

Abstract
Arylamines, serving as crucial building blocks in natural products and finding applications in multifunctional materials, are synthesized on a large scale via an electrophilic nitration/reduction sequence. However, the current methods for aromatic C–H amination have not yet attained the same level of versatility as electrophilic nitration. Here we show an extensively investigated transition metal-free and regioselective strategy for the amination of nitrobenzenes, enabling the synthesis of 4-nitro-N-arylamines through C(sp2)-H/N-H cross-coupling between electron-deficient nitroarenes and amines. Mechanistic studies have elucidated that the crucial aspects of these reactions encompass the generation of nitrogen radicals and recombination of nitrobenzene complex radicals. The C(sp2)-N bond formation is demonstrated to be highly effective for primary and secondary arylamines as well as aliphatic amines under mild conditions, exhibiting exceptional tolerance towards diverse functional groups in both nitroarenes and amines (>100 examples with yields up to 96%). Notably, this C(sp2)-H/N-H cross-coupling exhibits exclusive para-selectivity.
Similar content being viewed by others

Introduction
Arylamines, as structural units in many natural products, are widely applied in the synthesis of medicinal agents, agrochemicals, and multifunctional materials1,2,3,4,5. Hence, the construction of C-N bonds to deliver arylamines has become one of the basic transformations in both academia and industry6. Traditionally, transition metal-catalyzed C-N cross-coupling have revolutionized the field of synthetic chemistry (Fig. 1a). For several decades, Buchwald-Hartiwig7,8, Chan-Lam couplings9, and Ullmann amination10, dominated by Cu- and Pd-based catalytic systems, have revolutionized this area by coupling of amines with aryl halides, pseudohalides or arylboronic acids. Indeed, limitations of these reactions exist, such as the use of pre-functionalized arenes, strong base, and elevated temperatures. In recent years, transition-metal catalysis has also shown great advantage in realizing direct C-H/N-H cross-coupling to facilitate arenes C-H amination without needing preinstallation of a leaving group11,12,13,14,15,16,17,18.

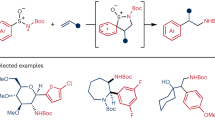
a Traditional transition metal-catalyzed C-N cross-coupling. b Prior work and our design: C-H amination of electron-poor nitroarenes via radical process. c This work: a transition metal-free, base/O2 mediated para-selective C-H/N-H amination of nitroarenes with complex amines.
However, these reactions largely require amines with directing groups and the use of stoichiometric sacrificial oxidants, and the undesired metal by-products remain an environmental concern19. Thus, direct amination of C–H bonds without activating or directing groups is considered as atom-economic and operationally efficient transformations. As a result, the pursuing of new strategies for direct C–H amination by C–H/N–H dehydrogenative coupling under mild metal-free conditions is certainly worthy.
Nitroarenes, as versatile building blocks, are obtained from the facile nitration of aromatic compounds and, thus, represent one of easy-to-access nitrogenous partners for the C-N coupling via radical process, which have received significant attention because of the step economy, ready availability, and easy manipulation. In addition, the direct transformation of the NO2 group has been an attractive option in cross-coupling chemistry20,21,22,23,24. Hence, synthetic methods to access privileged scaffold arylamines directly from nitroarenes are valuable.
In general, C-H amination of nitroarenes has shown uncommon through the formation of σH adducts by SNArH reaction, and simultaneously often afforded products with limited regioselectivity or predominant ortho-selectivity25,26,27,28. The development of new efficient strategies that rely on different functionalized coupling partners, particularly electron-poor nitroarenes, could enable access to synthetically challenging arylamines. More recently, direct radical C–H amination strategies have exhibited particular innovation29,30,31,32,33,34,35, but challenges remain with C–H amination of electron-poor nitroarenes due to the essence of the electron-deficient nitrogen radical. In 2019, Ritter36,37and Carreira38 independently reported the discovery of N-centered radical cations, which offer a direct pathway for the C-H amination of nitroarenes (Fig. 1b). However, polarity matching of radicals and arenes leads to a mixture of positional selectivity, resulting in the formation of ortho-, meta-, and para-substituted products. Thus, the approaches, realizing the straightforward site-selective synthesis of functionalized arylamines by using synthetically upstream nitroarenes with further efficient denitrative transformations of the NO2 group, are challenging but desirable20. For offering new options in step- and atom-economic organic synthesis, we expect that the direct C–H amination could be achieved directly from nitroarenes and simple amines for the C(sp2)-N coupling.
