
Abstract
Sympathoexcitation, under the regulatory control of the brain, plays a pivotal role in the etiology of hypertension. Within the brainstem, significant structures involved in the modulation of sympathetic nerve activity include the rostral ventrolateral medulla (RVLM), caudal ventrolateral medulla (CVLM), nucleus tractus solitarius (NTS), and paraventricular nucleus (paraventricular). The RVLM, in particular, is recognized as the vasomotor center. Over the past five decades, fundamental investigations on central circulatory regulation have underscored the involvement of nitric oxide (NO), oxidative stress, the renin-angiotensin system, and brain inflammation in regulating the sympathetic nervous system. Notably, numerous significant findings have come to light through chronic experiments conducted in conscious subjects employing radio-telemetry systems, gene transfer techniques, and knockout methodologies. Our research has centered on elucidating the role of NO and angiotensin II type 1 (AT1) receptor-induced oxidative stress within the RVLM and NTS in regulating the sympathetic nervous system. Additionally, we have observed that various orally administered AT1 receptor blockers effectively induce sympathoinhibition by reducing oxidative stress via blockade of the AT1 receptor in the RVLM of hypertensive rats. Recent advances have witnessed the development of several clinical interventions targeting brain mechanisms. Nonetheless, Future and further basic and clinical research are needed.

Similar content being viewed by others

Introduction
Sympathoexcitation plays a pivotal role in the pathogenesis of hypertension [1,2,3,4,5]. The brain’s regulation of the sympathetic nervous system involves the rostral ventrolateral medulla (RVLM), a renowned vasomotor center situated in the brainstem [6, 7]. The basal central sympathetic outflow is determined by inputs from baroreceptors, chemoreceptors, and visceral receptors, which are relayed through the nucleus tractus solitarius (NTS) [7,8,9,10,11,12] and the paraventricular nucleus (PVN) of the hypothalamus [8, 13, 14]. Notably, NTS stimulation inhibits neurons in the RVLM through the caudal ventrolateral medulla (CVLM) pathway [15]. (Fig. 1)

The neural network of nucleus tractus solitarius (NTS), paraventricular nucleus (PVN) of the hypothalamus, rostral ventrolateral medulla (RVLM), and caudal ventrolateral medulla (CVLM)
Over the past five decades, extensive investigations have explored the regulatory role of various factors in the brain’s modulation of sympathetic nerve activity, including nitric oxide (NO), angiotensin II type 1 (AT1) receptor-induced oxidative stress [16,17,18,19,20,21,22,23,24,25], and inflammation [26,27,28,29,30,31,32]. This review provides a comprehensive summary of the research on central circulatory regulation, shedding light on the underlying mechanisms of hypertension.
NO dysfunction in the brain causes hypertension with sympathoexcitation
NO is a crucial mediator of intracellular signaling in various tissues, including the central nervous system [33,34,35]. However, conflicting responses have been observed in acute experiments conducted under anesthetized conditions concerning the effects of NO in the RVLM and NTS [25, 36,37,38,39,40,41]. Chronic experiments employing in vivo techniques for gene transfer of endothelial NO synthase (eNOS) into the NTS of rats have demonstrated that elevated levels of NO within the NTS lead to a reduction in blood pressure accompanied by sympathoinhibition in both normotensive and hypertensive rats [21, 25, 42]. Additionally, our findings have indicated that NO within the RVLM elicits a prolonged depressor response associated with sympathoinhibition mediated by an augmented release of gamma-aminobutyric acid (GABA), an inhibitory amino acid, within the RVLM of normotensive and stroke-prone spontaneously hypertensive rats (SHRSP) [43, 44]. Moreover, NO within the RVLM improves impaired baroreflex control of heart rate in hypertensive rats [45]. These outcomes imply that dysfunctional NO signaling and subsequent disinhibition of the RVLM may contribute to sympathoexcitation in hypertensive rats.
Accumulating evidence from animal models of hypertension and human hypertensive subjects suggests that alterations in central neural pathways within the PVN, regulated by neuronal NO, modulate sympathetic outflow. Previous investigations have demonstrated that administering an NO donor, sodium nitroprusside, into the PVN leads to decreased sympathetic nerve activity, blood pressure, and heart rate in rats [46,47,48,49].
In light of these investigations, we can postulate that NO within the RVLM, NTS, and PVN induces a depressor response with sympathoinhibition and that NO dysfunction within the brain contributes to hypertension characterized by sympathoexcitation.
Oxidative stress in the brain causes hypertension with sympathoexcitation
Oxidative stress in the brain has been identified as a significant contributing factor to sympathoexcitation, complementing the role of NO. We quantified oxidative stress in the RVLM using the electron spin resonance or thiobarbituric acid-reactive substances method. Our findings revealed an elevation in oxidative stress levels within the RVLM of SHRSP [50,51,52,53,54,55,56,57,58] and spontaneously hypertensive rats (SHR) [59, 60]. Notably, we conducted microinjections of tempol, a membrane-permeable superoxide dismutase (SOD) mimetic, into the RVLM, resulting in reduced blood pressure and heart rate solely in SHRSP, while normotensive rats showed no response [50]. We also employed adenovirus vectors encoding the manganese SOD (MnSOD) gene to transfect the RVLM in SHRSP. The overexpression of MnSOD in the RVLM elicited decreased blood pressure, heart rate, and urinary norepinephrine excretion exclusively in SHRSP, with no effect observed in normotensive rats [50]. Moreover, our investigation revealed reduced SOD activity within the RVLM of SHRSP compared to normotensive rats, impairing the capability to scavenge superoxide anions [50]. These observations highlight the role of oxidative stress in the RVLM as a causative factor in sympathoexcitation, thereby contributing to the neural pathophysiology of hypertension in SHRSP. Additionally, we demonstrated that oxidative stress in the RVLM induces sympathoexcitation in various other hypertensive models, including salt-induced hypertension [59], dietary-induced hypertension [54, 61], and experimental jet lag [53]. These results align with previous studies conducted by other researchers [19, 20, 22, 62, 63]. Furthermore, in renovascular (two-kidney one-clip) hypertensive rats, sympathoexcitation is associated with oxidative stress in the RVLM and PVN [64,65,66]. These findings strongly support the notion that the upsurge in oxidative stress within the RVLM is a primary cause rather than a consequence of sympathoexcitation, ultimately leading to hypertension. Several investigations have also demonstrated that oxidative stress within the NTS or PVN contributes to hypertension via sympathoexcitation [67,68,69,70].
