
Abstract
Neotropical fishes exhibit remarkable karyotype diversity, whose evolution is poorly understood. Here, we studied genetic differences in 60 individuals, from 11 localities of one species, the wolf fish Hoplias malabaricus, from populations that include six different “karyomorphs”. These differ in Y-X chromosome differentiation, and, in several cases, by fusions with autosomes that have resulted in multiple sex chromosomes. Other differences are also observed in diploid chromosome numbers and morphologies. In an attempt to start understanding how this diversity was generated, we analyzed within- and between-population differences in a genome-wide sequence data set. We detect clear genotype differences between karyomorphs. Even in sympatry, samples with different karyomorphs differ more in sequence than samples from allopatric populations of the same karyomorph, suggesting that they represent populations that are to some degree reproductively isolated. However, sequence divergence between populations with different karyomorphs is remarkably low, suggesting that chromosome rearrangements may have evolved during a brief evolutionary time. We suggest that the karyotypic differences probably evolved in allopatry, in small populations that would have allowed rapid fixation of rearrangements, and that they became sympatric after their differentiation. Further studies are needed to test whether the karyotype differences contribute to reproductive isolation detected between some H. malabaricus karyomorphs.
Similar content being viewed by others


Introduction
The Neotropical region includes vast freshwater ichthyological biodiversity, with more than 6000 described species (Fricke et al. 2024), in line with the well-established tendency of the tropics to display higher biodiversity than other latitudes for multiple otherwise comparable environments (Hillebrand 2004). Recent evidence suggests that speciation rates have been affected by tectonic and climatic changes in the Neotropics during the geologically recent Neogene and Pleistocene periods (Meseguer and Condamine 2020; Hoorn et al. 2010; Garzón‐Orduña et al. 2014). These changes are likely to cause extinction of populations and create population subdivision (Albert and Reis 2011; Rull 2020). Indeed, allopatric speciation is often considered the major speciation process, with reproductive isolation evolving as a by-product of the genetic divergence of populations isolated by distance, geographic barriers, or through landscape features separating populations, combined with adaptation to different local environments (de Queiroz 2007).
The Characiformes fish group is particularly interesting as it is one of the largest freshwater fish orders, with approximately 3100 species (see Fricke et al. 2024). It includes the Erythrinidae family with only three genera, Hoplias Gill (1903); Hoplerythrinus Gill (1896) and Erythrinus Scopoli (1777), all with multiple species endemic to Central and South America. Among the 13 currently accepted Hoplias species, H. malabaricus, is widely distributed in several South American river basins (Oyakawa 2003). This species grows to a size of up to 60 centimetres; it is non-migratory and mainly inhabits still waters, but is also common in rivers. Sexual maturity is reached in the second year of life, and the growth rate is slow and constant (Barbieri 1989). Hoplias malabaricus spawns multiple times, in open nests, and shows parental care, with males aggressively protecting the nest alone or alongside the females (Araujo-Lima and Bitencourt, 2001; Prado et al. 2006).
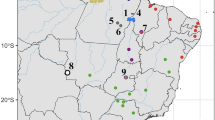
Hoplias malabaricus has at least seven distinct karyomorphs (Bertollo 2007; Cioffi et al. 2012), which could represent distinct isolated species (Fig. 1). Descriptions focus on the sex chromosomes, because they are most distinctive (Cioffi et al. 2013), but other chromosomes also differ between the karyomorphs (as illustrated in Fig. 1, methods section). While the sex chromosomes always indicate male heterogamety, some karyomorphs have differentiated XY pairs, while some do not, and some have sex chromosome-autosome fusions, resulting in different chromosome numbers and multiple sex chromosome systems. Based on previous cytogenetic and FISH analyses of the sex chromosomes, karyomorph B is derived from A, and karyomorph D from C (Bertollo et al. 2000), while the F and G karyomorphs are both derived from E (Bertollo et al. 2000; de Oliveira et al. 2018). However, these evolutionary histories were proposed based on cytogenetics only, and molecular data can provide more details and further understanding of the evolution of the H. malabaricus karyomorphs.
Previous studies have shown that the X chromosomes of the different karyomorphs are not homologs, and at least two sex chromosome turnovers have occurred (Cioffi et al. 2013). Karyomorph B has a heteromorphic XX/XY system, with a small and visibly heterochromatic male-specific chromosome (Born and Bertollo 2000). This system is probably derived from an ancestral karyomorph like the present A (Cioffi and Bertollo 2010), whose chromosomes are similar to those of karyotype B. FISH experiments using karyomorph B X chromosome probe showed that their sex chromosome pair is homologous, but is homomorphic (morphologically undifferentiated) in A, versus heteromorphic in B (Cioffi et al. 2011). The chromosome identified as the X in both karyomorphs C and D, is not homologous with the X in karyomorphs A and B (Cioffi et al. 2013). Karyomorph C has minor Y-X morphological differences (Cioffi and Bertollo 2010), but D has a Y-autosome fusion (supported by C-banding, whole chromosome painting, and comparative genomic hybridization, and repetitive DNA analyses, see Cioffi and Bertollo 2010), resulting in a large Y plus X1 and X2 chromosomes (Bertollo et al. 2000). The X of the homomorphic, but little studied, karyomorph E appears to represent yet another non-homologous chromosome, and this X seems to have given rise to the XX/XY and XX/XY1Y2 systems of karyomorphs F and G, respectively, (Cioffi and Bertollo 2010; de Freitas et al. 2018; de Oliveira et al. 2018). Karyomorph G is formed by an X-autosome fusion involving an X like that in karyomorph E, creating a large metacentric X and separating Y1 and Y2 chromosomes in males; in karyomorph F both X and Y chromosomes are fused with autosomes, resulting in a large XY metacentric pair, whose Y is similar in size to its X. Karyomorph E is not included in the present study since its geographic distribution within the Amazon River basin is very restricted, and recent collecting efforts failed to find new samples.
Sex chromosome homomorphism in karyomorphs A and E suggests that their (non-homologous) Ys have no extensive non-recombining regions, or that recombination has not been suppressed for a long enough evolutionary time for cytogenetic differentiation or genetic degeneration to evolve (including accumulation of repetitive sequences and/or losses of Y-linked genes and deletions of the region). In contrast, karyomorph B is strongly heteromorphic (due to shrinkage of the Y). C and F show minor Y-X morphology differences (Cioffi and Bertollo 2010; de Freitas et al. 2018), but each has persisted for a long enough evolutionary time for derived multiple sex chromosome systems (D and G, respectively) to have evolved.
Among these karyomorphs, some are widely distributed, while others are endemic to small geographic areas (See Fig. 1, Table 1 below). Although some are found sympatric with other karyomorphs, in the same habitat, potentially allowing mating between different karyomorphs, no hybrids have been detected (Scavone 1994; Bertollo et al. 1997; Lopes et al. 1998), except for a single case involving natural triploidy (Utsunomia et al. 2014). Moreover, the divergence between the cytochrome B sequences of two karyomorphs collected in a single geographic location (A and D) is 10.4%, similar to the divergence of 10.7% between H. microlepsis and H. malabaricus (Utsunomia et al. 2014). Both this cytochrome B divergence, and the extensive karyotypic evolution, suggest that the nominal species H. malabaricus is probably a species complex, with karyotypes representing reproductively isolated and independently evolving units (Bertollo et al. 2000).