Herein, we report a synthetic protocol leading to the efficient and highly regioselective synthesis of 4-nitro-N-arylamines, which have been widely found in optoelectronic fields39, advanced materials40, and starting materials of conducting functional polymers41 (Fig. 1c). Under mild transition metal-free conditions, selective C(sp2)-H/N-H cross-coupling is realized through nitrogen radicals of complex arylamines and especially aliphatic amines. Compared with arylamines, alkylamines with the enhanced instability of their corresponding nitrogen radicals, often show much more challenging related reactivities and tend to translate into highly stabilized α-N carbon-radicals33.
Results
Optimization and Scope Elucidation
Initially, 1,2,3,4-tetrahydroquinoline 1a-1 and nitrobenzene 2a were chosen as model substrates to investigate suitable conditions for the regioselective para-site cross-coupling. We then try to optimize the reaction conditions, and it turned out that the reaction was observed to proceed only in DMSO and DMF among the investigated solvents. To our delight, the reaction could deliver the expected coupling product 3a-1 in 71% yield under O2 with DMSO as solvent and tBuONa as base (Supplementary Table 1). Next, we found that indoline derivatives 1b also reacted well with nitrobenzene 2a to give exclusively the para-substituted products 3b. We began with the reaction of indoline 1b-1 and 2a for the optimization of reaction. Solvents and bases screening showed that DMSO and tBuOK were still the best solvent/base system for this transformation to give 3b-1 in 52 % yield. However, we found that when DMF was used as solvent and tBuOK (4.0 equiv.) as base, lowering the reaction temperature to −30°C could enhance the yield of 3b-1 to 85% (Supplementary Table 10).
With the established optimum conditions in hand, we explored the substrate scope of amines derivatives 1. The reaction exhibited compatibility with a range of 1,2,3,4-tetrahydroquinoline derivatives 1a and indoline derivatives 1b as summarized in Fig. 2. Consequently, nitrobenzene C-H amination products 3a-1 to 3b-24 were obtained with moderate to high yields (30-94%) and exclusive para-regioselectivity. The aryl rings of 1a and 1b exhibited excellent tolerance towards various substituents, including electron-donating groups (-Me, -OMe, -OiPr, -OH) and electron-withdrawing groups (-F, -Cl, -Br, -COOH), resulting in the anticipated products. The presence of a methyl group at C2-C6 (3b-2-3b-8) led to a slight decrease in yield, particularly when positioned at C3 (45%, 3b-3) and C5 (58%, 3b-5). Figure 2 demonstrates the inability of 1a-26 with substituents at C8 to yield the product. However, the reaction between indole derivative 7-methylindoline 1b-24, bearing a methyl group at C7, and compound 2a resulted in the formation of product 3b-24 with a low yield of 30%. The ester group in 1a-25 underwent hydrolysis to form the carboxyl group under alkaline conditions, leading to the synthesis of product 3a-23 with a yield of 79%. Furthermore, a moderate yield of 54% was also achieved for the formation of 3b-23 from indole 1b-23. The relative configuration of 3a-6 and 3b-1 was unambiguously assigned by X-ray analysis of single crystal.

a Scope of 1,2,3,4-tetrahydroquinoline derivatives 1a. Reaction conditions A: 1 (0.5 mmol), 2a (4.0 equiv.), tBuONa (3.0 equiv.), DMSO (3.0 mL), O2 (1.0 atm) and the reaction was conducted at 40 °C. b Scope of indoline derivatives 1b. Reaction conditions B: 1 (0.5 mmol), 2a (3.0 equiv.), tBuOK (4.0 equiv.), DMF (5.0 mL), O2 (1.0 atm) and the reaction was conducted at −30 °C. c Scope of aniline derivatives 1c. d Scope of aliphatic amines 1d. Reaction conditions C: 1 (0.2 mmol), 2a (4.0 equiv.), tBuOK (3.0 equiv.), DMSO (3.0 mL), O2 (1.0 atm) and the reaction was conducted at 40 °C. e Substrates with no products. Isolated yield.
In an effort to broaden the substrate scope for amines, we are delighted to observe that primary amines such as 1- and 2-naphthylamines (1c-1 and 1c-2), acyclic N-methylanilines with various substituents (-Me, -OMe, -halogen, -ethynyl, -1H-pyrrol-1-yl, -CF3, -OCF3, -SMe, -carbonyl) on the phenyl ring as well as N-methylpyridin-2-amine 1c-9 and diphenylamine 1c-10 can also participate in reactions with 2a to yield para-nitroaryl amines 3c-1-3c-19 in high yields (Fig. 2c). However, the reactions involving other arylprimary amines yielded unsatisfactory results, with the prominent formation of azobenzene compounds observed as ones of defined by-products. The reaction proceeds effectively even when the methyl group of N-methylaniline is substituted with various aliphatic chains or functional groups (3c-20-3c-24).
