
Abstract
Previous studies have shown that female offspring are resistant to fetal high-fat diet (HFD)-induced programming of heightened vascular contraction; however, the underlying mechanisms remain unclear. The present study tested the hypothesis that estrogen plays a key role in protecting females from fetal programming of increased vascular contraction induced by maternal HFD exposure. Pregnant rats were fed a normal diet (ND) or HFD (60% kcal from fat). Ovariectomy (OVX) and 17β-estradiol (E2) replacement were performed on 8-week-old female offspring. Aortas were isolated from adult female offspring. Maternal HFD exposure increased angiotensin II (Ang II)-induced contractions of the aorta in adult OVX offspring, which was abrogated by E2 replacement. The AT1 receptor (AT1R) antagonist losartan (10 μM), but not the AT2 receptor (AT2R) antagonist PD123319 (10 μM), completely blocked Ang II-induced contractions in both ND and HFD offspring. In addition, HFD exposure caused a decrease in endothelium-dependent relaxations induced by acetylcholine (ACh) in adult OVX but not OVX-E2 offspring. However, it had no effect on sodium nitroprusside (SNP)-induced endothelium-independent aorta relaxation in any of the six groups. Maternal HFD feeding increased AT1R, but not AT2R, leading to an increased AT1R/AT2R ratio in HFD-exposed OVX offspring, associated with selective decreases in DNA methylation at the AT1aR promoter, which was ameliorated by E2 replacement. Our results indicated that estrogen play a key role in sex differences of maternal HFD-induced vascular dysfunction and development of hypertensive phenotype in adulthood by differently regulating vascular AT1R and AT2R gene expression through a DNA methylation mechanism.
Similar content being viewed by others

Introduction
Obesity has become a major global health challenge due to health-related risk factors. The incidence of overweight and obesity among fertile women has been increasing over the past years. Growing evidence suggests that maternal obesity has adverse effects not only on mothers but also on their offspring. Excessive weight gain or obesity during pregnancy is associated with short- and long-term complications affecting the health of offspring and increases the risk of obesity, diabetes and cardiovascular disease in the offspring later in life [1, 2]. High-fat diet intake is a major risk factor for obesity development. Using a mature rat model of high-fat diet (HFD) consumption during pregnancy, our previous study showed that maternal HFD contributes to heightened vasoconstriction in adult male but not female offspring [3]. However, the mechanisms underlying the maternal HFD consumption-induced sex-dependent development of vascular dysfunctional phenotypes in adult offspring are not fully understood.
Herein, we present our findings of an investigation into whether postnatal ovarian hormones are responsible for the sex-specific resistance in heightened vasoconstriction observed in female offspring exposed to a maternal HFD. Epidemiological studies have shown that premenopausal women have a lower incidence of coronary heart disease than postmenopausal women or age-matched men [4], suggesting vascular benefits of estrogen. Previous studies have discovered that hormone changes after menopause may cause increased organ damage and cardiovascular diseases such as increased arterial stiffness, coronary diseases, and chronic heart failure [5]. Estrogens exert their effects by activating the estrogen receptors ERα, ERβ and GPR30, which initiate downstream signaling pathways and regulate gene expression and posttranslational modifications.
Chronic activation of the renin–angiotensin–aldosterone system (RAAS) promotes and perpetuates congestive heart failure, systemic hypertension, and chronic kidney disease [6, 7]. Angiotensin II (Ang II) acts on two receptors, the Ang II receptor type‐1 receptor (AT1R) and the Ang II type‐2 receptor (AT2R). The action of Ang II on AT1R leads to increased sodium retention and vasoconstriction. Overstimulation of AT1R is associated with numerous diseases, including hypertension, stroke, cardiac hypertrophy and renal diseases. However, the actions of AT2R stimulation are counterregulatory to those of AT1R, leading to anti‐inflammatory, antifibrotic, and vasodilatory effects. AT2R is the dominant receptor type in the fetus and plays a key role in development but is less relevant in normal adults. Chronic AT1R blockade is associated with an inverted vasomotor response to Ang II via AT2R-mediated NO production in hypertensive rats [8]. Given that the programming of Ang II receptors in response to fetal insults plays a critical role in cardiovascular dysfunction later in life, the purpose of the present study was to investigate whether estrogen normalizes maternal HFD-induced enhanced vasoconstriction by regulating Ang II receptors in adult female offspring.
Several molecular epigenetic mechanisms, including DNA methylation, histone modification and noncoding RNA activities, may play vital roles in the aberrant development of cardiovascular disease later in life. Differences in fetal gene expression within the uterus can contribute to epigenetic modifications that can cause permanent changes in organ structure, cell number, and metabolism [9]. DNA methylation is the major mechanism and occurs at the cytosine of the dinucleotide sequence CpG. DNA methylation in gene promoter regions is generally associated with repression of transcription, resulting in long-term shutdown of the associated gene. Previous studies have shown that epigenetic alterations of genes induced by suboptimal maternal nutrition/endocrine factors or a fetal low-protein diet can promote metabolic syndrome and the development of hypertension later in life [10,11,12]. Thus, the present study tested the hypothesis that estrogen normalizes maternal HFD-induced heightened vasoconstriction via epigenetic regulation of AT1aR and AT2R gene expression patterns through a DNA methylation mechanism.
