
Abstract
While intraspecific variation in aposematic signals can be selected for by different predatory responses, their evolution is also contingent on other processes shaping genetic variation. We evaluate the relative contributions of selection, geographic isolation, and random genetic drift to the evolution of aposematic color polymorphism in the poison frog Adelphobates galactonotus, distributed throughout eastern Brazilian Amazonia. Dorsal coloration was measured for 111 individuals and genetic data were obtained from 220 individuals at two mitochondrial genes (mtDNA) and 7963 Single Nucleotide Polymorphisms (SNPs). Four color categories were described (brown, blue, yellow, orange) and our models of frog and bird visual systems indicated that each color was distinguishable for these taxa. Using outlier and correlative analyses we found no compelling genetic evidence for color being under divergent selection. A time-calibrated mtDNA tree suggests that the present distribution of dorsal coloration resulted from processes occurring during the Pleistocene. Separate phylogenies based on SNPs and mtDNA resolved the same well supported clades, each containing different colored populations. Ancestral character state analysis provided some evidence for evolutionary transitions in color type. Genetic structure was more strongly associated with geographic features, than color category, suggesting that the distribution of color is explained by localized processes. Evidence for geographic isolation together with estimates of low effective population size implicates drift as playing a key role in color diversification. Our results highlight the relevance of considering the neutral processes involved with the evolution of traits with important fitness consequences.
Similar content being viewed by others

Introduction
The presence of intraspecific variation in aposematic signals provides opportunities to examine the relative contributions of selection, drift, and geographic isolation to the evolution of this trait (Richards-Zawacki et al. 2013; Gehara et al. 2013; Kang et al. 2017; Lawrence et al. 2019). Because aposematic signals such as contrasting and bright colors advertise the presence of toxins or other defenses, the signal is under balancing selection and therefore is usually consistent in order to be effective as a deterrent (Endler and Greenwood 1988). Nevertheless, geographic variation in selective pressures, including differences in the local assemblage of predators, changes in predator behavior and Batesian and Mullarian mimicry complexes, can result in different colors being fixed in different populations (Endler and Mappes 2004; Darst and Cummings 2006; Amézquita et al. 2017). Adaptive potential is influenced by the distribution of genetic variation, and less effective aposematic coloration can be maintained if the level of gene flow is sufficiently low between populations with different colors (Lawrence et al. 2019). Furthermore, if different phenotypes experience the same level of selection, and effective population sizes are small enough, then diversification through random genetic drift is possible in accordance with Wright’s shifting balance theory (Wright 1932; Rudh et al. 2007; Chouteau and Angers 2012; Comeault and Noonan 2011).
Neotropical poison frogs (family Dendrobatidae) possess aposematic coloration in combination with skin toxins, which likely evolved in tandem (Summers and Clough 2001). Many species show variation in aposematic coloration and for this reason they are popular models to study the evolution of this trait (Chouteau and Angers 2012). Color differentiation in poison frogs has been a recent and recurrent evolutionary process for those species investigated thus far, often with intra-specific divergence in colors traced back to no earlier than the Pleistocene, 11,500–2.6 million years before present (Noonan and Gaucher 2006; Santos and Cannatella 2011; Gehara et al. 2013).
The Pleistocene is characterized by rapid environmental change and this seems likely to have altered both levels of connectivity and predation pressures. Disturbance during the Pleistocene has been characterized for some poison frogs as fine scaled habitat fragmentation, rather than large-scale retraction to refugia (Noonan and Gaucher 2006). Habitat disturbance during the Pleistocene resulted in fine-scaled divergence in the northern Amazonian dyeing poison frog Dendrobates tinctorius, which in concert with selection by predators, resulted in the establishment of populations that are monomorphic for different colors (Noonan and Gaucher 2006; Comeault and Noonan 2011). The process of isolation can also facilitate color diversification by hybridization following secondary contact. For example, hybridization has been implicated as the process generating color variation in the Andean harlequin poison frog Oophaga histrionica (Medina et al. 2013). Secondary contact and hybridization may be further facilitated by the likelihood of rapid habitat restructuring in the Amazon region (Sefc et al. 2017).
Isolation can also result from behavioral differences, potentially facilitating color differences at finer spatial scales than the effects of geographic isolation. For example, in parts of the strawberry poison frog (Oophaga pumilio) distribution, genetic distances between populations are more strongly associated with dorsal coloration than with landscape features, and this was attributed to selection against dissimilar immigrants (Wang and Summer 2010). Furthermore, in O. pumilo, sexual selection, coupled with drift in female preference is another of several mechanisms that have been proposed to explain color diversification (Mann and Cummings 2012; Gehara et al. 2013). Jointly analyzing whether genetic structure is associated with present day environmental features, or aposematic coloration, can provide clues about isolating processes and the scale at which these occur (Wang and Summer 2010). In addition, patterns of genetic structure associated with populations of different color types can be used to help infer whether signal variation is a consequence of dispersal or color becoming fixed within a population following de novo mutation.
Geographic isolation and drift has resulted in the diversification of color phenotypes for several poison frog species. Physical features that impede gene flow and influence the population sizes of poison frogs include rivers, topgraphic barriers, forest density and soil type because of their inherent variation in the water holding capacity (Kaefer et al. 2013; Simões et al. 2014; Ferreira et al. 2018). Where the habitat for O. pumilio is fragmented, low population sizes, increased drift, perhaps in combination with reduced predation pressure has allowed new phenotypes to increase in frequency and be fixed by selection (Rudh et al. 2007). The role of drift in generating color polymorphism has also been shown in the poison dart frog Ranitomeya imitator. Here it was shown that selection was relaxed at a point along an altitudinal gradient and this allowed new color phenotypes to be generated by drift (Chouteau and Angers 2012).
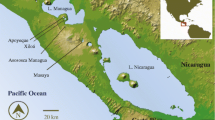
The Amazonian poison frog, Adelphobates galactonotus, varies in dorsal coloration throughout its distribution in eastern Amazonia, which comprises forest areas east of the Tapajós River to the Atlantic coast and south of the Amazon river to the southern boundary of the Amazon forest (Fig. 1; Hoogmoed and Avila-Pires 2012). Different dorsal colors (yellow, orange, red, blue, and brown) have not been found sympatrically and appear in a mosaic-like fashion across the species’ geographic range (Hoogmoed and Avila-Pires 2012). The variation in the dorsal coloration of A. galactonotus is intriguing because it is non-mimetic and populations containing individuals with different color types are found in close proximity and within areas of similar environmental conditions.

The sampling locations for Adelphobates galactonotus are shown with dots that are colored blue, yellow, orange, or brown to indicate the color category to which the frogs were assigned. The arrows point to images of A. galactonotus that are representative of the local dorsal color. The white dot represents a locality where color was not available because tissue was obtained from preserved museum specimens. The distribution of A. galactonotus in the regions sampled east and west of Xingu River are shown with yellow and green shading respectively
Here we use field-collected dorsal color measurements, mtDNA sequence data, and a data set of several thousand single nucleotide polymorphisms (SNPs) from A. galactonotus to test the following hypotheses: (1) the time of divergence between populations containing different dorsal colors is consistent with the timing of color diversification in other Neotropical poison frogs, (2) the same dorsal coloration evolved independently in different locations, and (3) that dorsal coloration of A. galactonotus is under selection.
Methods
Study area
We sampled Adelphobates galactonotus across most of its known geographic distribution in eastern Amazonia, south of the Amazon River, in the Brazilian state of Pará. This region contains two large southern tributaries of the Amazon, the Xingu and the Tocantins rivers (Fig. 1). Our sampling effort was concentrated to the west of the Tocantins River. Because of the difficulty of sampling within large tracts of forest, individuals were sampled either in close proximity (<15 km) to rivers, or to the few roads that are in the region. Visual appraisal suggests that structural elements of the forest do not vary systematically with respect to the presence of rivers or roads.
