

Abstract
While electrochemical N2 reduction presents a sustainable approach to NH3 synthesis, addressing the emission- and energy-intensive limitations of the Haber-Bosch process, it grapples with challenges in N2 activation and competing with pronounced hydrogen evolution reaction. Here we present a tandem air-NOx-NOx−-NH3 system that combines non-thermal plasma-enabled N2 oxidation with Ni(OH)x/Cu-catalyzed electrochemical NOx− reduction. It delivers a high NH3 yield rate of 3 mmol h−1 cm−2 and a corresponding Faradaic efficiency of 92% at −0.25 V versus reversible hydrogen electrode in batch experiments, outperforming previously reported ones. Furthermore, in a flow mode concurrently operating the non-thermal plasma and the NOx− electrolyzer, a stable NH3 yield rate of approximately 1.25 mmol h−1 cm−2 is sustained over 100 h using pure air as the intake. Mechanistic studies indicate that amorphous Ni(OH)x on Cu interacts with hydrated K+ in the double layer through noncovalent interactions and accelerates the activation of water, enriching adsorbed hydrogen species that can readily react with N-containing intermediates. In situ spectroscopies and density functional theory (DFT) results reveal that NOx− adsorption and their hydrogenation process are optimized over the Ni(OH)x/Cu surface. This work provides new insights into electricity-driven distributed NH3 production using natural air at ambient conditions.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
With an annual production of more than 175 million tonnes, ammonia (NH3) synthesized by the Haber-Bosch process at elevated temperatures and pressures has underpinned the population boom over the past century and is considered a promising carbon-neutral energy vector1,2. The huge energy consumption and carbon footprint of the Haber-Bosch process call for sustainable NH3 production methods that can operate under ambient conditions and make use of renewable energy sources3,4. One such alternative—electrochemical N2 reduction (eNRR)—has gained a surge of attention by its potential for decarbonizing NH3 production5,6,7,8. While encouraging, eNRR in aqueous solution generally encounters poor NH3 yield rate (~10−1 nmol s−1 cm−2) as a result of high activation barrier of stable N ≡ N bond and low solubility of N29. Additionally, severe hydrogen evolution reaction (HER) outcompetes eNRR, resulting in a Faradaic efficiency (FE) of less than 1%. Compared to eNRR, electrocatalytic NOx− reduction reactions (eNOx−RR, x = 2, 3) are more likely to occur because of their high solubility and lower activation energy of N = O bond (204 kJ mol−1) than that of N ≡ N bond (945 kJ mol−1)10,11,12. Furthermore, eNOx−RR is thermodynamically more favorable than HER and thus can proceed in a wide potential window without HER interference13. Therefore, efficient electrocatalytic synthesis of NH3 is expected to be achieved by converting stable N2 into more reactive NOx−, followed by an electrochemical reduction process14,15.
Inspired by natural lightning, non-thermal plasma (NTP)-enabled N2 oxidation reaction (pNOR) has been recently studied in the field of electrocatalytic NH3 synthesis16,17,18,19,20. For example, a spark discharge NTP was used to oxidize N2 into NOx, then the generated NOx species were captured to obtain an electrolyte containing NOx−, which underwent electrochemical reduction to synthesize NH319. Owing to its favorable NH3 selectivity, Cu-based catalysts are frequently employed in eNOx−RR19,20. Although eNOx−RR are not limited by reactant activation, the mismatch between fast electron transfer and sluggish proton supply over Cu restricts the NH3 synthesis efficiency in this pNOR-eNOx−RR tandem system. This arises due to the requirement of an alkaline solution for effective absorption of the NOx generated from pNOR. Within this alkaline environment, the H2O dissociation process on Cu, responsible for generating adsorbed hydrogen species (Had) involved in numerous deoxygenation and hydrogenation steps of NOx−, lags significantly behind the rapid NOx− consumption process21,22,23,24. Given that 9 and 7 protons are involved in eNO3−RR and eNO2−RR, respectively, a large overpotential is required to reach a dynamic equilibrium between the generation of Had and its timely consumption. Under such potentials, the competitive adsorption of H2O with NOx− on Cu will deteriorate the NH3 selectivity25. In addition, large overpotentials will lead to unsatisfactory energy efficiency for eNOx−RR due to more energy loss. In this context, improving the Had supply efficiency on the Cu surface at low overpotentials to meet the demand of the hydrogenation process is crucial but remains challenging.
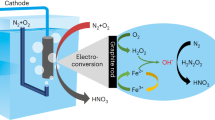
Herein, we demonstrate an efficient tandem NH3 synthesis route of pNOR-eNOx−RR by combining a spark discharge NTP for air-to-NOx conversion with Ni(OH)x/Cu-catalyzed NOx−-to-NH3 electroreduction. Kinetic isotopic effect (KIE) evaluation, electron paramagnetic resonance (EPR) measurement, molecular dynamics (MD) simulations, DFT, and in situ Raman spectra results reveal that the deposition of Ni(OH)x induces improved water activation and NOx− adsorption at the interface between Ni(OH)x and Cu. The enriched Had cater to the hydrogenation needs of nitrogenous intermediates on the Cu surface, enabling Ni(OH)x/Cu to achieve efficient eNOx−RR toward NH3 synthesis at low overpotentials. Further coupled with the optimized pNOR, the pNOR-eNOx−RR tandem system delivers a record NH3 yield rate of 3 mmol h−1 cm−2 with FE of 92% at −0.25 V vs. RHE in batch experiments, ranking among the highest performances reported to date. More significantly, in a flow mode concurrently operating the pNOR and the eNOx−RR using pure air as the feeding gas, a stale NH3 yield rate of ca. 1.25 mmol h−1 cm−2 is achieved over 100 h, culminating in the production of high-purity solid NH4Cl and liquid NH3 solution through an air stripping method. This work not only offers a strategy to develop superior eNOx−RR electrocatalysts but also provides insights for sustainable and distributed NH3 synthesis leveraging atmospheric nitrogen and renewable electricity.
Results and discussion
Catalyst synthesis and characterization
The synthetic procedure of the Ni(OH)x/Cu electrode is illustrated in Fig. 1a. Firstly, Cu foam was chemically oxidized by (NH4)2S2O8 in an alkaline solution and then calcinated in the air to obtain CuO nanowire array (NWA)26. Ni(OH)x was deposited on the CuO NWA surface by rinsing CuO NWA into a NiCl2 solution at open circuit potential (Supplementary Fig. 1)27,28. According to the Pourbaix diagrams of Cu and Ni, a cyclic voltammetry (CV) prereduction process within the potential range from −0.3 to 0.2 V in 1 M KOH with 0.1 M NO3− was then performed to obtain Ni(OH)x/Cu NWA (Supplementary Fig. 2). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images show that Ni(OH)x/Cu NWA well inherit the micrometer-long nanowire morphology from the CuO NWA and is evenly distributed on the Cu foam skeleton (Fig. 1b and Supplementary Figs. 3, 4). Element mapping images (Supplementary Figs. 5, 6) indicate the homogeneous distribution of Cu, Ni, and O over porous Ni(OH)x/CuO and Ni(OH)x/Cu nanowires. In the high-resolution TEM image of Ni(OH)x/Cu, a clear interface is observed between an amorphous layer and a crystalline phase (Fig. 1c). A lattice spacing of 0.208 nm is recognized in the crystalline zone and attributed to the (1 1 1) facet of Cu. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) combined with energy-dispersive X-ray spectroscopy further manifests the external coverage of nickel species on the Cu surface (Fig. 1d). As shown in Fig. 1e, the Raman signals appeared at 150, 218, 302, and 630 cm−1 which can be assigned as Cu oxides vanish after the CV prereduction process, indicating the reduction of Cu oxides to a metallic state29,30. Cu oxides reduction can be further confirmed by X-ray diffraction (XRD) patterns (Supplementary Fig. 7), Cu 2p spectra, and Cu LMM Auger spectra over Ni(OH)x/CuO and Ni(OH)x/Cu electrodes (Supplementary Fig. 8a, b).