A chief goal of the present study was to initiate a population genomic study of this neotropical fish, as a step towards developing this species complex as a model for understanding the contributions of different processes involved in its speciation, including geographic isolation and chromosomal rearrangements. Importantly, samples of at least 9 individuals of each sex per population indicate that different arrangements are fixed in different populations, with few exceptions (Table 1, below, summarizes this information along with the sample sizes used in the present study). The karyotype variability therefore does not represent within-population polymorphism such as the inversion polymorphisms in Diptera, including Drosophila and Coelopa, which often appear to be maintained by balancing selection (Schaeffer et al. 2003; Wright and Dobzhansky 1946; Kapun and Flatt 2019; Mérot et al. 2020). They more closely resemble intra-species differences between mice from different isolated islands (Britton-Davidian et al. 2000).
Recent studies of genetic diversity in H. malabaricus (Jacobina et al. 2018; Cardoso et al. 2018; Pires et al. 2021; Ferreira et al. 2021; Guimarães et al. 2022) mostly used populations without cytogenetic information and only small numbers of markers. Our analyses of high-throughput genotyping by sequencing data, provide the first genome-wide sequence information from populations with known karyotypes, and from the different geographic locations where they have been detected. The results described below yield evidence for geographic separation and the fixation of chromosome rearrangements, probably in small isolated populations, which, as discussed below, would not require a prolonged evolutionary time. This study also prepares the ground for future research to test whether the karyotype differences contribute to reproductive isolation, and, if so, whether the sex chromosome rearrangements play an important role in such diversification.
Material and methods
Specimen sampling and cytogenetic analysis
The collection sites, number, and sexes of the specimens investigated are shown in Table 1 and Fig. 1. Most of the samples were previously characterized cytogenetically, but two of the three samples of karyotype F listed in Table 1 are newly described here, using mitotic chromosomes obtained from kidney cells following Bertollo et al. (2015). Animals were collected with the authorization of the Brazilian environmental agency ICMBIO/SISBIO (license n°.48628-14) and SISGEN (A96FF09). Experiments followed ethical, and anesthesia conducts and were approved by the Ethics Committee on Animal Experimentation of the Universidade Federal de São Carlos (process number CEUA1853260315).

The colors indicate the different hydrographic basins in Brazil listed in the legend and the colored circles indicate the collection sites, with the corresponding karyomorphs found as: A blue, B pink, C yellow, D red, F green, and G black. The ellipse indicates a site with sympatry for both karyomorphs A and D. The ideograms on the right represent partial karyotypes of each karyomorph and their sex chromosomes. Pairs of karyomorphs with homologous sex chromosomes are boxed (see the text). Karyomorphs C and D differ from A and B by micro-rearrangements, though the details are not yet fully clear.
Sequencing and filtering
DNA was extracted from liver tissue for DArTseq sequencing (by Diversity Arrays Technology Pty Ltd, Australia). PstI and SbfI enzymes were used to digest DNA and enrich with sequences in lightly methylated regions, which should yield data enriched in non-repetitive sequences (Kilian et al. 2012) known as RADtags. Sequencing was carried out on the Illumina HiSeq 2500 platform. The raw data were processed using pyRAD v3.0.66 pipeline (Eaton 2014), according to the following procedure. We first trimmed the sequencing adapters and removed any sequences with more than five low quality (Q < 33) or undetermined (N) bases. After base calling, consensus sequences of the loci were clustered within each sample using USEARCH software (Edgar and Bateman 2010) to create “loci”, defined as short unannotated genomic regions that may include coding or non-coding regions. The sequences in each cluster were aligned with MUSCLE (Edgar 2004). Mean frequencies of heterozygosity and error rates were then estimated by using maximum likelihood (Lynch 2008). Following the pyRAD pipeline default settings (Eaton 2014), paralogs (which can also include high copy number DNA regions) were filtered by discarding consensus sequences containing one or more heterozygous sites shared across more than 3 individuals; as the sequences are short, a number of heterozygous sites exceeding this number are expected to be rare (as most variants are expected to be present at low frequencies), so this should largely restrict the subsequent analyses of diversity and divergence analyses to single-copy sequences, and minimize biases due to paralogs (see Jaegle et al. 2023). Consensus sequences were again clustered, this time across all samples, with USEARCH to find orthologs across the different samples and again aligned to identify and remove any further paralogs. The final clusters were filtered to exclude sequences shorter than 35 bp and exported in formats suitable for the downstream analyses.
A minimum coverage of 6 was required for statistical base calling at any site in a locus in a given individual. The similarity threshold for sequence clustering into loci was set to 0.88 (therefore, up to 12% nucleotide divergence was permitted between sequences from a given “locus”); this value would be high for pairwise differences within most species, but was chosen because it seemed likely that the samples represent multiple species (see Introduction). Only loci present in all individuals were kept, in order to allow comparisons of the same sequences between geographic populations or karyotypes/species. All other parameters were set to their default values in pyRAD.
Subsequent analyses were carried out with different outputs from the pyRAD pipeline, which we refer to as four data sets with the following numbers: (1) the genetic diversity and differentiation analyses described below used sequences from all individuals for each locus (from the pyRAD “.alleles” output). (2) A matrix of single nucleotide polymorphisms (SNPs) coded as 0 for homozygotes for the reference base, 1 for heterozygotes, and 2 for the alternative base homozygotes (from the .usnps.geno output). (3) A PHYLIP file with concatenated sequences used to generate a Maximum Likelihood phylogenetic tree. (4) The .vcf file used in the Principal Component Analysis (PCA), NeighborNet and STRUCTURE analyses, and D3 tests (see below).
Detection and exclusion of loci potentially under selection
Analyses for studying relationships require neutral or weakly selected variants. To test for markers with unusual levels of inter-population differentiation (either extremely low or high), which might reflect selective differences such as adaptation in some populations, we searched for outliers by BayeScan analysis (Foll and Gaggiotti 2008). This analysis used the SNPs in dataset (2) described in the previous section, pooled across all samples. The results presented below are based on analyses excluding these loci, but analyses including those loci yielded very similar results.