Sources of oxidative stress in the brain
Within the brain, various sources of oxidative stress, including nicotinamide adenine dinucleotide phosphate (NAD(P)H) oxidase, xanthine oxidase, uncoupled NOS, and mitochondria, activate the AT1 receptor/NAD(P)H oxidase pathway. This activation primarily induces oxidative stress in the RVLM of SHRSP [55, 71, 72]. The renin-angiotensin system in the brain is associated with augmented central sympathetic outflow [73,74,75,76,77,78,79]. Notably, it has been established that mitochondria-derived oxidative stress mediates angiotensin II-induced sympathoexcitation in the brain [80, 81]. Administration of exogenous angiotensin II into the RVLM elicits a pressor response through sympathoexcitation [82, 83]. In hypertensive rats, the inhibition of the AT1 receptor in the RVLM by AT1 receptor blockers induces sympathoinhibition [54,55,56, 59, 61, 72, 82]. Furthermore, the overexpression of MnSOD attenuates angiotensin II-induced pressor response associated with oxidative stress in the RVLM [71]. These findings collectively suggest that the AT1 receptor/NAD(P)H oxidase pathway is the primary source of oxidative stress in the RVLM of hypertensive rats. Additionally, Rac1, a small G protein involved in intracellular signaling pathways leading to NAD(P)H activation, requires lipid modifications to translocate from the cytosol to the cell membrane. Rac1 is associated with the activation of NAD(P)H oxidase [21, 23,24,25, 52, 80]. Inhibition of Rac1 through the transfection of adenovirus vectors encoding a dominant negative Rac1 into the RVLM or NTS results in decreased blood pressure, heart rate, and sympathetic nerve activity in SHRSP but not in normotensive rats [23, 80]. Moreover, blocking the translocation of Rac1 from the cytosol to the membrane in the RVLM of SHRSP induces sympathoinhibition by reducing NAD(P)H oxidase activity and oxidative stress [52]. These findings suggest that activating the AT1 receptor/NAD(P)H oxidase pathway, associated with Rac1, predominantly contributes to oxidative stress in the RVLM or NTS in hypertension.
Mechanisms of oxidative stress-induced sympathoexcitation in the brain
The interaction between superoxide and NO is pivotal, as a decrease in NO availability in the brain caused by superoxide may lead to sympathoexcitation. In SHRSP, a reduction in NO-mediated GABA release in the RVLM is partially involved in superoxide-induced sympathoexcitation [84]. Peroxynitrite is also critical in the relationship between superoxide and NO, as it exhibits excitotoxic effects [85, 86]. Reactive oxygen species and reactive nitrogen species can modulate the function of inducible nitric oxide synthase (iNOS) in a dose-dependent manner, and peroxynitrite diminishes both NO and superoxide production through enzymatic dysfunction of iNOS [85,86,87]. Furthermore, peroxynitrite contributes to the hypotensive effect of NO following the overexpression of endothelial nitric oxide synthase (eNOS) in SHR [88]. The role of peroxynitrite in the RVLM in regulating the sympathetic nervous system requires further investigation.
Glutamate, one of the excitatory amino acids in the RVLM, is recognized for its ability to induce robust sympathoexcitation [6, 7]. Our recent study demonstrated that oxidative stress influences the balance between glutamate and GABA in the RVLM of hypertensive rats [60]. These findings align with a previous report indicating that NAD(P)H oxidase-derived superoxide in the RVLM contributes to the angiotensin II-induced pressor response by enhancing presynaptic glutamate release [73]. We hypothesize that glutamate in the RVLM may play a role in oxidative stress-induced sympathoexcitation.
Furthermore, we have focused on elucidating the signal transduction pathways associated with oxidative stress. Activation of the AT1 receptor in the RVLM triggers caspase-3 activation through the Ras/mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway, which is implicated in sympathoexcitation in SHRSP [72]. In SHRSP, the activities of Ras, p38 MAPK, ERK, and caspase-3 are elevated in the RVLM compared to normotensive rats. Phosphorylation of the pro-apoptotic proteins Bax and Bad, leading to cytochrome c release from mitochondria, activates caspase-3 [72]. Conversely, phosphorylation of the anti-apoptotic protein Bcl-2 inhibits caspase-3 activation. Intracerebroventricular infusion of a caspase-3 inhibitor reduces blood pressure and heart rate while inducing sympathoinhibition in SHRSP but not in normotensive rats. ICV infusion of an AT1 receptor blocker also leads to sympathoinhibition and reduced activities of Ras, p38 MAPK, ERK, and caspase-3 in the RVLM of SHRSP [72]. These results are consistent with a prior report indicating that NAD(P)H oxidase-derived superoxide is responsible for p38 MAPK or ERK1/2 activation by angiotensin II in the RVLM [73]. Although the relationship between oxidative stress and kinase activation is bidirectional [89], these pathways likely exist downstream of the AT1 receptor in the RVLM of SHRSP and contribute to the elevation of blood pressure and sympathoexcitation.
Recently, there has been a focus on exploring additional central mechanisms of sympathoexcitation associated with oxidative stress, including perivascular macrophages in the brain [90, 91], the transcription factor nuclear factor kappa-B [92], and microglial cytokines [31]. Furthermore, our recent findings have demonstrated that neuron-astrocyte uncoupling mediated by AT1 causes sympathoexcitation in SHRSP receptor-induced oxidative stress in the RVLM [93, 94].