a Schematic diagram of the synthetic route for Ni(OH)x/Cu. b SEM image. c High-resolution TEM image of Ni(OH)x/Cu. d HAADF STEM-EDS mapping of Ni(OH)x/Cu. e Raman spectra of Ni(OH)x/CuO and Ni(OH)x/Cu. f Ni K-edge XANES and (g) Fourier-transformed EXAFS spectra of Ni foil, Ni(OH)x/Cu, and Ni(OH)2.
To detect the composition of the deposited nickel species over the Cu surface, we carried out various spectroscopic tests. In the high-resolution Ni 2p XPS spectra of Ni(OH)x/CuO and Ni(OH)x/Cu, two prominent peaks at bind energy of 855.9 and 873.4 eV can be assigned to Ni 2p3/2 and Ni 2p1/2 of Ni2+ spin-orbit doublets and those at 861.5 and 879.2 eV are ascribed to two accompanying satellites (Supplementary Fig. 8d)31. The O 1s spectra exhibit a clear peak at 531.5 eV, implying that nickel species exist as hydroxide (Supplementary Fig. 8c)32. Raman spectra of Ni(OH)x/CuO and Ni(OH)x/Cu nanowire were obtained by prolonging the Ni(OH)x deposition time to a detectable level (Supplementary Fig. 9). The bands at 457 and 500 cm−1 are attributable to lattice stretching modes of Ni-OH and Ni-O, respectively. The symmetric O-H stretching mode of the free external -OH group is observed at 3690 cm−1 33,34. Additionally, the selected area electron diffraction of Ni(OH)x/Cu only shows the polycrystalline nature of Cu without any detection of crystalline Ni(OH)2 (Supplementary Fig. 10), suggesting that the deposited nickel species exist in an amorphous structure. The fine structure of the deposited Ni(OH)x was investigated via X-ray adsorption spectroscopy. As shown in Fig. 1f and Supplementary Fig. 11, Ni(OH)x/Cu exhibits a very small edge shift in Ni K-edge X-ray absorption near edge structure (XANES) compared with the Ni(OH)2 reference, indicating the valence state of Ni in Ni(OH)x/Cu is slightly smaller than +2. The Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectrum (Fig. 1g) and fitting results (Supplementary Fig. 12 and Supplementary Table 1) show the presence of two distinct peaks at ~1.6 and ~2.7 Å, attributable to Ni-O and Ni-Ni bonds of Ni(OH)235,36. The nearest neighbor Ni-O and Ni-Ni bond distances of Ni(OH)x/Cu are 2.045 ± 0.001 Å and 3.113 ± 0.001 Å, respectively. These values closely match those of Ni-O and Ni-Ni bond distances in the Ni(OH)2 standard (2.050 ± 0.001 Å and 3.119 ± 0.007 Å). Yet, the coordination numbers (CN) of Ni-O (4.6 ± 0.3) and Ni-Ni (4.8 ± 0.7) are reduced compared to those of the crystalline Ni(OH)2 (CN of 6 for both Ni-O and Ni-Ni), which further confirms its disordered structure. Taken together, we conclude that the deposited nickel species on the Cu surface is amorphous Ni(OH)x, rather than Ni, NiO, or CuNi alloy. Other control samples, including Cu, Ni(OH)2, and Ni(OH)x/Cu with different Ni(OH)x deposition time, were also prepared, characterized, and evaluated (Supplementary Figs. 7, 13, and 14).
Evaluation of electrocatalytic performance in model electrolytes
Having established the structural properties of Ni(OH)x/Cu electrocatalyst, we then evaluated its eNO3−RR performance in a model electrolyte of 1 M KOH with 0.1 M NO3−. Given the presence of NO3− and NO2− in the pNOR-eNOx−RR system, and with NO2− serving as an intermediate in the eNO3−RR process, it is more representative to direct our focus onto eNO3−RR. The amounts of reactant (NO3−) and products (NH3 and NO2−) were quantified using ion chromatography and colorimetric methods, respectively (Supplementary Figs. 15−17). All the potentials were reported versus RHE unless otherwise stated (Supplementary Fig. 18).
The linear sweep voltammetry (LSV) polarization curves in Fig. 2a and Supplementary Fig. 19 reveal that, in the presence of NO3−, eNO3−RR exhibits greatly enhanced current density when compared to the HER LSV curve in the absence of NO3− over Cu, Ni(OH)2, and Ni(OH)x/Cu. Ni(OH)x/Cu shows both higher HER and eNO3−RR activities than those of the Cu electrode. At overpotentials less than −0.2 V, Cu shows no more than 60% of NH3 FE, which is consistent with previous reports that Cu exclusively catalyzes the conversion of NO3− to NO2− at low overpotential (Fig. 2b and Supplementary Fig. 20)12. In contrast, with a maximum FE of 91.6%, Ni(OH)x/Cu demonstrates superior NH3 yield rates than Cu and Ni(OH)2 counterparts after electrochemically active surface area (ECSA)-normalization, indicating the superior intrinsic activity of Ni(OH)x/Cu (Fig. 2b, c and Supplementary Figs. 21, 22). Figure 2d shows that the partial current density for NH3 production of Ni(OH)x/Cu reaches 639.6 mA cm−2 at −0.3 V, which is two times and three orders of magnitude higher than that of Cu and Ni(OH)2, respectively. Interestingly, the eNO3−RR performances of Ni(OH)x/Cu electrodes are varied with the coverage of deposited Ni(OH)x and exceed that of the Ni(OH)x/CuO electrode (Supplementary Figs. 23−25 and Supplymentary Table 2). The isotope labeling experiment using K15NO3 as the reactant was conducted to eliminate potential ammonium contamination from interfering with the results. As shown in Fig. 2e, when using 15NO3− as the nitrogen source, only doublet peaks of 15NH4+ are detected in the 1H nuclear magnetic resonance (NMR) spectra of the electrolyte without seeing any triple coupling peaks of 14NH4+. The batch NO3−-to-NH3 conversion capacity of Ni(OH)x/Cu was evaluated by prolonging the electrolysis time. With 99% selectivity of NO3−-to-NH3 and an overall FE of more than 98%, almost complete transformation can be achieved within 80 min (Supplementary Fig. 26). The appearance and disappearance of NO2− during the reaction indicate that it is an intermediate product and can be further reduced to NH3. Such an outstanding eNO3−RR activity of the Ni(OH)x/Cu was also assessed in the electrolytes with varied NO3− concentrations (Supplementary Fig. 27). As shown in Fig. 2f, Ni(OH)x/Cu exhibits over 80% FE of NH3 and over 0.75 of selectivity in the NO3− concentration range from 5 mM to 1 M, indicating NH3 synthesis is the main reaction. However, when the concentration diminishes to 1 mM, the FE drops to 70%, which could be attributed to the intensified HER at low NO3− concentration. At an extremely high NO3− concentration of 2 M, NH3 production will be unfavorable due to the insufficient hydrogenation of NO3−. The results from 15 independent experiments and 10 cyclic tests demonstrated good repeatability and stability of the Ni(OH)x/Cu catalyst (Supplementary Figs. 28, 29). In addition, the long-term stability of Ni(OH)x/Cu was also evaluated by chronopotentiometry in an H-type flow cell with continuous electrolyte flow to replenish the constantly consumed NO3− (Supplementary Fig. 30)12. The voltage for maintaining the current of 400 mA cm−2 remains stable over 25 h, and the FE of NH3 reaches a plateau of 90%, indicating the outstanding stability of the Ni(OH)x/Cu. SEM, XRD, and TEM results reveal that the structure and composition of Ni(OH)x/Cu remain intact after the long-term test (Supplementary Fig. 31). In addition, the eNO2−RR activity of Ni(OH)x/Cu also surpasses those of Cu and Ni(OH)2 (Supplementary Figs. 32, 33). Taken together, the Ni(OH)x/Cu catalyst can be used in the tandem pNOR-eNOx−RR system as an efficacious and stable catalyst for the eNOx−RR process.