Methods
Experimental animals
Eight-week-old Sprague‒Dawley rats were obtained from Guangdong Medical Laboratory Animal Center and housed under specific pathogen-free (SPF) conditions with controlled light (12 h) and dark (12 h) at 25 °C. The females were caged with proven males, and pregnancy was verified by the presence of sperm in the smear. Pregnant rats were randomly divided into two groups: the ND group fed a standard chow diet (4.3% fat, 20% protein, 49% carbohydrate) throughout gestation and the HFD group fed a high-fat diet (60% kcal fat, 20% protein, 20% carbohydrate, D12492) throughout gestation, as described previously [3]. At term, the dams were allowed to deliver, and the litters were standardized to 8 pups. All mothers were fed a normal chow diet after delivery. All offspring were fed a standard chow diet after birth and studied at 3 months of age. All animal experiments in the present study were approved in advance by the Institutional Animal Care and Use Committee of Guangzhou Medical University (No. GY2017-067, 10/17/2017).
Ovariectomy and estrogen replacement
Female offspring (8 weeks old) were anesthetized with isoflurane (2%-3%) and underwent either sham operation or bilateral ovariectomy (OVX). Seven days after OVX, 17β-estradiol (E2) pellets (1.5 mg for 90-day release; Innovative Research of America) were subcutaneously implanted in the lateral side of the neck for continuous release of hormones as previously reported [13, 14]. All rats were randomly divided into six groups: 1) ND-sham, 2) HFD-sham, 3) ND-OVX, 4) HFD-OVX, 5) ND-OVX-E2, and 6) HFD-OVX-E2.
Measurement of plasma E2 levels
Blood was collected from 3-month-old offspring via the jugular vein. Plasma was separated for measurement of E2 after centrifugation (3000 rpm) for 15 min. The plasma levels of E2 were determined using a commercial estradiol ELISA kit (Neobioscience, Shenzhen, China, EHC9025) according to the manufacturer’s instructions.
Contraction of aortic rings
Aortas with endothelial integrity were isolated from adult offspring and immersed in ice-cold Krebs-Henseleit solution (KHS). Each aorta was cleaned and cut into four rings (4 mm in length). The segments were immediately mounted in 10 mL organ baths containing KHS at 37 °C and equilibrated with a mixture of 95% O2 and 5% CO2, as described previously [3, 15]. The isometric force was recorded using a PowerLab 400TM data acquisition system (Software Chart, version 7.0, AD Instrument, MA, USA). The tissues were stretched to the optimal resting tension (1.5 g/mm2) and allowed to equilibrate for 1 h before starting the experiments. After equilibration, 120 mM KCl was used to contract the rings. The rings were then rinsed after reaching the smooth contraction amplitude. The contraction induced by the drugs was normalized to the KCl-elicited contraction. Arteries were stimulated with the cumulative addition of Ang II. In some experiments, arteries were pretreated with the AT1R inhibitor losartan (10 µM) or the AT2R inhibitor PD123,319 (10 µM) for 20 min and then exposed to increased concentrations of Ang II. For relaxation studies, endothelium-dependent responses to acetylcholine (ACh) or endothelium-independent responses to sodium nitroprusside (SNP) were also performed in rings precontracted with a submaximal concentration (1 μM) of phenylephrine (PE).
Western immunoblot analysis
Protein from aortas with endothelial integrity was isolated from tissues homogenized with lysis buffer (cell lysis buffer, CST) supplemented with protease and phosphatase inhibitors, as previously described [16, 17]. The homogenates were centrifuged at 4 °C for 15 min at 12000 × g, and the supernatants were collected. The protein concentrations were measured using a BCA protein assay kit (Thermo). The extracted proteins were separated on a 10% polyacrylamide gel with 0.1% SDS at 100 V for 90 min and transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, MA, USA). After blocking the membrane with a Tris-buffered saline solution containing 5% dry milk at room temperature for 1 h, the membrane was incubated with primary antibodies against AT1R (1:2000 dilution, ab124734, 41 kDa, Abcam) and AT2R (1:2000 dilution, ab92445, 41 kDa, Abcam) overnight at 4 °C. After washing, the membrane was incubated with secondary horseradish peroxidase-conjugated antibodies (1:5000 dilution CST, USA) and visualized with enhanced chemiluminescence reagents, and the blots were exposed to Hyperfilm. The band intensities were analyzed by densitometry (Bio-Rad image software). To ensure equal loading, the band intensities were normalized to those of GAPDH.
