
Abstract
Dietary intake of the heavy metal cadmium (Cd2+) is implicated in hypertension, but potassium supplementation reportedly mitigates hypertension. This study aims to elucidate the hypertensive mechanism of Cd2+. Vascular reactivity and protein expression were assessed in Cd2+-exposed rats for 8 weeks to determine the calcium-handling effect of Cd2+ and the possible signaling pathways and mechanisms involved. Cd2+ induced hypertension in vivo by significantly (p < 0.001) elevating systolic blood pressure (160 ± 2 and 155 ± 1 vs 120 ± 1 mm Hg), diastolic blood pressure (119 ± 2 and 110 ± 1 vs 81 ± 1 mm Hg), and mean arterial pressure (133 ± 2 and 125 ± 1 vs 94 ± 1 mm Hg) (SBP, DBP, and MAP, respectively), while potassium supplementation protected against elevation of these parameters. The mechanism involved augmentation of the phosphorylation of renal myosin light chain phosphatase targeting subunit 1 (MYPT1) at threonine 697 (T697) (2.58 ± 0.36 vs 1 ± 0) and the expression of p44 mitogen-activated protein kinase (MAPK) (1.78 ± 0.20 vs 1 ± 0). While acetylcholine (ACh)-induced relaxation was unaffected, 5 mg/kg b.w. Cd2+ significantly (p < 0.001) attenuated phenylephrine (Phe)-induced contraction of the aorta, and 2.5 mg/kg b.w. Cd2+ significantly (p < 0.05) augmented sodium nitroprusside (SNP)-induced relaxation of the aorta. These results support the vital role of the kidney in regulating blood pressure changes after Cd2+ exposure, which may be a key drug target for hypertension management. Given the differential response to Cd2+, it is apparent that its hypertensive effects could be mediated by myosin light chain phosphatase (MLCP) inhibition via phosphorylation of renal MYPT1-T697 and p44 MAPK. Further investigation of small arteries and the Rho-kinase/MYPT1 interaction is recommended.
Similar content being viewed by others

Introduction
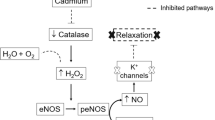
Cadmium (Cd2+) is considered one of the most significant naturally-occurring elemental pollutants in the environment that contribute to adverse health effects in humans and animals. It is toxic and nonbiodegradable, and exposure to Cd2+ reportedly causes oxidative damage, resulting in vascular dysfunction, reduced bioavailability of nitric oxide (NO), and hypertension [1]. Markers of oxidative stress include p44/42 mitogen-activated protein kinase (p44/42 MAPK; extracellular signal-regulated kinase 1 and 2, ERK1/2), the expression of which tends to be elevated. Hypertension is accompanied by oxidative stress; therefore, whether Cd2+ affects the expression of p44/42 MAPK has also been investigated. While the specific hypertensive mechanisms of Cd2+ have not yet been elucidated, they include a calcium ion-dependent pathway, interactions with calcium channels [2], renal oxidative stress [3], and endothelial nitric oxide synthase (eNOS) attenuation [1].
Potassium channels mediate vascular smooth muscle relaxation [4] via potassium efflux, which hyperpolarizes the membrane and balances sodium influx. Potassium reportedly reduces the blood pressure of normotensive animals and possibly alleviates or reduces blood pressure in the context of chronic hypertension [5]. This effect involves potassium efflux via inwardly rectifying potassium channels [6]. Modulating these and other potassium channels will therefore provide clues to the action of Cd2+ on the aorta. Cadmium has been implicated in enhancing potassium conductance [7], calcium entry into cells [8], and calcium channel blockade [9]. Evaluating the effects of potassium supplementation may therefore provide meaningful information about the role of calcium-handling in smooth muscle contraction and the mechanism by which Cd2+ induces hypertension.
Although regulation of smooth muscle contraction by myosin light chain phosphatase (MLCP) via phosphorylation of myosin light chain is a calcium-dependent process, smooth muscle contraction can also be regulated by calcium sensitization and desensitization processes, which alter the balance between MLCP and MLCK activity [10,11,12]. MLCP contains the MYPT1 subunit, which has two major Rho-associated kinase (Rho-kinase, ROCK) phosphorylation sites, namely, T697 and T855 in rats, and phosphorylation of MYPT1 at these sites regulates the activity of myosin. Binding of MYPT1 with Rho-kinase leads to the phosphorylation of MYPT1, which inhibits MLCP activity, allowing phosphorylation of the 20 kDa light chain (LC20) subunit and subsequent smooth muscle contraction. On the other hand, dephosphorylation of LC20 leads to smooth muscle relaxation. There is a paucity of research on the effect of Cd2+ on MYPT1, the threonine sites of MYPT1 that are phosphorylated by Rho-kinase (T697 and T855), and LC20. Consequently, this research sought to determine whether these factors are affected by Cd2+.
This study hypothesized that Cd2+ induces hypertension through activation of ERK pathways and inhibition of MLCP. The objective was to identify the mechanisms involved in Cd2+-induced hypertension in male Sprague-Dawley rats. This was achieved by assessing vascular reactivity and protein expression to determine the possible signaling pathways and mechanisms involved. The effects of potassium and its interaction with Cd2+ were also evaluated.
Methods
Ethical approval
The experiments were conducted in accordance with the guidelines and regulations of the Ethics Committee of the University Hospital of the West Indies/University of the West Indies Faculty of Medical Sciences (UHWI/UWI FMS), University of the West Indies, Mona (approval number AN 16, 12/13).
Experimental animals
Male Sprague-Dawley rats weighing 170–200 g were obtained from the Animal House, UWI, Mona, provided ad libitum access to standard rat chow diet and water, and kept on a 12 h light/dark cycle. The humidity and temperature were kept constant. The animals were acclimated to the laboratory conditions 1–2 weeks prior to starting experimental procedures. The animals were randomized into groups of 10 and treated for 8 weeks with Cd2+ (2.5 or 5 mg/kg b.w.), potassium (0.75%), or both Cd2+ and potassium. Normal controls received water only. Cadmium chloride was administered via gavage three times weekly, and potassium chloride was administered via drinking water. These concentrations were selected since 0.5–10 mg/kg b.w. Cd2+ has been shown to cause hypertension and significantly elevate blood pressure and MAP. Smoking (especially tobacco smoking) is one of the most important sources of Cd2+ in humans [1, 13]. In addition, at a higher concentration (100 mg/kg b.w.), Cd2+ has been shown by us and others to elevate MAP and systolic blood pressure (SBP) [14, 15]. At a concentration of 0.75%, potassium was previously shown by Omogbai et al. [16] to reduce blood pressure in normotensive and hypertensive rats; therefore, a similar concentration was used in this experiment. Weight was measured three times weekly using an Ohaus triple beam animal balance (Ohaus, NJ, U.S.A.), and blood pressure and heart rate (HR) were measured twice weekly using a CODA noninvasive blood pressure apparatus (Kent Scientific Corporation, CT, U.S.A.). SBP, diastolic blood pressure (DBP), and MAP were measured, and HR readings were taken automatically with a CODA machine. Pulse pressure (PP) was calculated as the difference between SBP and DBP. Prior to blood pressure measurements, rats were restrained in a rat restrainer and preconditioned for ~5 min. Fifteen to thirty inflation and deflation cycles were performed per rat per measurement session. The occlusion cuff was inflated to 250 mm Hg and deflated over 20 s. The volume-pressure recording (VPR) sensor cuff detected changes in the blood volume in the tail while the blood returned to the tail during deflation of the occlusion cuff. These measurements were performed by an investigator unblinded to the groups. The rats were sacrificed by cervical dislocation, and their thoracic aortas were excised for organ bath studies. The hearts were excised and weighed for analysis of the heart weight to body weight ratio. The aortas and mesenteric and renal arteries were harvested for western blot analysis and stored at −80 °C prior to use.