The successful reaction of various simple aliphatic amines, such as secondary amines pyrrolidines 3d-5-3d-11 with multiple functional groups, morpholine 3d-2, thiolmorpholine 3d-3 and 1-methylpiperazine 3d-4, primary amine cyclohexylamine 3d-2, as well as N-methyl-1-phenylmethanamine 3d-13, highlights their potential for achieving desired products. The unexpected product 4-nitro-N-phenylaniline 6 g was obtained with a yield of less than 20%. It appears that the formation of 6 g can be attributed to the reduction of 2a to aniline 5d within the system, followed by its subsequent reaction with 2a.
Unfortunately, certain amines failed to yield the anticipated products. Even when subjected to reactions in DMF at −50 °C and −10 °C respectively, 8-methyl-1,2,3,4-tetrahydroquinoline 1a-26 and 9H-carbazole 1a-27 only underwent oxidative decomposition. The reaction did not proceed from 1,2,3,4-tetrahydroisoquinoline 1a-28 and even under elevated temperatures exclusively afforded isoquinoline as the product via oxidative dehydrogenation. To our surprise, we didn’t get the product from dibutylamine 1d-14 but only 6 g with 15% yield.
Nitroarenes, as privileged scaffold of chemical synthesis, are of great significance in organic synthesis. Thus, the substrate scope of nitrobenzene derivatives 2 with amines 1 was explored. Under mild conditions, the reaction has delivered diverse arylamines 4a-1-4a-20 in moderate to high yields (30-96%), showing high functional group tolerance (Fig. 3). Exclusive para-selective regioselectivity was achieved from various electron-donating (-Me, -OMe, -SMe) and electron-withdrawing groups (-F, -Cl, -Br, -CN, -NO2, -CF3, -CONH2, -COOMe), either at ortho- or meta-position of the phenyl rings. It is interesting to see that we could obtain the products 4a-3 and 4b-3 in moderate yields from the reaction of 3-nitrobenzonitrile with the cyclic amines (1a-1 and 1b-1), while similar reaction with 1c-1 only gave the desired product 4c-3 in 30% yield at −30 °C. Product 4b-10 was not formed from 1b-10 and 1,2-dinitrobenzene 2k, while 4a-10 and 4c-10 were observed in moderate yields. The boric acid group was eliminated from nitroboric acid 2a-19 during the reaction, leading to the formation of product 3a-1, while the ester group underwent alkaline hydrolysis, resulting in the formation of a hydroxyl group (4a-20). Furthermore, the reaction is incompatible with functional groups such as ketone, alcohol, and carboxylic acid. The structures of 4a-7, 4b-8, 4b-9 and 4c-10 were unambiguously confirmed by single-crystal X-ray analysis.

a Reaction conditions A: 1 (0.5 mmol), 2 (4.0 equiv.), tBuONa (3.0 equiv.), DMSO (3.0 mL), O2 (1.0 atm) and the reaction was conducted at 40 °C. b Reaction conditions B: 1 (0.5 mmol), 2 (3.0 equiv.), tBuOK (4.0 equiv.), DMF (5.0 mL), O2 (1.0 atm) and the reaction was conducted at -30 °C. c Reaction conditions C: 1a-1 (0.2 mmol), 2 (4.0 equiv.), tBuONa (6.0 equiv.), DMSO (3.0 mL), O2 (1.0 atm) and the reaction was conducted at 40 °C. d Isolated yield.