a LSV curves of Cu, Ni(OH)2, and Ni(OH)x/Cu in 1 M KOH with and without 0.1 M NO3− under 400 r.p.m. without iR correction. Potential-dependent FEs of NH3 (b), NH3 yield rates (c), and NH3 partial current densities (d) over Cu, Ni(OH)2, and Ni(OH)x/Cu. e NMR spectra of the 14NH4+ and 15NH4+ references and the electrolyte after eNO3−RR over Ni(OH)x/Cu at −0.25 V vs. RHE using 14NO3− and 15NO3− as the nitrogen source. f The FE of NH3, NH3 yield rate, and selectivity (the ratio of the generated NH3 concentration [NH3] to the consumed NO3− concentration [NO3−]) over Ni(OH)x/Cu at −0.25 V vs. RHE at NO3− concentration in the range from 1 mM to 2 M. The error bars represent the standard deviation from at least three independent measurements.
Mechanism insight
In the alkaline media, the performance of eNO3−RR on Cu strongly dependent on Had that generated from water dissociation, which participate in both the deoxygenation and hydrogenation steps of adsorbed NO3− 22,23. Due to endothermic water adsorption and sluggish kinetics of water dissociation on Cu, eNO3−RR over Cu predominantly produces NO2− at low overpotentials (Supplementary Fig. 20)12. While at more negative overpotentials of HER region, high FE of NH3 can be obtained on Cu (Supplementary Fig. 34). Interestingly, the eNO3−RR performances of the Ni(OH)x/Cu electrodes show a correlation with their HER activities (Supplementary Fig. 23). Based on these observations, we hypothesize that the improved eNO3−RR performance on Ni(OH)x/Cu is due to its enhanced water activation.
We first performed kinetic isotopic effect (KIE) evaluation, a well-established method for studying reactions involving protons by substituting hydrogen with deuterium37,38,39. Cathode shifts are observed over the LSV curves of HER and eNO3−RR on Cu and Ni(OH)x/Cu because of the more sluggish dissociation kinetics of D2O than that of H2O (Fig. 3a, b)40. The Tafel slope for HER over Ni(OH)x/Cu is 175 mV/dec, which is smaller than that of Cu (275 mV/dec), suggesting that the Volmer step (H2O dissociation) is the rate-determining step in alkaline HER and more efficient H2O dissociation kinetics over Ni(OH)x/Cu (Supplementary Fig. 35)41. Additionally, the values of cathode shift for LSV cureves of HER and eNO3−RR over Ni(OH)x/Cu are smaller than those over Cu, indicating that the accelerated H2O dissociation kinetics makes the Ni(OH)x/Cu electrode less restricted by the isotope substitution (Supplementary Fig. 36).

Comparison of the LSV polarization curves over Cu and Ni(OH)x/Cu in electrolytes using H2O or D2O as the solvent without (a) and with (b) adding 0.1 M NO3−. c KIE of H/D for NH3 synthesis over Cu and Ni(OH)x/Cu and the corresponding FE of NH3 in the presence of 0.1 M NO3−. DMPO-involved EPR spectra of Cu (d) and Ni(OH)x/Cu (e) at different overpotentials in the absence of NO3−. f Effect of alkali metal cations on the eNO3−RR performance at –0.25 V vs. RHE over Cu and Ni(OH)x/Cu. Density profiles of Li+, Na+, K+, and Cs+ over the Cu (g) and the Ni(OH)x/Cu (h). i Gibbs free energy diagram of H2O adsorption and dissociation on Cu and Ni(OH)x/Cu surface in the presence of different alkali metal cation hydrates. The error bars represent the standard deviation from at least three independent measurements.
To gain further insights into the role of the activation of H2O in NO3− reduction, we carried out studies on the KIE of H/D over Cu and Ni(OH)x/Cu. The KIEs of H/D, defined as the ratio of NH3 yield rate in H2O and D2O, are calculated to be 5.15 and 1.96 for Cu and Ni(OH)x/Cu, respectively (Fig. 3c and Supplementary Fig. 37). These KIE values are characteristics of the primary kinetic isotope effect, implying that the H-OH bond breaking is involved in the rate-determining step for NO3− reduction to NH338. With a more sluggish proton supply, Cu only shows 23.4% of NH3 FE in the D2O media. By contrast, the smaller KIE value and the well-retained FE of NH3 (72.2%) over Ni(OH)x/Cu in D2O indicates that the eNO3−RR is less limited by H2O dissociation with the presence of surficial Ni(OH)x.
To compare the amount of Had on the surface of Cu and Ni(OH)x/Cu qualitatively, CV curves in the Ar-saturated 1 M KOH solution were recorded at a potential range of −0.3−0.05 V (Supplementary Fig. 38)40. The Had desorption peak of Ni(OH)x/Cu in the potential range of −0.2 ~ −0.1 V is more significant than that of Cu, suggesting the existence of more Had on the surface of Ni(OH)x/Cu. We further performed EPR measurement using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the Had trapping reagent to verify the generation of Had directly. A typical DMPO-H spin adduct signal pattern consisting of nine EPR peaks with an intensity ratio of 1:1:2:1:2:1:2:1:1 is observed in Fig. 3d, e13. The EPR signals of Ni(OH)x/Cu are much more intense than that of Cu, implying Ni(OH)x indeed improves Had formation. However, no signals of DMPO-H adduct are observed in the presence of 0.1 M NO3−, suggesting the generated Had is fast consumed by the hydrogenation process of adjacent N-containing intermediates, which is consistent with the results of the Had quenching test by adding DMPO (Supplementary Figs. 37, 39).
Note that anion-hydrated cation networks (Xδ−-M+(H2O)n, where n refers to the number of ionic hydrations) can be formed in the double layer through non-covalent Coulomb interactions between surface-adsorbed anionic species (e.g., F−, Sδ−) and hydrated cations in the electrolyte, thus enhancing H2O activation to generate Had38. To validate whether a similar promotion effect is involved in our reaction, we investigated the impact of different alkali metal cations in MOH and MNO3 (M = Li, Na, K, and Cs) electrolytes on eNO3−RR performance (Fig. 3f). Even though the impact of the alkali cation over Cu is very limited due to its weak interaction with hydrated cation, the NH3 yield rate over Ni(OH)x/Cu increases from 1.79 to 2.7 mmol h−1 cm−2 on changing the alkali cation from Li+ to K+, and decreases to 1.3 mmol h−1 cm−2 in the presence of Cs+. Since the cation hydrate’s structure and behavior can be varied with parameters like pH, potential on the electrode, and so on, classic molecular dynamics (MD) simulations were performed to gain a molecular perspective on this cation effect42,43. As displayed in Fig. 3g, h, the z-axial cation number density profiles from MD simulations revealed increasing peak intensity in the order of Li+ < Na+ < K+ < Cs+, indicating the greater willingness of large, weakly solvated cations to approach the electrode surface. The peak centers for Li+, Na+, K+, and Cs+ cations on the Ni(OH)x/Cu (6.4 Å, 6.9 Å, 4.9 Å, and 2.9 Å) are closer to the surface than those on Cu (6.8 Å, 7.2 Å, 5.5 Å, and 3 Å). In addition, the number densities corresponding to the highest peak for Li+, Na+, K+, and Cs+ on the Ni(OH)x/Cu (1.2, 1.3, 1.7, and 2.8) are larger than those of on the Cu (1.0, 1.1, 1.5, and 2.5). These results suggest that Ni(OH)x species can attract cations close to the electrified interface. We then calculated the Gibbs free energy of H2O adsorption and dissociation processes on Cu and Ni(OH)x/Cu in the presence of different alkali metal cation hydrates (Fig. 3i and Supplementary Figs. 40, 41). The water coordination numbers were set as 4, 5, and 7 for Li+, Na+, and K+ cations, respectively, based on the integration of cation−O (in H2O) radial distribution functions in Supplementary Fig. 42. Cs+ was excluded from the DFT calculations due to they mainly distribute at a distance of 3 Å from the surface, where they undergo physisorption or chemisorption on the catalyst surface and are expected to influence the eNO3−RR activity in a very different way (Supplementary Fig. 43). On the Cu surface, the Gibbs free energies for water adsorption (* → *H2O) in the presence of Li+(H2O)4, Na+(H2O)5, and K+(H2O)7 are −0.058 eV, −0.14 eV, and −0.282 eV, respectively, whereas the subsequent water dissociation process on Cu (*H2O → *OH-*H) delivers Gibbs free energy uphill of 0.205 eV, 0.436 eV, and 0.668 eV. These results demonstrate that water adsorption on the Cu surface becomes more favorable with the change of the cation from Li+ to K+, while the water dissociation process becomes more unfavorable. Thus, these two opposite influences eventually make the cation effect on the generation of Had on the Cu surface very limited. On the Ni(OH)x/Cu surface, the Gibbs free energies of water adsorption are −0.197 eV, −0.245 eV, and −0.359 eV in the presence of Li+(H2O)4, Na+(H2O)5, and K+(H2O)7, respectively. The subsequent water dissociation process on Ni(OH)x/Cu delivers Gibbs free energy uphill of 0.124 eV, 0.115 eV, and 0.104 eV. Compared with Cu, Ni(OH)x/Cu shows more Gibbs free energy downhill of the water adsorption process and less Gibbs free energy uphill of the water dissociation process in the presence of the same cation hydrate, indicating that the water adsorption and dissociation on Ni(OH)x/Cu are more favorable. Notably, the Gibbs free energy for water adsorption and the energy barrier for water dissociation on Ni(OH)x/Cu both decreases in the order of Li+ > Na+ > K+, indicating that the water adsorption and dissociation processes become more favorable with increasing cation size. This cation effect is well aligned with the experimentally observed cation-dependent HER activities (Supplementary Fig. 44).