Preparation of aortic rings for assessment of vascular responses
Chemicals were obtained from Sigma Chemical Co. (MO, U.S.A.) unless otherwise stated. Glucose, magnesium sulfate, and potassium dihydrogen phosphate were obtained from AnalaR BDH Chemicals Ltd. (Poole, England). The thoracic aorta was rapidly excised and placed in ice-cold, oxygenated modified physiological salt solution (PSS) containing (in mM) 119 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4·7H2O, 1.2 KH2PO4, 24.9 NaHCO3, and 11.1 glucose (pH 7.4). The aorta was cut transversely into 2–3 mm rings, and each was suspended in a Kent (Kent Scientific Corporation, CT, U.S.A.) or Radnoti (OX, UK) organ bath containing 10 ml of well-oxygenated (95% O2 and 5% CO2) PSS at 37 °C. A BSL Variable Force Transducer connected to an MP36 multichannel data acquisition system (Biopac Systems Inc., Goleta, CA, U.S.A.) was used to record isometric tension. The rings were equilibrated for 60 min under 1.0 g resting tension, which is optimal for inducing maximal contraction [17]. Relaxation of aortic rings precontracted with phenylephrine (Phe; 10−6 M) was induced by acetylcholine chloride (ACh; 10−6 M) to assess endothelial integrity, which considered at least 70% of maximal relaxation in response to ACh (modified from Almenara et al. [14], Angeli et al. [18], and Ok et al. [19]). Vasorelaxant responses of aortic rings precontracted with Phe (10−6 M) to cumulative concentrations of ACh (10−9 to 10−3 M) and sodium nitroprusside (SNP; 10−12 to 10−3 M) and their vasoconstrictor responses to Phe (10−9 to 10−3 M) were assessed.
Western blotting
Western blotting experiments were performed to measure the expression of proteins of interest according to previously established protocols [10]. Proteins were extracted using extraction buffer with freshly prepared dithiothreitol (DTT), and the extent of phosphorylation was determined via SDS-PAGE and western blotting using phospho-specific antibodies for MYPT1, pT697, pT855, LC20 (phos-tag SDS-PAGE), eNOS, p44/42 MAPK, p38 MAPK, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and actin. Relative expression levels were normalized to the expression of GAPDH or actin to account for loading differences and to ensure that differences in protein expression levels were not due to sample loading or protein transfer. For pT697 and pT855, Johnson et al. [10] found this approach to be more accurate for normalization than normalization to total protein levels, which involves stripping and reprobing of the blots with a pan-MYPT1 antibody and variable loss of proteins. The pressurized rat or mouse cerebral artery samples used by these authors were very tiny and yielded a very small amount of total protein, i.e., <1 µg from a single pressurized vessel segment. While some of our samples contained a greater amount of protein, assessment of the smallest mesenteric vessels required the use of a method that did not involve stripping and reprobing. For consistency, therefore, the same technique was employed for samples of other vessels. A similar concept that was also used by other researchers was applied for eNOS and MAPKs [14, 18, 20]. Proteins were quantified by western blotting on 10% mini gels (12.5% phos-tag acrylamide gel for LC20). A 3-step western blot technique [10] was employed for the following primary antibodies (1:200–1:2000): rabbit polyclonal anti-MYPT1 (Welsh Custom Antibody), anti-phospho-MYPT1 (T696) and anti-phospho-MYPT1 (T850) (Millipore, MA, U.S.A.; Cat# ABS45, RRID:AB_11212365 and Cat# 36-003, RRID:AB_310812), and anti-eNOS (1:1000–1:5000; Cell Signaling Technology, MA, U.S.A.; Cat# 9572, RRID:AB_329863). Biotin-conjugated goat anti-rabbit (1:5000–1:40,000; Millipore, MA, U.S.A.; Cat# AP132B, RRID:AB_92488) was used as the secondary antibody, and horseradish peroxidase (HRP)-streptavidin (1:2000; 1:100,000 for eNOS; Thermo Fisher Scientific, MA, U.S.A.; Cat# PI21126) was used as the tertiary antibody. The primary antibodies used for the 2-step procedure were anti-pan-MLC2 (1:2000; Santa Cruz Biotechnology, CA, U.S.A.; Cat# sc-48414, RRID:AB_2148042), anti-p38 and anti-p44/42 MAPK (ERK1/2) (1:1000; Cell Signaling Technology, MA, U.S.A.; Cat# 9212, RRID:AB_330713 and Cat# 9102, RRID:AB_330744), and anti-GAPDH (1:1000; Santa Cruz Biotechnology, CA, U.S.A.; Cat# sc-25778, RRID:AB_10167668), and rabbit polyclonal anti-pan actin (1:5000; Cytoskeleton, Inc., CO, U.S.A.; Cat.# AAN01-A, RRID:AB_10708070). HRP-conjugated goat anti-rabbit (1:5000 - 1:20,000; Millipore, MA, U.S.A.; Cat# 12-348, RRID:AB_390191) was used as the secondary antibody. Enhanced chemiluminescence (ECL) advance solution (Thermo Fisher Scientific, MA, U.S.A.) was used to develop the blots. Images were obtained using the Fujifilm LAS 3000 Mini Image Reader and Fuji Camera software v. 2.2 (GE Healthcare Biosciences, PA, U.S.A.) following exposure for 1–60 s. Densitometric analysis was conducted using the LAS 3000 Multi-gauge Image Reader v. 3.1 (GE Healthcare Biosciences, PA, U.S.A.).
Data analysis
The results are reported as the means ± standard errors of the mean (SEMs) using Origin Pro 7.0 software, with “n” indicating the number of animals used per experimental treatment. Blood pressure changes are expressed as the percentage of the baseline (preinjection) value. The effective dose at which 70% contraction or relaxation was obtained (ED70) was derived from graphs plotted with Origin Pro 7.0 software. The maximal and minimal responses (Emax and Emin) were determined from the dose-response curves, and the effective concentration that produced the half-maximal response (EC50) and sensitivity (pD2) were calculated using nonlinear regression analysis (Origin Pro 7 software). The sensitivity is expressed as the negative logarithm of the EC50, that is, pD2 = −log EC50. Densitometric analysis of the western blot images was conducted using Fujifilm LAS 3000 Multi-gauge Image Reader v. 3.1 software. Protein density was calculated as the ratio of normalized protein, actin, or GAPDH expression using Microsoft Excel 2016. The western blot ratios were normalized to the ratio of the control, which was set to 1, and a composite of blots was used to obtain data for each group. Statistical analysis was performed using IBM SPSS Statistics version 22 to determine the significance of the mean difference between groups via one-way analysis of variance (ANOVA). Levene’s test of homogeneity followed by the Games-Howell or Bonferroni post hoc test for multiple comparisons was performed, and a p < 0.05 was considered statistically significant. Graphs were plotted using Origin Pro 7.0 software.
Results
Effect of cadmium on blood pressure parameters
Cadmium elevates SBP, DBP, and MAP
Blood pressure was evaluated to determine whether Cd2+ induced hypertension. Cd2+ exposure (2.5 or 5 mg/kg b.w.) for three weeks was associated with a significant (p < 0.001) increase in mean SBP (160 ± 2 and 155 ± 1 vs 120 ± 1 mm Hg), which was maintained until the end of the study period (Fig. 1A and Supplementary Data, https://doi.org/10.6084/m9.figshare.8852351; URL: https://doi.org/10.6084/m9.figshare.8852351.v1). There was no significant difference between the two Cd2+-exposed groups.

The effect of cadmium and potassium on the mean systolic blood (A) diastolic blood (B), mean arterial (C), and pulse (D) pressures and heart rate (E) of male Sprague-Dawley rats. Animals were treated with cadmium (2.5 or 5 mg/kg b.w.) for 8 weeks. Cadmium significantly (p < 0.05) elevated blood pressures, and potassium ameliorated this effect. Cadmium significantly (p < 0.05) elevated pulse pressure after reducing it significantly (p < 0.05). *p < 0.01 vs the normal control; †p < 0.05 vs 2.5 mg/kg b.w. cadmium with potassium (0.75%); #p < 0.05 vs 5 mg/kg b.w. cadmium; **p < 0.05 vs the normal control and 5 mg/kg b.w. cadmium with potassium (0.75%); ¢p < 0.05 vs the normal control; §p < 0.01 vs all other groups; ‡p < 0.05 vs all other groups
Cd2+ exposure (2.5 or 5 mg/kg b.w.) for 2 weeks was also associated with significant (p < 0.001) increases in mean DBP (119 ± 2 and 110 ± 1 vs 81 ± 1 mm Hg) and MAP (133 ± 2 and 125 ± 1 vs 94 ± 1 mm Hg) (Fig. 1B, C and Supplementary Data, https://doi.org/10.6084/m9.figshare.8852351; URL: https://doi.org/10.6084/m9.figshare.8852351.v1). Compared to exposure to 5 mg/kg b.w. Cd2+, exposure to 2.5 mg/kg b.w. Cd2+ significantly (p < 0.05) elevated DBP and MAP at weeks 2, 5, 7, and 8 and weeks 2, 5, and 8, respectively.