Real-time RT‒PCR
Total RNA was extracted from aortic rings with endothelial integrity using the TRIzol protocol (Invitrogen), and reverse transcription was performed as previously described [18]. AT1aR, AT1bR and AT2R mRNA abundance was determined by real-time RT‒PCR using a StepOne™ real-time PCR system (Applied Biosystem, CA, USA) in a reaction mix containing 0.2 μM primers and the master mix provided in the Brilliant SYBR Green qPCR Master Mix (Takara). We used the following RT‒PCR protocol: 95 °C for 5 min followed by 40 cycles of 95 °C for 5 sec, 60 °C for 30 sec and 72 °C for 10 sec. GAPDH was used as an internal reference. PCR was performed in triplicate, and the threshold cycle numbers were averaged. The primers used were as follows: AT1aR (Gene ID: 24180), 5’-ggagaggattcgtggcttgag-3’ (forward) and 5’-ctttctgggagggttgtgtgat-3’ (reverse); AT1bR (Gene ID: 81638), 5’-atgtctccagtcccctctca-3’ (forward) and 5’-tgacctcccatctccttttg-3’ (reverse); and AT2R (Gene ID: 24182), 5’-caatctggctgtggctgactt-3’ (forward) and 5’-tgcacatcacaggtccaaag-3’ (reverse). The Ct value was defined as the PCR cycle number at which the fluorescence signal of the probe exceeded the background signal. The mRNA levels of the target genes were normalized to those of GAPDH to obtain the relative threshold cycle (ΔCt), and then the fold change in mRNA expression was calculated using the 2−ΔΔCt method.
Pyrosequencing for quantitative measurement of DNA methylation
We purified genomic DNA from the aortas of 3-month-old offspring using a QIAGEN DNeasy kit (69506, QIAGEN). After bisulfite conversion with a Qiagen EpiTect Bisulfite Kit (59104, QIAGEN), CpG methylation at the AT1aR and AT2R gene promoters was determined by pyrosequencing analysis (PyroMark Q96 ID, QIAGEN) according to the manufacturer’s instructions. The primers for pyrosequencing assays were designed using PyroMark Assay Design 2.0 software. The primer sequences are listed in Supplemental Table 1.
Statistical analysis
Concentration‒response curves were analyzed by computer-assisted nonlinear regression using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA) to obtain the pD2 (−logEC50) and the maximum response (Emax). The data are expressed as the means ± SDs. Statistical significance (P < 0.05) was determined by ANOVA followed by Neuman-Keuls post hoc testing or Student’s t-test, where appropriate.
Results
Plasma E2 levels
In the sham groups, maternal HFD consumption had no effect on the plasma E2 levels (52.33 ± 6.10 pg·mL−1 vs. 52.00 ± 5.21 pg·mL−1; n = 6) of adult female offspring. In the OVX groups, E2 levels were significantly decreased in both ND (9.67 ± 1.10 pg·mL−1; n = 6) and HFD (9.00 ± 1.80 pg·mL−1; n = 6) adult female offspring compared to the sham-group offspring. The E2 levels in both ND (67.00 ± 7.02 pg·mL−1; n = 6) and HFD (64.67 ± 6.98 pg·mL−1; n = 6) adult female offspring in the OVX-E2 groups were dramatically enhanced compared with those of offspring in the OVX groups.
Maternal HFD consumption enhanced Ang II-induced contraction of aortas in adult OVX offspring
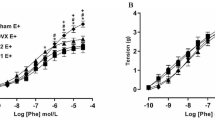
In sham-operated female offspring, consistent with our previous finding [3], prenatal HFD did not significantly alter Ang II-induced contraction of aortas (Fig. 1a and Table 1). However, in the OVX groups, maternal HFD significantly increased the maximal response (Emax), but not the pD2, of Ang II-induced contractions compared with those of ND adult offspring (Fig. 1b and Table 1). In the OVX-E2 groups, Ang II-induced contractions of the aorta were not significantly different between ND and HFD offspring, suggesting that the enhanced contractions in HFD-OVX offspring were abrogated by E2 replacement (Fig. 1c and Table 1).

Effect of maternal HFD consumption on angiotensin II (Ang II)-mediated contraction of aortas in adult female offspring that had been exposed in utero to a normal diet (ND) or high-fat diet (HFD). Ang II (a, b, c, n = 6/group)-induced contraction was analyzed in the aortas of sham, ovariectomized (OVX), and estrogen-treated ovariectomized (OVX-E2) adult female offspring that had been exposed in utero to a ND or HFD. ND, normal diet; HFD, high-fat diet. The pD2 values and the maximal response (Emax) are presented in Table 1. The values are the means ± SDs. The data were analyzed by two-way ANOVA. *P < 0.05, HFD vs. ND
Effect of losartan and PD123319 on Ang II–induced contractions in adult female offspring
To determine the receptor subtype of Ang II-induced vasoconstrictions, aortic rings were preincubated with losartan (AT1R inhibitor) or PD123319 (AT2R inhibitor). Losartan almost completely blocked Ang II-induced contractions in all six groups (Fig. 2a–f). Unlike losartan, PD123319 had no significant effect on Ang II-induced contractions among all six groups (Fig. 2a–f).

Effect of losartan and PD123319 on angiotensin II (Ang II)-induced contraction of aortas in adult female offspring. Ang II-induced contraction in the absence or presence of losartan or PD123319 (PD) was analyzed in aortas from female offspring that had been exposed in utero to a ND or HFD. a ND-sham group (n = 5/group). b HFD-sham group (n = 5/group). c ND OVX-group (n = 5/group). d HFD-OVX group (n = 5/group). e ND-OVX-E2 group (n = 5/group). f HFD-OVX-E2 group (n = 5/group). ND Normal diet, HFD High-fat diet. The values are the means ± SDs. The data were analyzed by two-way ANOVA. *P < 0.05. Ang II vs. Ang II + losartan
Maternal HFD consumption decreased endothelium-dependent relaxation in adult OVX offspring
To determine the effect of maternal HFD consumption on endothelium-dependent relaxation in adult female offspring, ACh-induced relaxation was examined (Fig. 3, left). In the sham groups, there were no significant differences in ACh-induced vasorelaxation between ND and HFD adult female offspring. However, ACh-induced relaxation of the aorta was markedly lower in HFD-OVX offspring than in ND-OVX offspring, which was reversed by E2 replacement (Fig. 3, left).