Tissue sampling
A total of 220 individual samples were obtained from fieldwork carried out from 2012 to 2014 and from the tissue collection of Museu Paraense Emilio Goeldi (MPEG), Belém, Brazil (Table 1). Fieldwork was conducted in three regions from eastern Amazonia: Caxiuanã–Anapu river basin, Tapajós-Jamanxim river basin, and Carajás (Fig. 1). Tissue samples consisted of toe clips, or muscle/liver tissue when voucher specimens were collected. Voucher specimens were deposited at the Osvaldo Rodrigues da Cunha Herpetological Collection of MPEG or at the Amphibian and Reptile Collection of Instituto Nacional de Pesquisas da Amazônia (INPA-H) in Manaus, Brazil. All tissue samples were stored in absolute ethanol prior to DNA extraction. We define a sampling locality as an area where all individuals were captured within 80 km of each other. These locations were consistently in continuous forest, without apparent biogeographic barriers (such as the presence of large rivers).
Color data collection
Adelphobates galactonotus has been collected at several of the sampling localities over a number of years and stored at MPEG (e.g., Caxiuanã-Anapu locality has samples collected over ~20 years). These collections consistently found the same single dorsal color at each of the localities (Table 1; Hoogmoed and Avila-Pires 2012). The dorsum of A. galactonotus most often appears to have consistent coloration, though there are instances of color patterns in the form of spots or flecks (Table 1). To describe color variation, we measured the color of 111 live specimens at 20 localities distributed in the Caxiuanã-Anapu, Tapajós-Jamanxim and Carajás regions (1–13 individuals used per locality; Table 1) with an Ocean Optics USB200 spectrometer (Ocean Optics, Inc., Dunedun, FL, USA), an Ocean Optics DH-2000 BAL light source and Spectra Suite® software. The spectrometer was calibrated for both white and black prior to the individual measurements being taken. All of these measurements were obtained in the laboratory under equivalent lighting conditions. Individual reflectance spectra were obtained by averaging and smoothing two dorsal measurements, one at the dorsal surface of the head, the other on the lower dorsum (smoothing parameters: span = 0.2, negative values fixed to 0). The averages of these spectra datasets were used to represent the dorsal coloration reflectance for each locality (Fig. 2).

Reflectance spectra for the dorsal coloration in four color categories: blue (a–e), brown (f), orange (g–i) and yellow (j). Images of live frogs representing these categories (correspondingly labeled a–j) are shown in Fig. 1. In each figure, the average reflectance spectrum value is shown with a solid black line while the shading shows the standard deviation. Figure titles provide information on the color category and sampling location where: East = east of the Xingu river; West = west of the Xingu river, and the localities are given using the codes provided in Table 1. At each locality, the number of individuals used for the measurement of reflectance spectra were as follows: OAL = 12; OAR = 20; BAR = 17; BRAR = 21; BAL = 13; BC = 2; OT = 4; YT = 7; BT = 6; BJ = 6
To categorize dorsal coloration and evaluate its variability, the mean reflectances were visualized and colorimetric variables (mean brightness, hue and chroma) from reflectance data were calculated (Endler 1990; Anderson and Prager 2006; Kemp et al. 2015) in the R package pavo (Maia et al. 2013). To test whether differences in dorsal coloration can be discriminated by conspecifics and potential predators (birds), we analyzed spectra with avian (tetrachromatic-V retina type) and frog (trichromatic) visual models using pavo (Vorobyev and Osorio 1998; Vorobyev et al. 2001). To model frog vision, we used the data from three cone classes of O. pumilio (Siddiqi et al. 2004). Specific predators of A. galactonotus are unknown, but birds are predators of many aposematic species including poison frogs (Lindström et al. 1999; Master 1999; Exnerová et al. 2008). Furthermore, in field experiments, birds were reported as the most frequent predators of artificial poison frog models (Noonan and Comeault 2009; Paluh et al. 2014; Rojas et al. 2015).
To test whether or not different color categories are perceived differently by conspecifics and birds, we estimated pairwise (localities) contrasts expressed in units of Just Noticeable Difference (JND). When JND values are above one it indicates that color distances would be discriminated by the visual system modeled (Siddiqi et al. 2004). All analyses were conducted in the R statistical computing environment (R Core Team 2014).
Mitochondrial sequence data
Total genomic DNA was extracted from preserved tissue samples using Promega® Wizard Extraction Kits, following the manufacturer’s guidelines. We amplified a 559 bp fragment of the Cytochrome Oxidase I (COI) mitochondrial gene from 133 individuals using primers CHMF4 and CHMR4 (Che et al. 2012) and a 402 bp fragment of the 16S rDNA from 176 individuals using the primers 16Sar and 16Sbr (Palumbi 1996). PCR cycles were as follows: 30 s at 92 °C followed by 35 cycles of 92 °C for 10 s, 50 °C for 35 s, and 72 °C for 90 s, with a final extension of 10 min at 72 °C. Amplicons were purified for sequencing with ExoSAP-IT solution, then subjected to EtOH/EDTA precipitation and sequenced in the forward direction using an ABI sequencer 3031x (Applied Biosystems®).
Development of SNPs
Genomic DNA was extracted from 186 tissue samples (Table 1) using the GeneCatchTM Blood & Tissue Genomic Mini Prep Kit (Epoch Life Science, Inc). Approximately 0.5 µg of DNA was sent to Diversity Arrays Technology Pty. Ltd. (Canberra, Australia—http://www.diversityarrays.com) where SNP discovery and genotyping was performed using the standard DartSeqTM protocol. DartSeqTM genotyping is a SNP-based genotyping-by-sequencing approach (Jaccoud et al. 2001; Kilian et al. 2012; Lambert et al. 2013) where sequencing is carried out on an Illumina platform (Sansaloni et al. 2011) to genotype thousands of SNPs across the genome (Petroli et al. 2012).
Library preparation
Template DNA was incubated in a 1X solution of Multi-CoreTM restriction enzyme (RE) buffer (Promega) at 37 °C for 2 h to check genomic DNA quality. Approximately 100 ng per µL of each sample was digested with a combination of PstI and SphI restriction enzymes. PstI and Sphl adapters and unique barcodes were ligated to each sample.
Each sample was amplified using PCR primers specific to barcode and adapter sequences. The PCR conditions were as follows: 1 min initial denaturation at 94 °C, followed by 30 cycles of 20 s denaturation (94 °C), 30 s annealing (58 °C) and 45 s extension (72 °C), and a final extension of 7 min at 72 °C. Using ~10 µL of each sample, all samples were pooled, diluted, and denatured using NaOH in preparation for hybridization to the flow cell. The library was sequenced on an Illumina HiSeq®2500 platform (single end read), resulting in 77 bp long fragments. A proportion of the samples (>40%) were processed again through the whole library preparation protocol and downstream analyses to create the set of technical replicates that were used to assess the reproducibility.
Quality control and initial SNP calling
Library sequences were converted to fastq format using the Illumina HiSeq®2500 software, and individuals were de-multiplexed based on the ligated barcode using customized scripts developed by DArT. Each read was assessed using Phred quality scores (Q-scores; Ewing and Green 1998) and any reads containing Q-scores < 25 were removed. All reads were checked against the DArT database and GenBank viral and bacterial sequences to assess for contamination. Following this primary workflow, SNPs were identified and called following the standard procedure in the DArT proprietary pipeline DArTSoft14TM (Diversity Arrays Technology). The workflow is technically similar to the STACKS pipeline (Catchen et al. 2013), but it differs by first calling sequence clusters from all pooled samples, prior to being called for each individual. All invariant sequence clusters were removed, and SNPs were called only if they were present in both homozygous and heterozygous forms across the whole data set. SNPs were retained in the data set if the average ratio of read depth between alleles ≤0.75, with reproducibility of 100% and a minimum read depth of five. We further filtered the dataset using the following criteria: We only included SNPs with a call rate of >98% (i.e., <2% missing data), where the average read depth for both alleles was >5 and the Minor Allele Frequency (MAF) > 0.05. We retained only a single SNP per fragment to avoid creating a dataset containing closely linked loci.
mtDNA analysis
Sequences were aligned using the ClustalW algorithm (Thompson et al. 1994) implemented in BioEdit 7.2.5 (version updated in November 2013, Hall 1999). Substitutions were checked with the original chromatographs. In order to evaluate the genealogical relationships among haplotypes and overall haplotype distributions, a haplotype network for COI was built from the resulting alignment using statistical parsimony (Templeton et al. 1992) with the software TCS 1.21 (Clement et al. 2000). Analyses of genetic diversity at 16S rDNA and COI regions were carried out for each sampling location and collectively for individuals sampled on each side of the Xingu river using DnaSP v.5.10.01 (Librado and Rozas 2009).