Dual effect of cadmium on pulse pressure
Exposure to Cd2+ (2.5 and 5 mg/kg b.w.) significantly (p < 0.05) reduced PP after 3 weeks (Fig. 1D). Exposure to 2.5 mg/kg b.w. Cd2+ was additionally associated with a significant (p < 0.05) decrease in PP after two weeks and a significant (p < 0.01) increase in PP at 6 weeks, at which point it peaked. The PP was, however, normalized during the last two weeks of treatment. Furthermore, the PP of rats exposed to 5 mg/kg b.w. Cd2+ rose from week 4 to the end of the study period, with a significant (p < 0.05 and 0.001) increase being observed at weeks 7 and 8. The difference in PP between rats exposed to 5 mg/kg b.w. Cd2+ and those exposed to 2.5 mg/kg b.w. Cd2+ was also significant (p < 0.05) at week 8. The data are provided in the Supplementary Data (https://doi.org/10.6084/m9.figshare.8852351; URL: https://doi.org/10.6084/m9.figshare.8852351.v1).
Cadmium elevates HR in rats
A general decline in HR was observed for all groups throughout the duration of the study (Fig. 1E and Supplementary Data, https://doi.org/10.6084/m9.figshare.8852351; URL: https://doi.org/10.6084/m9.figshare.8852351.v1). The highest mean HR of the normotensive group was 521 ± 7 beats per minute (bpm), and the lowest HR of this group was 469 ± 10 bpm. HR was not affected by exposure to 2.5 mg/kg b.w. Cd2+ (533 ± 8 to 452 ± 5 bpm) but was significantly (p < 0.05) elevated by exposure to 5 mg/kg b.w. Cd2+ (513 ± 7 to 485 ± 5 bpm) at weeks 3, 5, and 7. There was also a significant (p < 0.05) difference in HR between rats exposed to 5 mg/kg b.w. Cd2+ and those exposed to 2.5 mg/kg b.w. Cd2+ between weeks 5 and 7.
Potassium supplementation ameliorates the effects of cadmium on blood pressure
Potassium supplementation in normotensive rats significantly (p < 0.01) elevated SBP at week 5, but SBP was otherwise maintained within or close to the normal range. DBP and MAP were significantly (p < 0.05) elevated for the first 5 weeks (except week 2) in the potassium supplementation group compared to the normal controls but normalized within the last 3 weeks of treatment.
Potassium supplementation ameliorates the effects of cadmium on SBP, DBP, and MAP
SBP (p < 0.001 and 0.05), DBP (p < 0.05), and MAP (p < 0.05) were significantly attenuated in both Cd2+-treated groups (2.5 and 5 mg/kg b.w.) that received potassium supplementation compared to the groups that received Cd2+ alone, indicating that potassium supplementation ameliorated the effects of Cd2+ (2.5 and 5 mg/kg b.w.) (Fig. 1A–C). Compared with potassium supplementation in the presence of 5 mg/kg b.w. Cd2+, potassium supplementation in rats exposed to 2.5 mg/kg b.w. Cd2+ initially significantly (p < 0.05) lowered DBP and MAP. These parameters were, however, significantly (p < 0.01) elevated at week 7, and there was no difference between groups at the end of the study.
Potassium supplementation ameliorates the effects of cadmium on pulse pressure
Potassium supplementation significantly (p < 0.001 and 0.01, respectively) reduced PP in normotensive and Cd2+ (2.5 and 5 mg/kg b.w.)-treated rats at week 3 (Fig. 1D). Potassium supplementation in rats exposed to 5 mg/kg b.w. Cd2+ also significantly (p < 0.05) reduced PP at week 6. Potassium supplementation in rats exposed to 2.5 or 5 mg/kg b.w. Cd2+ prevented a further increase in PP, keeping it below or within the normal range, and therefore ameliorated the effects of 2.5 or 5 mg/kg b.w. Cd2+, respectively. Potassium supplementation in rats exposed to 5 mg/kg b.w. Cd2+ had a greater (p < 0.01) PP-lowering effect at week 6 than potassium supplementation in rats exposed to 2.5 mg/kg b.w. Cd2+. There was no significant difference in PP between normotensive rats that received potassium supplementation and 2.5 and 5 mg/kg b.w. Cd2+-treated rats that received potassium supplementation.
Potassium supplementation elevates HR and opposes the effects of cadmium
HR was significantly (p < 0.01) elevated at weeks 1, 4, and 5 in normotensive rats exposed to potassium supplementation but was normalized by progressive treatment (Fig. 1E). Potassium supplementation in rats exposed to either dose of Cd2+ (2.5 or 5 mg/kg b.w.) also significantly (p < 0.05 and 0.01) elevated HR (Fig. 1E). This increase was sustained by potassium supplementation in rats exposed to 2.5 mg/kg b.w. Cd2+ but normalized within the last two weeks of potassium supplementation in rats exposed to 5 mg/kg b.w. Cd2+. This increase in HR was also significant (p < 0.01) in rats exposed to 2.5 mg/kg b.w. Cd2+ or 5 mg/kg b.w. Cd2+ that received potassium supplementation compared to rats exposed to 2.5 or 5 mg/kg b.w. Cd2+. No significant difference in HR was observed between potassium-supplemented normotensive rats and potassium-supplemented rats exposed to 2.5 or 5 mg/kg b.w. Cd2+. A significant difference (p < 0.05 and 0.001) in HR was, however, observed between potassium-supplemented rats exposed to 2.5 mg/kg b.w. Cd2+ and potassium-supplemented rats exposed to 5 mg/kg b.w. Cd2+ at weeks 2 and 8.
Effect of cadmium on vascular reactivity
Cadmium attenuates Phe-induced contraction of rat aortic rings
Contraction of the aorta in response to increasing Phe concentrations was significantly (p < 0.001) attenuated by exposure to 5 mg/kg b.w. Cd2+ (79.42 ± 1.49 vs 95.52 ± 1.61%) but was unaffected by exposure to 2.5 mg/kg b.w. Cd2+ (Fig. 2A). Contraction in the presence of the lower dose of Phe was significantly (p < 0.05) lower than that in the presence of the higher dose of Phe (10−8 M). Exposure to Cd2+ did not affect the Emax or Emin response, the EC50, or sensitivity to Phe.

Effect of cadmium on phenylephrine-induced contraction (A) and acetylcholine- (B) and sodium nitroprusside-induced (C) relaxation of rat aortic rings. The higher dose of cadmium (5 mg/kg b.w.) significantly (p < 0.001) attenuated contraction, but potassium supplementation abolished this effect. Potassium treatment alone did not affect contraction. *p < 0.05, group 2 vs group 3; **p < 0.001, groups 1 and 3 vs group 6; †p < 0.05, group 4 vs group 6; §p < 0.001, group 1 vs group 3; ‡p < 0.05, group 3 vs group 6; #p < 0.05, group 1 vs group 5; €p < 0.05, group 1 vs group 6; ¢p < 0.05, group 2 vs group 1
Cadmium augments SNP- but not ACh-induced relaxation
SNP-induced relaxation was significantly (p < 0.05) augmented by exposure to 2.5 mg/kg b.w. Cd2+ (27.59 ± 3.56 vs 9.51 ± 2.07%) in response to a low dose of SNP (10−11 M), and the EC50 was significantly (p < 0.05) reduced (3.98 × 10−9 ± 8.02 × 10−10 vs 7.03 × 10−10 ± 2.94 × 10−10 M; Fig. 2C). SNP-induced relaxation was unaffected by exposure to 5 mg/kg b.w. Cd2+ (Fig. 2C). In addition, 2.5 mg/kg b.w. Cd2+ induced a significantly (p < 0.05) greater augmentation of SNP-induced relaxation than exposure to 5 mg/kg b.w. Cd2+ but did not affect the EC50. Furthermore, ACh (10−9 to 10−3 M) induced dose-dependent relaxation of Phe-contracted aortic rings (Fig. 2B), but relaxation and EC50 were unaffected by Cd2+ (2.5 or 5 mg/kg b.w.) administration. Compared to exposure to 2.5 mg/kg b.w. Cd2+, exposure to 5 mg/kg b.w. Cd2+ significantly (p < 0.05) augmented relaxation at a high dose of ACh (10−4 M) without affecting the EC50.