To uncover the reaction pathway of eNO3−RR to NH3, the reaction intermediates were monitored via in situ electrochemical attenuated total reflection surface-enhanced infrared spectroscopy (ATR-SEIRAS) (Supplementary Fig. 45). The absorption peak that appeared at 1190 cm−1 is assignable to -N-O- stretching vibration of hydroxylamine (NH2OH)44. We then performed DFT calculations for eNO3−RR toward NH3 on Cu and Ni(OH)x/Cu surface. The structural models in this work are shown in Supplementary Fig. 46. Figure 4a shows that all the Gibbs free energies of intermediates on the Ni(OH)x/Cu surface are smaller than those on the Cu surface, which means that the eNO3−RR activity on Ni(OH)x/Cu is superior. On pure Cu, the first NO3− adsorption step is the potential-determining step (PDS), of which the maximum free energy uphill reaches 0.49 eV. In contrast, Ni(OH)x/Cu with a free energy downhill of −0.27 eV is more likely to adsorb NO3−. The PDS is changed from the NO3− adsorption to the *NH2 protonation step (0.44 eV) upon introducing Ni(OH)x. Additionally, the hydrogenation process of *NO → *NHO is more likely to occur over Ni(OH)x/Cu (−0.63 eV) than Cu (0.13 eV), thus the reaction is facilitated over Ni(OH)x/Cu. The facilitated adsorption of intermediates on Ni(OH)x/Cu can be evidenced by the in situ Raman spectra, where the intensity of signals designated as the symmetric stretch of *NO3, N-O stretch with nitrito binding intermediates, and antisymmetric stretch of H-N-H in NH3 are more significant over Ni(OH)x/Cu (Fig. 4b and Supplementary Fig. 47)45.

a Gibbs free energy diagram of eNO3−RR on Cu and Ni(OH)x/Cu. b Potential-dependent in situ Raman contour maps of Cu and Ni(OH)x/Cu obtained during eNO3−RR in the potential range from 0.2 to −0.3 V vs. RHE. c Schematic illustration for the role of Ni(OH)x species in promoting water activation and N-containing intermediates hydrogenation on Cu.
Based on the above results and analysis, we propose that the Ni(OH)x species on Cu enrich hydrated cations (K+(H2O)n) near the catalyst surface in the double layer through noncovalent interactions. The near-surface H2O molecular can be facilely adsorbed and dissociated at the Ni(OH)x − Cu interface, forming Had to participate in the following hydrogenation steps of N-containing intermediates adsorbed on Cu toward NH3 (Fig. 4c).
Evaluation of the performance of air-to-NH3 conversion in the pNOR-eNOx −RR tandem system
In light of the above findings, we further evaluated the NH3 production performance of the pNOR-eNOx−RR tandem system with Ni(OH)x/Cu cathode using air as the source of nitrogen (Fig. 5a). In pNOR, we applied a 220 V source (AC) to initiate a neon-sign transformer to output a high voltage upon the spark discharge NTP (Supplementary Figs. 48, 49). After switching on the discharge, the color of the gas mixture turned brown, indicating the generation of NO2 (Supplementary Fig. 49c)19. The mass spectral signals of NO and NO2 in the air are almost undetectable yet become stronger after the discharge, indicating that NO and NO2 are indeed produced from the discharge process rather than from the feeding gas (Supplementary Fig. 50). Then, these produced NO and NO2 can evolve into NO2− and NO3− in the alkaline electrolyte through reactions as Eqs. (1, 2).

a Schematic of pNOR-eNOx−RR system and NH3 products collection. b Discharge time-dependent NH3 yield rate and FE over Ni(OH)x/Cu at −0.25 V for 30 min of electrolysis. c Comparison of the NH3 yield rate and FE of this work (red star) with the previous reports including eNRR (gray square), Li-mediated eNRR (blue sphere), and pNOR-eNOx−RR (yellow triangle). d A continuous run of the pNOR-eNOx−RR system at −0.25 V in a flow mode using pure air as the feeding gas.
The spark discharge conditions of pNOR, such as the ratio of air to O2, discharge distance, and gas flow rate, were then optimized (Supplementary Fig. 51). In addition, the pNOR process is stable for linearly producing NOx− (Supplementary Fig. 52). The failure of NH3 production by running either pNOR or eNOx−RR alone highlights the necessity of simultaneous operation of these two parts for NH3 synthesis (Supplementary Fig. 53). Moreover, using Ar as the feeding gas leads to no NH3 generation, confirming that N2 in the air is the N source. We then performed batch experiments to investigate the impact of the spark discharge time on the NH3 yield rate and corresponding FE (Supplementary Fig. 54). The solutions with different spark discharge time were applied as the afterward electrolyte. As shown in Fig. 5b, the NH3 yield increases rapidly as the discharge time increases from 10 to 30 min. Further expanding the discharge time to 120 min only leads to a small increment of NH3 yield rate from 2.7 to 3.0 mmol h−1 cm−2. The FEs of NH3 scatter around 90% when the discharge time exceeds 20 min, and a smaller NH3 FE of 83% is obtained under 10 min, maybe due to the competing HER reaction under low NOx− concentration. The NH3 production efficiency of our pNOR-eNOx−RR system surpasses other electrochemical NH3 synthesis alternatives such as eNRR, Li-mediated eNRR, and other reported pNOR-eNOx−RR works (Fig. 5c and Supplementary Table 3).