Several unresolved matters warrant attention. Firstly, the target cells of oxidative stress in the RVLM may vary. We have yet to ascertain which specific cell types, such as presympathetic neurons, interneurons, or axon terminals originating from NTS and PVN to RVLM, are affected by oxidative stress in the RVLM. Oxidative stress triggers calcium influx in neural cells, and the subsequent accumulation of calcium in mitochondria leads to sympathoexcitation associated with mitochondrial oxidative stress production [71], similar to what has been reported previously [81]. Furthermore, our investigations have revealed that oxidative stress in the RVLM enhances glutamatergic excitatory inputs while attenuating GABAergic inhibitory inputs from PVN to RVLM [60]. Interestingly, Chan et al. demonstrated that NAD(P)H oxidase-derived superoxide in the RVLM is implicated in the angiotensin II-induced pressor response by enhancing presynaptic glutamate release to RVLM neurons [73]. Our recent results suggest that the apoptosis of astrocytes might play a role in mediating oxidative stress-induced sympathoexcitation in the RVLM [72, 93, 94]. Secondly, the precise impact of oxidative stress on the electrophysiological characteristics of RVLM neurons remains incompletely understood. Kumagai et al. revealed that RVLM bulbospinal neurons in SHR experience depolarization and an increased firing rate in response to angiotensin II. Angiotensin II induces a sustained inward current and augments the frequency and amplitude of excitatory postsynaptic currents [95,96,97]. These angiotensin II-mediated responses in the RVLM could be associated with oxidative stress. Angiotensin II-induced oxidative stress can downregulate the expression of a voltage-gated potassium channel in the RVLM [98, 99]. Moreover, the AT1 receptor enhances the frequency of glutamate-sensitive spontaneous excitatory postsynaptic currents in the RVLM [73]. Further investigations are necessary to address these unresolved questions.
Effects of AT1 receptor blockers on oxidative stress in the brain and sympathetic nervous system
Peripherally administered AT1 receptor blockers can traverse the blood-brain barrier and effectively block AT1 receptors both within and outside the brain. However, the degree of receptor blocking action within the brain may vary among different AT1 receptor blockers [55, 61, 100,101,102,103]. Brain regions responsible for regulating the sympathetic nervous system, including the circumventricular organs outside the blood-brain barrier, exhibit a high density of AT1 receptors. As a result, peripherally administered AT1 receptor blockers can access these regions without being hindered by the blood-brain barrier and acting on AT1 receptors within the brain that lies behind the blood-brain barrier [104]. Systemic administration of AT1 receptor blockers also affects AT1 receptors within the brain, reducing blood pressure in hypertensive rats [23, 55, 61, 101, 105,106,107]. The actions of AT1 receptor blockers within the brain may partly depend on their lipophilicity and pharmacokinetics [100,101,102,103].
Furthermore, we have investigated the sympathoinhibitory effect achieved by reducing oxidative stress through inhibiting brain AT1 receptors, and our findings align with numerous previous studies [61, 100,101,102,103,104,105,106]. Orally administered telmisartan or olmesartan reduced blood pressure and demonstrated sympathoinhibition in SHRSP, accompanied by decreased oxidative stress within the brainstem, including the RVLM [23, 55, 107]. Notably, orally administered telmisartan exhibited a more remarkable ability to inhibit AT1 receptor-induced oxidative stress in the RVLM and induce sympathoinhibition in SHRSP compared to candesartan, despite producing similar blood pressure-lowering effects [55]. Similar outcomes were observed in obesity-induced hypertensive rats treated with telmisartan or losartan [61]. Therefore, it is plausible to suggest that orally administered AT1 receptor blockers can elicit sympathoinhibition through the blockade of AT1 receptors in the RVLM. Furthermore, the sympathoinhibitory effects of orally administered AT1 receptor blockers may not be uniform across the class.
Sympathoinhibition by targeting oxidative stress in the brain of hypertension
3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, commonly known as statins, are potent agents that inhibit cholesterol biosynthesis and have been shown to reduce blood pressure in hypertensive patients [108]. Furthermore, the potential sympathoinhibitory effects of statins have been demonstrated [109,110,111]. In animal studies, orally administered atorvastatin induces sympathoinhibition. It enhances impaired baroreflex sensitivity by decreasing oxidative stress through the inhibition of the AT1 receptor-NAD (P) H oxidase pathway and the up-regulation of MnSOD in the RVLM of SHRSP [51, 52, 110], which aligns with a previous report concerning the vasculature [112]. Additionally, orally administered atorvastatin can increase nitric oxide synthase in the brainstem of SHRSP [113]. Based on these findings, we hypothesize that statins can induce sympathoinhibition through the reduction of oxidative stress and activation of nitric oxide synthase in the RVLM.
Several calcium channel blockers have been established to cause sympathoinhibition by reducing oxidative stress in the RVLM of hypertensive rats. Orally administered amlodipine [114] or azelnidipine [58] induces sympathoinhibition by decreasing oxidative stress in the RVLM of SHRSP. We have verified that orally administered azelnidipine inhibits NAD (P) H oxidase activity and activates MnSOD in the RVLM of SHRSP [58]. Furthermore, a combination of atorvastatin and amlodipine [60] or olmesartan and azelnidipine [115] exhibits additive effects in sympathoinhibition through the reduction of oxidative stress in the RVLM.
Interestingly, calorie restriction [54] or exercise training [56] is crucial in inducing sympathoinhibition by reducing oxidative stress through the blockade of the AT1 receptor in hypertensive rats. These findings suggest that adipocytokines and insulin resistance may influence the AT1 receptor in the RVLM, leading to sympathoexcitation.