Estimating divergence times
To estimate the divergence times for A. galactonotus we used BEAST 1.8.0 (Drummond et al. 2012) to analyze 59 unique mtDNA haplotypes recovered from a concatenated database of 16S rDNA and COI sequences (totaling 962 bp). To construct a gene tree, sequences for 16S rDNA and COI were obtained from GenBank for A. castaneoticus, A. quinquevittatus and Phyllobates terribilis to use as outgroups (accession numbers DQ502058, DQ502157, DQ502234, DQ502780, DQ502861, and DQ502906). Sequences were aligned with the MUSCLE algorithm (Edgar 2004) as implemented in MEGA 6.06 (Tamura et al. 2013). The most probable substitution model for each region was estimated independently in jModeltest 2.1.7 (Guindon and Gascuel 2003; Darriba et al. 2012). We defined the approximate average rate of evolution for each region following Mueller (2006). As time calibration priors, we considered means and standard deviations previously estimated for the most recent common ancestor for A. galactonotus and A. qinquevittatus (10.0 ± 2.5 million years ago—m.y.a.) and A. galactonotus and A. castaneoticus (8.0 ± 2.1 m.y.a.) (Santos et al. 2009). We followed the recommendations provided by the software developers for BEAST 1.8.0 to select the parameters and priors for this analysis (Drummond et al. 2012). We used the following priors: partitions with unlinked substitution and clock models, evolutionary rates (the number of substitutions per site every 100 million years) for COI = 1.04 ± 0.27 and 16S = 0.70 ± 0.03 (Mueller 2006), a lognormal distribution and the tree prior was Yule. We applied a chain length of 80 million generations with samples taken every eight thousand generations. The first 10% of the trees were discarded as burn-in, resulting in 9000 sampled trees in the Markov Chain Monte Carlo (MCMC) run. The stationarity of the posterior distributions for all model parameters, including medians and 95% Highest Posterior Density intervals (HPD) of the nodes was verified on Tracer 1.6.0 (Rambaut et al. 2014). The final consensus tree was obtained from the MCMC output using Tree Annotator 1.8.0 (Drummond et al. 2012).
Constructing a population tree with SNP data
We generated a population tree with our SNP data using the Multispecies Coalescent (MSC) implemented by the SNAPP package (Bryant et al. 2012) and BEAST v. 2.4.7 (Drummond and Rambaut 2007; Bouckaert et al. 2014). The SNAPP analysis directly models incomplete sorting, which is expected in recently diverged species or populations, and can therefore be helpful in visualizing uncertainties around the majority-rule gene tree. For this analysis we randomly selected 2 individuals at each sampling location to reduce the computational time. Two independent runs were run for 10 million generations, sampling every 1000 generations. After stationarity was reached, we checked for convergence in Tracer and for consistency of the recovered topologies (Rambaut et al. 2014). The two runs were combined using LogCombiner, using a burnin of the first 3.5 million generations. The final tree was visualized in DensiTree v.2.2.6 where the individual lines represent trees sampled every 1000 generations (Bouckaert 2010).
Ancestral state analysis
To test whether evolutionary transitions in dorsal coloration occurred we conducted ancestral character reconstruction. Ancestral state reconstruction requires an accurate estimate of phylogenetic relationships, a model of trait evolution and accurate trait data (Duchêne and Lanfear 2015). Ancestral states were analyzed using the mtDNA consensus tree obtained using 16S rDNA and COI. To test whether this tree was influenced by incomplete lineage sorting, or undetected male gene flow we compared the topology with the population tree constructed using SNP data. Variation in dorsal coloration was collapsed into four character states according to the relative reflectance in short, medium, and long wave lengths, based on spectral data (in decreasing order: blue, yellow, and orange), and absolute light reflectance (brown); these color categories are distinguishable to a wide range of potential predators (Endler and Mappes 2004). A fifth state (patterned) was added for the outgroup species in the mitochondrial tree to indicate a general black dorsum crossed by white longitudinal stripes (A. quinquevittatus) or blotches (A. castaneoticus). A black dorsum with yellow/whitish dorsolateral stripes represents the ancestral color pattern in the genus Phyllobates, although the species we used here (P. terribilis) loses this pattern as an adult and becomes uniformly yellow (see Fig. S8 for images of these three outgroup species). The use of parsimony for ancestral state reconstruction can be biased by rapid evolution, a problem that can be solved using a maximum likelihood or a Bayesian approach (Cunningham et al. 1998). We preferred a maximum likelihood approach rather than stochastic character mapping by Bayesian inference because of the inherent difficulties in visually representing the results (Revell 2013). The reconstruction of ancestral color states was implemented with a continuous-time Markov model (Mk) in the R package ape (Paradis and Schliep 2018) and phytools (Revell 2012). The MK model assumes the random evolution of these color states and estimates the relative probability of each color category at each node. The analysis accounts for branch length, and transitions are independent of changes occurring elsewhere in the tree. Our model assumed that transitions occurred at equal rates for each color category. Equal transition rates were assumed because color type is highly variable in dendrobatid frogs and no directional changes were apparent.
Genetic structure estimated with SNP data
To describe patterns of genetic structure we performed a Principal Component Analysis (PCA) based on genotype frequency data using the R package adegenet (Jombart and Ahmed 2011). Genetic structure was further investigated using the software fastSTRUCTURE (Raj et al. 2014), a fast algorithm for the approximate inference of the model implemented by STRUCTURE (Pritchard et al. 2000). We ran fastSTRUCTURE using K values ranging from 1 to 10, the logistic prior and five-fold cross-validation. Analyses on the entire dataset revealed a clear partitioning between individuals east and west of the Xingu river. We then repeated the analyses separately for individuals sampled east and west of the Xingu river. We considered smaller values of K with prediction errors within one standard error of the lowest estimate as useful models of the data (Raj et al. 2014). Results from fastSTRUCTURE were plotted in R using the package pophelper.
We then tested whether geographic distance and the presence of rivers explained the observed patterns of genetic differentiation. We constructed a matrix of river distances by summing river widths that were measured where intersected by a straight line between sampling locations. Genetic divergence was estimated with Weir and Cockerham’s FST calculated using GenAlEX v. 6.5 (Peakall and Smouse 2012). Spatial analyses were performed separately for sampling locations east and west of the Xingu river. Three different maximum‐likelihood population‐effects (MLPE) mixed models were tested, using the R packages corMLPE and nlme (Clarke et al. 2002). The first model included geographic distance alone, the second river distance alone, and the third used both distance matrices as predictors. The intercepts, slopes, and random effect terms were estimated using restricted maximum likelihood. Because marginal R2 values are biased towards more complex models, the best model was selected using Akaike information criteria (AIC) and Bayesian information criteria (BIC), following Row et al. (2017).
To assess whether loci putatively under the influence of selection were affecting patterns of genetic structure, we ran all of the analyses a second time with the data set excluding outlier loci identified via FST outlier testing in OutFLANK (see below; Whitlock and Lotterhos 2015). In all cases our analyses produced very similar results and we present genetic structure analyses using the SNP data excluding FST outliers.
Effective population size
For each sampling location we used NeEstimator v. 2.1 (Do et al. 2014) to calculate effective population size by linkage disequilibrium (LD). In a closed population with random mating and unlinked loci, LD results from genetic drift, occurring at a rate inversely proportional to the Ne. The random mating model under LD was chosen, and a critical value of 0.02 and 0.05 was selected to represent the minimum allele frequency cut off (O’Leary et al. 2013; Trask et al. 2017). Confidence intervals for Ne estimates were obtained by jackknifing (Do et al. 2014).