Effect of potassium supplementation on vascular reactivity
Potassium supplementation attenuates Phe-induced contraction and ACh-induced relaxation in rat aortic rings
Vascular reactivity studies were conducted to evaluate the effect of potassium supplementation (0.75%) on the contraction and relaxation of the aorta to identify its mechanism of action. Potassium supplementation (0.75%; 100 mM) in normotensive rats did not affect Phe-induced contraction or ACh- and SNP-induced relaxation (Fig. 2A–C). Potassium supplementation in the presence of 5 mg/kg b.w. Cd2+ produced a dual and opposing effect on contraction based on the concentration of Phe. Initially, the contraction of potassium-treated aortic rings was significantly (p < 0.001) attenuated compared with that of both the normal control and aortic rings treated with 5 mg/kg b.w. Cd2+ and potassium-treated aortic rings exhibited significant (p < 0.01) elevation of the EC50 and reduced sensitivity. As the Phe dose increased, however, potassium supplementation in the presence of 5 mg/kg b.w. Cd2+ significantly (p < 0.05) augmented contraction, thereby nullifying the effect of 5 mg/kg b.w. Cd2+ (Fig. 2A). Potassium supplementation in the presence of either dose of Cd2+ (2.5 and 5 mg/kg b.w.) also significantly (p < 0.05) attenuated ACh-induced relaxation (10−7 to 10−6 M), producing a rightward shift in the response curve, but did not affect SNP (10−12 to 10−3 M)-induced relaxation (Fig. 2B, C). The EC50 of SNP-induced relaxation was, however, significantly (p < 0.05) reduced by exposure to potassium supplementation in the presence of 5 mg/kg b.w. Cd2+ (3.98 × 10−9 ± 8.02 × 10−10 vs 1.40 ± 0.63 × 10−10 M). There was no significant difference in the effect of potassium supplementation in the presence of 2.5 and 5 mg/kg b.w. Cd2+.
Effect of cadmium and potassium supplementation on protein expression
Cadmium increases total MYPT1 levels in the aorta without phosphorylating the phosphospecific sites of MYPT1
Exposure to 5 mg/kg b.w. Cd2+ significantly (p < 0.01) increased the total MYPT1 level in the aorta without affecting its phosphorylation at the phosphospecific sites T697 and T855 (Fig. 3A–C), which was only slightly elevated. There was no significant difference in the protein levels between the group exposed to 2.5 and the group exposed to 5 mg/kg b.w. Cd2+.

Effect of cadmium and potassium on the levels of MYPT1 (A) MYPT1 phosphorylated at T697 (B), and MYPT1 phosphorylated at T855 (C) in the thoracic aorta of male Sprague-Dawley rats. Cd2+ (5 mg/kg b.w.) significantly (p < 0.01) increased the total MYPT1 level. Potassium supplementation in the presence of Cd2+ significantly (p < 0.05) attenuated MYPT1 phosphorylated at T697 and MYPT1 phosphorylated at T855 levels. Expression levels were normalized to the level of GAPDH. Ratios were normalized to the ratio in the control group, which was set to 1. *p < 0.01 vs group 1; †p < 0.05 vs group 4; **p < 0.05 vs group 1; #p < 0.01 vs group 4; ‡p < 0.01 vs group 3. Exposure time (A–C): 10 s
Potassium supplementation attenuates the phosphorylation of MYPT1 at T697 and T855 in the aorta
Potassium supplementation in rats exposed to 2.5 or 5 mg/kg b.w. Cd2+ significantly attenuated the phosphorylation of MYPT1 at T697 (p < 0.05 and 0.001) and T855 (p < 0.01) in the aorta, thereby significantly (p < 0.05) opposing the effect of 5 mg/kg b.w. Cd2+ (Fig. 3B, C). The attenuation of MYPT1 phosphorylation at T697 was also significant (p < 0.01) compared to potassium-supplemented normotensive rats. There was no significant difference in the levels of protein expression between potassium-supplemented rats exposed to 2.5 and those exposed to 5 mg/kg b.w. Cd2+.
Cadmium did not affect protein levels, but potassium supplementation augmented MYPT1 expression and phosphorylation at T697 and T855 in the mesenteric arteries of Cd2+-exposed rats
Exposure to Cd2+ (2.5 and 5 mg/kg b.w.) and potassium supplementation (0.75%) in normotensive rats did not affect the protein levels of total MYPT1 or its phosphorylation at the major regulatory sites T697 and T855 (Fig. 4A–C). Total MYPT1 expression was, however, significantly (p < 0.01) augmented by potassium supplementation in rats exposed to 2.5 mg/kg b.w. Cd2+. Similarly, potassium supplementation in rats exposed to 5 mg/kg b.w. Cd2+ significantly (p < 0.05) augmented the levels of MYPT1 phosphorylated at T697 and T855 in the mesenteric artery (Fig. 4A–C). There was no significant difference between rats exposed to 2.5 and those exposed to 5 mg/kg b.w. Cd2+ or between them and their potassium-supplemented counterparts.

Effect of cadmium and potassium on the levels of MYPT1 (A), MYPT1 phosphorylated at T697 (B), and MYPT1 phosphorylated at T855 (C) in the mesenteric arteries of male Sprague-Dawley rats. Cd2+ and potassium alone did not affect protein levels, but potassium supplementation in the presence of Cd2+ significantly (p < 0.05) elevated MYPT1, MYPT1 phosphorylated at T697, and MYPT1 phosphorylated at T855 levels in the mesenteric arteries. Expression levels were normalized to the levels of GAPDH. Ratios were normalized to the ratio in the control group, which was set to 1. *p < 0.01 vs group 1; **p < 0.05 vs group 1. Exposure time (A–C): 4 s
Cadmium elevates total MYPT1 expression, MYPT1 phosphorylation at T697, and p44 MAPK expression in the renal artery
The significant (p < 0.01 and 0.001) elevation of total MYPT1 expression induced by exposure to 5 mg/kg b.w.Cd2+ compared to the control and exposure to 2.5 mg/kg b.w. Cd2+ was abolished (p < 0.01) by potassium supplementation (Fig. 5A). Phosphorylation of MYPT1 at its phosphorylation sites T697 and T855 was not affected by exposure to 5 mg/kg b.w. Cd2+, but phosphorylation of MYPT1 at T697 was significantly (p < 0.05) augmented (2.58 ± 0.36 vs 1 ± 0) by exposure to 2.5 mg/kg b.w. Cd2+ (Fig. 5B, C). This augmentation was prevented by potassium supplementation, as the level of MYPT1 phosphorylated at T697 was significantly (p < 0.05 and 0.01) attenuated in Cd2+ (5 mg/kg b.w.)-exposed rats that received potassium supplementation compared to both normotensive and Cd2+ (2.5 mg/kg b.w.)-exposed rats. Exposure to 5 mg/kg b.w. Cd2+ also significantly (p < 0.05) elevated (1.78 ± 0.20 vs 1 ± 0) total renal p44 MAPK levels (Fig. 5C). There was no significant difference in the effect of supplementation between the rats exposed to 2.5 mg/kg b.w. Cd2+ and those exposed to 5 mg/kg b.w. Cd2+ (Figs. 3–5).

Effect of cadmium and potassium on the levels of MYPT1 (A), MYPT1 phosphorylated at T697 (B), and p44/42 MAPK (C) in the renal arteries of male Sprague-Dawley rats. Cadmium (5 mg/kg b.w.) significantly elevated MYPT1 (p < 0.01) and p44 MAPK (ERK1) (p < 0.05) levels, while the lower dose (2.5 mg/kg b.w.) significantly (p < 0.01) elevated pT697 levels. Potassium supplementation in the presence of Cd2+ ameliorated the changes in the levels of MYPT1 and MYPT1 phosphorylated at T697 but not p44 MAPK levels. Cadmium and potassium did not affect eNOS, p38, or p42 MAPK levels. Expression levels were normalized to the level of GAPDH. Ratios were normalized to the ratio in the control group, which was set to 1. *p < 0.01 vs group 1; **p < 0.05 vs group 1; ‡p < 0.01 vs group 3; §p < 0.01 vs group 2. Exposure time (A–C): 4–30 s
Proteins unaffected by cadmium and potassium supplementation
LC20, eNOS, and MAPK (p38 and p42) levels were unaffected by all treatments in the arteries assessed (Fig. 5C and Supplementary Figs. 1D–G to 3D–G, https://doi.org/10.6084/m9.figshare.13489185; URL: https://doi.org/10.6084/m9.figshare.13489185.v2). Similarly, p44 MAPK levels in the aorta and mesenteric artery were unaffected (Supplementary Figs. 1G and 2G). The results related to eNOS levels are supported by previous findings of Nwokocha et al. [15] and Angeli et al. [18], both of which used much higher doses of Cd2+ (100 mg/kg b.w. and 10 µM, respectively).
Effect of cadmium and potassium supplementation on the heart weight to body weight ratio
Neither Cd2+ nor potassium supplementation affected the heart weight to bodyweight ratio.
Discussion
The objective of this study was to elucidate the mechanisms involved in Cd2+-induced hypertension. Assessments of vascular reactivity and protein expression were employed to determine the calcium-handling effect of Cd2+ and the possible signaling pathways and underlying mechanisms involved.