Inflammation in the brain exaggerates hypertension with sympathoexcitation
In the past decade, several investigations have proposed a link between brain inflammation and the occurrence of sympathoexcitation and hypertension. Haspula’s research indicates that neuroinflammation and heightened sympathetic activity are pivotal in amplifying sympathetic activity in individuals with hypertension [26]. Winklewski’s findings point to inflammation in the forebrain and hindbrain nuclei, which regulate the outflow of the sympathetic nervous system from the brain to the periphery, as an emerging concept in understanding the pathogenesis of neurogenic hypertension [27]. Waki’s study highlights the upregulation of proinflammatory molecules in endothelial cells of the microvasculature supplying the NTS in SHR compared to normotensive rats [28]. Interestingly, macrophage infiltration into vascular walls has been implicated in the development of hypertension by promoting vascular inflammation and endothelial dysfunction [29]. Iyonaga et al. demonstrated the involvement of brain perivascular macrophages in the pathogenesis of hypertension through enhanced sympathetic activation [30]. Similarly, Shi et al. revealed that activated microglia in the PVN release proinflammatory cytokines, contributing to neurogenic hypertension [31]. These collective findings strongly suggest a causal relationship between brain inflammation and hypertension accompanied by sympathoexcitation. However, our research has demonstrated that activated microglia with morphological alterations within the PVN is not implicated in the maintenance of established severe hypertension, and inflammation within the PVN cannot be considered a therapeutic target for established hypertension [32]. Therefore, brain inflammation likely exacerbates hypertension with sympathoexcitation rather than initiating its onset.
Perspectives
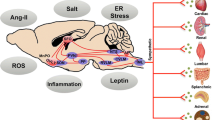
Figure 2 illustrates the underlying concept of modulating the sympathetic nervous system through the action of NO derived from the overexpression of eNOS and AT1 receptor-induced oxidative stress in the RVLM (Fig. 2). In managing subnormal sympathoexcitation in cardiovascular diseases, our investigations suggest that blocking the AT1 receptor in the RVLM is imperative, aligning with prior research [61, 100,101,102,103,104,105,106,107]. AT1 receptor blockers are extensively employed in treating hypertension [116]. Moreover, these blockers have been postulated to exert neuroprotective effects, reducing the occurrence of stroke and enhancing cognitive function [116, 117]. Notably, recent findings on renal nerve ablation in patients with resistant hypertension indicate the potential contribution of renal afferent nerves to elevated blood pressure [118,119,120]. These renal afferent nerves project directly to various regions within the central nervous system, including the NTS and the hypothalamus [121, 122]. In the phenol renal injury model of hypertension, which involves stimulating renal afferent nerves, oxidative stress mediates sympathoexcitation. Activation of the brain’s AT1 receptor and NAD(P)H oxidase occurs in this model. Previous research has proposed that increased production of oxidative stress and diminished expression of neuronal nitric oxide synthase may be implicated in this mechanism, resulting in alterations in brain cytokines [123, 124]. Therefore, it is reasonable to emphasize the AT1 receptor and the associated production of oxidative stress in the RVLM as crucial therapeutic targets for addressing abnormal sympathoexcitation in hypertension.

A schema shows our concept in regulating sympathetic nerve activity via nitric oxide and angiotensin II type 1 (AT1) receptor-induced oxidative stress in the rostral ventrolateral medulla (RVLM). GABA; γ-amino butyric acid
Conclusion
Considering our investigations and prior studies, we contend that the escalation of AT1 receptor-induced oxidative stress and the impairment of NO function in the brain, particularly in the RVLM, predominantly contribute to sympathoexcitation in hypertension [125]. Furthermore, it is plausible that targeting AT1 receptor-induced oxidative stress and NO dysfunction in the brain could offer therapeutic prospects for hypertension. However, additional, comprehensive research is warranted at both the fundamental and clinical levels.
References
Grassi G. Assessment of sympathetic cardiovascular drive in human hypertension: achievements and perspectives. Hypertension. 2009;54:690–7.
Grassi G. Sympathetic neural activity in human hypertension and related diseases. Am J Hypertens. 2010;23:1052–60.
Grassi G, Seravalle G, Quarti-Trevano F. The ‘neurogenic hypothesis’ in hypertension: current evidence. Exp Physiol. 2010;95:581–6.
Esler M. The 2009 Carl Ludwig lecture: pathophysiology of the human sympathetic nervous system in cardiovascular diseases: the transition from mechanisms to medical management. J Appl Physiol. 2010;108:227–37.
Mauo K, Lambert GW, Esler MD, Rakugi H, Ogihara T, Schlaich MP. The role of sympathetic nervous system activity in renal injury and end-stage renal disease. Hypertens Res. 2010;33:521–8.
Dampney RAL. Functional organization of central pathways regulating the cardiovascular system. Physiol Rev. 1994;74:323–64.
Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–46.
Pilowsky PM, Goodchild AK. Baroreceptor reflex pathways and neurotransmitters: 10 years on. J Hypertens. 2002;20:1675–88.
Sved AF, Ito S, Sved JC. Brainstem mechanisms of hypertension: role of the rostral ventrolateral medulla. Curr Hypertens Rep. 2003;5:262–8.
Dampney RAL, Polson JW, Potts PD, Hirooka Y, Horiuchi J. Functional organization of brain pathways subserving the baroreceptor reflex: studies in conscious animals using immediate early gene expression. Cell Mol Neurobiol. 2003;23:597–616.
Campos RR, Bergamschi CT. Neurotransmission alterations in central cardiovascular control in experimental hypertension. Curr Hypertens Rev. 2006;2:193–8.
Carlson SH, Wyss JM. Neurohormonal regulation of the sympathetic nervous system: new insights into central mechanisms of action. Curr Hypertens Rep. 2008;10:233–40.
Dampney RAL, Horiuchi J, Killinger S, Sheriff MJ, Tan PSP, McDowall LM. Long-term regulation of arterial blood pressure by hypothalamic nuclei: some critical questions. Clin Exp Pharm Physiol. 2005;32:419–25.
Coote JH. Landmarks in understanding the central nervous control of the cardiovascular system. Exp Physiol. 2007;92:3–18.
Agarwal SK, Gelsema AJ, Calaresu FR. Inhibition of rostral VLM by baroreceptor activation is relayed through caudal VLM. Am J Physiol. 1990;258:R1271–8.
Krukoff TL. The central action of nitric oxide in the regulation of autonomic functions. Brain Res Rev. 1999;30:52–65.
Patel K, Li YF, Hirooka Y. Role of nitric oxide in central sympathetic outflow. Exp Biol Med. 2001;226:814–24.
Zanzinger J. Role of nitric oxide in the neural control of cardiovascular function. Cardiovasc Res. 1999;43:639–49.