Sequence diversity
The DnaSPv.6.12.03 software (Rozas et al. 2017) was used to estimate two measures of nucleotide diversity per site, π and Watterson’s theta (θW), taking account of both variable and invariant sites, to reflect genome-wide diversity levels. This analysis used dataset (1) described above, and was not confined to a subset of sites, such as fourfold degenerate sites (as there is currently no annotated assembly for the species); instead, all site types were included. As many of our sequences are probably in non-coding regions, our diversity estimates should be comparable with those from synonymous sites. We also estimated values of Tajimaʼs D, and the similar indicator of variant frequencies (Δθ) defined as 1 − π/θW. This indicator is preferable to Tajimaʼs D, as it measures departures from the expected equilibrium neutral variant frequencies in a manner that is less affected by differences in sequence lengths (Jackson et al. 2017). Although our sample sizes are small, they are suitable for these measures, as larger sample sizes provide little additional information for nucleotide diversity (Pons and Chaouche 1995), and, importantly, our estimates are based on many diploid sequences genome-wide. We also estimated pairwise FST values between populations of karyomorphs (calculated as averages of the values obtained for each individual SNP separately), as well as the raw nucleotide divergence per site, Dxy, between such pairs of samples, and net divergence, corrected for variation within the samples analyzed (Da). Da best reflects the relative times when populations began to evolve independently (Nei 1975), and can be compared with relative times to common ancestry within samples, based on diversity estimates.
Analysis of population structure and introgression
To assess how DNA sequence diversity is distributed within and between karyomorphs (population structure), we carried out a Principal Component Analysis (PCA), using the ipyrad.pca tool (https://ipyrad.readthedocs.io/en/latest/API-analysis/cookbook-pca.html), followed by a STRUCTURE analysis (Pritchard et al. 2000), which applies a Bayesian approach to detect population structure. Both analyses used dataset (4). We used standard settings to perform five independent runs of 200,000 MCMC iterations after burn-in of 100,000 replicates with the maximum number of groups (K) set to eleven. The Ipyrad cookbook (https://ipyrad.readthedocs.io/en/master/API-analysis/cookbook-structure.html) was used to implement the STRUCTURE analysis and evaluate the best K value following the approach of Evanno et al. (2005).
To estimate a maximum likelihood phylogeny with dataset (3), we first carried out ModelTest-NG (Darriba et al. 2020) to determine the substitution model best fitting our data. The substitution model with the smallest Bayesian Information Criterion (BIC) was selected as the best model. We estimated the phylogenetic tree using MEGA v. 11.0.13 (Tamura et al. 2021) under the General Time Reversible model with the rate among sites defined as gamma distributed with invariant sites (GTR + G + I). The number of gamma categories was set to 6, and we performed 1000 bootstrap replicates with the same parameters. All other parameters were left at default.
To test for potential deviations from tree-like evolution of the populations, we also performed a NeighborNet network analysis with the SplitsTree software, which can detect potential conflicting signals based on parallel edges in the resulting graph (Bryant and Moulton 2004). We used a script deposited in Simon Martin’s GitHub (https://github.com/simonhmartin/genomics_general/blob/master/distMat.py) to calculate genetic distances between all individuals in our samples. To test for a statistically significant signal of non-tree-like structure, we used the D3 test (Hahn and Hibbins 2019). Like other D statistics, such as from ABBA-BABA tests (Martin et al. 2015), this relies on the prediction under incomplete lineage sorting that asymmetric gene tree topologies and branch lengths often reflect introgression. By using branch lengths, the D3 approach requires only three taxa to distinguish among topologies, rather than at least four for previous methods; under incomplete lineage sorting alone, the expectation of D3 is 0, but it can differ significantly from zero if gene flow occurs (Hahn and Hibbins 2019). Specifically, given a species tree for three taxa with a ((M:N);O) topology, introgression or mixing between two taxa, N and O, leads to more sequence trees having shorter pairwise distance between these two lineages, compared with taxa M and O, creating negative D3, whereas gene flow between taxa M and O produces positive values. We applied the D3 statistic to test for introgression in localities occurring in the same or in geographically close river basins (A4 and G1; A3, F2 and F3; F1 and B1; A2 and D1). To select the three localities required for each D3 test, for the purpose of implementing the analysis, we pooled different locations for which STRUCTURE indicated close genetic relationships, and assumed that the karyotypes are monophyletic and related to each other as described in the Introduction section (Fig. 1). We used a script designed for windows in an assembled genome implemented in Platt et al., (2021; https://github.com/nealplatt/sch_man_nwinvasion/blob/master/notebooks/10-summary_stats_and_selection.ipynb), modified so that the bootstrapping resampled 1000 random SNPs, since our sequences are not mapped to a genome assembly. The analysis generates Z-score tests of significance, and we considered values higher than 3 (positive or negative) to be significant. Both the NeighborNet and the D3 tests were carried out with dataset (4).
Results
Sequencing, filtering, and detection of markers in sequences under selection
Our sequencing (see Table 1 for the collection sites, number, and sexes of the specimens) yielded approximately 2 million reads per sample, with mean length ~88 bp. After filtering and removing sequencing adapters (see Methods), 6848 loci with sequence of at least 68 bp in all of our individuals (including a total of 14,105 SNPs) remained for analysis. We kept all 6848 loci for datasets (1) and (3), for datasets (2) and (4) we kept only the unlinked SNPs (6200 loci). BayeScan analysis of dataset (2) indicated that 73 loci (~1% of the polymorphic “loci” analyzed) might have been influenced by selection, rather than evolved neutrally (Tables S1; S2).
Population structure
Nine of the ten localities from which our eleven samples were obtained included only one karyomorph each (Table 1), and most karyomorphs were found in only a single geographic location, though A and F are geographically widespread. Multiple individuals of 5 of the 6 karyomorphs were sequenced (though B was represented by a single individual). Initial exploration of genome-wide sequence variation by PCA analysis indicated pronounced population structure (Fig. 2A), especially within karyomorph A, with the samples from localities in three different river basins forming three distinct groups; A1 and A2, from the same basin, were less differentiated and appear close to the B1 sample. Previous work using COI sequence data also suggested distinct lineages within karyomorph A (Jacobina et al. 2018). Karyomorphs C and D showed the expected low differentiation, and samples from karyomorphs F and G also clustered together, close to the A4 samples. In a maximum likelihood phylogeny based on the same data (see Methods), each karyomorph formed a monophyletic clade (Fig. S1). Congruent with the PCA results, the C + D and F + G groups again appeared with high bootstrap support, while karyotype A samples from different localities are scattered into other distinct groups.

A Principal component analysis with individuals from different karyomorphs represented by the same colors as in Fig. 1. B Structure barplots for K = 3 and K = 5. The top two rows show the barplots for iterations with K = 3, with those for 3 of the 5 iterations above those from the other 2 iterations; results for K = 5 are shown at the bottom. Each vertical bar represents an individual, whose karyomorphs and sampling locations, separated by black vertical bars, are shown above each barplot; the bar colors represent the population clusters into which the program classified each individual. C Violin plots of the Δθ values calculated for each population. For each sample displayed in the plot, the widths represent the number of loci with the Δθ value indicated on the left. The plots indicate the distribution and density of loci across Δθ values. The black diamonds indicate mean values for all loci, horizontal dashes indicate confidence intervals. D NeighborNet network analysis estimated by SplitsTree, confirming strong clustering of individuals into populations, and showing wide differences between the genotypes from different populations with karyotype A, and a pronounced split between two subsets of karyotype A4 individuals.