Effect of maternal HFD on acetylcholine (ACh)- or sodium nitroprusside (SNP)-induced relaxation of aortas in sham, ovariectomized (OVX), and estrogen-treated ovariectomized (OVX+E2) groups of female offspring that had been exposed in utero to a normal diet (ND) or high-fat diet (HFD). Aortic rings were pretreated with 1 µM phenylephrine (PE) and then subjected to cumulative addition of ACh (n = 5/group) or SNP (n = 5/group). a ACh-induced relaxation of aortas in sham offspring rat. b SNP-induced relaxation of aortas in sham offspring rat. c ACh-induced relaxation of aortas in OVX offspring rat. d SNP-induced relaxation of aortas in OVX offspring rat. e ACh-induced relaxations of aorta in OVX+E2 offspring rat. f SNP-induced relaxation of aortas in OVX+E2 offspring rat. The values are the means ± SDs. The data were analyzed by two-way ANOVA. *P < 0.05, HFD vs. ND
Unlike ACh-induced relaxation, SNP-induced endothelium-independent relaxation showed no marked difference compared with that of ND rats in any of the six groups (Fig. 3, right).
Maternal HFD consumption increased the aorta AT1R/AT2R ratio in adult OVX offspring
As shown in Fig. 4, AT1R protein levels and AT1aR mRNA abundance were significantly increased in aortas from HFD offspring compared with those from ND offspring (Fig. 4a, b). AT2R protein and mRNA abundance was also increased in the HFD group, resulting in no significant difference in the AT1R/AT2R ratio between the ND and HFD groups in adult sham offspring (Fig. 4a, b). In contrast to the findings in sham offspring, aorta AT1R protein and AT1aR mRNA levels, but not AT2R protein and mRNA levels, were dramatically increased in HFD-OVX offspring compared to ND-OVX offspring, resulting in a significant increase in the AT1R/AT2R ratio in adult OVX offspring (Fig. 4c, d). However, after estrogen replacement, the enhanced AT1R/AT2R ratio was restored to be equivalent to that in the sham group (Fig. 4e, f).

Effect of maternal HFD on AT1R and AT2R expression in the aortas of adult female offspring. AT1R and AT2R protein levels were determined by Western blotting in aortas from sham (a), OVX (c) and OVX + E2 (e) adult female offspring. AT1R and AT2R mRNA levels were determined by real-time RT‒PCR in aortas from sham (b), OVX (d) and OVX-E2 (f) adult female offspring. ND Normal diet, HFD High-fat diet. The values are the means ± SDs. The data were analyzed by Student’s t test. *P < 0.05, HFD vs. ND, n = 4–5/group
Effect of maternal HFD consumption on DNA methylation of the CpG locus at the AT1aR and AT2R promoters in adult OVX offspring
Previous studies have demonstrated that the DNA methylation pattern, which is set during development, is critical for the life‐long stability of tissue‐ and cell‐specific gene expression programs [19]. Previous studies have identified transcription factor-binding sites in AT1aR and AT2R gene promoters [20, 21]. As shown in Fig. 5a, maternal HFD consumption dramatically decreased the methylation levels of the CpG locus at the Sp1 transcription factor-binding site (−96), CREB binding site (−150), ERα and ERβ binding site (−484) of the AT1aR promoter in the aortas of adult OVX offspring compared with those of ND offspring. However, there was no significant difference in the methylation of the GATA-1 binding site (−809) at the AT1aR gene promoter region between ND offspring and HFD offspring (Fig. 5a). Unlike the case for AT1aR, maternal HFD consumption did not alter the methylation levels of the CpG locus at the CREB binding site (−444), GRE binding site (+11) or CpG site (−52) near the TATA box at the promoter of AT2R in the OVX groups (Fig. 5b).

Effect of maternal HFD on DNA methylation of the CpG locus at the AT1aR and AT2R promoters in aortas from adult ovariectomized (OVX) offspring. Aortic rings were freshly isolated from adult OVX offspring that had been exposed in utero to a high-fat diet or normal diet. DNA was isolated, and methylation levels were determined by pyrosequencing. a DNA methylation of the CpG locus at the AT1aR promoter. b DNA methylation of the CpG locus at the AT2R promoter. ND normal diet, HFD high-fat diet. The values are the mean ± SDs. The data were analyzed by Student’s t-test. *P < 0.05, HFD vs. ND, n = 5/group
Discussion
The findings of the present study emphasize the contributions of sex steroid hormones to the relationship between sex and maternal HFD-induced elevations in vasoconstriction in the offspring. The major findings of the present study were as follows: 1) maternal HFD consumption increased Ang II-induced vasoconstriction and decreased endothelium-dependent vasorelaxation in adult OVX offspring, which was abrogated by estrogen replacement; 2) maternal HFD increased aortic AT1R expression, but not AT2R expression, in adult OVX offspring, which was rescued by E2 replacement; and 3) the differential regulation of AT1aR and AT2R gene expression via a DNA methylation mechanism may have been involved in the development of maternal HFD-induced increases in vasoconstriction and a hypertensive phenotype later in life.