Mechanistic studies
Although a few nitroarene o- and p- aminations have been reported and were suggested to proceed by SNArH via the nucleophilic attack of the aminyl anion to thenitroarene, the mechanisms of these reactions should be studied in depth42,43. Although formally resembling an SNArH reaction, control experiments and radical clock experiments demonstrate that the reaction proceeds via a radical mechanism in the DMSO/tBuONa/O2 or DMF/tBuOK/O2 system. Further prove of the radical mechanism comes from the reaction of 4-phenyl-1,2,3,4-tetrahydroquinoline 5a with 2a, which gives the 1,4-di(4-nitrophenyl) substituted product 6a in 41% yield (Fig. 4a). Similar reaction of 3-methyl-4-phenyl tetrahydroquinoline 5b with 2a, on the other hand, affords only the product 6b in 75% yield, probably manifesting the sensibility of a radical reaction toward steric hindrance (Fig. 4b). At the same time, the reaction of 4-methyltetrahydroquinoline 5c with 2a could not proceed to give the 4-nitrophenyl product 6c (Fig. 4c). These facts clearly indicate that in the formation of product 6a, the process of deprotonation of the benzylic C-H bond followed by attack of the resulting carbanion to 2a is not involved. Meanwhile, a thermodynamic consideration showed that a proton transfer from the 4-benzylic C-H bond (pKa~33 in DMSO) to tetrahydroquinolinyl anion (pKa ~ 29.5 in DMSO)44 is not feasible. These results strongly disfavor a pathway involving nucleophilic attack by an aminyl anion but can be rationalized by a radical mechanism (Fig. 4d). Deprotonation/oxidation of 5a yielded the aminyl radical 6a-I, where thermodynamically favorable intramolecular 1,4-hydrogen atom shift from the 4-benzylic C-H bond (Bond dissociation energy (BDE) ~73 kcal/mol) to the 4-aminyl radical (BDE of C-H bond ~89 kcal/mol) furnished the 4-benzylyc radical 6a-II45. Recombination of 6a-II with 2a gave the primary product 6a-III, whose further similarly reaction with 2a led to the final product 6a. The reaction didn’t almost happen under N2 atmosphere after deoxygenation by lyophilization for five times (Fig. 4e, f). The reaction can give azobenzene 6d with aniline 5d from radical homo-coupling under standard conditions, indicating the nitrogen radical (Fig. 4g)46,47.

A Control experiments. a–d The essential role played by the N-H bonds of 1. e, f The crucial role of O2 in the reactions. g Radical homo-coupling of 5d. h The substitution of the nitro group with cyanide is not feasible. i Experiment in dark. j–l The impact of reactant stoichiometry and base type on the reaction. B Radical clock experiments. m The radical clock experiment of 5i with 2a and the possible process.
Considering the analogous characteristics shared by benzonitrile 5f and 2a, we subsequently investigated the reactivity of 5f with 1a-1. However, no product was obtained even under elevated temperatures (Fig. 4h). The comparison of critical data between their respective transition state intermediates is imperative for elucidating the role of the NO2 group. The NO2 group demonstrates a superior ability to stabilize negative charge in comparison to the CN group during C-N bond formation. Besides, both Mayer Bond Order (MBO) and electron density ρ of C…N bond in TS1 of 1a-1 surpass those in TS1c of 5f, indicating that TS1 has stronger C…N bond than TS1c, thereby accounting for the lower observed free energy in TS1 (Supplementary Fig. 12). It is noteworthy that this reaction is independent of visible light and can occur even under dark conditions (Fig. 4i). Moreover, revealing results have demonstrated that the specific alkali metal ions exert a substantial influence on reactions involving aliphatic amines. The reaction did not proceed as expected under standard conditions, resulting in the formation of an unexpected product 6e from 1d-1 (Fig. 4j). Upon modifying the standard conditions, another unexpected product 6 g was obtained in addition to 6e. It appears that the conversion of 2a into aniline 5d occurred, followed by a subsequent reaction between 5d and 2a to yield the desired para-substituted nitrobenzene C-H amination product 6 g (Fig. 4k). However, when tBuOK was used as the base instead of tBuONa (Fig. 4l), the reaction proceeded successfully to afford product 3d-1 along with compound 6g. To further validate the reaction mechanism, a radical clock experiment was conducted using N-cyclopropyl-3-methoxyaniline 5i and 2a to confirm the plausibility of a radical-mediated pathway (Fig. 4m). The expected product 6i-1 was determined by GC-MS in the system, resulting in the formation of 6i with a yield of 35%. We postulated that 5i could undergo SET to generate radical 5i-I, which subsequently decomposed into aniline 5j. The resulting aniline then engaged in a reaction with 2a to afford the desired product 6i. To elucidate the mechanistic pathway, control experiments were carried out. According to these experiment results, we further came to the conclusion that the reaction proceeds through a radical pathway (Supplementary Fig. 2).
To further investigate the reaction mechanism, confirmation experiments are designed by Electron Paramagnetic Resonance (EPR), further confirming a radical pathway (Fig. 5). When there wasn’t 2a, another competing reaction is that nitrogen radical tended to be easily transformed into aminoxyl radical (g = 2.0054, AN = 11.5 G) in the DMSO/tBuONa/O2 system through Korcek’s radical-trapping antioxidant (RTA)48, and was also obvious after reacting for 10 min. Furthermore, the absence of tBuONa or O2 resulted in no observation of any nitrogen radical, indicating that both the base and O2 are essential for the formation of nitrogen radical.

Reaction conditions: 1a-1 (0.5 mmol), tBuONa (3.0 equiv.), O2 (1.0 atm), react in DMSO (3.0 mL) for 10 min.