Tai MH, Wang LL, Wu KLH, Chan JYH. Increased superoxide anion in the rostral ventrolateral medulla contributes to hypertension in spontaneously hypertensive rats via interactions with nitric oxide. Free Rad Biol Med. 2005;38:450–62.
Peterson JR, Sharma RV, Davisson RL. Reactive oxygen species in the neuropathogenesis of hypertension. Curr Hypertens Rep. 2006;8:232–41.
Hirooka Y. Role of reactive oxygen species in the brainstem in neural mechanisms of hypertension. Auton Neurosci. 2008;142:20–4.
Campos RR. Oxidative stress in the brain and arterial hypertension. Hypertens Res. 2009;32:1047–8.
Hirooka Y, Sagara Y, Kishi T, Sunagawa K. Oxidative stress and central cardiovascular regulation: pathogenesis of hypertension and therapeutic aspects. Circ J. 2010;274:827–35.
Hirooka Y. Oxidative stress in the cardiovascular center has a pivotal role in the sympathetic activation in hypertension. Hypertens Res. 2011;34:407–12.
Hirooka Y, Kishi T, Sakai K, Takeshita A, Sunagawa K. Imbalance of central nitric oxide and reactive oxygen species in the regulation of sympathetic activity and neural mechanisms of hypertension. Am J Physiol. 2011;300:R818–26.
Haspula D, Clark MA. Neuroinflammation and sympathetic overactivity: mechanisms and implications in hypertension. Auton Neurosci. 2018;210:10–17.
Winklewski PJ, Radkowski M, Wszedybyl-Winklewska M, Demkow U. Brain inflammation and hypertension: the chicken or the egg? J Neuroinflammation. 2015;12:85.
Waki H, Gouraud SS, Maeda M, Raizada MK, Paton JFR. Contributions of vascular inflammation in the brainstem for neurogenic hypertension. Respir Physiol Neurobiol. 2011;178:422–8.
Rodriguez-Iturbe B, Pons H, Johnson RJ. Role of the immune system in hypertension. Physiol Rev. 2017;97:1127–64.
Iyonaga T, Shinohara K, Matsuura T, Hirooka Y, Tsutsui H. Brain perivascular macrophages contribute to the development of hypertension in stroke-prone spontaneously hypertensive rats via sympathetic activation. Hypertens Res. 2019;43:99–110.
Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q. Brain microglial cytokines in neurogenic hypertension. Hypertension. 2010;56:297–303.
Takesue K, Kishi T, Hirooka Y, Sunagawa K. Activation of microglia within the paraventricular nucleus of the hypothalamus is NOT involved in the maintenance of established hypertension. J Cardiol. 2017;69:84–8.
Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–802.
Talman WT, Dragon DN. Transmission of arterial baroreflex signals depends on neuronal nitric oxide synthase. Hypertension. 2004;43:820–4.
Talman WT. NO and central cardiovascular control: a simple molecule with a complex story. Hypertension. 2006;48:552–4.
Hirooka Y, Polson JW, Dampney RAL. Pressor and sympathoexcitatory effects of nitric oxide in the rostral ventrolateral medulla. J Hypertens. 1996;14:1317–24.
Zanzinger J, Seller H. Species differences in the distribution of nitric oxide synthase in brain stem regions that regulate sympathetic activity. Brain Res. 1997;764:265–8.
Chen SY, Mao SP, Chai CY. Role of nitric oxide on pressor mechanisms within the dorsomedial and rostral ventrolateral medulla in anesthetized cats. Clin Exp Pharm Physiol. 2001;28:155–63.
Morimoto S, Sasaki S, Miki S, Kawa T, Nakamura K, Itoh H, et al. Nitric oxide is an excitatory modulator in the rostral ventrolateral medulla in rats. Am J Hypertens. 2000;13:1125–34.
Tseng CJ, Liu HY, Lin HC, Ger LP, Tung CS, Yen MH. Cardiovascular effects of nitric oxide in the brain stem nuclei of rats. Hypertension. 1996;27:36–42.
Huang CC, Chan SH, Hsu KS. cGMP/protein kinase G-dependent potentiation of glutamatergic transmission induced by nitric oxide in immature rat rostral ventrolateral medulla neurons in vitro. Mol Pharm. 2003;64:521–32.
Sakai K, Hirooka Y, Matsuo I, Eshima K, Shigematsu H, Shimokawa H, et al. Overexpression of eNOS in NTS causes hypotension and bradycardia in vivo. Hypertension. 2000;36:1023–8.
Kishi T, Hirooka Y, Sakai K, Shigematsu H, Shimokawa H, Takeshita A. Overexpression of eNOS in the RVLM causes hypotension and bradycardia via GABA release. Hypertension. 2001;38:896–901.
Kishi T, Hirooka Y, Ito K, Sakai K, Shimokawa H, Takeshita A. Cardiovascular effects of overexpression of endothelial nitric oxide synthase in the rostral ventrolateral medulla in stroke-prone spontaneously hypertensive rats. Hypertension. 2002;39:264–8.
Kishi T, HIrooka Y, Kimura Y, Sakai K, Ito K, Shimokawa H, et al. Overexpression of eNOS in RVLM improves impaired baroreflex control of heart rate in SHRSP. Hypertension. 2003;41:255–60.
Vincent SR, Kimura H. Histochemical mapping of nitric oxide synthase in the rat brain. Neuroscience. 1992;46:755–84.
Horn T, Smith PM, McLaughlin BE, Bauce L, Marks GS, Pittman QJ, et al. Nitric oxide actions in the paraventricular nucleus: cardiovascular and neurochemical implications. Am J Physiol Regul Integr Comp Physiol. 1994;266:R306–13.
Zhang K, Mayhan WG, Patel KP. Nitric oxide within the paraventricular nucleus mediates changes in renal sympathetic nerve activity. Am J Physiol. 1997;273:R864–72.
Zheng H, katsurada K, Nandi S, Li Y, Patel KP. A critical role for the paraventricular nucleus of the hypothalamus in the regulation of the volume reflex in normal and various cardiovascular disease states. Curr Hypertens Rep. 2022;24:235–46.