The present study demonstrated that Ang II-induced contraction of the aorta in adult female offspring was not different between the ND and HFD-treated groups, which is consistent with our previous findings that maternal HFD consumption increases Ang II-induced vasocontraction in male but not female offspring [3]. It has been reported that there are sex differences in fetal programming of adult cardiovascular diseases in several animal models [22, 23]. Previous studies in rats have shown that fetal and neonatal nicotine exposure increases KCl- and norepinephrine-induced contractions of the aorta in male but not female offspring [24]. In addition, antenatal nicotine exposure causes aberrant vascular reactivity and increased blood pressure in adult male rat offspring in a sex-dependent manner [25]. Our previous studies have demonstrated that maternal HFD consumption causes a sex-dependent increase in vasoconstriction in adult male but not female rat offspring [3]. Of importance, similar to the finding in male offspring from the same model, we found that removal of ovaries (OVX), the main sources of circulating estrogen in females, significantly facilitated the development of increased Ang II-induced arterial contractions in HFD-treated rats compared with ND-treated rats, and this effect was abolished by estrogen replacement, suggesting that estrogen plays a vital role in protecting female offspring from the development of enhanced contractility in response to maternal HFD exposure. This finding is consistent with a previous study demonstrating that OVX rats fed a HFD exhibit vascular dysfunction [26]. Similar findings have been reported in a rat model of prenatal nicotine: estrogen protects female offspring from developmental programming of a hypertensive phenotype [15]. Estrogen replacement protects female offspring from maternal hypertension-induced sensitization to Ang II hypertension [27]. Oral estrogen reverses ovariectomy-induced morning surge hypertension in growth-restricted mice [28]. Estrogen administration decreases blood pressure and increases vascular conductance in ovariectomized rats [29]. These results suggest that the chronic absence of circulating estrogen may well be responsible for the observed differences in vascular dysfunction in HFD-OVX rats. The mechanisms underlying the estrogen-induced counteraction of maternal HFD-induced vasoconstriction are far more complex. Maternal HFD rats did not have significantly different E2 levels than ND rats in the sham, OVX, and OVX-E2 groups. Consistent with our findings, previous studies have also indicated that in utero adverse environmental exposure does not affect the circulating estrogen levels in adult female offspring [15, 23], suggesting that the protective role of estrogen may not be directly related to the level of estradiol but may be related to the effects of estradiol on other systems controlling vascular function in adult female HFD offspring. Estrogen may trigger downstream signaling pathways and lead to genomic and nongenomic vascular effects such as vasodilation and reduced vascular remodeling [30].
Our previous study demonstrated that the increased phenylephrine-induced aortic contraction in male HFD offspring may be primarily due to loss of endothelium-mediated vasodilation [3]. Since endothelium-dependent vasodilatation is a vital measurement of endothelial function, we examined ACh-induced relaxation of the aorta in adult female offspring. We found that maternal HFD consumption caused a nearly 50% reduction in vasorelaxation in response to the endothelium-dependent vasodilator ACh in adult OVX offspring, which was consistent with the findings of our previous study in adult male offspring [3]. However, there were no significant differences in the endothelium-independent vasodilator SNP-induced relaxation among all six groups. These findings suggested that maternal HFD caused impaired endothelium-dependent vasorelaxation. Previous studies have shown that adverse intrauterine environments may contribute to reductions in endothelium-dependent vasorelaxation in offspring in both human and animal models [25, 31]. Endothelial nitric oxide synthase (eNOS) is an important mediator of the effects of ACh on endothelial cells. A previous study showed that maternal nicotine exposure decreased eNOS activity, which was reversed by estrogen, resulting in altered NO-mediated vasodilation in a sex-dependent manner [15]. However, the role of the estrogen-mediated NO pathway in the sex difference of programmed vascular dysfunction is complex. Therefore, precise studies of the NO pathway are warranted in the future to further determine potential endothelium-dependent mechanisms.
One of the potential targets for estrogen regulation is the renin–angiotensin system (RAS). Growing evidence supports a vital role of RAS in the pathogenesis of cardiovascular dysfunction in a model of fetal programming [3, 13, 32]. Regulation of Ang II receptors by estrogen is one of the key mechanisms for sex differences in fetal programming of adult cardiovascular dysfunction [23]. The effects of estrogen on the RAS are complex and sometimes contradictory, but the overall effect appears to be a reduction in RAS activity. Ang II regulates vascular resistance through AT1R and AT2R. Activation of AT1R by Ang II induces vasoconstriction, whereas activation of AT2R opposes the classic actions of AT1R, resulting in vasodilation [33]. Estrogen treatment decreases hepatic AT1R protein levels in ischemia/reperfusion injury rats [34]. Estrogen reduces Ang II-induced acceleration of senescence in endothelial progenitor cells (EPCs) partially through downregulation of AT1R expression [35]. Previous studies have reported that in rat kidneys, ovariectomy decreases AT2R expression, which is reversed by estrogen replacement [36]. In addition, maternal nicotine exposure increases Ang II-induced vascular contraction and the blood pressure response in OVX offspring compared with those in the control offspring, which is associated with increased expression of vascular AT1R. Nevertheless, estrogen replacement can eliminate the effect of OVX on the nicotine-induced response [15]. Physiological levels of estrogen might promote the vasodilating effect of AT2R activation in the renal vasculature, while supraphysiological levels might promote vasoconstriction [37].