STRUCTURE supported these conclusions (Fig. 2B). The ΔK criterion (Evanno et al. 2005) suggested K = 3 as the most likely value (Fig. S2), again consistent with the results just described. All K = 3 results clustered A4 clustered with F1, F2, F3 and G1. In three of 5 runs (major K = 3 result), A3 clustered with C1 and D1, but in two cases (minor K = 3 result) it formed a separate group with A1, A2 and B1. Following the suggestions of Meirmans (2015) and Perez et al. (2018), we also evaluated the results for K = 5, where a small peak in ΔK is seen (Fig. S2). As with K = 3, karyotype C and D individuals remained clustered. Also, samples from A1, A2 and the single B1 individual grouped, separated from A3. The K = 5 results, however, yielded signs of possible admixture in localities A4, C1 and G1, while F1, F2, F3 and G1 individuals still clustered together, along with some A4 individuals.
Sequence differentiation and within-population diversity
If within-population diversity is low, differentiation will be high even if sequence divergence is low (Cruickshank and Hahn 2014; Charlesworth et al. 1997; Charlesworth 1997). We therefore estimated within-population nucleotide diversity. Except for population A4, discussed below, which has exceptionally high diversity, within-population nucleotide diversity values were all low (Table 2). The mean π was 0.12% per site, or 0.088% excluding A4. This is four or five times lower than the mean of 0.48% for comparable mostly single population estimates from 10 fish species in the compilation by Buffalo (2021) also based on all site types. Estimates using pooled samples of H. malabaricus A and F karyomorphs from different locations are indeed higher (0.69% and 0.31%, respectively, see Fig. 1 and Table 2 rows with values for pooled populations with the same karyomorph).
The low diversity in some individual collection sites might reflect recent bottlenecks. We therefore used the Δθ to visualize departures of variant frequencies from the expected distribution for neutral variants at equilibrium under mutation and genetic drift. The individual population samples are too small for reliable estimates of Δθ (or Tajima’s D) for individual loci, but the distributions of values across many loci are informative. Specifically, a population recovering from loss of diversity due to a recent bottleneck is expected to have an excess of low frequency variants, and a Δθ value > 0 (or negative Tajima’s D). Some H. malabaricus populations have small positive mean Δθ values (Table 2), consistent with recovery from bottlenecks (Fig. 2C); large positive Δθ values suggest an especially recent bottleneck in population A3. In contrast, the A4 sample shows a large negative Δθ value. Together with its exceptionally high nucleotide diversity, this suggests that diverged lineages that evolved in allopatry later became mixed within the A4 locality, leading to intermediate variant frequencies. It is important to note that negative values are also seen when all individuals from the same karyomorph (A or F) are pooled (Table 2).
Network analysis supports the conclusion that different populations with the morphologically undifferentiated sex chromosome karyotype A differ in their genotypes (Fig. 2D). Interestingly, the A3 locality was recovered in a star-like node together with A1, A2 and samples from karyomorphs B, C and D. This is in agreement with the different results (major and minor) obtained for K = 3 in STRUCTURE (Fig. 2B). In the A4 sample, it detects a pronounced split between individuals belonging to two very distinct lineages, supporting the conclusion above that this population’s high diversity reflects mixing of diverged populations. We cannot definitively identify the sources of these individuals, though PCA suggests similarities between the A4, G and F populations, and STRUCTURE (with K = 3 or 5) identifies some A4 individuals as close to those from population with karyotypes G and F and, in one case, C (Fig. 2A, B).
Based on these results, we tested for introgression using the D3 test with the following topologies that take account of differences in geographical distances or river basins (with the ((M;N);O) notation described in the Methods section): ((C1;D1);A2) to test between D1 and A2 in sympatry, ((A1-2-3;B1);F1) between F1 and B1 in the East Atlantic and São Francisco basin, ((F1;F2-3);A3) between A3 and F2-3 in the Tocantins-Araguaia basin, ((A4;A1-2-3);F + G) to test for introgression between A4 and the karyomorphs F and G in the Amazon river basin, ((G1;F1-2-3);A4) to test introgression between A4 and G1 (both from Amazon Basin). In order to infer which individuals from A4 were mixed and with which populations we also performed the following D3 tests: ((A4i;A1-2-3);F1), ((A4i;A1-2-3);F2), ((A4i;A1-2-3);F3), ((A4i;A1-2-3);G1) (with i being each individual from A4). The D3 test values can identify populations that have undergone introgression events, with positive values indicating events between the taxa indexed as M and O. Taking values higher than 3 (positive or negative) as statistically significant, introgression was detected only between the A4 and F + G groups (Table S3) and between A4 individuals and F or G groups (Table S4), in agreement with the STRUCTURE and Network results. As all the tests in Table S4 indicated introgression, it was not possible to infer with which groups each A4 individual was mixed. However, it is important to note that the D3 test, like other D-statistics can produce false positive results when substitution rates among groups are non-uniform (Amos 2020; Frankel and Ané 2023; Koppetsch et al. 2023) Since our results do not support monophyly of karyomorph A from different geographic localities (Figs. 2D, S1), we also tested a hypothesis with the topology ((A4; F + G); A1-2-3); this did not indicate significant introgression (Table S3).
Consistent with all these findings, pairwise FST values between sampling localities were high (Table 3), ranging from 0.190 for nearby populations of the same karyomorph to 0.825 between populations with different karyomorphs. Thirty-six of the fifty-five comparisons resulted in FST above 0.7 (although values for comparisons involving the single B1 population individual are unreliable). For the A4 population, however, all 10 comparisons yielded FST below 0.7, consistent with the other evidence suggesting the mixing of distinct populations with this karyomorph (which has high intra-karyomorph diversity).
The high diversity estimates when multiple karyomorph A and F population samples are pooled suggest that the allopatric populations of these karyomorphs are descended from ancestors much longer ago than the coalescence time within populations, allowing inter-population divergence. As explained above, the high FST between populations with other karyomorphs also reflects differentiation between samples of individuals from geographically separated population with generally low within-population diversity. For instance, the differentiation between A1 and A2 (the geographically closest karyomorph A populations from the Parana basin) versus other samples with karyomorph A, indicated by PCA and STRUCTURE analyses, probably largely reflects this effect of geographic distance. However, net divergence (Da) estimates, which correct for within-population diversity to reflect fixed differences between different populations, are also low for these comparisons, and for most others (between 0.5% and 1%, Table 4). Estimates were also low between the two karyomorph F populations with the closest geographic locations, F2 and F3, from the same river drainage. Overall, however, Da values, even among different A or F karyomorph populations, are nevertheless several times higher than the within-sample diversity values (π in Table 2). The highest divergence values involved the A3 and D1 samples, but the A3 population sequences differ from sequences from allopatric karyomorph A samples almost as much as from sequences from other karyomorphs, again consistent with PCA and STRUCTURE results.
Da is also high between A2 and the sympatric D1 sample, suggesting that the two karyomorphs are as isolated as most population pairs from geographically distant sites. Illustrating the difficulty of separating different processes, we also see that, despite the geographically separated location of the A4 sample, its high within-sample diversity reduces its net divergence from other karyotype A samples, compared with the values between the other populations. Overall, however, it is clear that divergence estimates between populations are 5 to 10 times higher than within-population pairwise sequence divergence (π values).
