

Abstract
Tricyclic furan derivatives with multiple chiral centers are ubiquitous in natural products. Construction of such tricyclic scaffolds in a stereocontrolled, step-economic, and atom-economic manner is a key challenge. Here we show a nickel-catalyzed highly enantioselective synthesis of hydronaphtho[1,8-bc]furans with five contiguous chiral centers via desymmetrization of alkynyl-cyclohexadienone by oxidative cyclization and following formal [4 + 2] cycloaddition processes. Alkynyl-cyclohexadienone was synthesized in one step from easily accessible phenols. This reaction represents excellent chemo-selectivity, regio-selectivity, diastereo-selectivity, and enantio-selectivity (single diastereomer, up to 99% ee). An extraordinary regioselectivity in the formal [4 + 2] cycloaddition step with enones revealed the diverse reactivity of the nickelacycle intermediate. Desymmetrization of alkynyl-cyclohexadienones via oxidative cyclization on nickel was supported by the isolation of a nickelacycle from a stoichiometric reaction. Enantioenriched tricyclic products contain various functional groups such as C=O and C=C. The synthetic utility of these products was demonstrated by derivatization of these functional groups.
Similar content being viewed by others

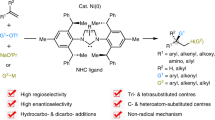

Introduction
Hydronaphtho[1,8-bc]furan rings with multiple chiral centers are a common structural motif in biologically active natural products (Fig. 1a)1,2,3,4, – 5. Such tricyclic structures are also found in key synthetic intermediates that are employed in a large number of sesquiterpenoids6,7,8,9. Owing to diverse biological activities and synthetic potentials associated with these fused tricyclic structures, a significant amount of attention has been paid to their enantioselective syntheses. Despite the existence of various stepwise stereoselective methods1,2,3,4,5,6,7,8,9, direct access to such tricyclic fused rings in a completely enantio-controlled, diastereo-controlled, step-economic, and atom-economic manner would be a remarkable development10,11,12,13. Recently, Alemán reported a straightforward method for the construction of tricyclic fused rings from a cyclohexadienone tethered alkenal by employing an organocatalyzed asymmetric desymmetrization strategy (Fig. 1b)14. In this process, desymmetrization step involved the intramolecular [4 + 2] cycloaddition of chiral dienamine with a diastereotropic enone constructed tricyclic fused rings with three chiral centers.

Hydronaphtho[1,8-bc]furans and their syntheses from phenols. a Representative examples of biologically active compounds. b Previous work employed desymmetrization by intramolecular [4+2] cycloaddition, catalyzed by organocatalyst. c Present work employed desymmetrization by oxidative cyclization and following intermolecular formal [4+2] cycloaddition process with nickel
We envisaged the enantioselective desymmetrization15,16,17,18,19 of alkynyl-cyclohexadienone via an intramolecular oxidative cyclization on nickel in the presence of a chiral ligand. Alkynyl-cyclohexadienone were synthesized in one step from easily accessible phenols. The oxidative cyclization of an enantiotropic enone with a tethered alkyne unit would form a tricyclic fused nickelacycle20,21,22,23,24 with three chiral centers, which could react with another olefin to yield a tricyclic product with the concomitant generation of two more chiral centers. Nickel(0)-catalyzed trimerization of an alkyne with two enones has been reported by Montgomery et al.25,26,27,28 and by us29, 30. Here, we report a nickel(0)-catalyzed enantioselective synthesis of chiral hydronaphtho[1,8-bc]furans with five contiguous chiral centers via desymmetrization of alkynyl-cyclohexadienone and following intermolecular formal [4 + 2] cycloaddition reaction processes.