For further understanding of the reaction mechanism, density functional theory (DFT) was calculated (Fig. 6). As mentioned above, the reaction gave azobenzene 6d with aniline 5d from radical homo-coupling under standard conditions, indicating the nitrogen radical (Fig. 4g). We then took 1a-1 as an example to illustrate the possible mechanism of corresponding N-radicals (denoted as 1a-1-radical). Firstly, 1a-1 was combined with tBuONa to form complex int1(1a-1), then proton transfer occurred via Ts1(1a-1) to give int2(1a-1). It should be noticed that G(Ts1(1a-1)) is slightly lower than G(int2(1a-1)) but E(Ts1(1a-1)) is higher than E(int2(1a-1)), indicating this step is barrierless (Fig. 6a). Afterwards tBuOH was released to generate int3(1a-1), and O2(triplet) then oxidized int3(1a-1) into int4(1a-1)(triplet), which is a diradical.

a Relative Gibbs free energies (in kcal·mol−1): The free energies of 1a-1, 2a, O2 (triplet), and tBuONa were set to 0.0 kcal·mol−1 as a reference. b Proposed reaction mechanism.
Finally, 1a-1-radical was obtained by releasing \({{{{{\rm{Na}}}}}} ^+{{{{{\rm{O}}}}}}_2^{\,\,\cdot-}\). It can be seen that the formation of 1a-1-radical is only 7.3 kcal/mol endothermic, and the reaction free energy barrier is quite low. In the tBuONa/DMSO/O2 system, 1a-1-radical can attack both para and ortho positions of NO2 of 2a, resulting two transition states TS1 and TS1’ correspondingly. ΔG(TS1) is lower than ΔG(TS1’), indicating that para position attack is more preferred, resulting the INT1. Next step is the aromatization reaction of INT1, and \({{{{{\rm{Na}}}}}} ^+{{{{{\rm{O}}}}}}_2^{\,\,\cdot-}\) can abstract the H atom via TS2, resulting the INT2(triplet) state. Thus, INT2 (triplet) was converted into INT2 (singlet), and then released both NaO2H and 3a-1. After all, a possible mechanism is proposed (Fig. 6b).
Practical application
Triphenylamine derivatives, as organic electroluminescent materials, are of great potential for various optoelectronic applications49,50,51,52. However, their practical applications are still hampered by lack of efficient and convenient synthetic methods. The approach, realizing the straightforward synthesis of functionalized arylamines by using synthetically upstream nitroarenes, offers powerful synthetic options to construct diverse chemical bonds often found in, for example, advanced materials and pharmaceuticals by further efficient denitrative transformations of the NO2 group in cross-coupling chemistry20,21,22,23,24. With the feasibility of practical application of the approach, we then evaluated the scalability of the reaction by performing a gram-scale reaction. To our delight, the scaled-up reaction kept the high reaction efficiency and gave 3c-10 (1.20 g) from 1c-10 (1.20 g, 7.1 mmol) in 70% yield (Fig. 7a). Especially, the direct utilization of the synthetically upstream nitroarenes leads to step- and atom-economic access to the formation of C-C, C-N, and C-H bonds (Fig. 7b).

a Scaled-up reaction. b Denitrative transformations of the NO2 group.
Discussion
In summary, we have successfully developed a direct C-H and N-H dehydrogenative coupling reaction between amines and nitroaromatic compounds in DMSO/tBuONa/O2 or DMF/tBuOK/O2 system to provide an efficient and versatile synthetic method for para-nitroarylamine derivatives. The reaction proceeds by aminyl radicals coupling mechanism with the environmentally benign O2 as an oxidant under mild and transition metal-free conditions. Further advantages include the good functional tolerance and wide substrate scope in regard to both amines and nitroarenes, and exclusive para-regioselectivity. Mechanistic studies have demonstrated that in the DMSO/tBuONa(tBuOK)/O2 system, O2 (triplet) could deliver nitrogen radicals as an oxidant from Na-amide, thereby providing a mild and versatile approach to accessing synthetically significant N-centered radicals. Meanwhile, electron acceptor compounds such as nitroarenes are prone to transform into anion radicals in this system. These may open new reaction pathways via radical-radical anion recombination. Further applications of this reaction system are underway in our laboratories.