Discussion
Genetic diversity, population structure, and genetic isolation
The results overall suggest that geographic isolation is a major contributor to genome-wide sequence divergence in H. malabaricus, as differentiation between populations is not restricted to ones with different karyomorphs, but is also pronounced for the two cases where we could compare different populations with the same karyomorph. Nevertheless, between-karyomorph net divergence and FST values place them in the “gray zone” defined by Roux et al. (2016); using their data estimated that median Da for reproductively isolated species is 5.7% (versus an inter-population, within-species value of 0.1%), and the respective median FST values are 0.006 and 0.287. The data are not extensive, and more data should be collected, but similar results were obtained for African cichlid fish (Weber et al. 2021).
Given the small Da values, it is surprising that the 7 different karyomorphs currently known (involving three non-homologous X chromosomes that must reflect sex chromosome turnovers, two separate sex chromosome-autosome fusions, and other changes involving the autosomes) have already evolved. However, estimates of rates of chromosome rearrangements are scanty, and most rates so far estimated are across large evolutionary distances, involving different genera, and may not correctly estimate the numbers of changes (e.g., Olmo 2005; Yoshida and Kitano 2021).
Fixation of chromosome rearrangements are expected to be rare events, because rearrangements are often disadvantageous when heterozygous (underdominance, reviewed in Lande 1984 and Mackintosh et al. 2023). Holocentric chromosomes may not experience this problem, and may thus have fast fixation rates of chromosome rearrangements, and indeed one of the best estimates is for Heliconius butterflies, with 10 chromosome fusions in the 6 million years since the split with its sister genus Eueides (Davey et al. 2016). Other genomic characteristics that may affect rates are currently not well understood.
Observations of many rearrangements can, in principle, be explained if selection favoring the rearrangements is strong enough to overcome such disadvantages, but this seems implausible when many changes have been documented in a group of populations of closely related species. It is more plausible that underdominant rearrangements can become fixed in small populations, since small population sizes are permissive for disfavored changes (Lande 1984). Our data tend to support this hypothesis, as they demonstrate that H. malabaricus populations are indeed isolated, and have low within-population diversity, implying small effective sizes. Isolation is also important in a species of the butterfly genus with rearranged chromosomes, Brenthis (Mackintosh et al. 2023), and in the house mouse (e.g., Britton-Davidian et al. 2007).
The only exception to the generally low within-population diversity in H. malabaricus is the A4 sample (whose within-population nucleotide diversity is still only 0.4%, see Table 2). Analyses described above suggest that this sample’s slightly higher diversity, compared with that of the other samples, reflects mixing of diverged A4 populations, which is plausible, given the wide geographic distribution of the A karyotype and A4 inter-population sequence differences. Indeed, our D3 tests showed a significant result for introgression involving locality A4 and karyomorphs F and G (Tables S3; S4), which are presumably not closely related to karyomorph A (Fig. 1). Mixing could occur after changes in river courses, which have been suggested for the Araguaia-Tocantins (Rossetti and Valeriano 2007) and the Amazon basins (Albert et al. 2018), from which our A karyotype populations were sampled. Moreover, some populations of this karyotype may have become extinct. These, or unsampled populations, could represent “ghost populations” (Slatkin 2005) that could have contributed to the mixed A4 population. An alternative hypothesis would be that karyomorph A is not monophyletic and that some individuals of population A4 are more related to karyomorphs F and G, as suggested by the phylogeny (Fig. S1). In this case there is no signal of introgression with A4 (Table S3).
The positive Δθ values (Table 2, and negative Tajima’s D in Fig. 2C) in most samples are consistent with their generally low diversity, and suggest that they are still recovering diversity after bottleneck events, so that variants are often still below equilibrium frequencies. Low diversity can explain the observed high between-population differentiation.
The clusters suggested by the population structure and network analyses (Fig. 2B, D), and our phylogeny, are generally consistent with the relationships proposed for these karyomorphs in Fig. 1. Specifically, karyomorph B is derived from A, and both PCA and network analyses suggested that population B1 is closely related to A1. Similarly, karyomorph D was thought to be derived from C (Bertollo et al. 2000), and these are close in the PCA and clustered by STRUCTURE. The F and G karyomorphs both derive from E (Bertollo et al. 2000; de Oliveira et al. 2018), and all our analyses clustered the F karyomorph localities and G1. As the sex chromosomes of karyomorph A are undifferentiated, this karyomorph could reflect an ancestral state before an extensive non-recombining region evolved (Bertollo et al. 2000).
The possible involvement of the sex chromosomes in speciation
The rearrangements between the karyomorphs might contribute to the genome-wide genetic isolation reflected in high FST values between different karyomorphs. Multiple sex chromosome systems such as those in this species complex are expected to lead to isolation. When species differ by such rearrangements, trivalents form in the heterozygotes, causing irregular segregation and unbalanced gamete production, potentially leading to post-zygotic isolation (reviewed in Zhang et al. 2021). The Hoplias malabaricus complex is therefore suitable for testing whether rearrangements contribute, and whether the sex chromosomes are especially important.
Moreover, other chromosome rearrangements, including autosomal ones, can also contribute to suppressing recombination, preventing introgression and reducing gene flow, albeit mainly in the rearranged genome regions (Machado et al. 2007; Yannic et al. 2009; McGaugh and Noor 2012; Ostberg et al. 2013), and acting along with other reproductive barriers (Ostevik et al. 2016).
The H. malabaricus sex chromosomes are also suitable system for estimating the sizes of genome regions affected by recent genome rearrangements such as sex chromosome-autosome fusions. These are interesting for asking whether the newly sex-linked arms have evolved suppressed recombination or continue to recombine with their non-fused autosomal counterparts. The physical sizes of such neo-sex chromosome regions are small in the few species so far studied, including the threespine stickleback, Gasterosteus aculeatus (Schultheiß et al. 2015) and great reed warbler, Acrocephalus arundinaceus (Ponnikas et al. 2022). Larger regions may be discovered when different systems are studied in the future.
The Hoplias malabaricus karyomorphs can contribute valuable data about rates of chromosomal evolution within evolutionary time scales that can be related to those likely to be involved in speciation. Such information, together with emerging estimates of karyotype evolution rates, can help understand the extent to which chromosome changes contribute to speciation. Genome assemblies will allow future investigations in H. malabaricus, along with studies of genomic patterns of introgression (e.g., Yamasaki et al. 2020). Two recent studies illustrate the value of identifying and studying markers on individual chromosomes in species complexes such as H. malabaricus. In a plant, Rumex hastatulus, neo-X chromosome SNPs showed significantly steeper clines than the genome-wide average in a hybrid zone between populations with and without an X-autosome fusion, suggesting that the neo-sex chromosome affects reproductive isolation between the cytotypes (Beaudry et al. 2022). In an experimental hybrid population between two recently diverged Drosophila species, D. nasuta and D. albomicans, whose sequence divergence is similar to that between H. malabaricus populations, an autosome is fused with the ancestral X and Y, and a block of overlapping inversions on the neo-sex chromosome stood out as the strongest barrier to introgression (Wang et al. 2022). Introgression of the neo-sex chromosome showed asymmetry, with female hybrids showing an excess of the D. albomicans neo-X, while males showed an excess of heterozygous genotypes (Wang et al. 2022). Even if artificial hybrids cannot be bred, population genomic approaches have the potential to detect the involvement of rearranged chromosomes in the speciation process, if complete chromosome sequence assemblies can be made.