Results
Reaction optimization
Prior to developing the reaction in an asymmetric fashion, achiral ligands were examined using cyclohexadienone-yne (1a) and 4-methoxychalcone (2a) for the model transformation to hydronaphtho[1,8-bc]furan 3aa (Fig. 2, see also Supplementary Table 1 for detail). 1,3-Bis-(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) proved to be an optimal ligand to deliver rac-3aa in 96% yield, whereas PCy3 failed to give any product. A single diastereomer of 3aa was obtained out of sixteen possible isomers. Moreover, it is remarkable that an extraordinary regioselectivity was observed in the formal [4 + 2] cycloaddition step, fixing two carbonyls at the 1,4-positions in 3aa, whereas 1,5-dicarbonyl compounds were obtained in reports of nickel(0)-catalyzed cycloaddition reactions20,21,22,23,24,25,26,27,28,29,30. Considering the efficiency of N-heterocyclic carbene (NHC) in this transformation, chiral (R,R)-NHCs, generated in situ by treating the corresponding imidazolinium salts with NaOtBu, were investigated to afford enantioenriched 3aa (Fig. 2). It is worth mentioning that despite much exploration of the use of chiral NHCs, there has been less reports with nickel-catalyzed reactions31,32,33,34,35,36,37,38,39,40. N-(2-Biphenyl)- (L*1) and N-(2-isopropylphenyl)- (L*2) substituted NHCs41 were ineffective to give 3aa. In a similar manner, N-(2,7-diisopropylnaphthyl)- (L*4)42 and N-(2,7-dicyclohexylnaphthyl) (L*5)34-substituted NHCs also failed to yield any products. However, N-2,6-diethylphenyl-substituted NHC (L*3) 43 successfully gave hydronaphtho[1,8-bc]furan 3aa in 13% yield with high enantioselectivity (94% ee). NHCs L*6 35 and L*7 34 furnished 3aa in moderate chemical yields (36 and 48%, respectively) with excellent enantioselectivities (98 and 89% ee, respectively). Given the excellent enantioselectivity with L*6, extensive effort was devoted to improving the yield. When the reaction was conducted at 60 °C using a lower concentration (0.02 M of 2a), 3aa was obtained in 74% yield with 98% enantioselectivity (see Supplementary Table 1 for detail).

Evaluation of Ligands. (R,R)-L*n·HBF4, NaOtBu, Ni(cod)2 (0.01 mmol each), 1a (0.12 mmol), 2a (0.10 mmol), and toluene (1.0 ml) are employed. Isolated yields are given and enantioselectivity was determined by SFC equipped with a chiral stationary phase. aYield and enantioselectivity was measured at 27% conversion in 3 days. b5 ml toluene was employed and reaction was conducted at 60 °C. c10 mol% IPr was used instead of chiral NHC salt and NaOtBu. d20 mol% PCy3 was used instead of chiral NHC salt and NaOtBu
Substrate scope
With the aforementioned optimal reaction conditions, we explored the scope of substrates (Fig. 3). A range of electron-rich and electron-deficient aryl-substituted enones 2 was examined with 1a. The reaction proceeded smoothly with 2b and 2c, giving 3ab and 3ac in 73 and 72% yields, respectively, with enantioselectivities of 98% each. Reaction with an enone containing 2-furyl group (2d) was also examined with 1a to afford 3ad in 72% yield with 99% ee. In contrast, the reaction of 1a with 1-aryl-2-buten-1-ones (2e–2i) gave 3ae–3ai in slightly lower yields (62–70%), albeit enantioselectivities remained excellent (94–97% ee). In these cases, about 5% of fully- intermolecular [2 + 2 + 2] cycloaddition products (3′) of an alkyne unit of dienone-yne (1a) with two enones (2e–2i) were observed (See Supplementary Fig. 10 for 3ae′). When ethyl group at R3 of an enone was introduced, a complex mixture was obtained. Next, alkynyl-cyclohexadienone substrates (1) were investigated by varying the substituents R1 and R2. Ethyl as well as 2-methoxyethyl-substituted alkynyl-cyclohexadienone (1b and 1c) gave 3bb, 3bj, 3ca, and 3ck with 2b, 2j, 2a, and 2k, respectively in good yields (60–73%) with excellent enantioselectivities (96–99%). Aryl and alkyl groups on alkynes were also examined. Phenyl-acetylene-substituted dienone 1d gave 3de in good yield with excellent enantioselectivity (70% yield and 99% ee). Electron-rich anisyl-group substrate 1e gave 3ec (71% yield and 98% ee) and 3ek (68% yield and 99% ee) with p-halogenated (F– and Cl–) chalcones 2c and 2k, respectively. No corresponding dehalogenated products were detected. An alkynyl-dienone 1f bearing an electron-deficient p-CO2EtC6H4– group gave 3fc with 2c in 60% yield with 98% ee, whereas 3ge was obtained in 71% yield with 96% ee from p-CF3-substituted 1g. An ethyl-group and a triethylsilyloxy-methyl-substituted alkynyl-cyclohexadienone (1h and 1i, respectively) gave corresponding tricyclic fused rings 3he, 3ic, and 3ik with 2e, 2c, and 2k, respectively, in good yields and enantioselectivities (69–77% yields, 95–98% ee). A N-tosyl analog of alkynyl-cyclohexadienone 1j failed to give the desired product 3jc with 2c under the present reaction conditions. This could have been due to coordination ability of tosyl group to nickel which inhibits the coordination of 2c 44, 45. The absolute configurations of all five chiral centers in 3 were assigned according to an analogy with 3ik, which was unambiguously determined by X-ray crystallographic analysis (Fig. 3). It also supports all the stereo selective and regioselective outcomes in 3.