Previous studies have shown that AT1R plays key roles in modulating physiological/pathophysiological conditions such as vasoconstriction, hypertension, atherosclerosis and impaired sodium excretion [38]. AT1R blockers regulate endothelial dysfunction by increasing the release of NO [39]. AT2R has been recognized to suppress the proliferation of vascular smooth muscle cells (VSMCs) [40] and promote vasorelaxation by activating eNOS or NO-cGMP production in endothelial cells (ECs) and VSMCs [41]. In the present study, we found that the AT1R blocker losartan, but not the AT2R blocker PD123319, almost completely blocked Ang II-induced contractions of the aorta in both the ND and HFD animals in all six groups, indicating a primary role of AT1R in Ang II-increased vascular tension. Our previous study demonstrated that maternal HFD caused an increase in the vascular AT1R/AT2R ratio in male but not female offspring [3], indicating that the effect of HFD on vascular AT1R/AT2R expression may be abrogated by sex hormones. Indeed, the present study showed that maternal HFD significantly increased AT1R, but not AT2R protein expression, contributing to a significant increase in the AT1R/AT2R ratio in the aortas of OVX offspring that was abrogated by estrogen replacement; this finding further supports an important role of estrogen in the prevention of the HFD-induced increase in the AT1R/AT2R ratio. In addition, aortic AT1aR mRNA expression, but not AT2R mRNA expression, was dramatically increased in HFD-OVX offspring, suggesting that the maternal HFD-induced alteration of AT1R protein levels is primarily regulated at the transcriptional level. Unlike for AT1R, the exact role of AT2R and the extent to which AT2R participates in the regulation of vasoconstriction remain unclear. AT2R stimulation promotes vasoconstriction in isolated mesenteric arteries from spontaneously hypertensive rats [42]. A previous study has also shown that AT1R antagonists induce an important rise in circulating Ang II and that AT2R might then be chronically overstimulated, participating in the maintenance of vasodilation, particularly since this effect is not easily desensitized [42]. Our previous study demonstrated that estrogen normalizes maternal HFD-induced cardiac hypertrophy in offspring by regulating AT2R [13]. Our previous study also showed that estrogen protects the heart from ischemic injury by regulating cardiac AT1R/AT2R expression patterns [43]. These studies demonstrate that the estrogen-induced counteraction of the maternal HFD-induced increase in the AT1R/AT2R ratio may be one of the vital mechanisms for the sex difference in the development of increased vasoconstriction and a hypertensive phenotype in later life.
During critical periods of fetal development, gene expression can be influenced by epigenetic mechanisms, and DNA methylation is one of the major mechanisms for epigenetic modification of gene expression. DNA methylation is a stable epigenetic modification that consists of the covalent binding of a methyl group to the 5′ carbon of cytosine and mostly occurs at CpG dinucleotide sequences in the mammalian genome [44]. In rodents, the AT1aR and AT2R genes contain a number of CG dinucleotides. Several CpG sites at transcription factor-binding sites in AT1aR and AT2R promoters have been demonstrated [21], including the −809, −484, −150, and −96 CpG loci and the −444, −52, +11 CpG loci, respectively. In the present study, the methylation levels at the −484, −150 and −96 CpG loci at the AT1aR promoter were significantly decreased in the aortas of adult HFD-OVX offspring compared with ND-OVX offspring, indicating that maternal HFD-induced upregulation of aortic AT1aR may primarily be due to hypomethylation of these CpG loci at the AT1aR promoter. Previous results have suggested that the effect of early treatment on DNA methylation is sex-, gene- and tissue-specific [45]. Unlike the the hypomethylation at specific CpG sites in the AT1aR promoter, maternal HFD consumption did not significantly alter the methylation levels at the −444, −52 and +11 CpG loci at the AT2R promoter in OVX offspring arteries, which led to no alteration of AT2R in OVX offspring, suggesting that the effect of maternal HFD consumption on DNA methylation is at least gene dependent. Further studies on the precise underlying mechanisms by which estrogen interacts with the Ang II receptor via DNA methylation patterns are warranted.
In summary, our previous studies have shown that maternal HFD consumption causes heightened vasoconstriction in adult male but not female rat offspring. The present study provides novel evidence that estrogen plays a key role in the sex difference in fetal programming of the adult hypertension phenotype. Maternal HFD consumption causes programming of vascular AT1aR gene expression by altering the methylation of specific CpG sites at the AT1aR promoter, contributing to the heightened vascular contractility in adult OVX but not OVX-E2 offspring. These findings further support an important role of estrogen in preventing HFD-induced increases in the AT1R/AT2R ratio in OVX offspring. Although it may be difficult to translate the present findings directly to humans, the possibility that fetal HFD exposure results in the programming of heightened vascular contractions in a sex-dependent manner provides insights into a mechanism worthy of investigation in humans. By improving understanding of the epigenetic molecular mechanisms underlying maternal HFD-induced fetal programming of vascular dysfunction, our findings may support early therapeutic intervention for cardiovascular diseases later in life.