Kishi T, Hirooka Y, Kimura Y, Ito K, Shimokawa H, Takeshita A. Increased reactive oxygen species in rostral ventrolateral medulla contributes to neural mechanisms of hypertension in stroke-prone spontaneously hypertensive rats. Circulation. 2004;109:2357–62.
Kishi T, Hirooka Y, Shimokawa H, Takeshita A, Sunagawa K. Atorvastatin reduced oxidative stress in the rostral ventrolateral medulla of stroke-prone spontaneously hypertensive rats. Clin Exp Hypertens. 2008;30:3–11.
Kishi T, Hirooka Y, Konno S, Sunagawa K. Sympathoinhibition induced by centrally administered atorvastatin is associated with alteration of NAD(P)H oxidase and Mn superoxide dismutase activity in rostral ventrolateral medulla of stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharm. 2010;55:184–90.
Kishi T, Sunagawa K. Experimental ‘jet lag’ causes sympathoexcitation via oxidative stress through AT1 receptor in the brainstem. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:1969–72.
Kishi T, Hirooka Y, Ogawa K, Konno S, Sunagawa K. Calorie restriction inhibits sympathetic nerve activity via anti-oxidant effect in the rostral ventrolateral medulla of obesity-induced hypertensive rats. Clin Exp Hypertens. 2011;33:240–5.
Kishi T, Hirooka Y, Sunagawa K. Sympathoinhibition caused by orally administered telmisartan through inhibition of AT(1) receptor in the rostral ventrolateral medulla. Hypertens Res. 2012;35:940–6.
Kishi T, Hirooka Y, Katsuki M, Ogawa K, Shinohara K, Isegawa K, et al. Exercise training causes sympathoinhibition through antioxidant effect in the rostral ventrolateral medulla of hypertensive rats. Clin Exp Hypertens. 2012;34:278–83.
Kishi T, Sunagawa K. Combination therapy of atorvastatin and amlodipine inhibits sympathetic nervous system activation and improves cognitive function in hypertensive rats. Circ J. 2012;76:1934–41.
Konno S, Hirooka Y, Araki S, Koga Y, Kishi T, Sunagawa K. Azelnidipine decreases sympathetic nerve activity via antioxidant effect in the rostral ventrolateral medulla of stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharm. 2008;52:555–60.
Koga Y, Hirooka Y, Araki S, Nozoe M, Kishi T, Sunagawa K. High salt intake enhances blood pressure increase during development of hypertension via oxidative stress in rostral ventrolateral medulla of spontaneously hypertensive rats. Hypertens Res. 2008;31:2075–83.
Nishihara M, Hirooka Y, Matsukawa R, Kishi T, Sunagawa K. Oxidative stress in the rostral ventrolateral medulla modulates excitatory and inhibitory inputs in spontaneously hypertensive rats. J Hypertens. 2012;30:97–106.
Konno S, Hirooka Y, Kishi T, Sunagawa K. Sympathoinhibitory effect of telmisartan through the reduction of oxidative stress in rostral ventrolateral medulla of obesity-induced hypertensive rat. J Hypertens. 2012;30:1992–9.
Fujita M, Ando K, Nagae A, Fujita T. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in salt-sensitive hypertension. Hypertension. 2007;50:360–7.
Nagae A, Fujita M, Kawarazaki H, Matsui H, Ando K, Fujita T. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in obesity-induced hypertension. Circulation. 2009;119:978–86.
Oliveira-Sales EB, Dugaich AP, Carillo BA, Abreu NP, Boim MA, Martins PJ, et al. Oxidative stress contributes to renovascular hypertension. Am J Hypertens. 2008;21:98–104.
Oliveira-Sales EB, Nishi EE, Carillo BA, Dolnikoff MS, Bergamaschi CT, Campos RR. Oxidative stress in the sympathetic premotor neurons contributes to sympathetic activation in renovascular hypertension. Am J Hypertens. 2009;22:484–92.
Oliveira-Sales EB, Colombari DSA, Davisson RL, Kasparov S, Hirata AE, Campos RR, et al. Kidney-induced hypertension depends on superoxide signaling in the rostral ventrolateral medulla. Hypertension. 2010;56:290–6.
Nishi EE, Almedia VR, Amaral FG, Simon KA, Futuro-Neto HA, Pontes RB, et al. Melatonin attenuates renal sympathetic overactivity and reactive oxygen species in the brain in neurogenic hypertension. Hypertens Res. 2019;42:1683–91.
Chan SH, Chan JY. Brain stem NOS and ROS in neural mechanisms of hypertension. Antioxid Redox Signal. 2014;20:146–63.
Xia WJ, Liu KL, Wang XM, Yang Y, Meng T, Qiao JA, et al. Hypothalamic paraventricular nucleus hydrogen sulfide exerts antihypertensive effects in spontaneously hypertensive rats via the Nrf2 pathway. Am J Hypertens. 2023 https://doi.org/10.1093/ajh/hpad012.
Niu LG, Sun N, Liu KL, Su Q, Qi J, Fu LY, et al. Genistein alleviates oxidative stress and inflammation in the hypothalamic paraventricular nucleus by activating the Sirt1/Nrf2 pathway in high salt-induced hypertension. Cardiovasc Toxicol. 2022;22:898–909.
Nozoe M, Hirooka Y, Koga Y, Araki S, Konno S, Kishi T, et al. Mitochondria-derived reactive oxygen species mediate sympathoexcitation induced by angiotensin II in the rostral ventrolateral medulla. J Hypertens. 2008;26:2176–84.
Kishi T, Hirooka Y, Konno S, Ogawa K, Sunagawa K. Angiotensin II type 1 receptor activated caspase-3 through ras/mitogen-activated protein kinase/extracellular signal-regulated kinase in the rostral ventrolateral medulla is involved in sympathoexcitation in stroke-prone spontaneously hypertensive rats. Hypertension. 2010;55:291–7.
Chan SH, Hsu KS, Huang CC, Wang LL, Qu CC, Chan JY. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced pressor effect via activation of p38 mitogen-activated protein kinase in the rostral ventrolateral medulla. Circ Res. 2005;97:772–80.