Data archiving
All data have been archived at Dryad: https://doi.org/10.5061/dryad.sbcc2frgc.
References
Albert JS, Reis ER (2011) Historical biogeography of Neotropical freshwater fishes. University of California Press, California
Albert JS, Val P, Hoorn C (2018) The changing course of the Amazon River in the Neogene: center stage for Neotropical diversification. Neotrop Ichthyol, 16
Amos W (2020) Signals interpreted as archaic introgression appear to be driven primarily by faster evolution in Africa. Royal Society Open. Science 7:191900
Araujo-Lima CARM, Bittencourt MM (2001) A reprodução e o início da vida de Hoplias malabaricus (Erythrinidae; Characiformes) na Amazônia Central. Acta Amaz 31(4):693–693. https://doi.org/10.1590/1809-43922001314697
Barbieri G (1989) Dinâmica da reprodução e crescimento de Hoplias malabaricus (Bloch, 1794) (Osteichthyes, Erythrinidae) da Represa do Monjolinho, São Carlos/SP. Rev Bras Zool 6(2):225–233. https://doi.org/10.1590/S0101-81751989000200006
Beaudry FE, Rifkin JL, Peake AL, Kim D, Jarvis-Cross M, Barrett SC, Wright SI (2022) Genomic signatures of hybridization on the neo-X chromosome of Rumex hastatulus. Mol Ecol 31:3708–3721
Bertollo LAC, Moreira-Filho O, Fontes MS (1997) Karyotypic diversity and distribution in Hoplias malabaricus (Pisces, Erythrinidae): Cytotypes with 2n = 40 chromosomes. Brazil J Genet 20:237–242
Bertollo LAC (2007) Chromosome evolution in the Neotropical Erythrinidae fish family: an overview. In: Pisano E, Ozouf-Costaz C, Foresti F, Kapoor BG (eds), Fish cytogenetics, 1st edn. Taylor & Francis, Enfield, p 195–213
Bertollo LAC, Born GG, Dergam JA, Fenocchio AS, Moreira-Filho O (2000) A biodiversity approach in the neotropical Erythrinidae fish, Hoplias malabaricus. Karyotypic survey, geographic distribution of cytotypes and cytotaxonomic considerations. Chromosome Res 8:603–613
Bertollo LAC, Cioffi MB, Moreira-Filho O (2015) Direct chromosome preparation from Freshwater Teleost Fishes. In: Ozouf-Costaz C, Pisano E, Foresti F, Almeida Toledo LF (eds), Fish cytogenetic techniques ray-fin fishes and chondrichthyans, 1st edn. CRC Press, Boca Raton. p 21–26
Blanco DR, Lui RL, Bertollo LAC, Margarido VP, Moreira Filho O (2010) Karyotypic diversity between allopatric populations of the group Hoplias malabaricus (Characiformes: Erythrinidae): evolutionary and biogeographic considerations. Neotrop Ichthyol 8:361–368
Born GG, Bertollo LAC (2000) An XX/XY sex chromosome system in a fish species, Hoplias malabaricus, with a polymorphic NOR-bearing X chromosome. Chromosome Res 8:111–118
Britton-Davidian J, Catalan J, da Graça Ramalhinho M, Ganem G, Auffray JC, Capela R, Biscoito M, Searle JB, da Luz Mathias M(2000) Rapid chromosomal evolution in island mice. Nature 403(6766):158–158. https://doi.org/10.1038/35003116
Britton-Davidian J, Catalan J, Lopez J, Ganem G, Nunes AC, Ramalhinho MG, Auffray JC, Searle JB, Mathias ML(2007) Patterns of genic diversity and structure in a species undergoing rapid chromosomal radiation: an allozyme analysis of house mice from the Madeira archipelago. Heredity 99(4):432–442. https://doi.org/10.1038/sj.hdy.6801021
Bryant D, Moulton V (2004) Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21(2):255–265
Buffalo V (2021) Quantifying the relationship between genetic diversity and population size suggests natural selection cannot explain Lewontin’s paradox. Elife 10. https://doi.org/10.7554/ELIFE.67509
Cardoso YP, Rosso JJ, Mabragaña E, González-Castro M, Delpiani M, Avigliano E et al. (2018) A continental-wide molecular approach unraveling mtDNA diversity and geographic distribution of the Neotropical genus Hoplias. PLoS One 8:e0202024
Charlesworth B, Nordborg M, Charlesworth D (1997) The effects of local selection, balanced polymorphism and background selection on equilibrium patterns of genetic diversity in subdivided populations. Genet Res 70:155–174
Charlesworth B (1997) Measures of divergence between populations and the effect of forces that reduce variability. Mol Biol Evol 15:538–543
Cioffi MB, Bertollo LAC (2010) Initial steps in XY chromosome differentiation in Hoplias malabaricus and the origin of an X1X2Y sex chromosome system in this fish group. Heredity 105:554–561
Cioffi MB, Liehr T, Trifonov V, Molina WF, Bertollo LAC (2013) Independent sex chromosome evolution in lower vertebrates: a molecular cytogenetic overview in the Erythrinidae fish family. Cytogenet Genome Res 141:186–194
Cioffi MB, Martins C, Bertollo LAC (2009) Comparative chromosome mapping of repetitive sequences. Implications for genomic evolution in the fish Hoplias malabaricus. BMC Genet 10:1–11
Cioffi MB, Moreira‐Filho O, Almeida‐Toledo LF, Bertollo LAC (2012) The contrasting role of heterochromatin in the differentiation of sex chromosomes: an overview from Neotropical fishes. J Fish Biol 80:2125–2139
Cioffi MB, Sánchez A, Marchal JA, Kosyakova N, Liehr T, Trifonov V et al. (2011) Whole chromosome painting reveals independent origin of sex chromosomes in closely related forms of a fish species. Genetica 139:1065–1072
Cruickshank TE, Hahn MW (2014) Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol Ecol 23:3133–3157
Darriba D, Posada D, Kozlov AM, Stamatakis A, Morel B, Flouri T (2020) ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol 37(1):291–294. https://doi.org/10.1093/molbev/msz189
Davey JW, Chouteau M, Barker SL, Maroja L, Baxter SW, Simpson F, Joron M, Mallet J, Dasmahapatra KK, Jiggins CD (2016) Major improvements to the Heliconius melpomene genome assembly used to confirm 10 chromosome fusion events in 6 million years of butterfly evolution. G3: Genes Genomes Genet 6(3):695–708. https://doi.org/10.1534/G3.115.023655/-/DC1
de Freitas NL, Al-Rikabi AB, Bertollo LAC, Ezaz T, Yano CF, de Oliveira EA et al. (2018) Early stages of XY sex chromosomes differentiation in the fish Hoplias malabaricus (Characiformes, Erythrinidae) revealed by DNA repeats accumulation. Curr Genom 19:216–226
de Oliveira EA, Sember A, Bertollo LAC, Yano CF, Ezaz T, Moreira-Filho O et al. (2018) Tracking the evolutionary pathway of sex chromosomes among fishes: characterizing the unique XX/XY1Y2 system in Hoplias malabaricus (Teleostei, Characiformes). Chromosoma 127:115–128
de Queiroz K (2007) Species concepts and species delimitation. Syst Biol 56:879–886
Eaton DA (2014) PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics 30:1844–1849
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/NAR/GKH340
Edgar RC, Bateman A (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/BIOINFORMATICS/BTQ461
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 8:2611–2620
Ferreira A, Ribeiro LB, Feldberg E (2021) Molecular analysis reveals high diversity in the Hoplias malabaricus (Characiformes, Erythrinidae) species complex from different Amazonian localities. Acta Amaz 51:139–144
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993
Frankel LE, Ané C (2023) Summary tests of introgression are highly sensitive to rate variation across lineages. Syst Biol 72, https://doi.org/10.1093/sysbio/syad056
Fricke R, Eschmeyer W, Van der Laan R (2024) Eschmeyer’s catalog of fishes: genera, species, references. (http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp). Accessed 10/04/2024.