References
Youngson NA, Uddin GM, Das A, Martinez C, Connaughton HS, Whiting S, et al. Impacts of obesity, maternal obesity and nicotinamide mononucleotide supplementation on sperm quality in mice. Reproduction 2019;158:169–79.
Sanli E, Kabaran S. Maternal obesity, maternal overnutrition and fetal programming: Effects of epigenetic mechanisms on the development of metabolic disorders. Curr Genomics. 2019;20:419–27.
Chen F, Cao K, Zhang H, Yu H, Liu Y, Xue Q. Maternal high-fat diet increases vascular contractility in adult offspring in a sex-dependent manner. Hypertens Res. 2021;44:36–46.
Wehbe Z, Nasser SA, El-Yazbi A, Nasreddine S, Eid AH. Estrogen and bisphenol a in hypertension. Curr Hypertens Rep. 2020;22:23.
Zilberman JM. Menopause: Hypertension and vascular disease. Hipertens Riesgo Vasc. 2018;35:77–83.
Ames MK, Atkins CE, Pitt B. The renin-angiotensin-aldosterone system and its suppression. J Vet Intern Med. 2019;33:363–82.
Almeida LF, Tofteng SS, Madsen K, Jensen BL. Role of the renin-angiotensin system in kidney development and programming of adult blood pressure. Clin Sci (Lond). 2020;134:641–56.
Cosentino F, Savoia C, De Paolis P, Francia P, Russo A, Maffei A, et al. Angiotensin II type 2 receptors contribute to vascular responses in spontaneously hypertensive rats treated with angiotensin II type 1 receptor antagonists. Am J Hypertens. 2005;18:493–99.
Neri C, Edlow AG. Effects of maternal obesity on fetal programming: Molecular approaches. Cold Spring Harb Perspect Med. 2015;6:a026591.
Desai M, Jellyman JK, Ross MG. Epigenomics, gestational programming and risk of metabolic syndrome. Int J Obes (Lond). 2015;39:633–41.
Ramzan F, Vickers MH, Mithen RF. Epigenetics, microRNA and Metabolic Syndrome: A Comprehensive Review. Int J Mol Sci. 2021;22:1–20.
Bogdarina I, Welham S, King PJ, Burns SP, Clark AJ. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ Res. 2007;100:520–26.
Chen F, Yu H, Zhang H, Zhao R, Cao K, Liu Y, et al. Estrogen normalizes maternal HFD-induced cardiac hypertrophy in offspring by regulating AT2R. J Endocrinol. 2021;250:1–12.
Theodorsson A, Hilke S, Rugarn O, Linghammar D, Theodorsson E. Serum concentrations of 17beta-estradiol in ovariectomized rats during two times six weeks crossover treatment by daily injections in comparison with slow-release pellets. Scand J Clin Lab Invest. 2005;65:699–705.
Xiao D, Huang X, Yang S, Zhang L. Estrogen normalizes perinatal nicotine-induced hypertensive responses in adult female rat offspring. Hypertension 2013;61:1246–54.
Li YX, Long DL, Liu J, Qiu D, Wang J, Cheng X, et al. Gestational diabetes mellitus in women increased the risk of neonatal infection via inflammation and autophagy in the placenta. Med (Baltim). 2020;99:e22152.
Xue Q, Chen F, Zhang H, Liu Y, Chen P, Patterson AJ, et al. Maternal high-fat diet alters angiotensin II receptors and causes changes in fetal and neonatal ratsdagger. Biol Reprod. 2019;100:1193–203.
Xue Q, Patterson AJ, Xiao D, Zhang L. Glucocorticoid modulates angiotensin II receptor expression patterns and protects the heart from ischemia and reperfusion injury. PLoS One. 2014;9:e106827.
Nemoda Z, Szyf M. Epigenetic alterations and prenatal maternal depression. Birth Defects Res. 2017;109:888–97.
Zhao Y, Zhu Q, Sun S, Qiu Y, Li J, Liu W, et al. Renal transplantation increases angiotensin II receptor-mediated vascular contractility associated with changes of epigenetic mechanisms. Int J Mol Med. 2018;41:2375–88.
Xiao D, Dasgupta C, Li Y, Huang X, Zhang L. Perinatal nicotine exposure increases angiotensin II receptor-mediated vascular contractility in adult offspring. PLoS One. 2014;9:e108161.
Goldstein JM, Handa RJ, Tobet SA. Disruption of fetal hormonal programming (prenatal stress) implicates shared risk for sex differences in depression and cardiovascular disease. Front Neuroendocrinol. 2014;35:140–58.
Ojeda NB, Grigore D, Robertson EB, Alexander BT. Estrogen protects against increased blood pressure in postpubertal female growth restricted offspring. Hypertension 2007;50:679–85.
Xiao D, Huang X, Lawrence J, Yang S, Zhang L. Fetal and neonatal nicotine exposure differentially regulates vascular contractility in adult male and female offspring. J Pharm Exp Ther. 2007;320:654–61.