Substrate Scope. Reaction was examined at 0.1−0.4 mmol scale. Isolated yields are given and enantioselectivity was determined by SFC equipped with a chiral stationary phase. ORTEP diagram of 3ik is shown with thermal ellipsoid at 30% probability level. Hydrogen atoms are omitted for clarity except those bound to the chiral carbon centers. Flack parameter for 3ik = 0.017(7)
Gram scale synthesis and transformations of product 3aa
To demonstrate applicability, a half-gram-scale reaction of 1a (0.53 g, 3.0 mmol) was carried out with 2a (0.6 g, 2.5 mmol) to afford 3aa in 73% yield and 98% ee. These enantioenriched tricyclic products could be useful synthetic intermediates for further transformations (Fig. 4). The methylene group of a tetrahydrofuran ring was oxidized with PCC46 to yield a butyrolactone scaffold (4, 90% yield), that is present in natural products and also is a key synthetic intermediate in many sesquiterpenoids (Fig. 1a)6,7,8,9. Epoxidation and Michael addition of an enone gave the corresponding functionalized products 5 and 6 as single diastereomers in 75 and 85% yields, respectively, whereas hydrogenation of an enonic C = C bond with H2/Pd(C) in ethyl acetate gave 7 in 89% yield. The enantioselectivity remained consistent in all these transformations.

Synthetic Transformation of 3aa. Isolated yields are given and enantioselectivity was determined by SFC equipped with a chiral stationary phase
Stoichiometric experiment
In order to gain deeper insight into a possible reaction mechanism, stoichiometric experiments were conducted in C6D6. An attempts to isolate a chiral nickelacycle corresponds to 1g using an optimal NHC L*6 and Ni(cod)2 was unsuccessful. 19F NMR spectra showed seven peaks, revealed the existence of several intermediates, in which one of them might be much more reactive leads to desired product in the presence of an enone. However, an η3-oxaallyl nickelacycle (8) was isolated in 99% yield when a stoichiometric reaction of 1a was conducted with IPr and Ni(cod)2. The molecular structure of 8 was confirmed by X-ray crystallography (Fig. 5). The 1H, 13C, and 2D NMR analyses of 8 demonstrated that its structure in solution was consistent with that observed in crystal lattice. The reaction of 8 with 2e gave rac-3ae in 95% isolated yield, which supported that desymmetrization by oxidative cyclization would play a key role in the present transformation.

Stoichiometric reaction with 1a. Isolated yield of 8 and rac-3ae are given. The molecular structure of η3-cyclohexadienyl Ni-complex 8 is disordered and one of the disordered structure is depicted for simplicity with thermal ellipsoid at 30% probability level. iPr groups and hydrogen atoms (except those bound to the chiral carbons) are omitted for clarity
A plausible reaction mechanism is drawn on the basis of the results of the stoichiometric experiment and previous reports (Fig. 6)20,21,22,23,24,25,26,27,28,29,30, 33,34,35,36,37,38,39,40,41,42. First, the intramolecular oxidative cyclization of 1 via the simultaneous coordination of an alkyne and an enantiotropic enone to the chiral Ni(0)/L* species gives a desymmetrized nickelacycle A, which would be in equilibrium with its η3-oxaallylnickel structure A′. Coordination of 2 to nickel center of A giving B, followed by insertion through a Ni–C sp 2 bond could form a thermodynamically favorable η3-oxaallylnickel structure either C or C′. Then, a subsequent reductive elimination could afford a tricyclic fused structure 3 as a single diastereomer with the regeneration of nickel(0) species.

A plausible reaction mechanism. Nickelacycle A is generated by enantioselective desymmetrization of 1 via oxidative cyclization. Subsequent insertion of an enone 2 to A gives a tricyclic product 3
In conclusion, a catalytic enantioselective method has been developed for the rapid construction of hydronaphtho[1,8-bc]furans with five contiguous chiral centers via desymmetrization of alkynyl-cyclohexadienone and formal [4 + 2] cycloaddition reaction with nickel. The synthetic utility of tricyclic products was also demonstrated. Isolation of desymmetrized η3-oxaallyl nickelacycle and subsequent reactions in the stoichiometric experiment revealed that desymmetrization by oxidative cyclization is the key in this transformation. Furthermore, unusual regioselectivity in the insertion step of enone revealed the diverse reactivity of an η3-oxaallyl nickel-complex. The developed strategy involving two steps from the easily accessible phenols, demonstrates a practical and step economic protocol to access synthetically valuable fused tricyclic frameworks bearing five consecutive chiral carbon centers with excellent enantioselectivities.