Signatures of selection
Divergent selection could result in significantly larger genetic distances than expected under neutrality for loci situated in parts of the genome responsible for the traits under selection. We evaluated this process by searching for FST outliers. For this approach we excluded localities with <5 individuals collected to ensure adequate representation of allele frequencies within each sampling locality. This resulted in data from 12 locations (with 5–26 individuals per location). Because of strong genetic divergence between populations separated by the Xingu River (see results), we conducted FST outlier analysis separately for populations east (K = 8 populations) and west (K = 4 populations) of the Xingu River. We used the software OutFLANK (Whitlock and Lotterhos 2015) and applied the default parameters for False Discovery Rate (FDR = 5%) and for the left and right trim factors of the FST distribution.
We also searched for signatures of selection on dorsal coloration using linkage disequilibrium (LD) network analyses developed by Kemppainen et al. (2015) and implemented in R package LDna. LD network clusters (single-outlier clusters, SOCs) represent networks of loci in high LD. Using loci from SOCs as an input for PCA, it is possible to discern whether these loci in LD represent population structure (if they group individuals by geography) or local adaptation (in which case they will group individuals by phenotype). This method has been successfully used to identify genomic regions under selection in distinct ecotypes of three-spined sticklebacks (Kemppainen et al. 2015) and parallel selection in ecotypes of Littorina saxatilis (Ravinet et al. 2016). LDna was first run using the data from all sampling localities and SNPs with frequencies >5%. This first analysis revealed that the strongest patterns of LD were associated with divergence between populations east and west of the Xingu river. Analyses were then run separately for populations on each side of the Xingu river. The two main parameters in LDna are |E|min (minimum number of edges in a cluster), and λ which represents the change in LD when two clusters merge. We retained only SOCs with a |E|min of ten and for which λ exceeded the median by a multiplier (φ) of five. These parameters were chosen interactively as outlined in the LDna tutorial and after consulting the developer of the software.
We looked for associations between color category and SNP genotypes using the latent factor mixed model approach implemented within the software LFMM (Frichot et al. 2013). The LFMM analysis considers genotypic matrix values as response variables in a linear regression model. The LFMM analysis accounts for the effects of neutral genetic structure by defining a number of latent factors (LF), estimated beforehand by genetic clustering analysis (Frichot et al. 2015). After accounting for genetic structure, significant associations between SNP allele frequencies and observed ecological (or morphological) variables may be interpreted as evidence for selection (Frichot and François 2015). Compared to other outlier and environmental association analysis methods, LFMM reduces the type I error (i.e., false positives) in datasets with complex underlying genetic structure (Frichot et al. 2013; de Villemereuil et al. 2014; Rellstab et al. 2015). We ran LFMM in R using the LEA package (Frichot and François 2015). The number of LFs was based on the number of sampling locations, justified on the basis of the observed genetic and phenotypic structure (east: LF = 8; west: LF = 4). The program was run for 10,000 iterations with the first 5000 iterations discarded as burn-in. This was repeated 5 times and median z-scores were combined across runs, and p-values were adjusted for multiple tests using the Benjamini-Hochberg procedure (False Discovery Rate (FDR) = 5%) as outlined within the LEA package (Frichot and François 2015). The median genomic inflation factor (GIF) was calculated across runs following Devlin and Roeder (1999) to further evaluate FDR. Successful control of FDR is indicated by a GIF close to, or slightly below 1.0 (Frichot et al. 2015). A p-value threshold of p < log10−6 was used to identify significant SNP-by-color associations.
DNA sequences containing SNPs identified as outliers or associating with color were ran through the BLASTN online database to search for matches with functionally relevant genes that may relate to color synthesis. We set the BLAST e-value acceptance threshold as 1 × 10−6 with a sequence identity of more than 70% (Benestan et al. 2016).
Results
Analysis of dorsal coloration
The dorsal coloration of Adelphobates galactonotus could be placed into four categories, yellow, orange, blue, and brown with no individuals found to have intermediate coloration (Figs. 1, 2 and Table 1). Consistent with Hoogmoed and Avila-Pires (2012), we never observed more than a single color category at a locality, with individuals exhibiting only slight variation in color and pattern. Although we can statistically partition the color variation into four color categories, there appears to be considerable geographic variation within each of these categories (Figs. 1, 2 and S1). In particular, the blue color category showed a great deal of variation, not only with respect to reflectance spectra, but also the pattern of dorsal coloration (Fig. 1).
Models of bird and frog vision suggests that both of these taxa can discriminate between dorsal coloration in the blue, yellow, orange, and brown categories (JND > 1, Fig. S2) with the birds being capable of detecting subtler differences (JND values were higher for birds).
Timing the divergence into different color categories
We identified 37 variable sites and 35 haplotypes for 16S rDNA and 42 variable sites and 34 haplotypes for COI. Haplotype networks built using statistical parsimony were similar for both mtDNA regions, hence we show only the network using COI (Fig. 3; see Table S1 for additional data). No association was found between color categories and the genetic structure inferred from mtDNA markers (e.g., some individuals in different color categories have the same haplotypes). However, there were no haplotypes in common for A. galactonotus sampled east and west of the Xingu River, suggesting that the river has been a long-term barrier to female mediated gene flow.

A haplotype network built using 559 bp of sequence from Cytochrome Oxidase I (COI) from 133 individuals. Circles denote individual haplotypes and circle size is proportional to haplotype frequency. Each circle is partitioned into colors to show the proportion of individuals in each dorsal coloration category (orange, yellow, brown, and blue) sharing that haplotype. The white color represents sequences obtained from preserved specimens where dorsal color could not be determined. Haplotypes from samples collected west of the Xingu River have background shading in yellow, while samples collected east of the Xingu River have a background shading of green. The number in each circle refer to number of individuals with that haplotype
The consensus tree based on mtDNA has well supported clades (bootstrap support >0.70) that show instances of populations with different dorsal coloration sharing a common ancestor (Fig. 4). Samples collected from regions west and east of the Xingu River form two well-supported basal clades. The most recent common ancestor for these clades is estimated to be late Pliocene, most probably around 4.8 million years ago (Fig. 4). This is broadly consistent with the timing for range expansions reported for poison frogs in the Amazon basin (<10.0 m.y.a.) by Santos et al. (2009). The genetic divergence associated with the present day distribution of color categories occurred more recently, during the Pleistocene.

A time calibrated phylogenetic tree obtained using BEAST with sequences from 16S rDNA and COI. The two major clades recovered correspond to samples collected east (green shading) and west (yellow shading) of the Xingu River. Error bars on the nodes represent the 95% highest posterior density interval for node ages. The posterior probability of nodes lower than 0.70 are omitted. The timing of divergence (m.y.a) can be gauged using the scale given beneath tree. Tips are colored according to dorsal coloration in the four color categories; blue, orange, yellow, and brown. White represents samples obtained from specimens of unknown color. The black tips correspond to outgroup sequences (Pt = Phyllobates terribilis; Aq = Adelphobates quinquevittatus; Ac = Adelphobates castaneoticus). Codes indicate the sampling localities described in Table 1. The first letter in each code refers to color (B = blue, Y = yellow, B = brown, O = orange, U = unknown). Summary statistics for the mtDNA data at each locality is detailed in Table S1
The population tree constructed by coalescence using SNP data with the software SNAPP showed a broadly similar topology as the consensus tree using mtDNA, there are several well supported clades, each with populations of different dorsal colorations (Fig. 5). The clade representing the basal split associated with the Xingu river, and two clades in the east, each have posterior probabilities of one and correspond to the three clades in the mtDNA consensus tree with bootstrap support >90. In addition, in the east, the three nodes associated with populations BAR-YMV and the two nodes associated with populations OA-BC each had posterior probability support of 1 (Fig. 5). Similarly, these same nodes are well supported in the mtDNA consensus tree with bootstrap support >0.88. This SNP based population tree demonstrates that incomplete lineage sorting, or male mediated gene flow, is not confounding our conclusion that populations with different colors share a common ancestor.