Methods
Reaction conditions A
A clean, oven-dried Schlenk tube with previously placed magnetic stir-bar was charged with 1,2,3,4-tetrahydroquinoline (66.6 mg, 0.5 mmol), nitrobenzene (246.3 mg, 2.0 mmol) and tBuONa (144.2 mg, 1.5 mmol) in dry DMSO (3 mL) solvent under argon atmosphere. The reaction was evacuated and back filled with O2 (1.0 atm) and this sequence was repeated for three additional times. The reaction mixture was vigorously stirred at 40°C and monitored by TLC. After the complete consumption of 1,2,3,4-tetrahydroquinoline, the reaction mixture was cooled to room temperature and then quenched with water (5 mL), diluted with ethyl acetate, and extracted with ethyl acetate (25 mL × 3). The combined organic phases were washed with brine (5 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by neutral Al2O3 column chromatography (PE: EtOAc = 50:1, v/v).
Reaction conditions B
A clean, oven-dried Schlenk tube with previously placed magnetic stir-bar was charged with nitrobenzene (184.7 mg, 1.5 mmol), tBuOK (224.5 mg, 2.0 mmol) in dry DMF (5 mL) solvent at room temperature under argon atmosphere. After the reaction mixture was stirred at −30 °C for 10 min, the reaction was evacuated and back filled with O2 (1.0 atm) and this sequence was repeated for three additional times. After indoline (59.6 mg, 0.5 mmol) was added, the reaction stirred at −30 °C and monitored by TLC. After the complete consumption of indoline, the reaction mixture was quenched with water (5 mL), diluted with ethyl acetate, and extracted with ethyl acetate (25 mL × 3). The combined organic phases were washed with brine (5 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by neutral Al2O3 column chromatography (PE: EtOAc = 40:1, v/v).
Reaction conditions C
A clean, oven-dried Schlenk tube with previously placed magnetic stir-bar was charged with pyrrolidine (27.3 mg, 0.3 mmol), tBuOK (109.8 mg, 0.90 mmol), nitrobenzene (147.6 mg, 1.20 mmol) in dry DMSO (3 mL) solvent at room temperature under argon atmosphere. The reaction was evacuated and back filled with O2 (1.0 atm) and this sequence was repeated for three additional times. The reaction mixture was vigorously stirred at 40°C and monitored by TLC. After complete conversion of pyrrolidine, the reaction mixture was restored to room temperature and then quenched with water. diluted with ethyl acetate, and extracted with ethyl acetate (25 mL × 3). The combined organic phases were washed with saturated NaCl aqueous solution. The combined organic phases were washed with brine (5 mL), dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (PE: EtOAc = 80:1, v/v).
Data availability
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2105132, 2105448, 2176176, 2176175, 2175172. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2105132 (3a-6), CCDC 2105448 (3b-1), CCDC 2176176 (4a-7), CCDC 2176175 (4b-8), CCDC 2175172 (4b-9) and CCDC 2181802 (4c-10). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. The data generated or analyzed during this study are included in this article and the supplementary information. The Cartesian coordinates are available from the Source Data. Details about materials and methods, experimental procedures, characterization data, computational details, and NMR spectra are available in the Supplementary Information. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Roughley, S. D. & Jordan, A. M. The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 54, 3451–3479 (2011).
Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 57, 10257–10274 (2014).
Goldberg, F. W., Kettle, J. G., Kogej, T., Perry, M. W. & Tomkinson, N. P. Designing novel building blocks is an overlooked strategy to improve compound quality. Drug. Discov. Today 20, 11–17 (2015).
Zhao, Y., Huang, B., Yang, C. & Xia, W. Visible-light-promoted direct amination of phenols via oxidative cross-dehydrogenative coupling reaction. Org. Lett. 18, 3326–3329 (2016).
Blakemore, D. C. et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394 (2018).
Ruiz-Castillo, P. & Buchwald, S. L. Applications of palladium-catalyzed C-N cross-coupling reactions. Chem. Rev. 116, 12564–12649 (2016).
Guram, A. S. & Buchwald, S. L. Palladium-catalyzed aromatic aminations with in situ generated aminostannanes. J. Am. Chem. Soc. 116, 7901–7902 (1994).
Paul, F., Patt, J. & Hartwig, J. F. Palladium-catalyzed formation of carbon-nitrogen bonds. reaction intermediates and catalyst improvements in the hetero cross-coupling of aryl halides and tin amides. J. Am. Chem. Soc. 116, 5969–5970 (1994).
Chan, D. M. T., Monaco, K. L., Wang, R.-P. & Winters, M. P. New N- and O-Arylations with Phenylboronic Acids and Cupric Acetate. Tetrahedron Lett. 39, 2933–2936 (1998).
Ullmann, F. Ber. Dtsch. Chem. Ges. 36, 2382–2384, doi:10.1002/cber.190303602174 (1903).& Ueber eine neue Bildungsweise von Diphenylaminderivaten.