Methods
General procedure for tricyclic product 3
To a screw cap vial in a glove box was added L*6·HBF4 (10 mol%) and NaOtBu (10 mol%) and toluene (5 ml). The suspension was allowed to stir at room temperature for 10 min and then Ni(cod)2 (10 mol%) was added. After further stirring for 10 min at room temperature was added a solution of alkynyl-cyclohexadienone (1, 0.24 mmol) and enone (2, 0.20 mmol) in toluene (5 ml). The reaction mixture was taken out of glove box and heated at 60 °C for 36 h with stirring. After cooling to room temperature, the mixture was filtered through celite and washed with Et2O. The filtrate was concentrated in vacuo and the residue was purified by silica gel flash chromatography (5–20% ethyl acetate in hexane) to afford the desired product 3.
Data availability
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 1523827 (3ik) and 1523828 (8). These data can be obtained free of charge from The CCDC via www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study are available within the article and its Supplementary Information file or from the authors upon reasonable request. For NMR spectra of the compounds in this article, see Supplementary Figs. 2–31.
References
Germain, J. & Deslongchamps, P. Total synthesis of (±)-momilactone A. J. Org. Chem. 67, 5269–5278 (2002).
Liu, X. & Lee, C.-S. Total synthesis of (−)-teucvidin. Org. Lett. 14, 2886–2889 (2012).
Jauch, J. et al. Total synthesis of azadirachtin—finally completed after 22 years. Angew. Chem. Int. Ed. 47, 34–37 (2008).
Nicolaou, K. C., Lim, Y. H. & Becker, J. Total synthesis and absolute configuration of the bisanthraquinone antibiotic BE-43472B. Angew. Chem. Int. Ed. 48, 3444–3448 (2009).
Schütz, J. et al. Synthesis and biological evaluation of 14-alkoxymorphinans. 17. highly δ opioid receptor selective 14-alkoxy-substituted indolo- and benzofuromorphinans. J. Med. Chem. 45, 5378–5383 (2002).
Ye, H., Deng, G., Liu, J. & Qiu, F. G. Expedient construction of the Ziegler intermediate useful for the synthesis of forskolin via consecutive rearrangements. Org. Lett. 11, 5442–5444 (2009).
Leclaire, M., Levet, R., Péricaud, F., Ricard, L. & Lallemand, J. Y. Studies on the preparation of the “Ziegler intermediate”, a key intermediate in the total synthesis of forskolin. Tetrahedron 52, 7703–7718 (1996).
Miyaoka, H., Yamanishi, M., Kajiwara, Y. & Yamada, Y. Total synthesis of cladocorans A and B: a structural revision. J. Org. Chem. 68, 3476–3479 (2003).
Hanessian, S., Boyer, N., Reddy, G. J. & Deschênes-Simard, B. Total synthesis of oidiodendrolides and related norditerpene dilactones from a common precursor: metabolites CJ-14,445, LL-Z1271γ, oidiolactones A, B, C, and D, and nagilactone F. Org. Lett. 11, 4640–4643 (2009).
Trost, B. M. et al. The atom economy–a search for synthetic efficiency. Science 254, 1471–1477 (1991).
Lautens, M., Klute, W. & Tam, W. Transition metal-mediated cycloaddition reactions. Chem. Rev. 96, 49–92 (1996).
Malacria, M. et al. Selective preparation of complex polycyclic molecules from acyclic precursors via radical mediated- or transition metal-catalyzed cascade reactions. Chem. Rev. 96, 289–306 (1996).
Trost, B. M. & Krische, M. J. Transition metal catalyzed cycloisomerizations. Synlett. 1998, 1–16 (1998).
Martín-Santos, C. et al. Highly enantioselective construction of tricyclic derivatives by the desymmetrization of cyclohexadienones. Angew. Chem. Int. Ed. 53, 8184–8189 (2014).
Kalstabakken, K. A. & Harned, A. M. Asymmetric transformations of achiral 2,5-cyclohexadienones. Tetrahedron 70, 9571–9585 (2014).