A Densitree from SNAPP analyses. The thin lines forming each branch represent individual trees sampled every 1000 generations. The populations west of the Xingu River were indistinguishable from each other. East of the Xingu River there are two well supported clades (each with a posterior probability of one), one clade includes individuals from locations on the right bank of the Anapu River, Monteverde and Altamira, and the other one includes individuals from locations on the left bank of the Anapu River, Caxiuanã Bay, and Serra Sul. The localities represented by the code given at each tip are provided in Table 1
Ancestral state analysis
The reconstruction of ancestral coloration in A. galactonotus provides some support for transitions of color within the two major clades, east and west of the Xingu river (Fig. 6). There are two instances of populations with frogs of yellow color having ancestors with high posterior probability for blue coloration, and one well supported instance of a population with yellow frogs having an orange colored ancestor in the western clade. The node in the mtDNA consensus tree representing the most recent common ancestor for the western clade, has high bootstrap support (0.99) and also a very high posterior probability of blue coloration. The tips of this clade represent populations with yellow, blue and orange coloration. In the eastern clade, there was strong support for a transition from blue to orange and orange to brown. For these two cases, there was also strong bootstrap support for the nodes associated with these transitions (blue to orange = 0.92 and orange to brown = 0.75) and the timing of these transitions was estimated to have occurred during the Pleistocene (Fig. 4). It can be seen that the ancestral colors could not be confidently assigned for several of the most recent common ancestors for population with different colors. The ancestral coloration for the eastern clade was also equivocal.

Reconstruction of ancestral coloration in Adelphobates galactonotus (Ag) according to four color categories: blue, yellow, orange, and brown. Circles represent individuals at the tips and at each node. The circles are colored to reflect each of the four color categories. The colored pie charts at each node denote the posterior probabilities for alternative reconstructed character states, empirically obtained from a continuous-time Markov model. Black denotes the generally dark dorsum of outgroup species: A. castaneoticus (Ac), A. quinquevittatus (Aq), and the ancestor of the genus Phyllobates (Pt)
Neutral genetic structure based on SNPs
We identified a total of 7963 SNPs after filtering. Principal Components Analysis (PCA) resulted in two axes (PC1 and PC2) summarizing nearly 70% of the total genetic variation. Two genetic clusters corresponded to samples collected from the west and east of the Xingu River (Fig. S5b). The PCAs carried out separately for samples collected from each side of the Xingu River did not show patterns of divergence associated with color (Fig. 7a, b), but there were clear signals of geographic structure, with individuals clustering according to sampling locations. West of the Xingu River, the major differentiation is between blue populations (from Moraes de Almeida and the Tapajós River) and orange and yellow populations from the Tapajós River.

Principal Component Analyses (PCA) based on SNP data from samples located east (a) and west (b) of the Xingu River. Data points are color-coded to reflect the color category. c The figures show the prediction error from five-fold cross-validation with fastSTRUCTURE analysis carried out separately for samples collected east and west of the Xingu River. Histograms represent individual ancestry proportions that were calculated using fastSTRUCTURE. Results are given for individuals west (d) and east (e) of the Xingu River for values of K ranging from two to four
Analysis of genetic clustering using fastSTRUCTURE suggested values of K that ranged from two to four (Fig. 7c–e) for both the east and west datasets. These genetic groupings contain populations of frogs with different categories of dorsal coloration, potentially reflecting the relatively recent diversification shown with mtDNA (Figs. 3 and 4). Nevertheless, limited gene flow among sample locations is shown by significant pairwise FST values between each of the sampling localities (east, N = 8; west, N = 4; Table S2). For localities with SNP data from a sufficient number of individuals, effective population sizes were low enough to suggest considerable levels of genetic drift (Ne ranged from 45–213), though large or infinite confidence intervals bounded most of these estimates (Table S4).
There was no evidence of isolation by geographic or river distance in populations sampled west of the Xingu River, though this is likely to be because only four populations were sampled. East of the Xingu river both geographic (p < 0.000001) and river distance (p < 0.005) explained part of the observed genetic structure. The model including both river and geographic distance had the lowest AIC and BIC and explained the majority of the variance (R2 = 0.57, Fig. 8, Table S5).

The variance explained by relationship between the fitted FST values from the best MLPE model (geographic + river distance) and observed FST among population pairs east of the Xingu river is shown in this scatter plot (R2 = 0.57)
Detection of outlier and color-associated SNPs
The FST outlier tests were carried out separately for samples collected east and west of the Xingu river. West of the river there were 13 SNPs (0.25 % of SNPs) identified as having significantly high FST values based on a model of neutral FST distributions, east of the river no FST outliers were detected. However, the model fit for the FST distributions using OutFLANK was poor for both analyses (see FST histograms: Figs. S3 and S4). This could be a result of strong genetic structure among many of the sampling localities (e.g., Fig. 6 and Table S2).
SNP-by-color association analyses using LFMM (Frichot et al. 2013) identified SNPs that were significantly associated with color, but the number of these SNPs differed east and west of the Xingu river (Table S3). The identity of SNPs that were associated with color also differed east and west of the Xingu River, with only 1.2% of these SNPs being in common. Furthermore, the color these SNPs were associated with differed when comparing associations found east and west of the Xingu. There were four SNPs associated with brown east of the Xingu that were associated with blue west of the Xingu, two SNPs associated with yellow in the east, were associated with orange in the west and two SNPs associated with yellow in the east, were associated with blue in the west. Of the 13 FST outlier loci identified west of the Xingu river using OutFLANK, four of these SNPs were also identified as significantly associated with color using LFMM.
Linkage Disequilibrium (LD) network analysis identified several single outlier clusters (SOCs) of loci in high LD, most of which were consistent with the genetic structure associated with geographic distance and the presence of rivers, and none associated with dorsal coloration. When we analyzed the whole dataset, six SOCs were identified (Fig. S5a). Four of these SOCs grouped individuals east and west of the Xingu river (Fig. S5a, c–f) and the remaining two SOCs were consistent with genetic structure between two regions east of the Xingu River (Altamira and Serra Sul, respectively; Fig. S5g–h). A separate analysis of data collected east of the Xingu river with LDNa identified nine SOCs (Fig. S6a), eight of which were associated with geographic structure and none grouped individuals according to coloration (Fig. S6). The analyses of individuals west of the Xingu river identified only one SOC, which grouped individuals according to sampling location (Fig. S7).
We found no significant matches to functional genes with DNA sequences identified as outliers or associated with color using LFMM.
Discussion
The dorsal coloration of the poison frog Adelphobates galactonotus can be categorized as yellow, orange, blue or brown, and individuals inhabiting the same region always belong to the same color category. Using both mtDNA and SNP data, we show that populations with different colors are grouped in well supported clades and share a common ancestor. There is no obvious geographic pattern to color type, and our time calibrated phylogeny suggests that the present day distribution of coloration is a result of processes that most likely took place during the Pleistocene. We have some evidence from ancestral character state analysis that dorsal coloration has transitioned from one color category to another, but the ancestral color type could not be determined for many nodes. There are several possible explanations for the present day distribution of dorsal coloration. Broadly, these include founding events and hybridization, or the effects of geographic isolation, where new mutations fixed by drift or selection might account for the color variation observed today.
Genetic divergence associated with the current distribution of colors occurred well within the Pleistocene, even allowing for the uncertainty around estimates of clade ages (Fig. 4). This was a period of high and repetitive climatic instability, especially in the eastern portion of the Amazon basin (Maslin and Burns 2000; Vonhof and Kaandorp 2011; Cheng et al. 2013). Such cyclic disturbances seem likely to have generated intermittent isolation and small population sizes through the contraction and expansion of adequate habitat, and the formation of new riverine barriers. Populations of A. galactonotus could be especially prone to isolation, partly because of their patchy distribution, a feature contributed to by their dependence on small water bodies for reproduction (Hoogmoed and Avila-Pires 2012). Our estimates of low contemporary effective population sizes (Ne ~ 50–250; Table S4) and significant pairwise FST estimates (Table S2) are compatible with generally low levels of gene flow among populations.