Cadmium induced hypertension within three weeks of administration by elevating mean SBP, DBP, and MAP, but the responses of the aorta to contractile and relaxant agents did not give credence to the mechanism. Consistent with our results, Angeli et al. [18] observed significantly elevated SBP; however, they found that responses to ACh and Phe were unaffected or reduced. Contrary to our findings, Almenara et al. [14] found an increased response to Phe. While the lower dose of Cd2+ (2.5 mg/kg b.w.) elevated blood pressure levels in our study, SNP produced a significant increase in sensitivity or responsiveness in the Cd2+-exposed rat aorta without significantly altering maximum relaxation. This is anomalous and may be attributed to hypertrophy [21], a compensatory mechanism in the aorta, or endothelial dysfunction, which affects endothelium-derived hyperpolarizing or relaxing factor (EDHF or EDRF) production, rather than NO since eNOS levels were unaffected and the eNOS/NO pathway was not impaired.
The significant reduction in the PP of the Cd2+ (2.5 mg/kg b.w., p < 0.05; 5 mg/kg b.w., p < 0.001)- and potassium (p < 0.001)-treated rats after three weeks of treatment may have been a compensatory response to the initial significant (p < 0.001) elevation of SBP, DBP, and MAP. There is an extreme paucity of research on the effect of Cd2+ on PP in humans as well as a lack of animal studies. Nevertheless, increased Cd2+ body burden was found by Schutte et al. [22] to be associated with lower PP. Other recent research has shown that PP is a better predictor of coronary heart disease risk than SBP and DBP in individuals over 50 years of age [23]. Assmann et al. [23] also reported that increases in SBP, DBP, and PP by 10 mm Hg are associated with a greater hazard ratio (~10%) of coronary heart disease. The significant elevation in PP seen following exposure to Cd2+ (2.5 and 5 mg/kg b.w.) may be a sign of aortic stiffening [24], which can lead to elevated blood pressure and hypertension. Furthermore, PP elevation is associated with a near-linear stroke volume (SV) increase [hence increased cardiac output (CO)], and when the SV is kept constant, increased central arterial stiffness results in elevation of SBP and reduction in DBP, thereby contributing to greater PP [25]. Based on the results, PP may have been influenced or affected by CO. The increase in blood pressure in response to exposure to 5 mg/kg b.w. Cd2+ was associated with the observed elevated HR, and elevation of HR increases CO; however, this does not rule out the contribution of the SV or total peripheral resistance (TPR), although Ozturk et al. [26] disagree.
The current findings prompted the analysis of protein levels in the aorta and the smaller, more contractile renal and mesenteric arteries. A dual mechanism of Cd2+-induced hypertension was observed in the renal artery. First, exposure to 2.5 mg/kg b.w. Cd2+ was found to be associated with a significant (p < 0.05) elevation of the phosphorylation of MYPT1 at T697. This is a novel finding and a strength of this study, and it shows a mechanism involving inhibition of MLCP. Such inhibition is also associated with an increase in the level of calcium sensitization of contractile elements [10]. Augmented phosphorylation of MYPT1 indicates the activity of the RhoA/ROCK pathway and correlates with hypertension [27]. In addition, changes in calcium sensitization and augmentation of calcium-sensitive RhoA/ROCK activity may precede vascular diseases such as hypertension [27]. The effect of Cd2+ on the phosphorylation of MYPT1 at T697 in the thoracic aorta and mesenteric or renal artery is undocumented, and this research sheds light on this area of smooth muscle research. Nevertheless, increased phosphorylation has been identified using other contractile agents in the mesenteric and renal arteries and other tissues, such as the protein phosphatase inhibitor calyculin A in the mesenteric artery, potassium in rat caudal arterial smooth muscle, and angiotensin II in the aorta [28,29,30]. A lack of phosphorylation in the aorta was observed [31, 32]. These results show that the myogenic response is tissue-dependent.
Second, exposure to 5 mg/kg b.w. Cd2+ significantly (p < 0.05) augmented the total renal p44 MAPK (ERK1) level. This indicates that oxidative stress occurred in renal tissue, which was not observed in the aorta or mesenteric artery. Cd2+ is associated with the production of reactive oxygen species (ROS) [33], which causes renal oxidative stress [3, 34], and MAPKs have been shown to mediate responses to ROS [35]. Furthermore, some tissues, including brain tissues, have been shown to utilize p44/42 MAPK to respond to stressful stimuli such as oxidative stress [36]. Hypertension or high blood pressure is associated with elevated ERK1/2 (p44/42 MAPK) levels [37], although this is disputed by Parenti et al. [38]. The significant elevation of p44 MAPK levels was consistent with the change in MYPT1 phosphorylation at T697 in renal tissue, which was also significantly (p < 0.05) elevated.
The higher dose of Cd2+ acted via another mechanism, the p44 MAPK pathway, to elevate blood pressure. It is not known exactly why both doses did not have a similar effect on both MYPT1 phosphorylation at T697 and p44 MAPK; however, given the tissue specificity and pressure dependence of the myogenic response [10], it can be assumed that the effects of the two concentrations of Cd2+ were based on the amount of pressure generated and the tissue affected. Cd2+ preferentially affects the kidneys. Thind [39] found that the kidneys of hypertensive and control rabbits had the highest concentration of Cd2+, followed by the mesenteric artery and then the aorta. Eum et al. [40] reported positive associations between SBP, DBP, and MAP and blood Cd2+ levels, an effect that was markedly enhanced with reduced kidney function. On the other hand, Staessen et al. [41] found no correlation between blood pressure and blood or urinary Cd2+ levels, except for a correlation between DBP and Cd2+ levels in men. In that study, the average SBP and DBP in men and women in areas of northeastern Belgium affected by Cd2+ (131.5 ± 14.2 mm Hg and 78.5 ± 8.3 mm Hg in men and 125.4 ± 15.9 mm Hg and 76.1 ± 7.8 mm Hg in women) were lower than the values obtained in the present study. Satarug et al. [42] pointed to the possibility that Cd2+ accumulates in the kidney and triggers responses involving heme oxygenase-1 (HO-1; an antioxidant enzyme) and metallothionein (a metal-binding protein) to combat the rise in blood pressure and protect against oxidative stress. Another action of Cd2+, as purported by Thind [39], involves significant dose-dependent reversible inhibition of angiotensin responses in the renal artery in normal rabbit aortic strips, and such inhibition was induced by application of 1 μg/ml Cd2+ for 4 h or greater concentrations of Cd2+ for a shorter period of time. Additionally, Broseghini-Filho et al. [43] reported that angiotensin I-converting enzyme is inhibited by Cd2+.
Hypertension was not observed in patients with Japanese itai-itai (it hurts-it hurts or ouch ouch) disease [44], but renal tubular dysfunction was evident or posed a significant mortality risk [45,46,47]. Elevated mean kidney (43.8 mg/kg in the renal cortex) and liver (5.3 mg/kg) Cd2+ concentrations in autopsied Jamaicans from areas with low soil Cd2+ concentrations (by the Jamaican standard) were second only to those in Japanese individuals [46]. Mean Cd2+ levels in the liver, pancreas, and thyroid (56, 44, and 43 mg/kg, respectively) of patients with itai-itai disease were more than five times higher than the reference concentrations (10, 9, and 9 mg/kg, respectively) [47]. The authors reported a clear indication that patients with itai-itai disease and residents of the Jinzu River basin of Toyama were exposed to extremely high levels of Cd2+. Interestingly, the mean Cd2+ levels in the kidney were much lower than the reference values, primarily in the renal cortex (31 vs 73 mg/kg; 23 vs 34 mg/kg in the medulla), and this decline was attributed to a loss of Cd2+ in the tubules arising from the clinical presence of renal tubular dysfunction. Cd2+-related renal biomarkers and an evident association between urinary Cd2+ and beta-2-microglobulin (β2-MG) levels were identified in the Jamaican study population, but the absolute concentrations were well below the critical limits for the onset of acute or chronic renal effects [48].
Cd2+ alone or in combination with potassium seems to exert dose- and tissue-dependent effects that are not well defined. Potassium chloride (KCl) induces translocation of RhoA to the membrane; RhoA/ROK activation; significant phosphorylation of MYPT1 at T697, MYPT1 at T855, and LC20; and sustained contraction of rat caudal arterial smooth muscle [29]. Seok et al. [31] reported augmentation of MYPT1 phosphorylation at T855 but not T697 in the rat aorta using KCl. One response of tissues undergoing oxidative stress is contraction, which facilitates the entry of more calcium into renal artery smooth muscle cells, although Lopin et al. [9] reported that Cd2+ blocks calcium channels. There was no quantitative measurement of calcium concentration; however, the results of the vascular studies indicated either a decrease in the calcium concentration in the presence of Cd2+ or a competitive effect [49]. It is also possible that, as in a study by Osol et al. [50], there is no change in intracellular calcium concentration when pressure increases.