Even with pure air for pNOR, the NOx− generation rate (4.05 mmol h−1) was still greater than the maximum NOx− consumption rate (3 mmol h−1) achievable with a 1-cm2 Ni(OH)x/Cu, so we conducted a 100-h test in a flow mode with simultaneous initiation of NTP and electrolysis using a pure air inlet (Supplementary Fig. 55). Figure 5d shows that in the initial stage, the current density and NH3 yield increases with the constant accumulation of NOx− (Supplementary Fig. 56). Although the charge-transfer resistance of the electrode in electrochemical impedance spectroscopies decreased gradually due to the increasing concentration of NOx−, the increasing solution resistance after 5 h of operation limited the increase in current density (Supplementary Fig. 57). Considering that the pH barely changes during the 100-h operation, the increasing solution resistance mainly be caused by the accumulation of NOx− with lower mole conductivities (NO3−: 71.44 S cm2 mol−1, NO2−: 71.7 S cm2 mol−1) than that of OH− (198.0 S cm2 mol−1) (Supplementary Figs. 58, 59). Eventually, the whole system delivers a steady current density of ca. 240 mA cm−2 and NH3 production rate of ca. 1.25 mmol h−1 cm−2, outperforming the results obtained over the Cu electrode (Supplementary Fig. 60). Note that the FE for NH3 production cannot be determined because the amounts of NO2− and NO3− under continuous reaction are imponderable. Our pNOR-eNOx−RR tandem system delivers an overall energy consumption of 18.36 MJ mol−1 NH3 and energy efficiency of 2.08%, which is comparable to a previous work by combining a non-thermal plasma bubble column reactor and Cu nanowire-catalyzed electrochemical conversion and much lower than a plasma-activated proton conducting solid oxide electrolyzer for NH3 synthesis (605 MJ mol−1 NH3) (Supplementary Note 1)20,46. Although this energy consumption value is still larger than that of the Haber-Bosch process (0.52−0.81 MJ mol−1 NH3), the application of the pNOR-eNOx−RR system can provide a new option for the development of decentralized NH3 synthesis, considering that the traditional Haber-Bosch process is only economically viable in large-scale production47.
We then applied an air stripping method to collect the NH3 product from the electrolyte and absorption solution due to the high NH3 vaper pressure in the alkaline media48. The stripped-out NH3 was either condensed into a concentrated NH3 aqueous solution or trapped by the acid solution to collect solid NH4Cl after performing rotary evaporation. Eventually, 69.8% and 23% of the produced NH3 was collected as NH4Cl (4.6956 g) and NH3 aqueous solution (63.8 mL, 0.77 wt%), respectively (Supplementary Fig. 61). The purity of liquid and solid products is confirmed by 1H NMR and XRD, respectively (Supplementary Fig. 62).
In summary, we present a tandem NH3 synthesis system that combines a spark discharge NTP-enabled N2-to-NOx conversion with electrocatalytic NOx−-to-NH3 transformation. The pNOR-eNOx−RR system with the Ni(OH)x/Cu cathode performs a high NH3 yield of 3 mmol h−1 cm−2 and corresponding FE of 92% using air and H2O as the sources of nitrogen and proton, respectively. It also exhibits excellent stability for long-term continuous operation. Experimental and theoretical results reveal that Ni(OH)x species on the Cu surface interact with hydrated K+ in the double layer, contributing to enhancing H2O adsorption and dissociation to form active Had at low overpotentials for the hydrogenation of nitrogenous intermediates on Cu. Meanwhile, Ni(OH)x/Cu shows optimized intermediates adsorption behavior, making the reaction proceed in the direction of NH3 formation. This work provides a perspective for the rational design of an electrocatalyst for eNOx−RR in alkaline conditions and offers one renewable-electricity-powered avenue for decentralized NH3 mass production using air at ambient conditions.
Methods
Preparation of Ni(OH)x/Cu NWA, Cu NWA, and Ni(OH)2 catalysts
In a typical procedure, a piece of 1 × 3 cm2 Cu foam was washed respectively with 1 M HCl, ethanol, and DI water for 10 min each to clean its surface. Then, the Cu foam was immersed in 30 mL aqueous solution containing 3 g NaOH and 0.68 g (NH4)2S2O8 for 20 min to obtain Cu(OH)2 NWA on the Cu skeleton. The synthesized Cu(OH)2 NWA was annealed in a muffle furnace at 200 °C for 3 h to convert it into a CuO NWA. Next, the CuO NWA was soaked in 0.1 M NiCl2·6H2O for 15, 30, and 45 min at open circuit potential to achieve varied surface coverage of hydroxide. After the deposition step, the electrode was rinsed with DI water thoroughly and dried at 60 °C in an oven. Finally, a cyclic voltammetry prereduction process was performed in the electrolyte from 0.2 V to −0.3 V for 10 cycles with a scan rate of 5 mV s−1 to fully reduce CuO to Cu. Cu NWA electrode was synthesized by performing the same procedure except for the deposition of Ni(OH)x. Ni(OH)2 was synthesized via a solvothermal method according to the reference49. Typically, 0.7 g of Ni(NO3)2 ∙ 6H2O was dissolved into 48 mL of ethanol under magnetic stirring, followed by adding 4.8 mL of oleylamine and 24 mL of ethanol in sequence. After being stirred for 30 min, the mixture was transferred to a 120 mL Teflon-lined stainless autoclave and heated to 190 °C for 16 h. Finally, the precipitates were washed with cyclohexane, DI water, and ethanol thoroughly and dried at 60 °C under vacuum.
Materials characterizations
The morphology was analyzed by field emission SEM (TESCAN MAIA3LMH) and TEM (Talos F200X). XRD patterns were recorded on a Shimadzu XRD-6100 with Cu Kα radiation. XPS spectra were collected on a Thermo Fisher ESCALAB Xi+ X-ray photoelectron spectrometer. All the XPS data were calibrated by shifting the C 1s peaks to 284.8 eV. The Raman spectra were measured on a Renishaw inVia Qontor Ramna microscope using laser excitation wavelength of 633 nm for copper and copper oxide detection and 532 nm for Ni(OH)x species detection. The concentration of nitrate in the electrolyte was quantified on a Thermo Scientific Dionex Integrion. The EPR measurements were performed on a JEOL JES-FA200 spectrometer. The 1H NMR spectra were measured on a AVANCE III HD 600 MHz NMR spectrometer. The mass spectra were collected on a GCMS-QP2020NX Shimadzu instrument. The contents of Ni and Cu elements in the Ni(OH)x/Cu samples were measured on a NexION 350D inductively coupled plasma mass spectrometer (ICP-MS).
XAFS measurement
The Ni K-edge XAFS spectra were collected at the 1W1B beam line of Beijing Synchrotron Radiation Facility, employing transmission mode for Ni foil and Ni(OH)2, while fluorescence mode was utilized for Ni(OH)x/Cu. Data processing and analysis were conducted using the Athena and Artemis software from the Demeter data analysis packages, incorporating the FEFF6 program for EXAFS fitting50,51. Energy calibration was performed referencing both standard and Ni foil measurements concurrently. To prepare the data, a linear function was subtracted from the pre-edge region, followed by normalization of the edge jump with Athena software. Subsequently, χ(k) data isolation involved subtraction of a smooth, third-order polynomial approximating the absorption background of an isolated atom. Fourier transformation of the k3-weighted χ(k) data followed, utilizing a HanFeng window function (Δk = 1.0). For EXAFS modeling, global amplitude EXAFS parameters (CN, R, σ2, and ΔE0) were derived via nonlinear fitting in Artemis software, incorporating least-squares refinement52. In the analysis of Ni(OH)2 EXAFS, the obtained amplitude reduction factor S02 value (0.954) was utilized to determine coordination numbers (CNs) in the Ni-O, Ni-Ni, and Ni-Cu scattering paths within the sample.