The present day distribution of dorsal coloration could be a result of de novo mutations for color becoming fixed, from founding events distributing pre-existing color types, or hybridization and rapid selection on standing genetic variation (e.g., Barrett and Schluter 2008; Sefc et al. 2017). Our data suggests that localized color transitions are a more likely explanation for the current color variation than founding events distributing preexisting color types. We have strong support from both SNP and mtDNA data that for several clades, populations with different dorsal coloration share a common ancestor (Figs. 4 and 5). We could also confidently infer transitions in dorsal coloration for at least a few lineages with ancestral character state analysis (Fig. 6). Alternately, if there was widespread movement of preexisting color variation, we expected that genetic divergence would generally be greater between populations with different categories of dorsal coloration compared with those of the same color category. Contrary to this, we show with both mtDNA and SNP data that genetic structure is more strongly associated with geographic distance and the presence of rivers, rather than color type (e.g., Figs. 7 and 8). Furthermore, localized color transitions by de novo mutations might explain the geographic variation in dorsal coloration that we observed within each color category, especially among populations with the blue category (Figs. 2 and S1). Although strong genetic evidence for hybridization is lacking, we can not discount it as a process generating color diversity, especially given that a brown color category appears intermediate to the blue and orange color categories on the right bank of the Anapu River (Fig. 7e). If the geographic distribution of dorsal coloration is at least partially a consequence of de novo mutations, or hybridization, the function of color as an aposematic signal suggests that selection has a role in driving these mutations to fixation.
In species with aposematic coloration, visually oriented predators are frequently thought to drive color diversification through natural selection (e.g., Jiggins et al. 2001). Our model of avian vision suggests that these predators can distinguish between the different color categories of A. galactonotus (Fig. S2). However, recent field experiments using A. galactonotus wax models failed to observe predatory bias toward particular colors (Rojas et al. 2015). It is possible that this result reflected the experimental design, because movement is relevant to prey detection by birds, which are considered predators of poison frogs (Paluh et al. 2014). It is also possible that the color variation reflects selective pressures that are not current, or that the variation is a result of other forms of selection. These could include environmental differences, such as the lighting and background color, or differing predator assemblages (Endler 1992). It has also been demonstrated that sexual selection can effectively and rapidly drive a trait to fixation (Tazzyman and Iwasa 2010; Rudh et al. 2011) and thereby result in polyphyletic evolution. However, sexual dichromatism in A. galactonotus is not obvious to a human observer, so if sexual selection drives color divergence, it has not resulted in the magnitude of dichromatism reported in species where color plays a role in mate choice (Bell and Zamudio 2012).
Evidence for selection in our genetic data set was limited. We did identify SNPs potentially associated with directional selection, but believe these results should be cautiously interpreted. This is partially due to the following two reasons: first, the generally high FST between sampling localities, though accounted for to some degree in our analyses, may still compromise model outcomes (see Whitlock and Lotterhos 2015). Second, in cases where different localities had frogs assigned to the same color category, different SNP identities were found to be associating with that color. Similarly, groups of loci in linkage disequilibrium were consistently associated with patterns of geographic structure rather than color. Future analyses could incorporate environmental variables and a more extensive sampling strategy to tease apart selection processes. In addition, a data set with higher density sequencing across the genome may be required, especially if a simple genetic mechanism underpins color in A. galactonotus.
Although the genetic mechanisms of color in anurans have received relatively little attention (Rudh et al. 2013), it is known that color is determined by three cell layers, the melanophores, the xantophores and the iridophores, each containing specific pigments (Hofreiter and Schöneberg 2010). Conceivably, color variation could be due to one, or a few point mutations related to genes involved in color determination. These genes could be responsible for the synthesis or destruction of pigments like pteridines or carotenoids, which could be responsible for the yellow/orange/brown coloration. If the carotenoids and pteridines are removed, these colors disappear and the guanine crystals deeper in the epidermis are the main determinants of the iridescent, bluish color (Bagnara and Matsumoto 2007; Bagnara et al. 2007).
The maintenance of populations with different colors in close proximity suggests mechanisms that prevent recombination. Our model of frog vision suggests that A. galactonotus can discriminate between each of the categories of dorsal coloration (Fig. S2). Therefore, a plausible mechanism preventing recombination is that lower hybrid fitness selected for assortative mating with respect to color type. Narrow hybrid zones across unbroken rainforest landscapes have been characterized in other Amazonian frogs (e.g., Brown and Twomey 2009; Simões et al. 2012) suggesting that in these cases, hybridization lowers fitness. Further work could test for the presence of reproductive character displacement in A. galactonotus by taking color measurements at increasing distances from potential contact zones of populations with frogs of different dorsal coloration. Here, the prediction is that color contrast between the two populations will increase as the proximity between the two populations decreases (e.g., Lambert et al. 2013). Assortative mating that maintains reproductive isolation upon secondary contact is potentially a mechanism generating biodiversity both in the Amazon, and elsewhere, yet evidence for this process remains limited (Boul et al. 2007). More generally, further knowledge of the processes resulting in spatial variation in trait variation is needed to test whether polymorphism is typically a precursor to speciation (McLean and Stuart-Fox 2014). The distribution of dorsal color variation and genetic variation that we describe here for A. galactonotus provides an ideal opportunity to further test these hypotheses.
Data availability
The mtDNA sequences can be accessed in GenBank (accession code KU597806 -KU597873). All mtDNA sequences, genotypes for SNP loci and summary statistics (call rate, heterozygosity, read depth, and reproducibility) are accessible from the Dryad Digital Repository (https://datadryad.org/stash/share/3fnquEBQT_q4p7bIH-A1WITH5yEVb4XYZdNj2XQLp1c). Trimmed sequence reads used to generate our SNP data set are held at Diversity Arrays Technology Pty. Ltd. (Canberra, Australia) and can be made available.
References
Amézquita A, Ramos Ó, González MC, Rodríguez C, Medina I, Simões PI et al. (2017) Conspicuousness, color resemblance, and toxicity in geographically diverging mimicry: the pan-Amazonian frog Allobates femoralis. Evolution 71:1039–1050
Anderson S, Prager M (2006) Quantifying colors. In: McGraw KJ, Hill GE (eds) Bird Coloration, Vol I. Harvard University Press, Cambridge, MA, pp 41–89
Bagnara JT, Fernandez PJ, Fujii R (2007) On the blue coloration of vertebrates. Pigment Cell Res 20:14–26
Bagnara JT, Matsumoto J (2007) Comparative anatomy and physiology of pigment cells in nonmammalian tissues. In: Norlund J, Boissy R, Hearing V, King R, Oetting W, Ortonne JP (eds) The pigmentary system: physiology and pathophysiology, Blackwell Publishing Ltd, Oxford, UK, pp 11–59
Barrett RDH, Schluter D (2008) Adaptation from standing genetic variation. Trends Ecol Evol 23:8–44
Bell RC, Zamudio KR (2012) Sexual dichromatism in frogs: natural selection, sexual selection and unexpected diversity. Proc Biol Sci 279:4687–4693
Benestan L, Quinn BK, Maaroufi H, Laporte M, Clark FK, Greenwood SJ et al. (2016) Seascape genomics provides evidence for thermal adaptation and current-mediated population structure in American lobster (Homarus americanus). Mol Ecol 25:5073–5092
Bouckaert RR (2010) DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 26:1372–1373
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D et al. (2014) BEAST 2: a software platform for bayesian evolutionary analysis. PLoS Comput Biol 10:1–6
Boul KE, Funk WC, Darst CR, Cannatella DC, Ryan MJ (2007) Sexual selection drives speciation in an Amazonian frog. Proc Biol Sci 274:399–406
Brown JL, Twomey E (2009) Complicated histories: three new species of poison frogs of the genus Ameerega (Anura: Dendrobatidae) from north-central Peru. Zootaxa: 2049:1–38
Bryant D, Bouckaert R, Felsenstein J, Rosenberg NA, Roychoudhury A (2012) Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol Biol Evol 29:1917–1932
Catchen J, Hohenlohe Pa, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Che J, Chen H-M, Yang J-X, Jin J-Q, Jiang K, Yuan Z-Y et al. (2012) Universal COI primers for DNA barcoding amphibians. Mol Ecol Resour 12:247–258
Cheng H, Sinha A, Cruz FW, Wang X, Edwards RL, D’Horta FM et al. (2013) Climate change patterns in Amazonia and biodiversity. Nat Commun 4:1411
Chouteau M, Angers B (2012) Wright’s shifting balance theory and the diversification of aposematic signals PLoS ONE 7:e34028
Clarke RT, Rothery P, Raybould AF (2002) Confidence limits for regression relationships between distance matrices: Estimating gene flow with distance. J Agric Biol Environ Stat 7:361–372
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Comeault AA, Noonan BP (2011) Spatial variation in the fitness of divergent aposematic phenotypes of the poison frog, Dendrobates tinctorius. J Evol Biol 24:1374–1379
Cunningham CW, Omland KE, Oakley TH (1998) Reconstructing ancestral character states, a critical reappraisal. TREE 13:361–366
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772
Darst CR, Cummings ME (2006) Predator learning favors mimicry of a less-toxic model in poison frogs. Nature 440:208–211
Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55:997–1004
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Drummond AJ, Rambaut A (2007) Bayesian evolutionary analysis by sampling trees. In: BMC Evolut Biol, Vol 7, 7:214.