The administration of potassium by itself prevented the increase in total MYPT1 expression in the aorta and renal artery that was seen in the Cd2+ (5 mg/kg b.w.)-exposed group. Potassium supplementation with Cd2+ also had an ameliorating effect, as it maintains blood pressure within or near the normal range, hence conferring a protective effect. To verify the protective effect of potassium on Cd2+-induced hypertension, a comparison was made between the Cd2+-exposed groups and their Cd2+-exposed counterparts that received potassium supplementation. Phosphorylation of MYPT1 at T697 was attenuated by potassium supplementation in the presence of 2.5 mg/kg b.w. Cd2+ in the renal artery and by potassium supplementation in the presence of both doses of Cd2+ in the aorta. Augmented phosphorylation of MYPT1 at the phosphospecific sites T697 and T855 was also reported by Cho et al. [28]. This effect is different from what was observed in the aorta. The implication is that the interaction between Cd2+ and potassium inhibited MLCP activity in the mesenteric arteries. Furthermore, this finding demonstrates the ability of Cd2+ to interact with other metals to induce hypertension in blood vessels other than the aorta. The augmentation of total MYPT1 expression implies that MLCP activity was facilitated and LC20 activity was dampened. Inhibition of LC20 phosphorylation by the isoflavones genistein and daidzein in the rat aorta was observed by Seok et al. [31].
We expected LC20 phosphorylation to explain the phosphorylation of the ROK phosphospecific sites on MYPT1 and hence MLCP inhibition; however, this was not the case. Indeed, increased contraction should be reflected by LC20 phosphorylation; however, the tissues were not challenged prior to being frozen. This unfortunately may have contributed to the lack of phosphorylation in this study. It is possible that the contraction at a basal pressure level in the unchallenged arteries was insufficient to induce changes in the level of phosphorylation. Challenging the tissues with a contractile agent such as Phe and then flash-freezing them may have led to better results in this study. Moreno-Domínguez et al. [51] reported that elevation in pressure from 80 to 120 mm Hg was associated with a reduction in the diameter of cerebral and skeletal muscle arterioles. Furthermore, the size of the resistance arteries served as a limiting factor in analyzing changes in the concentration of phosphorylated or expressed proteins associated with ROK signaling [52]. Pressurized cerebral and skeletal muscle arterioles were constricted at all pressures, LC20 was phosphorylated between basal pressures of 10 and 120 mm Hg, and there was no additional increase in LC20 phosphorylation between pressures of 80 and 120 mm Hg [51, 53]. The researchers further reported that actin cytoskeleton reorganization was the only mechanism generating force since a reduction in the G-actin pool was the only biochemical parameter that changed [51]. The SBP recorded in the tail vein in this study exceeded 140 mm Hg towards the end of the study. This elevated pressure may have also contributed to the lack of phosphorylation of LC20 in the renal artery and other blood vessels assessed, as in studies by Moreno-Domínguez et al. [51, 53].
The concentrations of Cd2+ used in this study (7.5 or 15 mg/kg b.w./week; 1.1 or 2.1 mg/kg/day) were above the provisional tolerable weekly intake (PTWI) for humans but below the level toxic to humans. The PTWI for humans is 2.5 and 7 µg/kg b.w. (0.0025 and 0.007 mg/kg b.w.) according to the European Union maintained by the European Food Safety Authority (EFSA) Panel on Contaminants in the Food Chain (CONTAM) [54] and the World Health Organization (WHO) [55], respectively. While exposure to Cd2+ orally does not result in reproductive and developmental toxicity, acute exposure to high doses ranging from 1500 to 8900 mg (20 to 30 mg/kg) results in death in humans (although rare) and abdominal pain, diarrhea, and vomiting associated with gastrointestinal (GI) irritation [56]. These GI symptoms may also occur at doses above 15 mg/kg, but the lowest emetic dose is reportedly 0.07 mg/kg, while fatalities have occurred at doses as low as 20–30 mg/kg [57]. Chronically ingested Cd2+ seems to target the kidneys, such as in Toyama prefecture and in the case of Japanese itai-itai disease [46].
Based on the results obtained, not all members of the MAPK family were stimulated in response to Cd2+-induced hypertension. Other signaling pathways may be involved, such as c-Jun N-terminal kinase 1 (JNK1), another stress-responding MAPK [38], pathway; the angiotensin II or renin-angiotensin-aldosterone system, RAAS [58]; and the RhoA/ROCK pathway [59]. The results of this study suggest that elevation of blood pressure was mediated via p44 MAPK (ERK1) and hence activation of the ERK1/2 signaling pathway, phosphorylation of renal MYPT1 at T697, and inhibition of MLCP. Although these results support the hypothesis of the study, there are other causative agents and risk factors for hypertension, including dietary salt consumption, high body weight, and alterations in the renin-angiotensin system [3, 60].
Investigation of the effect of ERK1/2 inhibition may provide more useful information to confirm this finding that renal p44 MAPK (ERK1) is involved in Cd2+-induced hypertension. In addition, experiments geared at directly assessing ROS or oxidative stress levels are warranted. Assessment of the possible correlation between increased oxidative stress and blood pressure may also be warranted since ERK1/2 (p44/42 MAPK) exerts pleiotropic effects that may be affected under diseased conditions. In light of the role of the kidney in blood pressure regulation and given that T697 is a Rho-kinase phosphorylation site leading to smooth muscle contraction [61], further investigation of small arteries and the Rho-kinase/MYPT1 interaction may provide additional useful information regarding the mechanism of Cd2+-induced hypertension. In addition, to bolster the findings of this study and validate the mechanism, immunoblotting experiments with phospho-p38 and p44/42 MAPK antibodies, as well as intervention studies involving the use of Rho-kinase and MAPK inhibitors, should be performed primarily in aortic and renal artery rings since Rho-kinase and MAPK inhibition have been shown to reduce blood pressure [30].
Potassium supplementation played a protective role in the arteries assessed and ameliorated the effects of Cd2+ (2.5 and 5 mg/kg b.w.). The ameliorating effect was mediated via activation of MLCP by attenuation of the phosphorylation of aortic MYPT1 at T697 and T855. In addition, potassium supplementation ameliorated the effects of 2.5 mg/kg b.w. Cd2+ by attenuating renal MYPT1 phosphorylation at T697, as suggested by Ruta et al. [8], and elevating total mesenteric MYPT1 levels. These results imply that MLCP is activated and the potential for LC20 dephosphorylation is increased, favoring smooth muscle relaxation.
The concentration of potassium (0.75%) used in this experiment represents high dietary potassium intake equivalent to 100 mEq. While potassium at this concentration has been shown to hyperpolarize cell membranes and reduce the blood pressure of normotensive and hypertensive animals, it is also linked to depolarization of membranes [16, 62]. Interestingly, reduced relaxation in response to ACh was observed when potassium was administered in the presence of Cd2+. This may have been due to phosphorylation of MYPT1 at the phosphospecific sites T697 and T855, which is similar to the findings of Cho et al. [28]. This is opposite the effect seen in the aorta and implies that the interaction between these two elements inhibits MLCP activity in the mesenteric arteries. This result also implies that Cd2+ can interact with other metals to induce hypertension in blood vessels other than the aorta. This could also be due to activation of voltage-gated calcium channels and the resultant influx of cytosolic calcium and activation of contractile machinery, which also involves activation of calcium-calmodulin-dependent MLCK, MLC phosphorylation, and contraction [16]. The effects of Cd2+ and potassium on blood pressure opposed each other when they acted individually; however, when they were applied in combination, the effects were similar to those of potassium alone, possibly because the influence of potassium was stronger.
Conclusion
This study set out to induce hypertension with Cd2+, assess the vascular response to pharmacological drugs, and perform western blotting to evaluate protein levels in rat tissues to identify the mechanism of Cd2+-induced hypertension. Hypertension was induced by elevating SBP, DBP, and MAP, as shown by other researchers [14, 18]. The effect of Cd2+ was associated with a dual mechanism in the renal artery: inhibition of MLCP via phosphorylation of MYPT1 at T697 and activation of ERK signaling pathways via augmentation of p44 MAPK (ERK1) expression. Potassium ameliorated the effects of Cd2+ by activating MLCP via attenuation of the phosphorylation of MYPT1 in the aorta and renal artery and increasing total mesenteric MYPT1 expression. Therefore, given the prevalence of hypertension and the findings of this study, more drug therapies that target renal tissue and alleviate oxidative stress are needed. Knowledge of the specific pathway may lead to novel therapeutic strategies for the management or prevention of hypertension and hence reduce the mortality and morbidity associated with blood pressure elevation and other associated cardiovascular complications.