Zeng, X.-P., Cao, Z.-Y., Wang, Y.-H., Zhou, F. & Zhou, J. Catalytic enantioselective desymmetrization reactions to all-carbon quaternary stereocenters. Chem. Rev. 116, 7330–7396 (2016).
He, Z.-T. et al. Efficient access to bicyclo[4.3.0]nonanes: copper-catalyzed asymmetric silylative cyclization of cyclohexadienone-tethered allenes. Angew. Chem. Int. Ed. 54, 14815–14818 (2015).
Fukui, Y. et al. Tunable arylative cyclization of 1,6-enynes triggered by rhodium(III)-catalyzed C−H activation. J. Am. Chem. Soc. 136, 15607–15614 (2014).
Clarke, C., Incerti-Pradillos, C. A. & Lam, H. W. Enantioselective nickel-catalyzed anti-carbometallative cyclizations of alkynyl electrophiles enabled by reversible alkenylnickel E/Z isomerization. J. Am. Chem. Soc. 138, 8068–8071 (2016).
Tasker, S. Z., Standley, E. A. & Jamison, T. F. Recent advances in homogeneous nickel catalysis. Nature 509, 299–309 (2014).
Ohashi, M., Hoshimoto, Y. & Ogoshi, S. Aza-nickelacycle key intermediate in nickel(0)-catalyzed transformation reactions. Dalton Trans. 44, 12060–12073 (2015).
Hoshimoto, Y., Ohashi, M. & Ogoshi, S. Catalytic transformation of aldehydes with nickel complexes through η2 coordination and oxidative cyclization. Acc. Chem. Res. 48, 1746–1755 (2015).
Standley, E. A., Tasker, S. Z., Jensen, K. L. & Jamison, T. F. Nickel catalysis: synergy between method development and total synthesis. Acc. Chem. Res. 48, 1503–1514 (2015).
Jackson, E. P. et al. Mechanistic basis for regioselection and regiodivergence in nickel-catalyzed reductive couplings. Acc. Chem. Res. 48, 1736–1745 (2015).
Seo, J., Chui, H. M. P., Heeg, M. J. & Montgomery, J. Novel chemoselectivity and stereochemical aspects of nickel-catalyzed [2+2+2] cycloadditions. J. Am. Chem. Soc. 121, 476–477 (1999).
Montgomery, J. & Savchenko, A. V. Nickel-catalyzed cyclizations of alkynyl enones with concomitant stereoselective tri- or tetrasubstituted alkene introduction. J. Am. Chem. Soc. 118, 2099–2100 (1996).
Montgomery, J., Oblinger, E. & Savchenko, A. V. Nickel-catalyzed organozinc-promoted carbocyclizations of electron-deficient alkenes with tethered unsaturation. J. Am. Chem. Soc. 119, 4911–4920 (1997).
Chevliakov, M. V. & Montgomery, J. A stereodivergent approach to (−)-α-kainic acid and (+)-α-allokainic acid utilizing the complementarity of alkyne and allene cyclizations. J. Am. Chem. Soc. 121, 11139–11143 (1999).
Ogoshi, S., Nishimura, A. & Ohashi, M. Nickel-catalyzed [2+2+2] cycloaddition of two enones and an alkyne. Org. Lett. 12, 3450–3452 (2010).
Nishimura, A., Ohashi, M. & Ogoshi, S. Nickel-catalyzed intermolecular [2+2] cycloaddition of conjugated enynes with alkenes. J. Am. Chem. Soc. 134, 15692–15695 (2012).
Kumar, R., Hoshimoto, Y., Yabuki, H., Ohashi, M. & Ogoshi, S. Nickel(0)-catalyzed enantio- and diastereoselective synthesis of benzoxasiloles: ligand-controlled switching from inter- to intramolecular aryl-transfer process. J. Am. Chem. Soc. 137, 11838–11845 (2015).
Kumar, R. et al. Nickel(0)/N-heterocyclic carbene-catalyzed asymmetric [2+2+2] cycloaddition of two enones and an alkyne: Access to cyclohexenes with four contiguous stereogenic centers. Org. Lett. 17, 6018–6021 (2015).
Kumar, R. et al. Nickel-catalyzed enantioselective synthesis of cyclobutenes via [2+2] cycloaddition of α,β-unsaturated carbonyls with 1,3-enynes. Synthesis 48, 2789–2794 (2016).
Hayashi, Y., Hoshimoto, Y., Kumar, R., Ohashi, M. & Ogoshi, S. Nickel(0)-catalyzed intramolecular reductive coupling of alkenes and aldehydes or ketones with hydrosilanes. Chem. Commun. 52, 6237–6240 (2016).