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973
Duchêne S, Lanfear R (2015) Phylogenetic uncertainty can bias the number of evolutionary transitions estimated from ancestral state reconstruction methods. J Exp Zool (Mol Dev Evol) 324:517–524
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 5:113
Endler JA (1990) On the measurement and classification of colors in studies of animal colour patterns. Biol J Linn Soc 41:315–332
Endler JA (1992) Signals, signal directions and the direction of evolution. Am Nat 139:125–153
Endler JA, Greenwood JJD (1988) Frequency-dependent predation, crypsis and aposematic coloration [and Discussion]. Philos Trans R Soc B Biol Sci 319:505–523
Endler JA, Mappes J (2004) Predator mixes and the conspicuousness of aposematic signals. Am Nat 4:532–547
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:186–194
Exnerová A, Svádová K, Fouá P, Fučíková E, Ježová D, Niederlová A et al. (2008) European birds and aposematic Heteroptera: review of comparative experiments. Bull Insectol 61:163–165
Ferreira A, Jehle R, Stow AJ, Lima AP (2018) Soil and forest structure predicts large scale patterns of occurrence and local abundance of a widespread Amazonian frog. PeerJ 26. https://doi.org/10.7717/peerj.5424
Frichot E, François O (2015) LEA: an R package for landscape and ecological association studies (B O’Meara, Ed.). Methods Ecol Evol 6:925–929
Frichot E, Schoville SD, Bouchard G, Francois O (2013) Testing for associations between loci and environmental gradients using latent factor mixed models. Mol Biol Evol 30:1687–1699
Frichot E, Schoville SD, De Villemereuil P, Gaggiotti OE, François O (2015) Detecting adaptive evolution based on association with ecological gradients: orientation matters! Heredity 115:22–28
Gehara M, Summers K, Brown JL (2013) Population expansion, isolation and selection: novel insights on the evolution of color diversity in the strawberry poison frog. Evol Ecol 27:797–824
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hofreiter M, Schöneberg T (2010) The genetic and evolutionary basis of colour variation in vertebrates. Cell Mol Life Sci 67:2591–603
Hoogmoed MS, Avila-Pires TCS (2012) Inventory of color polymorphism in populations of Dendrobates galactonotus (Anura: Dendrobatidae), a poison frog endemic to Brazil. Phyllomedusa 11:95–115
Jaccoud D, Peng K, Feinstein D, Kilian a (2001) Diversity arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Res 29:E25
Jiggins CD, Naisbit RE, Coe RL, Mallet J (2001) Reproductive isolation caused by colour pattern mimicry. Nature 411:302–305
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP. Data Bioinform 27:3070–3071
Kaefer IL, Tsuji-Nishikido BM, Mota EP, Farias IP, Lima AP (2013) The early stages of speciation in Amazonian forest frogs: phenotypic conservatism despite strong genetic structure. Evol Biol 40:228–245
Kang C, Sherratt TN, Kim YE, Shin Y, Moon J, Song U et al. (2017) Differential predation drives the geographical divergence in multiple traits in aposematic frogs. Behav Ecol 28:1122–1130
Kemp DJ, Herberstein ME, Fleishman LJ, Endler JA, Bennett ATD, Dyer AG et al. (2015) An integrative framework for the appraisal of coloration in nature. Am Nat 185:705–724
Kemppainen P, Knight CG, Sarma DK, Hlaing T, Prakash A, Maung Maung YN et al. (2015) Linkage disequilibrium network analysis (LDna) gives a global view of chromosomal inversions, local adaptation and geographic structure. Mol Ecol Resour 15:1031–1045
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, et al. (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. In: Pompanon F, Bonin A (eds) Data production and analysis in population genomics SE - 5, Methods in Molecular Biology. Berlin, German: Springer, Vol 888, pp 67–89
Lambert SM, Geneva AJ, Luke Mahler D, Glor RE (2013) Using genomic data to revisit an early example of reproductive character displacement in Haitian Anolis lizards. Mol Ecol 22:3981–95
Lawrence JP, Rojas B, Fouquet A, Mappes J, Blanchette A, Saporito RA, Bosque RJ, Courtois EA, Noonan BP (2019) Weak warning signals can persist in the absence of gene flow. PNAS 38:19037–19045
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lindström L, Alatalo R, Mappes J, Riipi M, Vertainen L (1999) Can aposematic signals evolve by gradual change? Nature 397:249–251
Maia R, Eliason CM, Bitton P-P, Doucet SM, Shawkey MD (2013) pavo: an R package for the analysis, visualization and organization of spectral data (A Tatem, Ed.). Methods Ecol Evol 4:906–913
Mann ME, Cummings ME (2012) poison frog colors are honest signals of toxity, particularly for bird predators. Am Nat 179:E1–E14
Maslin MA, Burns SJ (2000) Reconstruction of the Amazon Basin effective moisture availability over the past 14,000 years. Science (80-) 20:2285–2287
Master TL (1999) Predation by Rufous Motmot on Black-and-Green Poison Dart Frog. Wilson Bull 111:439–440
McLean CA, Stuart-Fox D (2014) Geographic variation in animal colour polymorphisms and its role in speciation. Biol Rev 89:860–873
Medina I, Wang IJ, Salazar C, Amézquita A (2013) Hybridization promotes color polymorphism in the aposematic harlequin poison frog, Oophaga histrionica. Ecol Evol 3:4388–400
Mueller RL (2006) Evolutionary rates, divergence dates, and the performance of mitochondrial genes in Bayesian phylogenetic analysis. Syst Biol 55:289–300
Noonan BP, Comeault AA (2009) The role of predator selection on polymorphic aposematic poison frogs. Biol Lett 5:51–54
Noonan BP, Gaucher P (2006) Refugial isolation and secondary contact in the dyeing poison frog Dendrobates tinctorius. Mol Ecol 15:4425–4435
O’Leary SJ, Hice LA, Feldheim KA, Frisk MG, McElroy AE, Fast MD et al. (2013) Severe inbreeding and small effective number of breeders in a formerly abundant marine fish. PLoS ONE 8:2–9
Paradis E, Schliep K (2018) ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35:526–528
Paluh DJ, Hantak MM, Saporito RA (2014) A test of aposematism in the dendrobatid poison frog oophaga pumilio: the importance of movement in clay model experiments. J Herpetol 48:249–254
Palumbi SR (1996) Nucleic acids II: the polymerase chain reaction. In: Hillis DM, Moritz C, Mable BK eds. Molecular systematics. Sinauer Associates, Inc, Sunderland, Massachusetts, pp 205–247
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinform 28:2537–2539
Petroli CD, Sansaloni CP, Carling J, Steane DA, Vaillancourt RE, Myburg AA et al. (2012) Genomic characterization of DArT markers based on high-density linkage analysis and physical mapping to the Eucalyptus genome. PLoS ONE 7:e44684
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Core Team (2014) A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Raj A, Stephens M, Pritchard JK (2014) FastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197:573–589
Rambaut A, Suchard M, Xie D, Drummond A (2014). Tracer v1.6
Ravinet M, Westram A, Johannesson K, Butlin R, André C, Panova M (2016) Shared and nonshared genomic divergence in parallel ecotypes of Littorina saxatilis at a local scale. Mol Ecol 25:287–305
Rellstab C, Gugerli F, Eckert AJ, Hancock AM, Holderegger R (2015) A practical guide to environmental association analysis in landscape genomics. Mol Ecol 24:4348–4370
Revell LJ (2012) phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol 3:217–223
