism. Exp. Mol. Med. 54, 377–392 (2022).
Prinz, P. & Stengel, A. Control of food intake by gastrointestinal peptides: mechanisms of action and possible modulation in the treatment of obesity. J. Neurogastroenterol. Motil. 23, 180–196 (2017).
Gribble, F. M. & Reimann, F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu. Rev. Physiol. 78, 277–299 (2016).
Tang-Christensen, M. et al. Central administration of GLP-1-(7-36) amide inhibits food and water intake in rats. Am. J. Physiol. 271, R848–R856 (1996).
Dockray, G. J. Cholecystokinin and gut-brain signalling. Regul. Pept. 155, 6–10 (2009).
Krieger, J. P. et al. Knockdown of GLP-1 receptors in vagal afferents affects normal food intake and glycemia. Diabetes 65, 34–43 (2016).
Garvey, W. T. et al. Two-year effects of semaglutide in adults with overweight or obesity: the STEP 5 trial. Nat. Med. 28, 2083–2091 (2022).
Coskun, T. et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol. Metab. 18, 3–14 (2018).
Titos, I. et al. A gut-secreted peptide suppresses arousability from sleep. Cell 186, 1382–1397.e1321 (2023).
Zhao, Y. et al. Hedgehog-mediated gut-taste neuron axis controls sweet perception in Drosophila. Nat. Commun. 13, 7810 (2022).
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974).
Esposito, G., Di Schiavi, E., Bergamasco, C. & Bazzicalupo, P. Efficient and cell specific knock-down of gene function in targeted C. elegans neurons. Gene 395, 170–176 (2007).
Kamath, R. S. & Ahringer, J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30, 313–321 (2003).
Srinivasan, S. et al. Serotonin regulates C. elegans fat and feeding through independent molecular mechanisms. Cell Metab. 7, 533–544 (2008).
Au, V. et al. CRISPR/Cas9 methodology for the generation of knockout deletions in Caenorhabditis elegans. G3 9, 135–144 (2019).
Arribere, J. A. et al. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198, 837–846 (2014).
Dickinson, D. J., Pani, A. M., Heppert, J. K., Higgins, C. D. & Goldstein, B. Streamlined genome engineering with a self-excising drug selection cassette. Genetics 200, 1035–1049 (2015).
Sawin, E. R., Ranganathan, R. & Horvitz, H. R. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26, 619–631 (2000).
Nawa, M. et al. Reduced expression of BTBD10, an Akt activator, leads to motor neuron death. Cell Death Differ. 19, 1398–1407 (2012).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Madeira, F. et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50, W276–W279 (2022).
Acknowledgements
This work was supported by research grants to S.S. from the NIH/NIDDK (R01 DK124706) and NIH/NIA (R01 AG056648). Strains were provided by the National Bioresource Project (Japan) and the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). We thank Dr. Anthony Perez of the Srinivasan lab for his critical comments on the manuscript. C.L. was supported by a Dorris Scholar Award from the Dorris Neuroscience Center, The Scripps Research Institute. Elements of Figs. 1a, 7a and Supplementary Fig. 1a were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
C.L., Conceptualization, Resources, Data curation, Formal analysis, Investigation, Methodology, Writing - Original Draft, Writing - Review and Editing; A.K., Formal analysis, Investigation; N.S, Resources; N.L., Formal analysis, Investigation, Methodology; A.S., Formal analysis, Investigation; S.S., Conceptualization, Resources, Supervision, Funding acquisition, Methodology, Writing - Original Draft, Writing - Review and Editing, Project administration. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yu Xue, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, CC., Khan, A., Seban, N. et al. A homeostatic gut-to-brain insulin antagonist restrains neuronally stimulated fat loss. Nat Commun 15, 6869 (2024). https://doi.org/10.1038/s41467-024-51077-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51077-3
- Springer Nature Limited