Hu L, Zhu DN, Yu Z, Wang JQ, Sun ZJ, Yao T. Expression of angiotensin type 1 (AT1) receptor in the rostral ventrolateral medulla in rats. J Appl Physiol. 2002;92:2153–61.
Reja V, Goodchild AK, Phillips JK, Pilowsky PM. Upregulation of angiotensin AT1 receptor and intracellular kinase gene expression in hypertensive rats. Clin Exp Pharm Physiol. 2006;33:690–5.
Leenen FHH. Brain mechanisms contributing to sympathetic hyperactivity and heart failure. Circ Res. 2007;101:221–3.
Huang BS, Leenen FHH. The brain renin-angiotensin-aldosterone system: a major mechanism for sympathetic hyperactivity and left ventricular remodeling and dysfunction after myocardial infarction. Curr Heart Fail Rep. 2009;6:81–8.
Zucker IH, Schultz HD, Patel KP, Wang W, Gao L. Regulation of central angiotensin type 1 receptors and sympathetic outflow in heart failure. Am J Physiol. 2009;297:H1557–66.
Dupont AG, Brouwers S. Brain angiotensin peptides regulate sympathetic tone and blood pressure. J Hypertens. 2010;28:1599–610.
Nozoe M, Hirooka Y, Koga Y, Sagara Y, Kishi T, Engelhardt JF, et al. Inhibition of Rac1-derived reactive oxygen species in nucleus tractus solitarius decreases blood pressure and heart rate in stroke-prone spontaneously hypertensive rats. Hypertension. 2007;50:62–8.
Zimmerman MC, Zucker IH. Mitochondrial dysfunction and mitochondria-produced reactive oxygen species: new aspects for neurogenic hypertension? Hypertension. 2009;53:112–4.
Hirooka Y, Potts PD, Dampney RAL. Role of angiotensin II receptor subtypes in mediating the sympathoexcitatory effects of exogenous and endogenous angiotensin peptides in the rostral ventrolateral medulla. Brain Res. 1997;772:107–14.
Dampney RAL, Tan PSP, Sheriff MJ, Fontes MAP, Horiuchi J. Cardiovascular effects of angiotensin II in the rostral ventrolateral medulla: the push-pull hypothesis. Curr Hypertens Rep. 2007;9:222–7.
Shinohara K, Hirooka Y, Kishi T, Sunagawa K. Reduction of nitric oxide-mediated γ-amino butyric acid release in the rostral ventrolateral medulla is involved in superoxide-induced sympathoexcitation of hypertensive rats. Circ J. 2012;76:2814–21.
Zielonka J, Zielonka M, Sikora A, Adamus J, Joseph J, Hardy M, et al. Global profiling of reactive oxygen and nitrogen species in biological systems: high-throughput real-time analysis. J Biol Chem. 2012;287:2984–95.
Zanzinger J. Mechanisms of action of nitric oxide in the brain stem: role of oxidative stress. Auton Neurosci. 2002;98:24–7.
Sun J, Druhan LJ, Zweler JL. Reactive oxygen and nitrogen species regulate inducible nitric oxide synthase function shifting the balance of nitric oxide and superoxide production. Arch Biochem Biophys. 2010;494:130–7.
Kung LC, Chan SH, Wu KL, Ou CC, Tai MH, Chan JY. Mitochondrial respiratory enzyme complexes in rostral ventrolateral medulla as cellular targets of nitric oxide and superoxide interaction in the antagonism of antihypertensive action of eNOS transgene. Mol Pharm. 2008;74:1319–32.
Rhee SG. Cell signaling, H2O2, is a necessary evil for cell signaling. Science. 2006;312:1882–3.
Yu Y, Zhang ZH, Wei SG, Serrats J, Weiss RM, Felder RB. Brain perivascular macrophages and the sympathetic response to inflammation in rats after myocardial infarction. Hypertension. 2010;55:652–9.
Hirooka Y. Brain perivascular macrophages and central sympathetic activation after myocardial infarction: heart and brain interaction. Hypertension. 2010;55:610–1.
Cardinale JP, Sriramula S, Mariappan N, Agarwal D, Francis J. Angiotensin II-induced hypertension is modulated by nuclear factor-kB in the paraventricular nucleus. Hypertension. 2011;59:113–21.
Kishi T. Disruption of central antioxidant property of nuclear factor erythroid 2-related factor 2 worsens circulatory homeostasis with baroreflex dysfunction in heart failure. Int J Mol Sci. 2018;19:646.
Kishi T. Heart failure is a disruption of dynamic circulatory homeostasis mediated by the brain. Int Heart J. 2016;57:145–9.
Kumagai H, Oshima N, Matsuura T, Iigaya K, Imai M, Onimaru H, et al. Importance of rostral ventrolateral medulla neurons in determining efferent sympathetic nerve activity and blood pressure. Hypertens Res. 2012;35:132–41.
Matsuura T, Kumagai H, Kawai A, Onimaru H, Imai M, Oshima N, et al. Rostral ventrolateral medulla neurons of neonatal Wister-Kyoto and spontaneously hypertensive rats. Hypertension. 2002;40:560–5.
Oshima N, Kumagai H, Onimaru H, Kawai A, Pilowski PM, Iigaya K, et al. Monosynaptic excitatory connection from the rostral ventrolateral medulla to sympathetic preganglionic neurons revealed by simultaneous recording. Hypertens Res. 2008;31:1445–54.
Wu KL, Chan SH, Chan JY. Neuroinflammation and oxidative stress in the rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation. 2012;9:212.
Gao L, Li Y, Schultz HD, Wang WZ, Finch M, Smith LM, et al. Downregulated Kv4.3 expression in the RVLM as a potential mechanism for sympathoexcitation in rats with chronic heart failure. Am J Physiol. 2010;298:H945–55.
Wang JM, Tan J, Leenen FHH. Central nervous system blockade by peripheral administration of AT1 receptor blockers. J Cardiovasc Pharm. 2003;41:593–9.