Electrochemical measurements
All the electrochemical measurements in this study were conducted using a CH Instruments 660E Potentiostat at room temperature. Nitrate reduction was performed in a commercial gas-tight H-type cell separated by a Nafion 117 cation exchange membrane (Dupont). In a typical three-electrode system, the synthesized Cu, Ni(OH)x/Cu, and Ni(OH)2 electrodes were used as the working electrode, while a graphite rod electrode and a Hg/HgO electrode were used as the counter and reference electrodes, respectively. For the Ni(OH)2 electrode, a catalyst ink was prepared by mixing 2 mg of Ni(OH)2 with 750 mL of isopropanol, 200 mL of distilled water, and 50 μL of Nafion solution (5 wt%). The mixture was sonicated for 30 min to form a homogeneous ink. Then, 100 μL of the catalyst ink was drop-cast onto a carbon paper electrode with an active area of 1 × 1 cm2. The synthesized Cu and Ni(OH)x/Cu were directly applied as the working electrode. The copper foam electrodes were sealed with silicone rubber to ensure a geometric area of 1 × 1 cm2. In this work, the recorded potentials against Hg/HgO were converted to the RHE scale. It was calibrated by performing CV in the high purity hydrogen saturated 1 M KOH electrolyte with two Pt electrodes as the working and counter electrodes. CVs were run at a scan rate of 1 mV s−1. The average value of the two potentials corresponding to zero current is taken as the thermodynamic potential of the hydrogen electrode reaction. 25 mL aqueous solution of 1 M KOH containing 0.1 M NO3− was used as the electrolyte in both the cathode and anode chambers. Chronoamperometry tests were performed under different biases at a stirring rate of 400 revolutions per minute. Ar was injected into the electrolyte for 10 min before testing to expel the oxygen and continuously pumped into the electrochemical cell during the test. A 0.01 M H2SO4 absorption solution was needed to collect NH3 in the exhaust gas since NH3 is volatile in an alkaline solution. All the batch experiments were tested for 30 min, and the electrolyte and absorption solution were taken out for quantitative analysis. The linear scanning voltammetry (LSV) curves were collected at a scan rate of 5 mV s−1. The long-term stability test was performed using the chronopotentiometry method at a current density of 400 mA cm−2 for 25 h in a homemade flow cell system at an electrolyte flow rate of 5 mL min−1. The electrochemical impedance spectroscopy was recorded with a frequency range of 0.1 Hz to 100 KHz and an amplitude of 5 mV.
The ECSA was calculated by Eq. (3).
where Cdl is the double-layer capacitance, and Cs is the specific capacitance. Cdl was determined by the CV scanning at the non-Faradaic potential range at different scan rates. The slope of the plot of half the difference between anodic and cathodic currents versus scan rate was taken as Cdl. The Cs value of 40 μF cm−2 was used in this study.
Ion concentration detection
NH4 + quantification
The yield of ammonia in the electrolyte was detected by the indophenol blue method. First, 2 mL of the diluted post-electrolysis electrolyte was mixed with 2 mL of 1 M NaOH solution containing 5 wt% salicylic acid and 5 wt% sodium citrate. Then, 1 mL of 0.05 M NaClO and 0.2 mL of 1 wt% sodium nitroferricyanide (C5FeN6Na2O) were added. The mixture was shaken for 30 s and then placed in the dark for 2 h to complete the color reaction. The ammonia concentration was measured using the UV-vis spectrometer to determine the absorbance at 650 nm. To quantify the amount of NH3, quantified NH4Cl was dissolved in 1 M KOH to obtain a series of standard solutions. The calibration curve was obtained by linear fitting.
NO2 − quantification
A color reagent was made by adding 4.0 g of p-aminobenzenesulfonamide, 0.2 g of N-(1-naphthyl) ethylenediamine dihydrochloride, and 10 mL of phosphoric acid (85 wt% in water) into 50 mL of DI water. The electrolyte was first diluted to the detection range. After adding 0.1 mL of the color reagent, the mixture was rested for 20 min. The absorbance at 540 nm was recorded using the UV-vis spectrometer. A concentration-absorbance curve was calibrated using standard NaNO2 solutions with a series of concentrations. The nitrite concentration was calculated based on the recorded absorbance and the calibration curve.
NO3 − quantification
The concentration of nitrate was quantitatively detected by ion chromatography. A concentration-peak area curve was calibrated using standard NaNO3 solutions with a series of engagements. The nitrate concentration was calculated based on the recorded peak area and the calibration curve.
NMR determination of ammonium
The produced ammonium in isotope-labeling experiments was detected via 1H nuclear magnetic resonance (NMR, 600 MHz) using DMSO-d6 as a solvent. Typically, the pH of the as-obtained solution was adjusted to 2 using 3 M HCl. Then, 0.4 mL of the solution was mixed with 50 μL of DMSO-d6 and 50 μL of 10 mM maleic acid.
EPR spectroscopy experiments
EPR spectra were obtained using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the Had trapping reagent. Before testing, the electrolyte was saturated with Ar to avoid the oxidation of DMPO. Then, DMPO was added to the cathode electrolyte to reach a concentration of 30 mmol L−1. Each EPR spectra was collected after 10 min electrolysis at −0.25 V vs. RHE under Ar-bubbling.
Calculation of FE, partial current density, NH3 yield rate, and selectivity
The FE of production of NH3 or NO2− was calculated with the Eq. (4).
where C represents the NH3 concentration in the electrolyte (mol L−1), V is the electrolyte volume in the cathode chamber (L), F is the Faraday constant (96485 C mol−1), Q is the total charge consumed on the electrode during electrolysis (C), n is the consumed electron number, which is 8 for producing one NH3 and 2 for NO2−.
Considering that both NO2− and NO3− contributed to the NH3 production in the pNOR-eNOx−RR system, the FE for NH3 production was calculated by Eq. (5).
where C1 and C2 were obtained by Eqs. (6, 7).
The partial current density for producing NH3 was calculated by Eq. (8).
where A is the electrode geometric area, and t is the electrolysis time.
The ammonia yield rate was calculated by Eq. (9).
The selectivity of consumed NO3− to NH3 was calculated by Eq. (10).
where Cammonia is the concentration of generated NH3 and ΔCnitrate is the concentration change of NO3−.
In situ Raman and in situ ATR-SEIRAS
In situ Raman spectra were conducted on a Renishaw inVia Qontor Ramna microscope using a 633 nm solid laser as an excitation source. The measurement was carried out in a homemade reactor with the glassy carbon electrode, Hg/HgO, and Pt wire as the working electrode, reference electrode, and counter electrode, respectively. The catalyst ink was obtained by mixing isopropanol, water, and Nafion solution, and the catalyst stripped by ultrasonic on a copper foam electrode. The working electrode was prepared by drop-casting the catalyst ink onto the glassy carbon electrode. The electrolyte of 1 M KOH and 0.1 M KNO3 was pumped with Ar for 30 min before testing. Raman spectra were collected under different potentials. Each potential was applied for at least 5 min before spectra collection to ensure a steady-state condition of the catalyst surface. During the test, the objective was protected by a Teflon film and immersed in the electrolyte. Water was filled into the gap between the objective and the Teflon protection film to avoid the interference of the air bubble.
In situ ATR-SEIRAS was performed on Thermo-Fisher Nicolet iS50 equipped with a liquid nitrogen-cooled HgCdTe (MCT) detector and VeeMax III ATR accessory. A silicon semi-cylindrical prism coated with gold and catalyst was used as the working electrode. The Hg/HgO electrode and Pt wire were used as the reference and counter electrodes, respectively. ATR-SEIRAS was recorded by stepwise switching the potential from 0.2 V to −0.4 V (vs. RHE). The spectra collected at open circuit voltage were used for the background subtraction. All ATR-SEIRAS measurements were acquired by averaging 64 scans at a spectral resolution of 4 cm−1.
pNOR-eNOx −RR continuous test and NH3 products collection
The spark discharge NTP was generated by a high voltage power supply (HB-C06, Foshan Hongba Electronics Co., Ltd, China), enabled by a manual contacting voltage regulator (TDGC2-0.5KVA, Zhejiang Chengqiang Electric Co., Ltd, China). The spark discharge NTP reactor consisted of a “T”-shaped glass tube with an inner diameter of 2 mm and two stainless-steel tubes with an inner diameter of 0.5 mm for gas inlet and plasma generation. Pure air was used as the feeding gas. 300 mL and 1000 mL of 1 M KOH were used as electrolytes in the cathode and anode chambers, respectively. The produced NOx was adsorbed in an external electrolyte storage tank, in which the electrolyte was circulated through a peristaltic pump. Two adsorption tanks each filled with 100 mL of 0.2 M HCl were applied at the gas outlet to collect volatile NH3. The pNOR-eNOx−RR continuous test was initiated by running the NTP and electrocatalysis simultaneously. After the 100-h continuous test, 0.5 mol KOH was added in the mixture of the electrolyte and the adsorption solution to increase its alkalinity. The NH3 was separated by air-stripping at a gas flow rate of 30 sccm for 12 h in an oil bath of 70 °C. To obtain solid NH4Cl product, the gas outlet stream was purged into 200 mL of 1 M HCl, which was then treated with a rotary evaporator to collect the NH4Cl powder. To obtain a concentrated NH3 aqueous solution, the NH3 vapor in the gas outlet stream was condensed into NH3 (aq) through a graham condenser.