Garzón‐Orduña IJ, Benetti‐Longhini JE, Brower AV (2014) Timing the diversification of the Amazonian biota: butterfly divergences are consistent with Pleistocene refugia. J Biogeogr 41:1631–1638
Gill TN (1903) Note on the fish genera named Macrodon. Proc US Natl Mus 26:1015–1016
Gill TN (1896) The differential characters of characinoid and erythrinoid fishes. Proc US Natl Mus 18:205–209
Guimarães KLA, Lima MP, Santana DJ, de Souza MFB, Barbosa RS et al. (2022) DNA barcoding and phylogeography of the Hoplias malabaricus species complex. Sci Rep 12:5288
Hahn MW, Hibbins MS (2019) A three-sample test for introgression. Mol Biol Evol 36:2878–2882
Hillebrand H (2004) On the generality of the latitudinal diversity gradient. Am Nat 163:192–211
Hoorn C, Wesselingh FP, Ter Steege H, Bermudez MA, Mora A, Sevink J et al. (2010) Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science 330:927–931
Jackson BC, Campos JL, Haddrill PR, Charlesworth B, Zeng K (2017) Variation in the intensity of selection on codon bias over time causes contrasting patterns of base composition evolution in Drosophila. Genome Biol Evol 9(1):102–123. https://doi.org/10.1093/GBE/EVW291
Jacobina UP, Lima SMQ, Maia DG, Souza G, Batalha-Filho H, Torres RA (2018) DNA barcode sheds light on systematics and evolution of neotropical freshwater trahiras. Genetica 146:505–515
Jaegle BR, Pisupati LM, Soto-Jiménez R, Burns FA, Rabanal et al. (2023) Extensive sequence duplication in Arabidopsis revealed by pseudo-heterozygosity. Genome Biol 24:44. https://doi.org/10.1186/s13059-023-02875-3
Kapun M, Flatt T (2019) The adaptive significance of chromosomal inversion polymorphisms in Drosophila melanogaster. Mol Ecol 28(6):1263–1282. https://doi.org/10.1111/MEC.14871
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H et al (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. In: Pompanon F, Bonin A (eds) Data production and analysis in population genomics, 1st edn. Human Totowa, New Jersey p 67–89
Koppetsch T, Malinsky M, Matschiner M (2023) bioRxiv, 2023.05.21.541635; https://doi.org/10.1101/2023.05.21.541635
Lande R (1984) The expected fixation rate of chromosomal inversions. Evolution 38:743–752
Lande R (1984) The expected fixation rate of chromosomal inversions. Evolution 38(4):743–752. https://doi.org/10.1111/J.1558-5646.1984.TB00347.X
Lopes PA, Alberdi AJ, Dergam JA, Fenocchio AS (1998) Cytotaxonomy of Hoplias malabaricus (Osteichthyes, Erythrinidae) in the Aguapey River (Province of Corrientes, Argentina). Copeia 2:485–487
Lynch M (2008) Estimation of nucleotide diversity, disequilibrium coefficients, and mutation rates from high-coverage genome-sequencing projects. Mol Biol Evol 25(11):2409–2419
Machado CA, Haselkorn TS, Noor MA (2007) Evaluation of the genomic extent of effects of fixed inversion differences on intraspecific variation and interspecific gene flow in Drosophila pseudoobscura and D. persimilis. Genetics 175:1289–1306
Mackintosh A, Vila R, Laetsch DR, Hayward A, Martin SH, Lohse K (2023) Chromosome fissions and fusions act as barriers to gene flow between Brenthis Fritillary butterflies. Mol Biol Evol 40(3). https://doi.org/10.1093/MOLBEV/MSAD043
Martin SH, Davey JW, Jiggins CD (2015) Evaluating the use of ABBA-BABA statistics to locate introgressed loci. Mol Biol Evol 32:244–257. https://doi.org/10.1093/MOLBEV/MSU269
McGaugh SE, Noor MA (2012) Genomic impacts of chromosomal inversions in parapatric Drosophila species. Philos Trans R Soc Biol Sci 367:422–429
Meirmans PG (2015) Seven common mistakes in population genetics and how to avoid them. Mol Ecol 24:3223–3231
Mérot C, Llaurens V, Normandeau E, Bernatchez L, Wellenreuther M(2020) Balancing selection via life-history trade-offs maintains an inversion polymorphism in a seaweed fly. Nat Commun 11(1):1–11. https://doi.org/10.1038/s41467-020-14479-7
Meseguer SA, Condamine FL (2020) Ancient tropical extinctions at high latitudes contributed to the latitudinal diversity gradient. Evolution 74:1966–1987
Nei M (1975) Molecular population genetics and evolution. North Holland. Press, Amsterdam
Olmo E (2005) Rate of chromosome changes and speciation in reptiles. Genetica 125(2–3):185–203. https://doi.org/10.1007/S10709-005-8008-2/METRICS
Ostberg CO, Hauser L, Pritchard VL, Garza JC, Naish KA (2013) Chromosome rearrangements, recombination suppression, and limited segregation distortion in hybrids between Yellow stone cutthroat trout (Oncorhynchus clarkii bouvieri) and rainbow trout (O. mykiss). BMC Genom 14:570
Ostevik K, Andrew R, Otto S, Rieseberg L (2016) Multiple reproductive barriers separate recently diverged sunflower ecotypes. Evolution 70:2322–2335
Oyakawa OT (2003) Family Erythrinidae (Trahiras). In: Reis RE, Kullander SO, Ferraris Junior CJ (eds) Checklist of the freshwater fishes of South and Central America, EDIPUCRS, Porto Alegre, p 515–526
Perez MF, Franco FF, Bombonato JR, Bonatelli IA, Khan G, Romeiro‐Brito et al. (2018) Assessing population structure in the face of isolation by distance: are we neglecting the problem? Divers Distrib 24:1883–1889
Pires WMM, Barros MC, Fraga EC (2021) DNA Barcoding unveils cryptic lineages of Hoplias malabaricus from Northeastern Brazil. Braz J Biol 81:917–927
Platt RN, Le Clec’h W, Chevalier FD, McDew‐White M, LoVerde PT et al. (2021) Genomic analysis of a parasite invasion: colonization of the Americas by the blood fluke Schistosoma mansoni. Mol Ecol 8:2242–2263
Ponnikas S, Sigeman H, Lundberg M, Hansson B (2022) Extreme variation in recombination rate and genetic diversity along the Sylvioidea neo‐sex chromosome. Mol Ecol 13:3566–3583
Pons O, Chaouche K (1995) Estimation, variance and optimal sampling of gene diversity II. Diploid locus. Theor Appl Genet 9:122–130
Prado CPA, Gomiero LM, Froehlich O (2006) Spawning and parental care in Hoplias malabaricus (Teleostei, Characiformes, Erythrinidae) in the Southern Pantanal, Brazil. Braz J Biol 66(2 B):697–702. https://doi.org/10.1590/S1519-69842006000400013