Xiao D, Huang X, Yang S, Zhang L. Antenatal nicotine induces heightened oxidative stress and vascular dysfunction in rat offspring. Br J Pharm. 2011;164:1400–9.
Li CM, Dong XL, Fan XD, Wu JH, Wang QH, Tian XL, et al. Aqueous extract of danshen (Salvia miltiorrhiza Bunge) protects ovariectomized rats fed with high-fat diet from endothelial dysfunction. Menopause 2013;20:100–9.
Xue B, Beltz TG, Guo F, Johnson AK. Sex differences in maternal gestational hypertension-induced sensitization of angiotensin II hypertension in rat offspring: The protective effect of estrogen. Am J Physiol Regul Integr Comp Physiol. 2018;314:R274–R281.
Haskell SE, Peotta V, Reinking BE, Zhang C, Zhu V, Kenkel EJ, et al. Oral oestrogen reverses ovariectomy-induced morning surge hypertension in growth-restricted mice. Clin Sci (Lond). 2016;130:613–23.
Hernandez I, Delgado JL, Diaz J, Quesada T, Teruel MJ, Llanos MC, et al. 17beta-estradiol prevents oxidative stress and decreases blood pressure in ovariectomized rats. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1599–605.
Smiley DA, Khalil RA. Estrogenic compounds, estrogen receptors and vascular cell signaling in the aging blood vessels. Curr Med Chem. 2009;16:1863–87.
Fan L, Lindsley SR, Comstock SM, Takahashi DL, Evans AE, He GW, et al. Maternal high-fat diet impacts endothelial function in nonhuman primate offspring. Int J Obes (Lond). 2013;37:254–62.
Moritz KM, Mazzuca MQ, Siebel AL, Mibus A, Arena D, Tare M, et al. Uteroplacental insufficiency causes a nephron deficit, modest renal insufficiency but no hypertension with ageing in female rats. J Physiol. 2009;587:2635–46.
Chen Z, Wang L, Ke J, Xiao D. Effects of estrogen in gender-dependent fetal programming of adult cardiovascular dysfunction. Curr Vasc Pharm. 2019;17:147–52.
Li W, Li D, Sun L, Li Z, Yu L, Wu S. The protective effects of estrogen on hepatic ischemia-reperfusion injury in rats by downregulating the Ang II/AT1R pathway. Biochem Biophys Res Commun. 2018;503:2543–48.
Imanishi T, Hano T, Nishio I. Estrogen reduces angiotensin II-induced acceleration of senescence in endothelial progenitor cells. Hypertens Res. 2005;28:263–71.
Hilliard LM, Sampson AK, Brown RD, Denton KM. The “his and hers” of the renin-angiotensin system. Curr Hypertens Rep. 2013;15:71–9.
Safari T, Nematbakhsh M, Evans RG, Denton KM. High-dose estradiol-replacement therapy enhances the renal vascular response to angiotensin II via an AT2-receptor dependent mechanism. Adv Pharm Sci. 2015;2015:682745.
Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: Regulation, signalling and function. Blood Press. 2003;12:70–88.
Konukoglu D, Uzun H. Endothelial Dysfunction and Hypertension. Adv Exp Med Biol. 2017;956:511–40.
Tang B, Ma S, Yang Y, Yang D, Chen J, Su X, et al. Overexpression of angiotensin II type 2 receptor suppresses neointimal hyperplasia in a rat carotid arterial balloon injury model. Mol Med Rep. 2011;4:249–54.
Kawai T, Forrester SJ, O’Brien S, Baggett A, Rizzo V, Eguchi S. AT1 receptor signaling pathways in the cardiovascular system. Pharm Res. 2017;125:4–13.
You D, Loufrani L, Baron C, Levy BI, Widdop RE, Henrion D. High blood pressure reduction reverses angiotensin II type 2 receptor-mediated vasoconstriction into vasodilation in spontaneously hypertensive rats. Circulation 2005;111:1006–11.
Xue Q, Xiao D, Zhang L. Estrogen regulates Angiotensin II receptor expression patterns and protects the heart from ischemic injury in female rats. Biol Reprod. 2015;93:6.
Pei F, Wang X, Yue R, Chen C, Huang J, Huang J, et al. Differential expression and DNA methylation of angiotensin type 1A receptors in vascular tissues during genetic hypertension development. Mol Cell Biochem. 2015;402:1–8.
Wang T, Lian G, Cai X, Lin Z, Xie L. Effect of prehypertensive losartan therapy on AT1R and ATRAP methylation of adipose tissue in the later life of highfatfed spontaneously hypertensive rats. Mol Med Rep. 2018;17:1753–61.
Acknowledgements
We would like to thank Guiping Zhang for the technical guidance.
Funding
This study was supported by the Natural Science Foundation of Guangdong Province (2018A030313719) and the High-level University Construction Fund of Guangdong Province (06-410-2107243).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, F., Zhao, R., Zhang, H. et al. Estrogen normalizes maternal HFD-induced vascular dysfunction in offspring by regulating ATR. Hypertens Res 45, 1743–1753 (2022). https://doi.org/10.1038/s41440-022-01002-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-022-01002-2
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Effect of estrogen on fetal programming in offspring from high-fat-fed mothers
Hypertension Research (2022)