Revell LJ (2013) Two new graphical methods for mapping trait evolution on phylogenies. Methods Ecol Evolution 4:754–759
Richards-Zawacki CL, Yeager J, Bart HPS (2013) No evidence for differential survival or predation between sympatric color morphs of an aposematic poison frog. Evol Ecol 27:783–795
Rojas D, Stow A, Amézquita A, Simões PI, Lima AP (2015) No predatory bias with respect to colour familiarity for the aposematic Adelphobates galactonotus (Anura: Dendrobatidae). Behaviour 152:1637–1657
Row JR, Knick ST, Oyler-McCance SJ, Lougheed SC, Fedy BC (2017) Developing approaches for linear mixed modeling in landscape genetics through landscape-directed dispersal simulations. Ecol Evol 7:3751–3761
Rudh A, Rogell B, Hȍglund J (2007) Non-gradual variation in color morphs of the strawberry poison frog Dendrobates pumilio: genetic and geographical isolation suggests a role for selection in maintaining polymorphism. Mol Ecol 16:4284–4294
Rudh A, Breed MF, Qvarnström A (2013) Does aggression and explorative behaviour decrease with lost warning coloration? Biol J Linn Soc 108:116–126
Rudh A, Rogell B, Håstad O, Qvarnström A (2011) Rapid population divergence linked with co-variation between coloration and sexual display in strawberry poison frogs. Evolution 65:1271–1282
Sansaloni C, Petroli C, Jaccoud D, Carling J, Detering F, Grattapaglia D et al. (2011) Diversity Arrays Technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc 5:P54
Santos JC, Cannatella DC (2011) Phenotypic integration emerges from aposematism and scale in poison frogs. Proc Natl Acad Sci USA 108:6175–6180
Santos JC, Coloma LA, Summers K, Caldwell JP, Ree R, Cannatella DC (2009) Amazonian amphibian diversity is primarily derived from late Miocene Andean lineages. PLoS Biol 7:0448–0461
Sefc KM, Mattersdorfer K, Zielgelbecker A, Neuhuttler N, Steiner O, Goessler O, Koblmuller (2017) Shifting barriers and phenotypic diversification by hybridization. Ecol Lett 20:651–662
Siddiqi A, Cronin TW, Loew ER, Vorobyev M, Summers K (2004) Interspecific and intraspecific views of color signals in the strawberry poison frog Dendrobates pumilio. J Exp Biol 207:2471–2485
Simões PI, Lima AP, Farias IP (2012) Restricted natural hybridization between two species of litter frogs on a threatened landscape in southwestern Brazilian Amazonia. Conserv Genet 13:1145–1159
Simões PI, Stow A, Hödl W, Amézquita A, Farias IP, Lima AP (2014) The value of including intraspecific measures of biodiversity in environmental impact surveys is highlighted by the Amazonian brilliant-thighed frog (Allobates femoralis). Trop Conserv Sci 7:811–828
Summers K, Clough ME (2001) The evolution of coloration and toxicity in the poison frog family (Dendrobatidae). Proc Natl Acad Sci USA 98:6227–6232
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis Version 6.0. Mol Biol Evol 30:2725–2729
Tazzyman SJ, Iwasa Y (2010) Sexual selection can increase the effect of random genetic drift—a quantitative genetic model of polymorphism in Oophaga pumilio, the strawberry poison-dart frog. Evolution 64:1719–1728
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132:619–633
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Trask AE, Bignal EM, McCracken DI, Piertney SB, Reid JM (2017) Estimating demographic contributions to effective population size in an age-structured wild population experiencing environmental and demographic stochasticity. J Anim Ecol 86:1082–1093
de Villemereuil P, Frichot É, Bazin É, François O, Gaggiotti OE (2014) Genome scan methods against more complex models: when and how much should we trust them? Mol Ecol 23:2006–2019
Vonhof HB, Kaandorp RJG (2011). Climate variation in Amazonia during the neogene and the quaternary. In: Hoorn C, Wesselingh F (eds) Amazonia: landscape and species evolution, Wiley Online Books. Wiley-Blackwell Publishing Ltd: Oxford, UK, pp 199–210
Vorobyev M, Brandt R, Peitsch D, Laughlin S, Menzel R (2001) Colour thresholds and receptor noise: behaviour and physiology compared. Vis Res 41:639–653
Vorobyev M, Osorio D (1998) Receptor noise as a determinant of receptor thresholds. Proc R Soc B Biol Sci 265:351–358
Wang IJ, Summers K (2010) Genetic structure is correlated with phenotypic divergence rather than geographic isolation in the highly polymorphic strawberry poison-dart frog. Mol Ecol 19:447–458
Whitlock MC, Lotterhos KE (2015) Reliable detection of loci responsible for local adaptation: inference of a null model through trimming the distribution of FST. Am Nat 186:S24–S36
Wright S (1932) The roles of mutation, inbreeding, cross breeding and selection in evolution. In: Proceedings of the 6th International Congress on Genetics 1:356–366
Acknowledgements
We thank the Editor and two anonymous reviewers for insightful comments that greatly improved this manuscript. Estação Científica Ferreira Penna and ICMBio provided valuable support and facilities at ICMBio Caxiuanã Station. R.M. Brabo, P.M. Brabo, M.R. da Silva, J.M.P. dos Santos, J.L.F. da Costa, E.P de Souza, J.N.B. Carneiro, A.P. Farias, M.F.M. Pedroso, J.A. da Costa Filho, G. O. da Silva, C.A. Lopes for field assistance. Instituto Chico Mendes de Conservação da Biodiversidade (ICMBio) provided collection permits (# 36135-1). Financial support was provided by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (Project 472198/2011-4) and Programa Ciência sem Fronteiras (Project 401327/2012-4). Support was also provided by a PVE fellowship to AS (312315/2012-0), SWE fellowship to DR (200292/2014-5), a PDJ fellowship for PIS (151409/2013-7), PhD fellowship from CNPq (141886/2012-9) to DR, and a BEV fellowship from CNPq (170211/2012-6) to AA, a research fellowship (312674/2013-9) to TCSAP. PM receives an Academy of Finland postdoctoral fellowship (grant # 316294).
Author information
Authors and Affiliations
Contributions
DR, AS, and APL designed the project. DR, APL, AS, and PIS collected samples. TCSA-P and MSH contribued useful discussion. TCSA-P, MSH and YOCB contributed samples and useful discussion. DR and AA measured, analyzed and interpreted the color data. AS, PIS, ILK, PM and RD analyzed and interpreted genetic data. DR and AS wrote the manuscript with contributions from all co-authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Rojas, D., Lima, A.P., Momigliano, P. et al. The evolution of polymorphism in the warning coloration of the Amazonian poison frog Adelphobates galactonotus. Heredity 124, 439–456 (2020). https://doi.org/10.1038/s41437-019-0281-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-019-0281-4
- Springer Nature Switzerland AG
This article is cited by
-
Developments in the study of poison frog evolutionary ecology I: social interactions, life history and habitat use across space and ontogeny
Evolutionary Ecology (2024)
-
Geographic Drivers of Genetic and Plumage Color Diversity in the Blue-Crowned Manakin
Evolutionary Biology (2023)
-
Contrasting environmental drivers of genetic and phenotypic divergence in an Andean poison frog (Epipedobates anthonyi)
Heredity (2022)
-
Selection and localised genetic structure in the threatened Manauense Harlequin Frog (Bufonidae: Atelopus manauensis)
Conservation Genetics (2022)
-
Evolutionary history and genetic connectivity across highly fragmented populations of an endangered daisy
Heredity (2021)