References
Yoopan N, Watcharasit P, Wongsawatkul O, Piyachaturawat P, Satayavivad J. Attenuation of eNOS expression in cadmium-induced hypertensive rats. Toxicol Lett. 2008;176:157–61.
Marchetti C. Role of calcium channels in heavy metal toxicity. ISRN Toxicol. 2013;2013:184360 https://doi.org/10.1155/2013/184360.
Abarikwu SO, Njoku R, Lawrence CJ, Charles IA, Ikewuchi JC. Rutin ameliorates oxidative stress and preserves hepatic and renal functions following exposure to cadmium and ethanol. Pharm Biol. 2017;55:2161–9. https://doi.org/10.1080/13880209.2017.1387575.
Jackson WF. Potassium channels in regulation of vascular smooth muscle contraction and growth. Adv Pharm. 2017;78:89–144. https://doi.org/10.1016/bs.apha.2016.07.001.
Poorolajal J, Zeraati F, Soltanian AR, Sheikh V, Hooshmand E, Maleki A. Oral potassium supplementation for management of essential hypertension: a meta-analysis of randomized controlled trials. PLoS One. 2017;12:e0174967–83. https://doi.org/10.1371/journal.pone.0174967.
Salihi AB. Activation of inward rectifier potassium channels in high salt impairment of hydrogen sulphide-induced aortic relaxation in rats. Physiol Pharm. 2016;19:263–73.
Nesovic-Ostojic J, Cemerikic D, Dragovic S, Milovanovic A, Milovanovic J. Low micromolar concentrations of cadmium and mercury ions activate peritubular membrane K+ conductance in proximal tubular cells of frog kidney. Comp Biochem Physiol A Mol Integr Physiol. 2008;149:267–74. https://doi.org/10.1016/j.cbpa.2007.12.006.
Ruta LL, Popa VC, Nicolau I, Danet AF, Iordache V, Neagoe AD, et al. Calcium signalling mediates the response to cadmium toxicity in Saccharomyces cerevisiae cells. FEBS Lett. 2014;588:3202–12.
Lopin KV, Thevenod F, Page JC, Jones SW. Cd2+ block and permeation of Cav3.1 (α-1G) T-type Ca2+ channels: candidate mechanism for Cd2+ influx. Mol Pharm. 2012;82:1183–93.
Johnson RP, El-Yazbi AF, Takeya K, Walsh EJ, Walsh MP, Cole WC. Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J Physiol. 2009;587:2537–53.
Kitazawa T, Somlyo AP. Modulation of Ca2+ sensitivity by agonists in smooth muscle. Adv Exp Med Biol. 1991;304:97–109. https://doi.org/10.1007/978-1-4684-6003-2_10.
Murahashi T, Fujita A. Kitazawa T. Ca2+ -induced Ca2+ desensitization of myosin light chain phosphorylation and contraction in phasic smooth muscle. Mol Cell Biochem. 1999;190:91–8.
Ashraf MW. Levels of heavy metals in popular cigarette brands and exposure to these metals via smoking. Sci World J. 1–5, 2012. https://doi.org/10.1100/2012/729430.
Almenara CCP, Broseghini-Filho GB, Vescovi MVA, Angeli JK, de Oliveira Faria T, Stefanon I, et al. Chronic cadmium treatment promotes oxidative stress and endothelial damage in isolated rat aorta. PLoS One. 2013;8:e68418.
Nwokocha CR, Baker A, Douglas D, McCalla G, Nwokocha M, Brown PD. Apocynin ameliorates cadmium-induced hypertension through elevation of endothelium nitric oxide synthase. Cardiovasc Toxicol. 2013;13:357–63. https://doi.org/10.1007/s12012-013-9216-0.
Omogbai EK, Ozolua RI, Ebeigbe AB. Effects of potassium adaptation on blood pressure and pressor responses in normotensive and renal hypertensive Wistar rats. Methods Find Exp Clin Pharm. 2005;27:5–10. https://doi.org/10.1358/mf.2005.27.1.875430.
Bautista-Ortega J, Stallone JN, Ruiz-Feria CA. Effects of arginine and antioxidant vitamins on pulmonary artery reactivity to phenylephrine in the broiler chicken. Poult Sci. 2013;92:1062–72. https://doi.org/10.3382/ps.2012-02472.
Angeli JK, Cruz Pereira CA, de Oliveira Faria T, Stefanon I, Padilha AS, Vassallo DV. Cadmium exposure induces vascular injury due to endothelial oxidative stress: the role of local angiotensin II and COX-2. Free Radic Biol Med. 2013;65:838–48.
Ok SH, Lee HL, Yu J, Park J, Shin IW, Lee Y, et al. Lipid emulsion attenuates acetylcholine-induced relaxation in isolated rat aorta. Biomed Res Int. 2015;2015:871545–53. https://doi.org/10.1155/2015/871545.
Dang N, Pang S, Song H, An L, Ma X. Inactivation of mitogen-activated protein kinase signalling pathway reduces caspase-14 expression in impaired keratinocytes. Iran J Basic Med Sci. 2016;19:28–33.
Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res. 1977;41:19–26.
Schutte R, Nawrot T, Richart T, Thijs L, Roels HA, Van Bortel LM, et al. Arterial structure and function and environmental exposure to cadmium. Occup Environ Med. 2008;65:412–9. https://doi.org/10.1136/oem.2007.035576.
Assmann G, Cullen P, Evers T, Petzinna D, Schulte H. Importance of arterial pulse pressure as a predictor of coronary heart disease risk in PROCAM. Eur Heart J. 2005;26:2120–6. https://doi.org/10.1093/eurheartj/ehi467.
Reymond P, Westerhof N, Stergiopulos N. Systolic hypertension mechanisms: effect of global and local proximal aorta stiffening on pulse pressure. Ann Biomed Eng. 2012;40:742–9. https://doi.org/10.1007/s10439-011-0443-x.
Kim DH, Yoo JY, Lee SY, Kim YJ, Lee SR, Park SY. Effects of pulse pressure alterations on cardiac output measurements derived from analysis of arterial pressure waveform. Anesth Pain Med. 2016;11:280–4. https://doi.org/10.17085/apm.2016.11.3.280.
Ozturk IM, Buyukakilli B, Balli E, Burak C, Gunes S, Semra Erdogan S. Determination of acute and chronic effects of cadmium on the cardiovascular system of rats. Toxicol Mech Methods. 2009;19:308–17.
Crestani S, Webb RC, da Silva-Santos JE. High-salt intake augments the activity of the RhoA/ROCK pathway and reduces intracellular calcium in arteries from rats. Am J Hypertens. 2017;30:389–99. https://doi.org/10.1093/ajh/hpw20.
Cho YE, Ahn DE, Morgan KG, Lee YH. Enhanced contractility and myosin phosphorylation induced by Ca2+-independent MLCK activity in hypertensive rats. Cardiovasc Res. 2011;91:162–70. https://doi.org/10.1093/cvr/cvr043.
Mita M, Tanaka H, Yanagihara H, Nakagawa J, Hishinuma S, Sutherland C, et al. Membrane depolarization-induced RhoA/Rho-associated kinase activation and sustained contraction of rat caudal arterial smooth muscle involves genistein-sensitive tyrosine phosphorylation. J Smooth Muscle Res. 2013;49:26–45. https://doi.org/10.1540/jsmr.49.26.
Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, et al. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res. 2003;92:411–8. https://doi.org/10.1161/01.RES.0000059987.90200.44.
Seok YM, Baek I, Kim YH, Jeong YS, Lee IJ, Shin DH, et al. Isoflavone attenuates vascular contraction through inhibition of the RhoA/Rho-kinase signalling pathway. J Pharm Exp Ther. 2008;326:991–8. https://doi.org/10.1124/jpet.108.138529.
Seok YM, Azam MA, Okamoto Y, Sato A, Yoshioka K, Maeda M, et al. Enhanced Ca2+-dependent activation of phosphoinositide-3-kinase class IIα isoform-Rho axis in blood vessels of spontaneously hypertensive rats. Hypertension. 2010;56:934–41. https://doi.org/10.1161/HYPERTENSIONAHA.110.160853.
Kim S, Cheon HS, Kim SY, Juhnn YS, Kim YY. Cadmium induces neuronal cell death through reactive oxygen species activated by GADD153. BMC Cell Biol. 2013;14:4–12. https://doi.org/10.1186/1471-2121-14-4.