Paudyal, M. P. et al. Dirhodium-catalyzed C-H arene amination using hydroxylamines. Science 353, 1144–1147 (2016).
Park, Y., Kim, Y. & Chang, S. Transition metal-catalyzed C-H amination: scope, mechanism, and applications. Chem. Rev. 117, 9247–9301 (2017).
Wang, H.-W. et al. Ligand-promoted rhodium(III)-catalyzed ortho-C-H amination with free amines. Angew. Chem. Int. Ed. 56, 7449–7453 (2017).
Zu, C., Zhang, T., Yang, F., Wu, Y. & Wu, Y. Copper (II)-catalyzed direct amination of 1-naphthylamines at the C8 site. J. Org. Chem. 85, 12777–12784 (2020).
Zhou, C. et al. Metal-free, redox-neutral, site-selective access to heteroarylamine via direct radical-radical cross-coupling powered by visible light photocatalysis. J. Am. Chem. Soc. 142, 16805–16813 (2020).
Kanemoto, K., Horikawa, N., Hoshino, S., Tokoro, Y. & Fukuzawa, S. I. Copper-catalyzed single C-H amination of 8-aminoquinoline-directed ferrocenes. Org. Lett. 23, 4966–4970 (2021).
Singh, H., Sen, C., Suresh, E., Panda, A. B. & Ghosh, S. C. C-H amidation and amination of arenes and heteroarenes with amide and amine using Cu-MnO as a reusable catalyst under mild conditions. J. Org. Chem. 86, 3261–3275 (2021).
Mandler, M. D. et al. Amination of nitro-substituted heteroarenes by nucleophilic substitution of hydrogen. Org. Lett. 24, 7643–7648 (2022).
Miyamoto, H. et al. Effective method to remove metal elements from pharmaceutical intermediates with polychelated resin scavenger. Org. Process Res. Dev. 19, 1054–1061 (2015).
Muto, K., Okita, T. & Yamaguchi, J. Transition-metal-catalyzed denitrative coupling of nitroarenes. ACS Catal. 10, 9856–9871 (2020).
Matsushita, N., Kashihara, M., Formica, M. & Nakao, Y. Pd-catalyzed etherification of nitroarenes. Organometallics 40, 2209–2214 (2021).
Kashihara, M. & Nakao, Y. Cross-coupling reactions of nitroarenes. Acc. Chem. Res. 54, 2928–2935 (2021).
Zou, D. et al. SET activation of nitroarenes by 2-azaallyl anions as a straightforward access to 2,5-dihydro-1,2,4-oxadiazoles. Nat. Commun. 12, 7060–7069 (2021).
Sil, S. et al. Reduced-phenalenyl-based molecule as a super electron donor for radical-mediated C–N coupling catalysis at room temperature. J. Am. Chem. Soc. 144, 22611–22621 (2022).
Ma̧kosza, M. & Białecki, M. Nitroarylamines via the vicarious nucleophilic substitution of hydrogen: amination, alkylamination, and arylamination of nitroarenes with sulfenamides. J. Org. Chem. 63, 4878–4888 (1998).
Khutorianskyi, V. V., Sonawane, M., Pošta, M., Klepetářová, B. & Beier, P. Oxidative nucleophilic aromatic amination of nitrobenzenes. Chem. Commun. 52, 7237–7240 (2016).
Wróbel, Z. K. Simple synthesis of N-Aryl-2-nitrosoanilines in the reaction of nitroarenes with aniline anion derivatives. Synthesis 2010, 3865–3872 (2010).
Bradley, W. & Robinson, R. 166. Kationoid reactivity of aromatic compounds. Part I. J. Chem. Soc. 1932, 1254–1263 (1932).
Alvarez, E. M. et al. Site-selective electrochemical arene C–H amination. J. Am. Chem. Soc. 146, 3591–3597 (2024).
Li, J. et al. Photocatalytic C–N bond construction toward high-value nitrogenous chemicals. Chem. Commun. 59, 14341–14352 (2023).
Gillespie, J. E., Morrill, C. & Phipps, R. J. Regioselective radical arene amination for the concise synthesis of ortho-Phenylenediamines. J. Am. Chem. Soc. 143, 9355–9360 (2021).
Svejstrup, T. D., Ruffoni, A., Juliá, F., Aubert, V. M. & Leonori, D. Synthesis of arylamines via aminium radicals. Angew. Chem. Int. Ed. 56, 14948–14952 (2017).
Ruffoni, A. et al. Practical and regioselective amination of arenes using alkyl amines. Nat. Chem. 11, 426–433 (2019).