Tsuchihashi T, Kagiyama S, Matsumura K, Abe I, Fujishima M. Effects of chronic oral treatment with imidapril and TCV-116 on the responsiveness to angiotensin II in ventrolateral medulla of SHR. J Hypertens. 1999;17:917–22.
Nishimura Y, Ito T, Hoe KL, Saavedra JM. Chronic peripheral administration of the angiotensin II AT1 receptor antagonist candesartan blocks brain AT1 receptors. Brain Res. 2000;871:29–38.
Gohlke P, Weiss S, Jansen A, Wienen W, Stangier J, Rascher W, et al. AT1 receptor antagonist telmisartan administered peripherally inhibits central responses to angiotensin II in conscious rats. J Pharm Exp Ther. 2001;298:62–70.
McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, McAllen RM, et al. The brain renin-angiotensin system: location and physiological roles. Int J Biochem Cell Biol. 2003;35:901–18.
Leenen FHH, Yuan B. Prevention of hypertension by irbesartan in Dahl S rats relates to central angiotensin II type 1 receptor blockade. Hypertension. 2001;37:981–4.
Lin Y, Matsumura K, Kagiyama S, Fukuhara M, Fujii K, Iida M. Chronic administration of olmesartan attenuates the exaggerated pressor response to glutamate in the rostral ventrolateral medulla of SHR. Brain Res. 2005;1058:161–6.
Araki S, Hirooka Y, Kishi T, Yasukawa K, Utsumi H, Sunagawa K. Olmesartan reduces stress in the brain of stroke-prone spontaneously hypertensive rats assessed by an in vivo ESR method. Hypertens Res. 2009;32:1091–6.
Golomb BA, Dimsdale JE, White HL, Ritchie JB, Criqui MH. Reduction in blood pressure with statins: Results from the USCD Statin Study, a randomized trial. Arch Intern Med. 2008;168:721–7.
Sinski M, Lewandowsk J, Ciarka A, Bidiuk J, Abramczyk P, Dobosiewicz A, et al. Atorvastatin reduced sympathetic activity and increased baroreceptor reflex sensitivity in patients with hypercholesterolemia and systemic arterial hypertension. Kardiol Pol. 2009;67:613–20.
Kishi T, Hirooka Y. Sympathoinhibitory effects of atorvastatin in hypertension. Circ J. 2010;74:2552–3.
Siddiqi L, Joles JA, Oey PL, Blankestijn PJ. Atorvastatin reduced sympathetic activity in patients with chronic kidney disease. J Hypertens. 2011;29:2176–80.
Wassmann S, Laufs U, Muller K, Konkol C, Ahlbory K, Baumer AT, et al. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2002;22:300–5.
Kishi T, Hirooka Y, Mukai Y, Shimokawa H, Takeshita A. Atorvastatin causes depressor and sympathoinhibitory effects with upregulation of nitric oxide synthase in stroke-prone spontaneously hypertensive rats. J Hypertens. 2003;21:379–86.
Hirooka Y, Kimura Y, Nozoe M, Sagara Y, Ito K, Sunagawa K. Amlodipine-induced reduction of oxidative stress in the brain is associated with sympatho-inhibitory effects in stroke-prone spontaneously hypertensive rats. Hypertens Res. 2006;29:49–56.
Shinohara K, Hirooka Y, Ogawa K, Kishi T, Yasukawa K, Utsumi H, et al. Combination therapy of olmesartan and azelnidipine inhibits sympathetic activity associated with reducing oxidative stress in the brain of hypertensive rats. Clin Exp Hypertens. 2012;34:456–64.
Iwanami J, Mogi M, Iwai M, Horiuchi M. Inhibition of the renin-angiotensin system and target organ protection. Hypertens Res. 2009;32:229–37.
Mogi M, Horiuchi M. Effects of angiotensin II receptor blockers on dementia. Hypertens Res. 2009;32:738–40.
Krum H, Schlaich MP, Whitbourn R, Sobotka P, Sadowski J, Bartus K, et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet. 2009;373:1275–81.
DiBona GF, Esler M. Translational medicine: the antihypertensive effect of renal denervation. Am J Physiol. 2010;298:R245–53.
Simplicity HTN-2 Investigators. Renal sympathetic denervation in patients with treatment-resistant hypertension (The Simplicity HTN-2 Trail): a randomized controlled trial. Lancet. 2010;376:1903–9.
Calaresu FR, Ciriello J. Renal afferent nerves affect discharge rate of medullary and hypothalamic single units in cat. J Auton Nerv Syst. 1981;3:311–20.
Stella A, Golin R, Genovesi S, Zanchetti A. Renal reflexes in the regulation of blood pressure and sodium excretion. Can J Physiol Pharm. 1987;65:1536–9.
Ye S, Zhong H, Campese VM. Oxidative stress mediates the sympathetic nerve activity in the phenol renal injury model of hypertension. Hypertension. 2006;48:309–15.
Campese VM, Shaohua Y, Huiquin Z. Oxidative stress mediates angiotensin II-dependent stimulation of sympathetic nerve activity. Hypertension. 2005;46:533–9.
Kishi T. Regulation of the sympathetic nervous system by nitric oxide and oxidative stress in the rostral ventrolateral medulla: 2012 academic conference award from the Japanese Society of Hypertension. Hypertens Res. 2013;36:845–51.
Acknowledgements
A Grant-in-Aid for Scientific Research supported this study from the Japan Society for the Promotion of Science (15590757, 17590745, 19390231, and 22790709) and, in part, a Kimura Memorial Foundation Research Grant. I want to appreciate Prof. Yoshitaka Hirooka, Kenji Sunagawa, and Akira Takeshita significantly. Furthermore, I express special thanks to Satomi Konno.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kishi, T. Clarification of hypertension mechanisms provided by the research of central circulatory regulation. Hypertens Res 46, 1908–1916 (2023). https://doi.org/10.1038/s41440-023-01335-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-023-01335-6
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Reactive oxygen species in hypertension
Nature Reviews Cardiology (2024)
-
Gestationally administered RAS modulators reprogram endotoxic cardiovascular and inflammatory profiles in adult male offspring of preeclamptic rats
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)