DFT calculation
The first-principles calculations were carried out with DFT implemented in the Vienna ab-initio simulation package (VASP). The generalized gradient approximation (GGA) with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional, the projector augmented wave (PAW) pseudopotential for the core electrons, and a plane-wave basis with the kinetic cut-off energy of 500 eV was applied in all calculations. We performed Brillouin-zone integrations using Monkhorst-Pack grids with (4 × 6 × 1) mesh for the structure optimization. The energy (converged to 1.0 × 10−5 eV atom−1) and force (converged to −0.05 eV Å−1) were set as the convergence criterion for geometry optimization. A vacuum space of 20 Å along the Z direction was used to separate the interaction between the neighboring slabs. Cu was represented by (1 1 1) terminated slab because it represents the low-energy surface53. Three molecular units of Ni(OH)2 were deposited on the Cu (1 1 1) surface and optimized. We used the Hubbard U correction in the implementation of Dudarev et al. with a U value of 6.2 for Ni54. To assess the role of alkali cations, we introduce in the simulation cell with solvated alkali cations (with 4, 5, and 7 H2O in Li+, Na+, and K+ coordination shell, respectively), fixing their z-coordinate to distances of 6.8, 7.2, and 5.5 Å from the Cu surface and 6.4, 6.9, and 4.9 Å from the Ni(OH)x/Cu surface55,56. The adsorption energy of reaction intermediates was defined as:
where EM-Sub, ESub, and EM represent the energies of an adsorbed system, a clean substrate, and an adsorbate, respectively. The Gibbs free energies variations were calculated by:
where ∆ZPE and ∆S represent the change of zero point energy and entropy, respectively.
The H2O dissociation energy was calculated by:
where * designates a surface adsorbed specie.
Given that it is difficult to directly calculate the energy of charged NO3‒, the adsorption free energy of NO3‒ (ΔG(*NO3)) was calculated with the assistance of the gaseous HNO3 (Supplementary Fig. 63 and Supplementary Note 2)57,58.
Molecular dynamics (MD) simulation
MD simulation boxes with dimensions of around 40 × 40 × 100 Å3 are created, in which the solid substrate is the Cu or the Ni(OH)x/Cu, while 1.1 M Li+, 1.1 M Na+, 1.1 M K+, or 1.1 M Cs+ aqueous solution is placed on the solid crystal for comparison. Periodic boundary conditions (PBCs) are imposed in the two orthogonal (x and y) directions to mimic infinite planar Cu crystalline substrate, while a wall-boundary condition is applied in the out-of-plane (z) direction of the substrate. The forcefield parameters of as-investigated systems are taken from the literature59. For the non-bonded atomic interactions in the system, the 12-6 Lennard-Jones potential with a cutoff distance of 10.0 Å is applied to describe the van der Waals (vdW) forces between atoms, while the standard Coulomb potential is utilized to mimic the electrostatic interactions that is evaluated by the particle−particle particle−mesh (PPPM) algorithm. To satisfy an imposed voltage of −0.25 V vs. RHE across the systems along the z-direction, the charges of each Cu atom are computed at each timestep using the constant potential fix in the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS)60. Prior to MD simulations, energy minimizations are firstly performed to relax the configuration of as-investigated systems with energy and force tolerances of 0.0001 Kcal/mol and 0.0001 Kcal/(mol·Å), respectively. Then, MD simulations with 1,000,000 timesteps are carried out to further relax the systems at temperature of 300 K under canonical (NVT) ensemble, in which the temperature is maintained by the Nose-hoover thermostat. Finally, production MD simulations with 1,000,000 timesteps are performed to capture the structural behaviors of ions and water in the vicinity of the surface of Cu-based substrates. During the whole MD simulations, the Cu-based substrates is frozen. The dynamics of atoms in the solution are based on the classical Newton’s motion, in which the velocity-Verlet algorithm with a timestep of 1.0 fs is applied to integrate the classic Newton’s equation. All the MD simulations are implemented using the LAMMPS package.
Data availability
The data supporting the plots within this paper and other study findings are available from the corresponding author upon request. Source data are provided with this paper.
Code availability
The codes that support the findings of this study are available from the corresponding author on request.
References
Ammonia technology roadmap. IEA. https://www.iea.org/reports/ammonia-technology-roadmap (2021).
MacFarlane, D. R. et al. A roadmap to the ammonia economy. Joule 4, 1186–1205 (2020).
Chen, J. G. et al. Beyond fossil fuel-driven nitrogen transformations. Science 360, eaar6611 (2018).
Foster, S. L. et al. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 1, 490–500 (2018).
Jiang, H., Chen, G.-F., Savateev, O. & Wang, H. Visualizing the reaction interface of lithium-mediated nitrogen fixation. Joule 7, 253–256 (2023).
Du, H. L. et al. Electroreduction of nitrogen with almost 100% current-to-ammonia efficiency. Nature 609, 722–727 (2022).
Li, Y. et al. Advanced electrocatalysis for energy and environmental sustainability via water and nitrogen reactions. Adv. Mater. 33, e2000381 (2021).
Klein, C. K. & Manthiram, K. Sustainable ammonia synthesis: Just around the corner? Joule 6, 1971–1973 (2022).
Suryanto, B. H. R. et al. Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia. Nat. Catal. 2, 290–296 (2019).
Xu, H., Ma, Y., Chen, J., Zhang, W. X. & Yang, J. Electrocatalytic reduction of nitrate - a step towards a sustainable nitrogen cycle. Chem. Soc. Rev. 51, 2710–2758 (2022).
Yang, J. et al. Potential-driven restructuring of Cu single atoms to nanoparticles for boosting the electrochemical reduction of nitrate to ammonia. J. Am. Chem. Soc. 144, 12062–12071 (2022).
Chen, F. Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759–767 (2022).
Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036–7046 (2020).
Meng, S.-L. et al. Cobaloxime: selective nitrite reduction catalysts for tandem ammonia synthesis. Energy Environ. Sci. 16, 1590–1596 (2023).
Wang, Y. et al. Enhanced nitrate-to-ammonia activity on copper-nickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702–5708 (2020).
Wu, A. et al. Direct ammonia synthesis from the air via gliding arc plasma integrated with single atom electrocatalysis. Appl. Catal. B Environ. 299, 120667 (2021).
Meng, Z. et al. Efficient ammonia production beginning from enhanced air activation. Adv. Energy Mater. 12, 2202105 (2022).
Li, L. et al. Efficient nitrogen fixation to ammonia through integration of plasma oxidation with electrocatalytic reduction. Angew. Chem. Int. Ed. 60, 14131–14137 (2021).
Ren, Y. et al. Microscopic-level insights into the mechanism of enhanced NH3 synthesis in plasma-enabled cascade N2 oxidation-electroreduction system. J. Am. Chem. Soc. 144, 10193–10200 (2022).
Sun, J. et al. A hybrid plasma electrocatalytic process for sustainable ammonia production. Energy Environ. Sci. 14, 865–872 (2021).
Fan, K. et al. Active hydrogen boosts electrochemical nitrate reduction to ammonia. Nat. Commun. 13, 7958 (2022).
Fang, J. Y. et al. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 13, 7899 (2022).
Liu, H. et al. Efficient electrochemical nitrate reduction to ammonia with copper-supported rhodium cluster and single-atom catalysts. Angew. Chem. Int. Ed. 61, e202202556 (2022).