Chaulagain, M. R., Sormunen, G. J. & Montgomery, J. New N-heterocyclic carbene ligand and its application in asymmetric nickel-catalyzed aldehyde/alkyne reductive couplings. J. Am. Chem. Soc. 129, 9568–9569 (2007).
Sato, Y., Hinata, Y., Seki, R., Oonishi, Y. & Saito, N. Nickel-catalyzed enantio- and diastereoselective three-component coupling of 1,3-dienes, aldehydes, and silanes using chiral N-heterocyclic carbenes as ligands. Org. Lett. 9, 5597–5599 (2007).
Ahlin, J. S. E., Donets, P. A. & Cramer, N. Nickel(0)-catalyzed enantioselective annulations of alkynes and arylenoates enabled by a chiral NHC ligand: efficient access to cyclopentenones. Angew. Chem. Int. Ed. 53, 13229–13233 (2014).
Donets, P. A. & Cramer, N. Ligand-controlled regiodivergent nickel-catalyzed annulation of pyridones. Angew. Chem. Int. Ed. 54, 633–637 (2015).
Ahlin, J. S. E. & Cramer, N. Chiral N-heterocyclic carbene ligand enabled nickel(0)-catalyzed enantioselective three-component couplings as direct access to silylated indanols. Org. Lett. 18, 3242–3245 (2016).
Ho, C.-Y., Chan, C.-W. & He, L. Catalytic asymmetric hydroalkenylation of vinylarenes: electronic effects of substrates and chiral N-heterocyclic carbene ligands. Angew. Chem. Int. Ed. 54, 4512–4516 (2015).
Seiders, T. J., Ward, D. W. & Grubbs, R. H. Enantioselective ruthenium-catalyzed ring-closing metathesis. Org. Lett. 3, 3225–3228 (2001).
Luan, X. et al. Matching the chirality of monodentate N-heterocyclic carbene ligands: a case study on well-defined palladium complexes for the asymmetric α-arylation of amides. Org. Lett. 10, 5569–5572 (2008).
Matsumoto, Y., Yamada, K. & Tomioka, K. C2 symmetric chiral NHC ligand for asymmetric quaternary carbon constructing copper-catalyzed conjugate addition of Grignard reagents to 3-substituted cyclohexenones. J. Org. Chem. 73, 4578–4581 (2008).
Ogoshi, S., Ikeda, H. & Kurosawa, H. Formation of an aza-nickelacycle by reaction of an imine and an alkyne with nickel(0): oxidative cyclization, insertion, and reductive elimination. Angew. Chem. Int. Ed. 46, 4930–4932 (2007).
Hoshimoto, Y., Ohata, T., Sasaoka, Y., Ohashi, M. & Ogoshi, S. Nickel(0)-catalyzed [2+2+1] carbonylative cycloaddition of imines and alkynes or norbornene leading to γ-lactams. J. Am. Chem. Soc. 136, 15877–15880 (2014).
Bonadies, F. & Bonini, C. Oxidation of active methylene compounds by pyridinium chlorochromate. Synth. Commun. 18, 1573–1580 (1988).
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (A) (No. JP25708018) and (B) (No. JP15K17824), Scientific Research on Innovative Areas (Nos. JP15H00943 and JP15H05803) from MEXT and by JST, Advanced Catalytic Transformation Program for Carbon Utilization (ACT-C, Grant Number JPMJCR12Y6), Japan. Y.H. acknowledges support from the Frontier Research Base for Global Young Researchers, Osaka University, on the program of MEXT. We sincerely thank Prof. Dr. Norimitsu Tohnai, Graduate School of Engineering, Osaka University, Japan for collecting X-ray data of 3ik. We also thank Prof. Dr. Mary Grellier, Laboratoire de Chimie de Coordination, CNRS, Toulouse, France for discussion on mechanistic part.
Author information
Authors and Affiliations
Contributions
S.O. and R.K. conceived and designed the synthetic routes. R.K. and Y.H. prepared the manuscript, and edited by all other authors. R.K. and E.T. carried out the experiments. M.O. analyzed the X-ray data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kumar, R., Hoshimoto, Y., Tamai, E. et al. Two-step synthesis of chiral fused tricyclic scaffolds from phenols via desymmetrization on nickel. Nat Commun 8, 32 (2017). https://doi.org/10.1038/s41467-017-00068-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-00068-8
- Springer Nature Limited