Boursalian, G., Ham, W., Mazzotti, A. & Ritter, T. Charge-transfer-directed radical substitution enables para-selective C–H functionalization. Nat. Chem. 8, 810–815 (2016).
Romero, N. A., Margrey, K. A., Tay, N. E. & Nicewicz, D. A. Site-selective arene C-H amination via photoredox catalysis. Science 349, 1326–1330 (2015).
Ham, W. S., Hillenbrand, J., Jacq, J., Genicot, C. & Ritter, T. Divergent late‐stage (Hetero)aryl C−H amination by the pyridinium radical cation. Angew. Chem. Int. Ed. 58, 532–536 (2019).
D’Amato, E. M., Börgel, J. & Ritter, T. Aromatic C–H amination in hexafluoroisopropanol. Chem. Sci. 10, 2424–2428 (2019).
Rössler, S. L. et al. Pyridyl radical cation for C−H amination of arenes. Angew. Chem. Int. Ed. 58, 526–531 (2019).
Pocker, Y. & Spyridis, G. T. Electrostatic Modulation by Ionic Aggregates: Charge Transitions in Solutions of Lithium Perchlorate-Diethyl Ether. J. Am. Chem. Soc. 124, 7390–7394 (2002).
Ma, Y. et al. Fluorescent zinc(II)-based metal–organic frameworks for nitroaromatics sensing. N. J. Chem. 42, 5162–5167 (2018).
Asahara, H. & Nishiwaki, N. Tailor-made synthesis of N, N, 2,6-Tetrasubstituted 4-nitroanilines by three-component ring transformation of dinitropyridone. Eur. J. Org. Chem. 2015, 1203–1206 (2015)
Mąkosza, M. Reactions of nucleophiles with nitroarenes: multifacial and versatile electrophiles. Chem. Eur. J. 20, 5536–5545 (2014).
Błaziak, K., Danikiewicz, W. & Mąkosza, M. How does nucleophilic aromatic substitution really proceed in nitroarenes? Computational prediction and experimental verification. J. Am. Chem. Soc. 138, 7276–7281 (2016).
Bordwell, F. G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 21, 456–463 (1988).
Bordwell, F. G., Zhang, X.-M. & Cheng, J.-P. Bond dissociation energies of the N-H bonds in anilines and in the corresponding radical anions. equilibrium acidities of aniline radical cations. J. Org. Chem. 58, 6410–6416 (1993).
Grirrane, A., Corma, A. & García, H. Gold-catalyzed synthesis of aromatic Azo compounds from anilines and nitroaromatics. Science 322, 1661–1664 (2008).
Cai, S. et al. Room temperature activation of oxygen by monodispersed metal nanoparticles: oxidative dehydrogenative coupling of anilines for azobenzene syntheses. ACS Catal. 3, 478–486 (2013).
Jensen, R. K., Korcek, S., Zinbo, M. & Gerlock, J. L. Regeneration of amine in catalytic inhibition of oxidation. J. Org. Chem. 60, 5396–5400 (1995).
Mishra, S. & Singh, A. K. Optical sensors for water and humidity and their further applications Coord. Chem. Rev. 445, 214063 (2021).
Gu, Y., Son, S. U., Li, T. & Tan, B. Low-cost hypercrosslinked polymers by direct knitting strategy for catalytic applications. Adv. Funct. Mater. 31, 2008265 (2021).
Farokhi, A., Shahroosvand, H., Monache, G. D., Pilkington, M. & Nazeeruddin, M. K. The evolution of triphenylamine hole transport materials for efficient perovskite solar cells. Chem. Soc. Rev. 51, 5974–6064 (2022).
Liu, X., Zhu, C. & Tang, B. Z. Bringing inherent charges into aggregation-induced emission research. Acc. Chem. Res. 55, 197–208 (2022).
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21861026, H.C. and 22075123, H.C.). Especially, we are very grateful to Prof. Dr. Jianhua Xu at the Institute of Chemistry and Chemical Engineering, Nanjing University, for his suggestions on the reaction mechanism.
Author information
Authors and Affiliations
Contributions
Z.Z. designed and carried out the experiments, and did the theoretical calculations, and analyzed the data and wrote the manuscript. S.Y. carried out the experiments under the supervision of Z.Z. and H.C. B.J., R.Y., T.Z., and L.S. carried out part of the experiments. S.W. analyzed the EPR data. A.L. and H.C. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Z., Yue, S., Jin, B. et al. Para-selective nitrobenzene amination lead by C(sp2)-H/N-H oxidative cross-coupling through aminyl radical. Nat Commun 15, 4186 (2024). https://doi.org/10.1038/s41467-024-48540-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48540-6
- Springer Nature Limited