Li, X., Shen, P., Li, X., Ma, D. & Chu, K. Sub-nm RuOx clusters on Pd metallene for synergistically enhanced nitrate electroreduction to ammonia. ACS Nano 17, 1081–1090 (2023).
Li, P. et al. Pulsed nitrate-to-ammonia electroreduction facilitated by tandem catalysis of nitrite intermediates. J. Am. Chem. Soc. 145, 6471–6479 (2023).
Wu, Y. et al. Converting copper sulfide to copper with surface sulfur for electrocatalytic alkyne semi-hydrogenation with water. Nat. Commun. 12, 3881 (2021).
Danilovic, N. et al. Enhancing the alkaline hydrogen evolution reaction activity through the bifunctionality of Ni(OH)2/metal catalysts. Angew. Chem. Int. Ed. 51, 12495–14498 (2012).
Auer, A. et al. Interfacial water structure as a descriptor for its electro-reduction on Ni(OH)2-modified Cu(111). ACS Catal. 11, 10324–10332 (2021).
Singhal, A. et al. Copper(I) oxide nanocrystals – one step synthesis, characterization, formation mechanism, and photocatalytic properties. Eur. J. Inorg. Chem. 2013, 2640–2651 (2013).
Debbichi, L., Marco de Lucas, M. C., Pierson, J. F. & Krüger, P. Vibrational properties of CuO and Cu4O3 from first-principles calculations, and Raman and Infrared spectroscopy. J. Phys. Chem. C. 116, 10232–10237 (2012).
Luo, M. et al. Hydroxide promotes carbon dioxide electroreduction to ethanol on copper via tuning of adsorbed hydrogen. Nat. Commun. 10, 5814 (2019).
Wang, W. et al. Vacancy-rich Ni(OH)2 drives the electrooxidation of amino C-N bonds to nitrile C≡N Bonds. Angew. Chem. Int. Ed. 59, 16974–16981 (2020).
Louie, M. W. & Bell, A. T. An investigation of thin-film Ni-Fe oxide catalysts for the electrochemical evolution of oxygen. J. Am. Chem. Soc. 135, 12329–12337 (2013).
Bantignies, J. L. et al. New insight into the vibrational behavior of nickel hydroxide and oxyhydroxide. J. Phys. Chem. C. 112, 2190–2201 (2008).
Dinh, C.-T. et al. Multi-site electrocatalysts for hydrogen evolution in neutral media by destabilization of water molecules. Nat. Energy 4, 107–114 (2018).
Yu, X. et al. Hydrogen evolution reaction in alkaline media: alpha- or beta-nickel hydroxide on the surface of platinum? ACS Energy Lett. 3, 237–244 (2017).
Deng, W., Zhang, P., Seger, B. & Gong, J. Unraveling the rate-limiting step of two-electron transfer electrochemical reduction of carbon dioxide. Nat. Commun. 13, 803 (2022).
Ma, W. et al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper. Nat. Catal. 3, 478–487 (2020).
Shah, A. H. et al. The role of alkali metal cations and platinum-surface hydroxyl in the alkaline hydrogen evolution reaction. Nat. Catal. 5, 923–933 (2022).
Ji, K. et al. Electrocatalytic hydrogenation of 5-hydroxymethylfurfural promoted by a Ru1Cu single-atom alloy catalyst. Angew. Chem. Int. Ed. 61, e202209849 (2022).
Wang, X., Xu, C., Jaroniec, M., Zheng, Y. & Qiao, S. Z. Anomalous hydrogen evolution behavior in high-pH environment induced by locally generated hydronium ions. Nat. Commun. 10, 4876 (2019).
Rossmeisl, J. et al. Realistic cyclic voltammograms from ab initio simulations in alkaline and acidic electrolytes. J. Phys. Chem. C. 124, 20055–20065 (2020).
Deng, G.-H., Zhu, Q., Rebstock, J., Neves-Garcis, T. & Baker, L. R. Direct observation of bicarbonate and water reduction on gold: understanding the potential dependent proton source during hydrogen evolution. Chem. Sci. 14, 4523–4531 (2023).
Pérez-Gallent, E., Figueiredo, M. C., Katsounaros, I. & Koper, M. T. M. Electrocatalytic reduction of nitrate on copper single crystals in acidic and alkaline solutions. Electrochim. Acta 227, 77–84 (2017).
Butcher, D. P. & Gewirth, A. A. Nitrate reduction pathways on Cu single crystal surfaces: Effect of oxide and Cl−. Nano Energy 29, 457–465 (2016).
Sharma, R. K. et al. Plasma activated electrochemical ammonia synthesis from nitrogen and water. ACS Energy Lett. 6, 313–319 (2021).
Hollevoet, L. et al. Towards green ammonia synthesis through plasma-driven nitrogen oxidation and catalytic reduction. Angew. Chem. Int. Ed. 59, 23825–23829 (2020).
Yuan, M.-H., Chen, Y.-H., Tsai, J.-Y. & Chang, C.-Y. Ammonia removal from ammonia-rich wastewater by air stripping using a rotating packed bed. Process Saf. Environ. 102, 777–785 (2016).
Liu, G. et al. Promoting active species generation by plasmon-induced hot electron excitation for efficient electrocatalytic oxygen evolution. J. Am. Chem. Soc. 138, 9128–9136 (2016).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Zabinsky, S. I., Rehr, J. J., Ankudinov, A., Albers, R. C. & Eller, M. J. Multiple-scattering calculations of x-ray-absorption spectra. Phys. Rev. B 52, 2995–3009 (1995).
Liang, C. et al. Pd/NiSix: an efficient and stable acid-ORR electrocatalyst. Chem. Catal. 4, 100849 (2024).
Tran, R. et al. Surface energies of elemental crystals. Sci. Data 3, 160080 (2016).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Xi, C. et al. Distribution of alkali cations near the Cu (111) surface in aqueous solution. J. Mater. Chem. A 8, 24428 (2020).
Ye, C., Dattila, F., Chen, X., López, N. & Koper, M. T. M. Influence of cations on HCOOH and CO formation during CO2 reduction on a PdMLPt(111) electrode.J. Am. Chem. Soc. 145, 19601–19610 (2023).
Wan, H., Bagger, A. & Rossmeisl, J. Electrochemical nitric oxide reduction on metal surfaces. Angew. Chem. Int. Ed. 60, 21966–21972 (2021).
Wang, Y., Zhou, W. Z., Jia, R., Yu, Y. & Zhang, B. Unveiling the activity Origin of a Copper-based Electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350–5354 (2020).
Huang, B. et al. Cation- and pH-dependent hydrogen evolution and oxidation reaction kinetics. JACS Au 1, 1674–1687 (2021).
Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117, 1–19 (1995).
Acknowledgements
This work was supported by the National Key R&D Program of China (2020YFA0710000, G.Y.), Joint Funds of the National Natural Science Foundation of China (U22A20391, G.Y.), National Natural Science Foundation of China (Grant No. 22078256, G.Y.), Innovation Capability Support Program of Shaanxi (NO. 2023-CX-TD-26, G.Y.), and the Programme of Introducing Talents of Discipline to Universities (B23025, G.Y.).
Author information
Authors and Affiliations
Contributions
W.L. designed and carried out the synthesis, characterizations and catalytic reactions, analyzed the data, and wrote the original draft. M.X. performed the DFT calculations. C.Z. carried out catalytic reactions. B.C. designed and drew the schematic diagrams. J.C. optimized the non-thermal plasma working conditions. H.L. and H.O. reviewed and revised the manuscript. G.Y. contributed significantly to the analysis of the data, supervision of the project, manuscript preparation, and funding acquisition. All the authors commented on the manuscript and have given approval for the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, W., Xia, M., Zhao, C. et al. Efficient ammonia synthesis from the air using tandem non-thermal plasma and electrocatalysis at ambient conditions. Nat Commun 15, 3524 (2024). https://doi.org/10.1038/s41467-024-47765-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-47765-9
- Springer Nature Limited