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rossetti DF, Valeriano MM (2007) Evolution of the lowest amazon basin modeled from the integration of geological and SRTM topographic data. Catena 70:253–265
Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N, Bierne N (2016) Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLOS Biol 14(12):e2000234. https://doi.org/10.1371/JOURNAL.PBIO.2000234
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE et al. (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302
Rull V (2020) Neotropical diversification: historical overview and conceptual insights. Neotropical diversification: patterns and processes. Springer, Cham, p 13–49
Santos U, Völcker CM, Belei FA, Cioffi MB, Bertollo LAC, Paiva SR et al. (2009) Molecular and karyotypic phylogeography in the Neotropical Hoplias malabaricus (Erythrinidae) fish in eastern Brazil. J Fish Biol 75(9):2326–2343
Scavone MD (1994) Sympatric occurrence of two karyotypic forms of Hoplias malabaricus (Pisces, Erythrinidae). Cytobios 80:223–227
Schaeffer SW, Goetting-Minesky MP, Kovacevic M, Peoples JR, Graybill JL, Miller JM, Kim K, Nelson JG, Anderson WW (2003) Evolutionary genomics of inversions in Drosophila pseudoobscura: evidence for epistasis. Proc Natl Acad Sci USA 100(14):8319–8324. https://doi.org/10.1073/PNAS.1432900100/SUPPL_FILE/2900FIG4.JPG
Schultheiß R, Viitaniemi HM, Leder EH (2015) Spatial dynamics of evolving dosage compensation in a young sex chromosome system. Genome Biol Evol 7(2):581–590
Scopoli GA (1777) Introductio ad historiam naturalem: sistens genera lapidum, plantarum, et animalium hactenus detecta, caracteribus essentialibus donata in tribus divisa, subinde ad leges naturae. Gerle
Slatkin M (2005) Seeing ghosts: the effect of unsampled populations on migration rates estimated for sampled populations. Mol Ecol 14(1):67–73
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027
Utsunomia R, Pansonato Alves JC, Paiva LRS, Costa Silva GJ, Oliveira C, Bertollo LAC et al. (2014) Genetic differentiation among distinct karyomorphs of the wolf fish Hoplias malabaricus species complex (Characiformes, Erythrinidae) and report of unusual hybridization with natural triploidy. J Fish Biol 85(5):1682–1692
Wang S, Nalley MJ, Chatla K, Aldaimalani R, MacPherson A, Wei KHC et al. (2022) Neo-sex chromosome evolution shapes sex-dependent asymmetrical introgression barrier. Proc Natl Acad Sci 119(19):e2119382119
Weber AAT, Rajkov J, Smailus K, Egger B, & Salzburger W (2021) Speciation dynamics and extent of parallel evolution along a lake-stream environmental contrast in African cichlid fishes. Sci Adv 7(45). https://doi.org/10.1126/SCIADV.ABG5391
Wright S, Dobzhansky T (1946) Genetics of natural populations. Xii. Experimental reproduction of some of the changes caused by natural selection in certain populations of Drosophila Pseudoobscura. Genetics 31(2):125. https://doi.org/10.1093/GENETICS/31.2.125
Yamasaki YY, Kakioka R, Takahashi H, Toyoda A, Nagano AJ, Machida Y et al. (2020) Genome-wide patterns of divergence and introgression after secondary contact between Pungitius sticklebacks. Philos Trans R Soc B 375(1806):20190548
Yannic G, Basset P, Hausser J (2009) Chromosomal rearrangements and gene flow over time in an inter-specific hybrid zone of the Sorex araneus group. Heredity 102(6):616–625
Yoshida K, Kitano J (2021) Tempo and mode in karyotype evolution revealed by a probabilistic model incorporating both chromosome number and morphology. PLOS Genet 17:e1009502. https://doi.org/10.1371/JOURNAL.PGEN.1009502
Zhang S, Lei C, Wu J, Zhou J, Xiao M, Zhu S et al. (2021) Meiotic heterogeneity of trivalent structure and interchromosomal effect in blastocysts with Robertsonian translocations. Front Genet 12:161
Acknowledgements
MBC was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (Proc. no 302449/2018-3) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (Proc. 2023/00955-2). FHSS was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (Proc. 2019/25009-7). MFP was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (Proc. 2017/10240-0). TE was partially supported by an Australian Research Council Discovery Grant DP200101406 led by Erik Wapstra, Tariq Ezaz, Cristopher Burridge and Oleg Simakov. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brasil (CAPES)-Finance Code 001. This study was supported by INCT - Peixes, funded by MCTIC/CNPq (proc. 405706/2022-7). The authors declare no conflicts of interest. We thank Dr. Simon Martin (University of Edinburgh) for advice and assistance with analyses of population structure and introgression.
Author information
Authors and Affiliations
Contributions
FHSS, MFP, and MBC conceptualized the study. Sampling and formal analysis were executed by FHSS, MFP, DC, PHNF, and MBC. FHSS and MFP wrote the first draft of the manuscript with input from DC, LACB, and TE, and all authors contributed to subsequent revisions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Animals were collected with the authorization of the Brazilian environmental agency ICMBIO/SISBIO (license n°.48628-14) and SISGEN (A96FF09). Experiments followed ethical, and anesthesia conduct and were approved by the Ethics Committee on Animal Experimentation of the Universidade Federal de São Carlos (process number CEUA1853260315).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Rui Faria.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Souza, F.H.S., Perez, M.F., Ferreira, P.H.N. et al. Multiple karyotype differences between populations of the Hoplias malabaricus (Teleostei; Characiformes), a species complex in the gray area of the speciation process. Heredity (2024). https://doi.org/10.1038/s41437-024-00707-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41437-024-00707-z
- Springer Nature Switzerland AG