Adi PJ, Burra SP, Vataparti AR, Matcha B. Calcium, zinc and vitamin E ameliorate cadmium-induced renal oxidative damage in albino Wistar rats. Toxicol Rep. 2016;3:591–7. https://doi.org/10.1016/j.toxrep.2016.07.005.
Scott AJ, O’Dea KP, O’Callaghan D, Williams L, Dokpesi JO, Tatton L, et al. Reactive oxygen species and p38 mitogen-activated protein kinase mediate tumour necrosis factor α-converting enzyme (TACE/ADAM-17) activation in primary human monocytes. J Biol Chem. 2011;286:35466–76. https://doi.org/10.1074/jbc.M111.277434.
Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, et al. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100:1813–21. https://doi.org/10.1172/JCI119709.
Nowak G, Clifton GL, Godwin ML, Bakajsova D. Activation of ERK1/2 pathway mediates oxidant-induced decreases in mitochondrial function in renal cells. Am J Physiol Ren Physiol. 2006;291:F840–55. https://doi.org/10.1152/ajprenal.00219.2005.
Parenti A, Cui XL, Hopfer U, Ziche M, Douglas JG. Activation of MAPKs in proximal tubule cells from spontaneously hypertensive and control Wistar-Kyoto rats. Hypertension. 2000;35:1160–6.
Thind GS. Role of cadmium in human and experimental hypertension. J Air Pollut Control Assoc. 1972;22:267–70. https://doi.org/10.1080/00022470.1972.10469637.
Eum K-D, Lee M-S, Paek D. Cadmium in blood and hypertension. Sci Total Environ. 2008;407:147–53. https://doi.org/10.1016/j.scitotenv.2008.08.037.
Staessen JA, Kuznetsova T, Roels HA, Emelianov D, Fagard R. Exposure to cadmium and conventional and ambulatory blood pressures in a prospective population study. Public health and environmental exposure to cadmium study group. Am J Hypertens. 2000;13:146–56. https://doi.org/10.1016/s0895-7061(99)00187-9.
Satarug S, Nishijo M, Lasker JM, Edwards RJ, Moore MR. Kidney dysfunction and hypertension: role for cadmium, P450 and haem oxygenases? Tohoku J Exp Med. 208: 179–202. https://doi.org/10.1620/tjem.208.179.
Broseghini-Filho GB, Almenara CC, Vescovi MV, Faria Tde O, Vassallo DV, Angeli JK, et al. Acute Cadmium exposure reduces the local angiotensin I converting enzyme activity and increases the tissue metal content. Biol Trace Elem Res. 2015;166:149–56. https://doi.org/10.1007/s12011-015-0250-6.
Nakagawa H, Nishijo M. Environmental cadmium exposure, hypertension and cardiovascular risk. J Cardiovasc Risk. 1996;3:11–7. https://doi.org/10.1177/174182679600300103.
Aoshima K. Itai-itai disease: renal tubular osteomalacia induced by environmental exposure to cadmium—historical review and perspectives. Soil Sci Plant Nutr. 2016;62:319–26. https://doi.org/10.1080/00380768.2016.1159116.
Lalor GC, Rattray R, Williams N, Wright P. Cadmium levels in kidney and liver of Jamaicans at autopsy. West Indian Med J. 2004;53:76–80.
Nishijo M, Nakagawa H, Suwazono Y, Nogawa K, Kido T. Causes of death in patients with Itai-itai disease suffering from severe chronic cadmium poisoning: a nested case-control analysis of a follow-up study in Japan. BMJ Open. 2017;7:e015694 https://doi.org/10.1136/bmjopen-2016-015694.
Wright PR, Rattray R, Lalor G, Hanson R. Minimal health impact from exposure to diet-sourced cadmium on a population in central Jamaica. Environ Geochem Health. 2010;32:567–81. https://doi.org/10.1007/s10653-010-9318-6.
Shirran SL, Barran PE. The use of ESI-MS to probe the binding of divalent cations to calmodulin. J Am Soc Mass Spectrom. 2009;20:1159–71. https://doi.org/10.1016/j.jasms.2009.02.008.
Osol G, Brekke JF, McElroy-Yaggy K, Gokina NI. Myogenic tone, reactivity, and forced dilatation: a three-phase model of in vitro arterial myogenic behaviour. Am J Physiol Heart Circ Physiol. 2002;283:H2260–7. https://doi.org/10.1152/ajpheart.00634.2002.
Moreno-Domínguez A, El-Yazbi AF, Zhu HL, Colinas O, Zhong XZ, Walsh EJ, et al. Cytoskeletal reorganization evoked by Rho-associated kinase- and protein kinase C-catalyzed phosphorylation of cofilin and heat shock protein 27, respectively, contributes to myogenic constriction of rat cerebral arteries. J Biol Chem. 2014;289:20939–52. https://doi.org/10.1074/jbc.M114.553743.
El-Yazbi AF, Abd-Elrahman KS. ROK and arteriolar myogenic tone generation: molecular evidence in health and disease. Front Pharm. 2017;8:87. https://doi.org/10.3389/fphar.2017.00087.
Moreno-Domínguez A, Colinas O, El-Yazbi A, Walsh EJ, Hill MA, Walsh MP, et al. Ca2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J Physiol. 2013;591:1235–50. https://doi.org/10.1113/jphysiol.2012.243576.
European Food Safety Authority (EFSA) Panel on Contaminants in the Food Chain (CONTAM). Scientific opinion on tolerable weekly intake for cadmium. EFSA J. 2011;9:1975–93. https://doi.org/10.2903/j.efsa.2011.1975.
World Health Organization. Safety evaluation of certain contaminants in foods: cadmium-impact assessment of different maximum limits. Prepared by the Sixty-fourth meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA). WHO Food Additives Series 55. 2006;82:156–203.
Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological profile for cadmium. ATSDR/U.S. Public Health Service, ATSDR/TP-88/08, Atlanta, US 1989.
Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Cadmium. Draft for public comment. US Department of Health and Human Services. Atlanta, US, 2008.
Xu Q, Liu Y, Gorospe M, Udelsman R, Holbrook NJ. Acute hypertension activates mitogen-activated protein kinases in arterial wall. J Clin Invest. 1996;97:508–14.
Chrissobolis S, Sobey CG. Evidence that Rho-kinase activity contributes to cerebral vascular tone in vivo and is enhanced during chronic hypertension: comparison with protein kinase C. Circ Res. 2001;88:774–9.
González J, Valls N, Brito R, Rodrigo R. Essential hypertension and oxidative stress: new insights. World J Cardiol. 2014;6:353–66. https://doi.org/10.4330/wjc.v6.i6.353.
Sutherland C, MacDonald JA, Walsh MP. Analysis of phosphorylation of the myosin-targeting subunit of myosin light chain phosphatase by Phos-tag SDS-PAGE. Am J Physiol Cell Physiol. 2016;310:C681–91. https://doi.org/10.1152/ajpcell.00327.2015.
Karaki H, Urakawa N, Kutsky P. Potassium-induced contraction in smooth muscle. Jpn J Smooth Muscle Res. 1984;20:427–44.
Acknowledgements
The authors would like to express gratitude to Cindy Sutherland, Aaron Cull, Drs. Ryan Mills, Alejandro Moreno-Domínguez, Olaia Colinas, Emma Walsh, Hai-Lei Zhu, and Anthony Liwa for assistance with western blotting and members of Basic Medical Sciences, UWI, for technical assistance and help with the organ bath experiment.
Funding
This work was funded by grants obtained from the Mona Campus Committee for Research and Publications and Graduate Awards of the School for Graduate Studies and Research, University of the West Indies.
Author information
Authors and Affiliations
Contributions
CRN conceptualized and designed the study and WCC, GM, and CC made some contributions to the methods. Material preparation and data collection and analysis were performed by GM, with western blot assistance from CC. The first draft of the paper was written by GM, and all authors commented on and edited subsequent versions of the paper. All authors contributed to the correction and finalization of the paper. GM and CRN acquired funding for the study. CRN and WCC provided resources for the study. CRN and PDB supervised the entire study. WCC supervised the western blot experiment.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
McCalla, G., Brown, P.D., Cole, W.C. et al. Cadmium-induced hypertension is associated with renal myosin light chain phosphatase inhibition via increased T697 phosphorylation and p44 mitogen-activated protein kinase levels. Hypertens Res 44, 941–954 (2021). https://doi.org/10.1038/s41440-021-00662-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-021-00662-w
- Springer Nature Singapore Pte Ltd.